

Abstract
The genome sequence of Streptomyces coelicolor contains three open reading frames (sco1441, sco2687, and sco6655) that encode proteins with significant (>40%) amino acid identity to GTP cyclohydrolase II (GCH II), which catalyzes the committed step in the biosynthesis of riboflavin. The physiological significance of the redundancy of these proteins in S. coelicolor is not known. However, the gene contexts of the three proteins are different, suggesting that they may serve alternate biological niches. Each of the three proteins was overexpressed in Escherichia coli and characterized to determine if their functions are biologically overlapping. As purified, each protein contains 1 molar equiv of zinc/ mol of protein and utilizes guanosine 5′-triphosphate (GTP) as substrate. Two of these proteins (SCO 1441 and SCO 2687) produce the canonical product of GCH II, 2,5-diamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate (APy). Remarkably, however, one of the three proteins (SCO 6655) converts GTP to 2-amino-5-formylamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate (FAPy), as shown by UV-visible spectrophotometry, mass spectrometry, and NMR. This activity has been reported for a GTP cyclohydrolase III protein from Methanocaldococcus jannaschii [Graham, D. E., Xu, H., and White, R. H. (2002) Biochemistry 41, 15074–15084], which has no amino acid sequence homology to SCO 6655. Comparison of the sequences of these proteins and mapping onto the structure of the E. coli GCH II protein [Ren, J., Kotaka, M., Lockyer, M., Lamb, H. K., Hawkins, A. R., and Stammers, D. K. (2005) J. Biol. Chem. 280, 36912–36919] allowed identification of a switch residue, Met120, which appears to be responsible for the altered fate of GTP observed with SCO 6655; a Tyr is found in the analogous position of all proteins that have been shown to catalyze the conversion of GTP to APy. The Met120Tyr variant of SCO 6655 acquires the ability to catalyze the conversion of GTP to APy, suggesting a role for Tyr120 in the late phase of the reaction. Our data are consistent with duplication of GCH II in S. coelicolor promoting evolution of a new function. The physiological role(s) of the gene clusters that house GCH II homologues will be discussed.
A substantial role for duplication in evolution of new function was proposed (1–3) long before the recent explosion of genome sequencing and annotation projects revealed the pervasiveness of intragenomic duplications in bacterial genomes. Gene duplication is now widely acknowledged to contribute to evolution of new protein functions by releasing the selective pressure for maintenance of existing biological roles. A detailed understanding of the catalytic repertoire spanned by duplicated genes is of considerable interest to understanding mechanisms of functional evolution.
Of the ~8000 proteins encoded in the genome of Streptomyces coelicolor A3 (2) nearly 41% have intragenomic homologues (4, 5). We have been intrigued by the presence of three open reading frames in the genome of S. coelicolor, each of which has been annotated as GTP cyclohydrolase II (GCH II),1 which catalyzes the first committed step in the biosynthesis of riboflavin. The presence of multiple GCH II homologues is not unique to S. coelicolor; in fact, Streptomyces avermitilis also harbors at least three open reading frames with substantial amino acid identity (40% over the GCH II domain) to the Escherichia coli GCH II protein. Notably, only one of the orfs is located in a riboflavin biosynthesis cluster.
GCH II belongs to a class of proteins that catalyze cleavage of carbon-8 of GTP to produce structurally related but distinct products (see Figure 1). GCH I catalyzes the conversion of GTP to dihydroneopterin triphosphate, which conversion of GTP to 2,5-diamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate (APy), which is a precursor to flavin cofactors (8, 9). GCH III catalyzes the conversion of GTP to 2-amino-5-formylamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate (FAPy) (10) whose physiological role remains to be established. These three classes of proteins share no detectable sequence homology. Furthermore, GCH I and GCH II, whose three-dimensional structures are known, adopt different folds (11, 12). However, the common theme that unifies these proteins is that each appears to produce FAPy, either as a product (as in GCH III) (10) or as an intermediate (as in GCH I and II) (13, 14). Therefore, although apparently unrelated structurally, they share the ability to hydrolyze the N9-C8 bond (GCH I, GCH II, and GCH III) and/or the N7-C8 bond (GCH I and GCH II) of GTP. As mentioned, only one of the three intragenomic homologues of GCH II in S. coelicolor appears to be involved in the biosynthesis of riboflavin, leading one to suggest that the tendency for the additional homologues to be retained reflects a distinct role for these proteins in the physiology of Streptomyces.
FIGURE 1.

Reactions catalyzed by GCH I, GCH II, and GCH III proteins.
One may envision three explanations for intragenomic duplication of GCH II. First, since GCH II catalyzes the committed step in the biosynthesis of riboflavin, the duplication may reflect a need for the cell to regulate riboflavin production in response to different growth conditions. Second, one may envision that APy is the building block for a biological molecule other than riboflavin; a duplicate copy of GCH II could fulfill the increased need for APy. In both cases the duplicate copy is functionally identical but may be regulated differently either at the gene or protein level. The third, and perhaps the most provocative, explanation is that these proteins do not produce APy at all. In this scenario, the GCH II homologue(s) would have evolved new function by exploiting the existing GCH II catalytic machinery. Such a protein, when studied in the context of homologues within the same organism could provide one with valuable insights into the parameters that constrain evolution of function.
Our aim, therefore, is to address a simple question: do the apparent GCH II homologues present in S. coelicolor catalyze identical chemical transformations, or has the presence of genetic redundancy freed a subset of these proteins from functional constraints that are dictated by cellular need for riboflavin? The extensive sequence similarities between these proteins suggest that, if indeed one or more have diverged functionally, one may be able to understand the mechanism of evolution of function in a large class of proteins. The first step toward addressing the hypothesis is characterization of the proteins that are encoded by the multiple orfs that encode proteins with homology to GCH II in the S. coelicolor genome. Although we have focused our initial studies on S. coelicolor, understanding the biological significance of these proteins will illuminate the path toward understanding the physiological roles of GCH II-like proteins in other organisms as well.
In this publication we show that each of the three homologues of GCH II in S. coelicolor utilizes GTP as a substrate; however, the product of one of the enzymes differs from that of the other two. Moreover, we show that site-directed mutation of a single amino acid in the active site of the protein that produces the alternate product switches the chemistry to that catalyzed by the “parental” GCH II protein. Finally, we examine the sequence contexts of these orfs to glean physiological function for these proteins. Our data support a model whereby the duplication of GCH II in S. coelicolor has permitted functional evolution to take place in this family of proteins.
MATERIALS AND METHODS
Materials
The Q-Sepharose (fast flow), butyl-Sepharose 4 (fast flow), Sephacryl S-300 (high resolution), and DEAE-Sephadex resins that were used in the purifications were from Amersham Biosciences. Tris(hydroxymethyl)aminomethane (Tris) was from EMD Sciences. Guanosine 5′-triphosphate (sodium salt) (GTP) was from Sigma. Solutions of GTP were prepared in 0.1 M potassium phosphate buffer (pH 6.8), and aliquots were frozen at −80 °C. Protease inhibitor cocktail Complete (–EDTA) was from Roche. Pfu DNA polymerase was from Stratagene, and deoxynucleotide triphosphates used in PCR were from Invitrogen. The restriction enzymes NdeI, NcoI, and HindIII were from New England Biolabs. The cell lines XL1-Blue, DH5α, and BL21(DE3) were from Stratagene, Invitrogen, and Novagen, respectively. LB medium refers to Lenox broth, which was prepared from 10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl per liter. The plasmids pET28a and pET29a were from Novagen. The pGEM T-Easy was purchased from Promega. PCR reactions were carried out on an Eppendorf MasterCycler gradient. UV-visible spectra were acquired on an Agilent 8453 diode array spectrophotometer fitted with an 8-position cell changer. The temperature of the samples was controlled with a circulating water bath. Mass spectral data were obtained using a ThermoFinnigan Deca XPPlus mass spectrometer using electrospray ionization.
Overexpression of Recombinant Proteins
Details on isolation of S. coelicolor genomic DNA and preparation of all constructs used in this study may be found in Supporting Information. In this study we utilized DNA constructs using the pET29a vector. The plasmids containing the desired gene were transformed into BL21(DE3) cells for overexpression purposes. A single colony from an overnight plate of the strain grown on LB medium containing 34 µg/mL kanamycin was placed in 0.1 L of LB containing 34 µg/mL kanamycin and grown at 37 °C overnight. The overnight culture served as starter for six 1 L cultures of LB in 2.8 L Fernbach flasks. In addition to 34 µg/mL kanamycin, the cultures overexpressing SCO 1441, SCO 2687, and SCO 6655 also contained 0.1 mM ZnCl2. The cultures were grown to OD600nm ~ 1, and expression of the protein was induced by addition of 0.1 mM IPTG. Cells were harvested after 4 h, frozen in liquid nitrogen, and stored at −80 °C until the purification.
Purification of SCO 1441
All purification steps were carried out at 4 °C. Cells (14.5 g) obtained as described above were resuspended into 40 mL of 0.02 M Tris-HCl (pH 8) containing 0.5 mM DTT and Complete (–EDTA) protease inhibitor mixture. The suspension was sonified in 30 s bursts (50% power; Branson 450 D sonifier) with 1 min rests in between each burst. The temperature of the suspension was not allowed to increase beyond ~12 °C. After a total of 7 min of cell disruption, the suspension was spun at 32000g for 45 min to obtain a cleared lysate. The lysate was loaded onto a Q-Sepharose column (2.5 × 14.5 cm), which had been equilibrated with 0.02 M Tris-HCl (pH 8) containing 0.5 mM DTT. The column was washed with 0.09 L of the equilibration buffer, and the protein was eluted with a 0.8 L linear gradient of 0.5 M KCl in the same buffer. Fractions containing GCH II activity were pooled. Enzymatic activity was monitored spectrophotometrically as described below. To this solution (47 mL) was added an equal volume of a solution containing 2 M ammonium sulfate in 0.02 M Tris-HCl (pH 8) over 20 min. The precipitate was collected by centrifugation at 32000g for 20 min, and the pellet was dissolved in 9 mL of 0.02 M Tris-HCl (pH 8) containing 0.5 mM DTT. The solution was desalted on a Sephacryl S-300 column (2.6 × 60 cm) which had been equilibrated with 0.02 M Tris-HCl (pH 8) buffer containing 0.5 mM DTT. Fractions containing activity were pooled, concentrated with Amicon centrifugal concentrators (YM-10 membrane), and frozen at −80 °C until further use. We have since streamlined the purification by replacing the protease inhibitor cocktail with 1 mM phenylmethanesulfonyl fluoride and eliminated the size exclusion step. The properties of the protein that is obtained from the modified purification are similar to those obtained with the purification outlined here.
Purification of SCO 6655, SCO 2687, and E. coli GCH II
E. coli cells overexpressing the recombinant protein (~20 g) prepared as described previously were thawed in 0.1 L of 0.02 M Tris-HCl (pH 8) which had been supplemented with Complete (–EDTA) protease inhibitor mixture. The cell suspension was sonified, and cleared cell lysates were obtained as described for SCO 1441. The lysates were chromatographed on a Q-Sepharose column essentially as described for SCO 1441. Fractions containing enzyme were identified by enzymatic activity assays (as described below) and pooled. To each pool was added an equal volume of solution containing 2 M ammonium sulfate, 0.02 M Tris-HCl (pH 8), and 0.5 mM DTT over 20 min. The resulting solution was applied to a butyl-Sepharose (fast flow) column (2.5 × 15 cm) that had been equilibrated with 0.02 M Tris-HCl (pH 8), 0.5 mM DTT, and 1 M ammonium sulfate. The protein was eluted with a linear gradient (1 L) from 1 to 0 M ammonium sulfate. Fractions containing the desired enzymatic activity were pooled, concentrated with an Amicon pressure concentrator (YM-10 membrane, 10000 molecular weight cutoff), and applied to a Sephacryl S-300 size exclusion column (2.5 × 60 cm). The protein was eluted with 0.02 M Tris-HCl (pH 8) containing 0.5 mM DTT. Fractions containing enzymatic activity were concentrated using centrifugal concentrators (Amicon, YM-10 membranes) and frozen at −80 °C until further use. The procedure outlined here describes our initial purification. We have since streamlined the purification by replacing the protease inhibitor cocktail with 1 mM phenylmethanesulfonyl fluoride and eliminated the size exclusion step. The properties of the protein obtained by the modified purification are similar to those obtained with the purification outlined here.
Zinc Metal Ion Determination
ICP-MS analysis of the recombinant proteins was carried out by Garratt-Callahan Co. (Burlingame, CA).
Protein Concentration Determination
Each of the purified recombinant proteins was quantified using a BCA protein assay kit (Pierce) with bovine serum albumin as standard.
Spectrophotometric Monitoring of the GTP Cyclohydrolase Reactions Catalyzed by SCO1441, SCO 2687, and SCO 6655
The reactions generally contained 0.1 M Tris-HCl (pH 8.0), 5 mM MgCl2, 0.5 mM DTT, and 0.1 mM GTP as substrate. Reactions were initiated by addition of the substrate, and absorption spectra were obtained at intervals after the addition. Rates were calculated from linear portions of the A299nm (for SCO 1441 and SCO 2695) or A275nm (for SCO 6655) versus time plots using difference extinction coefficients of ε299nm = 9040 and ε273nm = 4700 M−1 cm−1, respectively. The difference extinction coefficients were calculated, in separate experiments, from absorbance spectra that were obtained after complete conversion of GTP to products and are comparable to previously reported values (15).
3,4-Dihydroxy-2-butanone 4-Phosphate (DHBP) Synthase Activity of SCO 1441, SCO 2687, and SCO 6655
Each of the proteins was assayed for DHBP synthase activity essentially as described previously (16). Color was developed for 30 min at room temperature by addition of 0.4 mL of saturated creatine and 0.2 mL of a 35 mg/mL solution of Rnaphthol in 1 M NaOH. Absorbances were measured at 525 nm. A standard curve was generated using 2,3-butanedione.
Preparation and Purification of Reaction Products Produced by SCO 1441, SCO 2687, SCO 6655, and E. coli GCH II
GTP (5 mg) was dissolved in 3 mL of 0.1 M Tris-HCl (pH 8) containing 0.5 mM DTT and 5 mM MgCl2. The reactions were initiated by the addition of 0.15 unit of the desired enzyme, and reactions proceeded at room temperature for 60 min. The reactions mixtures were frozen at −80 °C and purified as needed. The products were purified by anion exchange at 4 °C as follows. Approximately 0.7 mL of the reaction mixture was applied at a flow rate of 1 mL/min to a 1 × 20 cm column of DEAE-Sephadex (40–125 µm dry bead size) that had been equilibrated with a buffer containing 0.05 M NH4HCO3 (pH 7.8) and 0.2 mM DTT. The column was washed with 15 mL of the loading buffer and developed with a linear gradient of 0.05–0.6 M NH4HCO3 in 60 mL. Fractions containing the product which eluted at ~0.3 M NH4HCO3 were pooled, frozen, and lyophilized. The lyophilized product was stored at −80 °C until needed.
Mass Spectral Analysis of Products
Products purified as described above were diluted in water to 50–250 µg/mL and analyzed on a LCQ Deca XPPlus electrospray ionization mass spectrometer. The sample was introduced using a syringe pump at a rate of 10–20 µL/min and detected in the negative ion mode using 300 °C desolvation temperature.
NMR Analysis of the Product of SCO 6655
To facilitate the analysis, uniformly 13C- and 15N-labeled GTP (Sigma) was utilized as substrate of SCO 6655 as described above. The product was purified as described for the unlabeled product. Negative ion mass spectrometry indicated a mass of 395, which as expected is 15 amu greater than that of the unlabeled material. Lyophilized samples containing ~2 mg of the purified SCO 6655 product were dissolved in 0.6 mL of D2O or in 0.6 mL of 90% H2O/10% D2O (50 mM CD3-CO2Na/HCl, pH 4.5). NMR spectra were acquired at 25 °C on a Bruker DRX-600 instrument operating at a 1H frequency of 600.13 MHz, using a Nalorac 5 mm HCN single-axis gradient probe. The 1H-decoupled 13C spectrum in D2O was acquired at 25 °C on a Bruker DRX-500 instrument operating at a 1H frequency of 499.68 MHz, using a Bruker 5 mm dual 13C/1H probe. 2D spectra were acquired with 750 FIDs and processed using Felix2000 software (Accelerys, San Diego, CA). Data were apodized using a sine-bell (DQF-COSY) or cosine-bell (all others) window function in both dimensions and zero-filled to a final data matrix of 2048 (F2) × 1024 (F1) real data points. 2D HSQC (17) and DQF-COSY (18) data are shown in the Supporting Information, along with experimental details. 2D HMBC (19) experiments used a gradient-selected magnitude mode pulse sequence with no low-pass filter and no decoupling during acquisition. The evolution delay was optimized for a long-range coupling of 10 Hz, and for 90% H2O samples the solvent peak was suppressed with 1.5 s of presaturation (γB1/2π = 26 Hz). For the {1H–15N} HMBC experiment (data not shown) a 13C 180° pulse was used in the center of the evolution time.
Site-Directed Mutagenesis of SCO 6655
A QuickChange (Stratagene) mutagenesis kit was used to introduce Met120Tyr into the pVBD13 construct that contains the gene for SCO6655. The mutagenesis reactions were carried out according to the manufacturer’s protocol. The template for the reactions was pVBD13, and mutagenic primers (forward, 5′-CCAAACTGCGCGCATATGCGTTGCAGGCG-3′, and reverse, 5′-GTTTGACGCGCGTATACGCAACGTCCGC-3′) were designed with an NdeI restriction endonuclease site (in italic) at the site of the mutation (underlined) to facilitate screening. The PCR reactions contained 10% (v/v) DMSO since sco6655 has a high G + C content (~70%). The annealing temperature was 58 °C. The presence of the desired mutation and lack of spurious changes were confirmed by sequencing the sco6655 gene.
RESULTS
Identification of Open Reading Frames with Homology to GCH II in the S. coelicolor Genome
A BLAST search of the genome of S. coelicolor (4) reveals three open reading frames encoding proteins (SCO 1441, SCO 2687, and SCO 6655) with ≥40% sequence identity with the GCH II protein of E. coli (B1277). The ClustalW alignments (Figure 2) of these proteins reveal that each of the proteins retains the three cysteine residues in the CX2GX7CXC motif that has been shown, by biochemical and structural studies with the E. coli homologue, to bind the active site zinc metal ion (12, 20, 21). Unlike SCO 6655 and SCO 2687 which appear to contain only a GCH II domain, SCO 1441 is bifunctional; a search of the conserved domain database (22) with SCO 1441 reveals homology with Pfam 0925 (GCH II) and 0926 (DHBP synthase) domain families. In the alignment in Figure 2 only the GCH II domain (starting at amino acid 200) is shown. By contrast, the conserved domain search with SCO 2687 and SCO 6655 reveals only a Pfam 0925 (GCH II) domain.
FIGURE 2.

Alignment of the E. coli GCH II protein with the proteins that are encoded by three open reading frames that have been annotated as GCH II proteins. The sequence accession numbers are B1277 for the E. coli protein and SCO 1441, SCO 2687, and SCO 6655 for the S. coelicolor proteins. SCO 1441 is a bifunctional protein with an N-terminal DHBP synthase and a C-terminal GCH II domain. For simplicity only the C-terminal region of the protein (starting from amino acid 200) is shown. The boxes denote the cysteine residues that have been shown to be essential for the activity of the E. coli GCH II (20, 21) and coordinate the catalytic zinc divalent metal ion (12). The molecular switch residue that dictates the fate of GTP as shown in SCO 6655 is shown in bold.
The physiological functions of SCO 1441 can be assigned on the basis of analysis of its genome context (see Table S1 in the Supporting Information for orfs housed near each homologue). SCO 1441 appears near genes for biosynthesis of riboflavin (SCO 1440 and SCO 1443); furthermore, it is fused to DHBP synthase, which is also involved in the biosynthesis of riboflavin. These observations strongly support a role for SCO 1441 as the first enzyme in the biosynthesis of riboflavin in S. coelicolor. Biochemical data presented below show that the protein has both GTP cyclohydrolase II and DHBP synthase activities, confirming the assignment. The two additional GCH II homologues are housed in distinct gene contexts, and we will speculate on the role of these clusters in the Discussion.
The presence of three orfs with homology to GCH II in S. coelicolor, in disparate regions of the chromosome and in widely different gene contexts, suggests that these proteins play nonoverlapping physiological roles. To address their roles the three homologues were overexpressed in E. coli and characterized.
Each GCH II Homologue of S. coelicolor Is a Zinc-Containing Metalloprotein
The E. coli GCH II protein has been shown to contain an essential zinc metal ion (20, 21). Site-directed mutations of cysteines 54, 65, or 67, which occur within a conserved CX2GX7CXC motif, lead to loss of catalytic activity (21). The zinc metal ion is located 3.8 Å from carbon-8 of the purine ring, implicating it in the catalytic activity of this protein (12). Each of the GCH II homologues in S. coelicolor contain ~1 equiv of zinc per monomer, as would be expected from the presence of the CX2GX7CXC motif in the sequence of the proteins (Table 1).
Table 1.
Specific Activity and Zinc Metal Ion Content of GCH II Proteins
| protein | specific activitya [µmol min−1 (mg of protein)−1] | turnover no. (min−1) | Zn/monomer |
|---|---|---|---|
| SCO 1441 | 0.038 ± 0.003 | 1.8 ± 0.1 | 0.9 |
| SCO 2687 | 0.13 ± 0.03 | 3.1 ± 0.6 | 1.0 |
| SCO 6655 | 0.18 ± 0.03 | 4.3 ± 0.6 | 1.1 |
| Met120Tyr SCO 6655 | 0.05 ± 0.01 | 1.2 ± 0.3 | 0.85 |
| E. coli GCH II | 0.08 ± 0.01 | 1.7 ± 0.2 | NDb |
Values are from an average of three determinations.
ND, not determined.
Spectrophotometric Analysis of TurnoVer of SCO 1441, SCO 2687, and SCO 6655 with GTP as Substrate
We first assessed whether SCO 1441, SCO 6655, and SCO 2687 utilize GTP as a substrate. In these experiments, the purified protein was incubated with Mg2+ and DTT, and UV-visible spectra of the mixtures were recorded at various times after addition of GTP (Figure 3). The incubation mixtures exhibit a time-dependent decrease in GTP absorbance at 253 nm and appearance of a new feature at either 299 nm [SCO 1441 (Figure 3A) and SCO 2687 (Figure 3B)] or 274 nm [SCO 6655 (Figure 3C)].
FIGURE 3.

Spectral changes (A–C) and difference spectra (D) observed with SCO 1441, SCO 2687, and SCO 6655 in the presence of GTP. Each of the assay mixtures contained 0.1 M Tris-HCl (pH 8.0), 5 mM MgCl2, 0.5 mM DTT, and 0.1 mM GTP in a total volume of 1 mL at 25 °C. The contents of the cuvettes were blanked in the absence of GTP. Spectra were acquired at various times after addition of GTP. The protein concentrations were (A) 7.5 µM SCO 1441, (B) 5.3 µM SCO 2687, and (C) 2.5 µM SCO 6655. The insets show that in each case the rate of the reaction is linear with the concentration of the protein. The difference spectra shown in (D) for SCO 1441 (−), SCO 2687 (•••), and SCO 6655 (---) were obtained by subtracting the spectrum of GTP acquired in the absence of protein from the final spectra that were obtained with each protein. The arrows denote increases and decreases in spectral features.
Difference spectra calculated from the spectral changes observed with each are shown in Figure 3D. The spectral changes observed with SCO 1441 and SCO 2687 are consistent with formation of APy, the canonical product of GCH II. Moreover, the difference absorbance spectra and difference extinction coefficients observed for the product of these proteins (9040 M−1 cm−1) are comparable to that reported for APy (15). By contrast, the difference spectrum that is observed with SCO 6655 is significantly different (compare difference traces in Figure 3D) from those that are observed with SCO 1441 and SCO 2687. In fact, the UV-visible spectrum of the product of SCO 6655 is similar to that reported for 2-formylamino-5-amino-6-hydroxy-4-(ribosylamino)pyrimidine 5′-phosphate (FAPy), which has an absorbance maximum at 274 nm (13).
The initial rates of formation of the new spectral features are proportional to the concentration of each enzyme (see insets in Figure 3). The maximal rates observed are summarized in Table 1. The turnover number with the recombinant E. coli GCH II obtained in our laboratory is comparable to those in the literature (8, 23).
Analysis of the Enzymatic Products of the Three S. coelicolor GCH II Homologues
In the initial phase of the analysis, reaction products produced by each of the three proteins were analyzed by ion-pairing HPLC with GTP, GDP, and GMP as standards. In each case, a product forms at the expense of GTP and with retention times that are consistent with the presence of one phosphate (data not shown); the product that was produced by each protein in the reaction mixtures was purified by anion-exchange chromatography, lyophilized, and analyzed by mass spectrometry. In addition, each protein also produced GMP in the course of turnover with GTP, consistent with GTPase activity previously demonstrated with GCH II from E. coli (14).
The negative ion mode mass spectra that were obtained with each are shown in Figure 4. The products of SCO 1441 and SCO 2687 show the expected peak at m/z = 352 (Figure 4B,C), corresponding to APy. The product of SCO 6655, however, has a peak at m/z = 380 (Figure 4D), which is consistent with FAPy. The [M + 23] peak that is observed in the mass spectra of SCO 1441 and SCO 2687 is from a sodium adduct. As a control we isolated the product produced by recombinant E. coli GCH II; the mass spectrum of this compound (Figure 4A) is identical to that of SCO 1441 and SCO 2687. Therefore, these results unambiguously demonstrate that the product of SCO 6655 is distinct from that of SCO 1441 and SCO 2687. The product of SCO 6655 has the molecular mass expected of FAPy and not APy, despite the substantial sequence identity that exists between SCO 6655 and other GCH II proteins.
FIGURE 4.

Mass spectral analysis of the products of E. coli GCH II (A), SCO 1441 (B), SCO 2687 (C), and SCO 6655 (D) proteins. The products were isolated as described in Materials and Methods. The mass spectrum of the product of E. coli GCH II is shown for comparison. In some cases, a [M− + Na+] complex is also observed.
To unambiguously assign the structure of the product of SCO 6655, the enzyme was incubated with [15N5-13C10]-GTP, and the product was isolated and analyzed. The mass of the molecular ion for this compound increased 15 amu relative to that of the unlabeled material (data not shown). The mass of the compound and UV-visible spectrum observed with the product of SCO 6655 are consistent with formation of FAPy. However, one may envision that the ring opening catalyzed by SCO 6655 may leave the formyl group attached to the N9 and not N7; such a compound would have the same mass spectrum as FAPy, and therefore, the mass spectral data do not unambiguously establish the structure.
An NMR assignment of [15N5-13C10]-FAPy generated by SCO 6655 was undertaken to confirm the identity of the product. A partial assignment of the 13C spectrum of FAPy (positions 1′–5′ and 8) has been published (13); the 13C, 1H, and 15N chemical shifts for the product of SCO 6655 are summarized in Table 2–Table 4. The spectra used in the assignments included 1H and 13C 1D spectra, 2D {1H–13C} and {1H–15N} HSQC (17), {1H–15N} and {1H-13C} MBC (19), and DQF-COSY (18); representative examples of these spectra are shown in the Supporting Information (Figures S1 – S5). FAPy has been shown to exist in four forms that differ in the stereochemistry of the formyl moiety (cis or trans) and the configuration of the base relative to the anomeric carbon of the ribose (α or β) (13). In this paper the α/β and cis/trans ratios were 1.2–1.9 and 7–10, respectively. The {1H,13C}-HSQC spectrum (Figure S4) that was observed with uniformly 15N,13C-labeled FAPy from reaction of SCO 6655 is similar to that observed previously (13). Long-range heteronuclear correlations were observed from H7α to C8α, H7β to C8β, and H9α to C6α (data not shown). DQF-COSY correlations were observed from H9 to H1′ for all four forms of FAPY (α-cis, β-cis, α-trans, and β-trans).
Table 2.
13C Chemical Shift and Assignments for [15N5,13C10]-FAPy in 2H2O
| 13C chemical shift (ppm)b | |||
|---|---|---|---|
| structure | carbona | α | β |
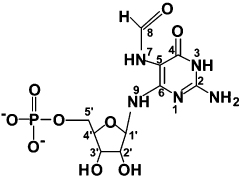 |
2 | 154.99 br s | |
| 4 | 162.15 br d | ||
| (93) | |||
| 5 | 89.30 ddd | ||
| (78, 85, 18c) | |||
| 6 | 160.66 dd | 161.48 dd | |
| (74, 20c) | (74, 20c) | ||
| 8, cis | 165.89 d | 166.23 d | |
| (13.3c) | (13.5c) | ||
| 8, trans | 171.02 d | 171.11 d | |
| (13.9c) | (13.7c) | ||
| 1′ | 81.93 dd | 85.30 dd | |
| (39, 11c) | (44, 12c) | ||
| 2′ | 71.93 t | 73.92 dd | |
| (38) | (44, 34) | ||
| 3′ | 70.70 t | 71.37 t | |
| (38) | (38) | ||
| 4′ | 81.71 ddd | 83.20 ddd | |
| (35, 41, 8d) | (35, 41, 8d) | ||
| 5′ | 64.75 d | 64.92 dd | |
| (43) | (43, 5d) | ||
For clarity, the numbering scheme shown here reflects that of the pyrimidine product. The structure, as drawn, corresponds to the β, cis configuration of FAPy.
The values in parentheses are the coupling constants to 13C nuclei (in Hz) except as indicated by superscripts. Chemical shifts are referenced to internal CH3OH standard at 49.50 ppm.
The values correspond to the coupling constant to 15N.
The value represents the coupling constant to 31P.
Table 4.
1H and 15N Chemical Shifts and Assignments for [15N5,13C10]-FAPy in 90% D2O/10% H2O
| 1H chemical shift (ppm)a | 15N chemical shift (ppm)b | |||
|---|---|---|---|---|
| position | α | β | α | β |
| 2-NH2, cis | 6.373 d (91) | 6.346 d (91) | 75.60 | 75.40 |
| 2-NH2, trans | 6.376b | 6.347b | 75.02 | 74.83 |
| 7, cis | 8.687 br d (96) | 8.658 br d (95) | 114.66 | 114.70 |
| 7, trans | 8.111b | 8.195b | 113.75 | 113.10 |
| 9, cis | 6.493 d (91) | 6.878 d (92) | 95.03 | 98.33 |
| 9, trans | 6.591 d (91) | 7.128 d (93) | 95.96 | 98.89 |
The numbers in parentheses are the 15N coupling constants (in Hz).
Chemical shift from the 15N-decoupled HSQC spectrum.
The location of the formyl group at N7 was confirmed by two sets of long-range heteronuclear correlations. First, the 13C-HMBC spectrum (Figure 5) shows a cross-peak between the formyl proton (doublet at 8.2 ppm) and a 13C resonance centered at 89.3 ppm and not the resonances due to the 1′-position of the ribose, which occur at 81.9 and 85.3 ppm for the α and β anomers, respectively. The assignment of the 89.3 ppm resonance to the C5 position of the pyrimidine ring is consistent with previous studies on related molecules (24, 25). Furthermore, there is no evidence in the spectrum of interaction between the formyl proton and the resonances due to C6 of the pyrimidine ring (160.7 and 161.5 ppm for the α and β, respectively). Second, the observation of a proton assigned to N9 and homonuclear correlation from this proton to the H1′ proton (DQF-COSY, Figure S5) as well as a long-range heteronuclear correlation to the C6 carbon ({1H-13C} HMBC, data not shown) are additional evidence for localization of the formyl moiety at N7.
FIGURE 5.

Expanded region of the 2D {1H-13C} HMBC spectrum of FAPy in 2H2O showing cross-peaks between the formyl proton (H8) and the 13C resonance at position 5 of the base. Horizontal lines show the 13C shifts of C6 (base) and C1′ (sugar), where cross-peaks would be expected if the formyl group had been located at N9 instead of N7.
The data presented in this paper clearly show that SCO 6655 is functionally distinct from GCH II. The GTP cyclohydrolase proteins have historically been named on the basis of the reactions that they catalyze. The naming of SCO 6655 is problematic: from an activity standpoint the protein should be named GCH III; however, from the point of view of sequence homology the protein is best described as GCH II like. Therefore, we favor naming this protein GCH II/III to reflect the fact that it has features in common with both classes.
SCO 1441 Contains an ActiVe RibB Domain
SCO 1441 is annotated as a bifunctional protein with an N-terminal region that codes for DHBP synthase and a C-terminal region that has homology to GCH II. In the previous section SCO 1441 was shown to be a functional GCH II. SCO 1441 was found to catalyze formation of DHBP at 3 ± 0.3 min−1, which is comparable to the turnover number of the E. coli protein (26). In contrast, no DHBP synthase activity was detected with SCO 2687 or SCO 6655 under similar conditions. Therefore, of the three GCH II homologues of S. coelicolor only SCO 1441 is a bifunctional protein that contains domains for production of both DHBP and APy.
Molecular Switch That Controls the Fate of GTP in SCO 6655
The differing fates of GTP observed with SCO 6655 relative to SCO 1441 and SCO 2687 are intriguing in the context of the substantial sequence identity that each shares with the E. coli GCH II. The recent X-ray crystal structure of the E. coli GCH II shows several residues that are in close proximity of the GMP-CPP substrate analogue that was cocrystallized with the protein (12). In the structure, Cys 54, 65, and 67 ligate the active site zinc metal ion, consistent with the biochemical data that show these residues to be essential (21). The hydroxyl oxygen of Tyr105 is 4.3 Å from N7 of the GMP-CPP, and due to its close proximity this residue has been postulated to be involved in the catalytic cycle of the protein (see Figure 6). However, sequence alignments show that the corresponding residue in SCO 6655 is a methionine (Met120). Intriguingly, a Met is present at the analogous position in a homologue of SCO 6655 from S. avermitilis (27) which is localized in a similar gene context. However, a Tyr occupies this position in all GCH II proteins that have been shown to catalyze the formation of APy.
FIGURE 6.

Active site of E. coli GCH II protein from ref 12. The protein was cocrystallized with the substrate analogue GMP-CPP. This figure was prepared using PyMol (51) and the PDB structure 2bz0.
To determine if the differing fate of GTP in SCO 6655 can be linked to Met120, a Met120Tyr site-directed mutation was introduced in SCO 6655. The protein was purified and assayed for activity with GTP. The difference spectral changes that are observed when this protein is incubated with GTP are shown in Figure 7 and are compared to the spectral changes that are observed with the wild-type SCO 6655 and SCO 2687 proteins under comparable conditions. The difference spectrum of the reaction with this variant is identical to that of SCO 2687.Moreover, the variant catalyzes the conversion of GTP to APy at a rate comparable to those observed with SCO 1441 and SCO 2687 (see Table 1). The formation of APy by the Met120Tyr variant of SCO 6655 was confirmed by mass spectrometry and comparison to the product isolated from the reaction of wild-type SCO 2687 with GTP (data not shown). Therefore, the Met120Tyr variant of SCO 6655 acquires the ability to produce APy. These results show that introduction of a single mutation can explain the functional diversification in GCH II homologues of S. coelicolor.
FIGURE 7.

Difference spectra observed with SCO 6655, SCO 2687, and the Met120Tyr variant of SCO 6655. Each assay mixture contained 0.1 M Tris-HCl (pH 8.0), 5 mM MgCl2, 0.5 mM DTT, and 85 µM GTP in a total volume of 0.8 mL at 25 °C. Protein concentrations were 1.4, 4.3, and 4.8 µM for SCO 6655, SCO 2687, and the Met120Tyr variant of SCO 6655, respectively. The raw UV-visible spectra that were obtained in this experiment were similar to those shown in Figure 3 (data not shown). Spectral differences were calculated from the initial and final spectra obtained immediately and 30 min after addition of substrate.
A mechanism for GCH II that is consistent with the experimental and structural data is shown in Figure 8. We hypothesize that the zinc metal ion activates a water molecule for attack at C8 of the substrate leading to formation of FAPy in the first half-reaction. In the second half-reaction, an additional water molecule is thought to be involved in hydrolysis of the formyl group from FAPy leading to formation of APy. Our data showing that the Met120Tyr variant of SCO 6655 produces APy instead of FAPy strongly support the notion that the tyrosine plays a key role in the second half-reaction but that it has little or no effect on the first. This is the key difference between the mechanism presented in this paper and others in the literature (12). An important corollary of our hypothesis is that the Tyr residue found in the enzymes that catalyze conversion of GTP to APy, when mutated to Met, should lead to a change in the fate of GTP (toward formation of FAPy).
FIGURE 8.

Proposed catalytic mechanism for GCH II. For simplicity, the hydrolysis of the pyrophosphate moiety, which occurs at a distant location, is not shown. The hydrolysis of C8 of GTP to generate APy has been proposed to occur in two half-reactions. Our data support the notion that the Tyr residue (not shown) in the active site of the protein (see Figure 6) acts in the second half-reaction.
Although GCH I and GCH II share little sequence similarity, there is an interesting parallel between the effect of site-directed mutation of residues near the N7 of the substrate and the activity of the corresponding proteins. In the X-ray crystal structures of the E. coli and Thermus thermophilus GCH I proteins (28) a histidine residue (His179 and His177, respectively) is found near N7 of the substrate. This position is analogous to that of Tyr105 in the E. coli GCH II structure. Interestingly, the His179 Ala, Asp, Phe, Lys, Asn, Gln, Arg, and Ser site-directed variants of the E. coli GCH I produce no detectable quantities of dihydroneopterin triphosphate (13) but acquire the ability to produce FAPy. The His179 residue in GCH I, therefore, has been postulated to be involved in the hydrolysis of FAPy to APy in the course of conversion of GTP to dihydroneopterin triphosphate (29). We note that the Met → Tyr site-directed variation in the analogous position of SCO 6655 results in a protein that catalyzes conversion of GTP to APy. Therefore, despite the ethereal sequence similarities between GCH I and GCH II, both utilize similar strategies for hydrolysis of C8 of GTP to produce formate. Therefore, the C8 cleavage activity of GCH I and GCH II may represent a case for convergent evolution.
DISCUSSION
The results in this paper show that the three genes annotated as GCH II in S. coelicolor do not have physiologically overlapping functions. The most striking finding from these studies is the observation that a single amino acid appears to be responsible for the fate of GTP. In this section, we will speculate on the biological role(s) of the three previously uncharacterized proteins annotated as GCH II in S. coelicolor.
Physiological Role(s) of Three GCH II Homologues in S. coelicolor
We were surprised to find initially three orfs with significant sequence similarity to GCH II in the genome sequence of S. coelicolor. The biochemical data shown in this paper confirm that at least one (SCO 6655) has new catalytic function that is different from that of the GCH II. The gene contexts of these proteins in S. coelicolor suggest that, indeed, the physiological functions of these proteins are not overlapping.
Closer examination of the sequence contexts for these genes revealed that only one of the corresponding proteins, SCO 1441, could be involved in the biosynthesis of riboflavin. Most (but not all) of the proteins that are required for the conversion of GTP to riboflavin are present in this cluster. The only protein that is absent is a riboflavin-specific deaminase/reductase. Two homologues of the deaminase/reductase, however, are present elsewhere in the genome of S. coelicolor. One of the homologues is located just downstream of SCO 2687, and the role of this protein will be described below. Nevertheless, it is reasonable to posit that the cluster that bears SCO 1441 is involved in the biosynthesis of riboflavin.
The biological role of the cluster bearing SCO 2687 has been tentatively assigned by comparison to a cluster of proteins in Burkholderia glumae responsible for formation of toxoflavin (30, 31). In retrospect, this is perfectly reasonable and explains the presence of GCH II and the riboflavin-specific deaminase/reductase in this cluster. Trace radiolabel experiments carried by Levenberg and colleagues (32, 33) in the 1970s with the producing strain, Pseudomonas cocovenenans, suggest that GTP is a precursor to toxoflavin and that in the course of biosynthesis of this molecule C8 of GTP is lost whereas C2 is retained. Recent phylogenetic evidence suggests that P. cocovenenans should be reclassified as Burkholderia cocovenenans (van Damme et al. 1960) comb. nov (34, 35). To our knowledge, S. coelicolor has not been shown to produce toxoflavin. However, an unspecified strain of Streptomyces has been shown to produce a molecule, xanthothricin, which is identical to toxoflavin (36, 37), suggesting that at least some strains of Streptomyces probably encode a toxoflavin biosynthetic pathway. In this paper, we show that the GCH II protein from the cluster that bears SCO 2687 is capable of removing C8 of GTP. Biosynthesis of toxoflavin also requires deamination of the remaining pyrimidine ring, a reaction that has been postulated to occur early in the biosynthetic pathway (33), and explains the need for a riboflavin-specific deaminase in this cluster (SCO 2688). The additional carbon and nitrogen atoms that are present in the azapteridine ring of toxoflavin have been shown by radiotracer experiments to be derived from glycine (32) and assembled into the azapteridine by an unknown pathway. In summary, SCO 2687, SCO 2688, SCO 2692, and SCO 2695 correspond toxB, toxE, toxC, and toxD genes that have been described in the toxoflavin biosynthetic pathway of B. glumae (30, 31).
The physiological role of the cluster that bears SCO 6655 has been more elusive. A cluster of four genes has been shown recently to be involved in the biosynthesis of queuosine in Bacillus subtilis (38). The cluster that bears SCO 6655 has one protein in common with the queuosinecontaining cluster. Both appear to have a homologue of 6-pyruvoyltetrahydropterin synthase (PTPS), which catalyzes the conversion of neopterin triphosphate to 6-pyruvoyltetrahydropterin. The biosynthesis of biopterin requires GCH I, 6-pyruvoyltetrahydropterin synthase, and sepiapterin reductase. We find no evidence of a sepiapterin reductase protein in the genome of S. coelicolor; however, the absence of a gene is not evidence for the lack of the pathway. Interestingly, a homologue of PTPS is also present in E. coli (annotated as ygcM) (39). Woo et al. (39) have shown that YgcM has an alternate biopterin side-chain cleavage activity. On the basis of sequence alignments, the Streptomyces SCO 6650 homologue lacks an essential active site cysteine side chain which has been proposed, on the basis of structural and site-directed studies, to be required for activity (40) but retains an aspartate residue that in the structure forms two hydrogen bonds to the pyrimidine ring of the substrate and is likely an important binding determinant for the substrate. Therefore, we postulate that the PTPS in the SCO 6655 cluster of S. coelicolor catalyzes transformation(s) involving a purine substrate, though the identity of the substrate for these proteins remains to be established.
The clusters bearing SCO 6655 in S. coelicolor differ in that the queuosine biosynthetic cluster also houses a protein that has homology to the radical SAM family of proteins and an nitrile reductase that has been shown to catalyze the conversion of preQ0 to preQ1 (38, 41). Both of these proteins are absent from the cluster of genes in S. coelicolor. We should note, in passing, that queuosine has a 7-deazapurine structure and that Streptomyces have been shown to make a large number of 7-deazapurine-containing secondary metabolites (42–47).
Multiple GCH II Proteins Are Present in Many Genomes
The duplication of GCH II-like proteins appears to be widespread. A recent PSI-BLAST search of genomes in the NCBI database using the E. coli GCH II protein (B1277) as the query suggests that multiple homologues (≥2) of GCH II are present in >100 genomes, including a number of pathogenic organisms (S. D. Morrison and V. Bandarian, unpublished observation). In at least one case to our knowledge, the GCH II protein has been implicated in imparting to the organism pathogenic characteristics (48–50), such as hemolysis and involvement in iron acquisition. A broader examination of the role of GCH II protein duplication in these and other organisms, in light of the studies presented in this paper, is of interest.
Summary
A detailed analysis of the gene clusters that bear the three GCH II homologues that are examined in this publication suggests that the physiological roles of these proteins are not overlapping. The presence of duplicate copies of the protein has presumably removed the physiological imperatives for retention of the parental activity leading to evolution of new function. Furthermore, we show that evolution of a new activity in the GCH II proteins appears to require a single amino acid change. This is remarkable and leads to two provocative questions: how many of the genes that are annotated in genome databases as participating in known chemical transformations have, in fact, evolved to catalyze related but distinctly different transformations, and to what extent has nature exploited shutting down of halfreactions carried out in the active site of an enzyme to evolve pathways for formation of new compounds? Additional studies geared toward understanding the mechanism of action of GCH II, addressing the physiological role(s) of SCO 6655 and SCO 2687 and the cluster of genes that bear them, and the biological role(s) of duplicated GCH II homologues in nature are topics of ongoing investigation in the laboratory.
Table 3.
1H Chemical Shift and Assignments for [15N5,13C10]-FAPy in 2H2O
| 1H chemical shift (ppm)b | ||
|---|---|---|
| protona | α | β |
| 8, cis | 8.252 ddd (199, 15,c7) | 8.224 ddd (199, 15,c7) |
| 8, trans | 7.869 | 7.840 |
| 1′, cis | 5.906 d (168) | 5.688 d (165) |
| 1′, trans | 5.913 | 5.675 |
| 2′ | 4.287 d (151) | 4.204 d (148) |
| 3′ | 4.284 d (151) | 4.268 d (153) |
| 4′ | 4.074 d (149) | 4.069 d (149) |
| 5′ | 3.883 d (146) | 3.851 d (146) |
Numbers refer to protons attached to positions indicated in the structure from Table 2.
The values in parentheses are the coupling constants to 13C nuclei (in Hz) except as indicated by superscripts. Chemical shifts are referenced to the residual HOD signal at 4.755 ppm.
The values correspond to the coupling constant to 15N.
ACKNOWLEDGMENT
V.B. acknowledges numerous helpful discussions with Dr. Matthew Cordes on protein structural and functional evolution.
Footnotes
This research is supported (in part) by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund (to V.B.). Additional support for V.B. from the NIH (Grant GM 72623) is gratefully acknowledged. J.E.S. is supported by an NIH predoctoral training grant (GM 08804).
Abbreviations: APy, 2,5-diamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate; DQF-COSY, double quantum filtered correlation spectroscopy; GCH II, GTP cyclohydrolase II; GTP, guanosine 5′-triphosphate; DHBP, 3,4-dihydroxy-2-butanone 4-phosphate; FAPy, 2-amino-5-formylamino-6-ribosylamino-4(3H)-pyrimidinone 5′-phosphate; HMBC, heteronuclear multiple bond correlation; HSQC, heteronuclear single-quantum correlation; PCR, polymerase chain reaction; PTPS, 6-pyruvoyltetrahydropterin synthase.
SUPPORTING INFORMATION AVAILABLE
Experimental procedures detailing the preparation of S. coelicolor genomic DNA, the cloning of the genes for the proteins that were discussed, primer sequences, genome contexts of SCO 1441, SCO 2687, and SCO 6655, and 13C, 1H, 2D {1H-13C} HSQC (17), {1H-15N} HMBC (19), and DQF-COSY (18) spectra used for the structural assignment of FAPy. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Horowitz NH. On the Evolution of Biochemical Synthases. 1945;31:153–157. doi: 10.1073/pnas.31.6.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohno S. Evolution by gene duplication. New York: Springer-Verlag; 1970. [Google Scholar]
- 3.Jensen RA. Enzyme recruitiment in evolution of new function. Annu. Rev. Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 4.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 5.Gevers D, Vandepoele K, Simillon C, Van de Peer Y. Gene duplication and biased functional retention of paralogs in bacterial genomes. Trends Microbiol. 2004;12:148–154. doi: 10.1016/j.tim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 6.Burg AW, Brown GM. The biosynthesis of folic acid. VI. Enzymatic conversion of carbon atom 8 of guanosine triphosphate to formic acid. Biochim. Biophys. Acta. 1966;117:275–278. doi: 10.1016/0304-4165(66)90180-2. [DOI] [PubMed] [Google Scholar]
- 7.Burg AW, Brown GM. The biosynthesis of folic acid. 8. Purification and properties of the enzyme that catalyzes the production of formate from carbon atom 8 of guanosine triphosphate. J. Biol. Chem. 1968;243:2349–2358. [PubMed] [Google Scholar]
- 8.Foor F, Brown GM. Purification and properties of guanosine triphosphate cyclohydrolase II from Escherichia coli. J. Biol. Chem. 1975;250:3545–3551. [PubMed] [Google Scholar]
- 9.Foor F, Brown GM. GTP cyclohydrolase II from Escherichia coli. Methods Enzymol. 1980;66:303–307. doi: 10.1016/0076-6879(80)66473-8. [DOI] [PubMed] [Google Scholar]
- 10.Graham DE, Xu H, White RH. A member of a new class of GTP cyclohydrolases produces formylaminopyrimidine nucleotide monophosphates. Biochemistry. 2002;41:15074–15084. doi: 10.1021/bi0268798. [DOI] [PubMed] [Google Scholar]
- 11.Auerbach G, Herrmann A, Bracher A, Bader G, Gutlich M, Fischer M, Neukamm M, Garrido-Franco M, Richardson J, Nar H, Huber R, Bacher A. Zinc plays a key role in human and bacterial GTP cyclohydrolase I. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13567–13572. doi: 10.1073/pnas.240463497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren J, Kotaka M, Lockyer M, Lamb HK, Hawkins AR, Stammers DK. GTP cyclohydrolase II structure and mechanism. J. Biol. Chem. 2005;280:36912–36919. doi: 10.1074/jbc.M507725200. [DOI] [PubMed] [Google Scholar]
- 13.Bracher A, Fischer M, Eisenreich W, Ritz H, Schramek N, Boyle P, Gentili P, Huber R, Nar H, Auerbach G, Bacher A. Histidine 179 mutants of GTP cyclohydrolase I catalyze the formation of 2-amino-5-formylamino-6-ribofuranosylamino-4(3H)-pyrimidinone triphosphate. J. Biol. Chem. 1999;274:16727–16735. doi: 10.1074/jbc.274.24.16727. [DOI] [PubMed] [Google Scholar]
- 14.Schramek N, Bracher A, Bacher A. Biosynthesis of riboflavin. Single turnover kinetic analysis of GTP cyclohydrolase II. J. Biol. Chem. 2001;276:44157–44162. doi: 10.1074/jbc.M107306200. [DOI] [PubMed] [Google Scholar]
- 15.Bracher A, Schramek N, Bacher A. Biosynthesis of pteridines. Stopped-flow kinetic analysis of GTP cyclohydrolase I. Biochemistry. 2001;40:7896–7902. doi: 10.1021/bi010322v. [DOI] [PubMed] [Google Scholar]
- 16.Picollelli MA, Viitanen PV, Jordan DB. Spectrophotometric determination of 3,4-dihydroxy-2-butanone-4-phosphate synthase activity. Anal. Biochem. 2000;287:347–349. doi: 10.1006/abio.2000.4867. [DOI] [PubMed] [Google Scholar]
- 17.Willker W, Leibfritz D, Kerssebaum R, Bermel W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993;31:287–292. [Google Scholar]
- 18.Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wuthrich K. Improved spectral resolution in COSY H-1-NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 19.Hurd RE, John BK. Gradient-enhanced proton-detected heteronuclear multiple-quantum coherence spectroscopy. J. Magn. Reson. 1991;91:648–653. [Google Scholar]
- 20.Kaiser J, Schramek N, Eberhardt S, Puttmer S, Schuster M, Bacher A. Biosynthesis of vitamin B2. An essential zinc ion at the catalytic site of GTP cyclohydrolase II. Eur. J. Biochem. 2002;269:5264–5270. doi: 10.1046/j.1432-1033.2002.03239.x. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser J, Schramek N, Puttmer S, Schuster M, Eberhardt S, Rebelo J, Auerbach G, Bader G, Bracher A, Nar H, Hosl C, Fischer M, Huber R, Bacher A. Essential zinc ions at the catalytic sites of GTP cyclohydrolases. In: Chapman S, Perham R, Scrutton N, editors. Flavins and flavoproteins. Berlin: Rudolf Weber; 2002. pp. 241–246. [Google Scholar]
- 22.Marchler-Bauer A, Anderson JB, Cherukuri PF, De Weese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Marchler GH, Mullokandov M, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Yamashita RA, Yin JJ, Zhang D, Bryant SH. CDD: a conserved domain database for protein classification. Nucleic Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bacher A, Richter G, Ritz H, Eberhardt S, Fischer M, Krieger C. Biosynthesis of riboflavin: GTP cyclohydrolase II, deaminase and reductase. Methods Enzymol. 1997;280:382–389. doi: 10.1016/s0076-6879(97)80129-2. [DOI] [PubMed] [Google Scholar]
- 24.Coxon B, Fatiadi AJ, Sniegoski LT, Hertz HS, Schaffer R. A novel acylative degradation of uric acid. Carbon-13 nuclear magnetic resonance studies of uric acid and its degradation products. J. Org. Chem. 1977;42:3132–3140. doi: 10.1021/jo00439a006. [DOI] [PubMed] [Google Scholar]
- 25.Cho BP. Structure of oxidatively damaged nucleic acid adducts: pH dependence of the 13C NMR spectra of 8-oxoguanosine and 8-oxoadenosine. J. Magn. Reson. 1993;31:1048–1053. [Google Scholar]
- 26.Richter G, Volk R, Krieger C, Lahm HW, Rothlisberger U, Bacher A. Biosynthesis of riboflavin: cloning, sequencing, and expression of the gene coding for 3,4-dihydroxy-2-butanone 4-phosphate synthase of Escherichia coli. J. Bacteriol. 1992;174:4050–4056. doi: 10.1128/jb.174.12.4050-4056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka Y, Nakagawa N, Kuramitsu S, Yokoyama S, Masui R. Novel reaction mechanism of GTP cyclohydrolase I. High-resolution X-ray crystallography of Thermus thermophilus HB8 enzyme complexed with a transition state analogue, the 8-oxoguanine derivative. J. Biochem. (Tokyo) 2005;138:263–275. doi: 10.1093/jb/mvi120. [DOI] [PubMed] [Google Scholar]
- 29.Rebelo J, Auerbach G, Bader G, Bracher A, Nar H, Hosl C, Schramek N, Kaiser J, Bacher A, Huber R, Fischer M. Biosynthesis of pteridines. Reaction mechanism of GTP cyclohydrolase I. J. Mol. Biol. 2003;326:503–516. doi: 10.1016/s0022-2836(02)01303-7. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Kim JG, Kang Y, Jang JY, Jog GJ, Lim JY, Kim S, Suga H, Nagamatsu T, Hwang I. Quorum sensing and the LysR-type transcriptional activator ToxR regulate toxoflavin biosynthesis and transport in Burkholderia glumae. Mol. Microbiol. 2004;54:921–934. doi: 10.1111/j.1365-2958.2004.04338.x. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki F, Sawada H, Azegami K, Tsuchiya K. Molecular characterization of the tox operon involved in toxoflavin biosynthesis of Burkholderia glumae. J. Gen. Plant Pathol. 2004;70:97–107. [Google Scholar]
- 32.Levenberg B, Linton SN. On the biosynthesis of toxoflavin, an azapteridine antibiotic produced by Pseudomonas cocovenenans. J. Biol. Chem. 1966;241:846–852. [PubMed] [Google Scholar]
- 33.Levenberg B, Kaczmarek DK. Enzymic release of carbon atom 8 from guanosine triphosphate, an early reaction in the conversion of purines to pteridines. Biochim. Biophys. Acta. 1966;117:272–275. doi: 10.1016/0304-4165(66)90179-6. [DOI] [PubMed] [Google Scholar]
- 34.Zhao N, Qu C, Wang E, Chen W. Phylogenetic evidence for the transfer of Pseudomonas cocovnenans (van Damme et al. 1960) to the genus Burkholderia as Burkholderia cocovnenans (van Damme et al. 1960) comb. nov. Int. J. Syst. Bacteriol. 1995;45:600–603. doi: 10.1099/00207713-45-3-600. [DOI] [PubMed] [Google Scholar]
- 35.Coenye T, Holmes B, Kersters K, Govan JR, Vandamme P. Burkholderia cocovenenans (van Damme et al. 1960) Gillis et al. 1995 and Burkholderia vandii Urakami et al. 1994 are junior synonyms of Burkholderia gladioli (Severini 1913) Yabuuchi et al. 1993 and Burkholderia plantarii (Azegami et al. 1987) Urakami et al. 1994, respectively. Int. J. Syst. Bacteriol. 1999;49:37–42. doi: 10.1099/00207713-49-1-37. Part 1. [DOI] [PubMed] [Google Scholar]
- 36.Latuasan HE, Berends W. On the origin of the toxicity of toxoflavin. Biochim. Biophys. Acta. 1961;52:502–508. doi: 10.1016/0006-3002(61)90408-5. [DOI] [PubMed] [Google Scholar]
- 37.Daves GD, Jr, Robins RK, Cheng CC. The total synthesis of toxoflavin. J. Am. Chem. Soc. 1961;83:3904–3905. [Google Scholar]
- 38.Reader JS, Metzgar D, Schimmel P, de Crecy-Lagard V. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J. Biol. Chem. 2004;279:6280–6285. doi: 10.1074/jbc.M310858200. [DOI] [PubMed] [Google Scholar]
- 39.Woo HJ, Hwang YK, Kim YJ, Kang JY, Choi YK, Kim CG, Park YS. Escherichia coli 6-pyruvoyltetrahydropterin synthase ortholog encoded by ygcM has a new catalytic activity for conversion of sepiapterin to 7,8-dihydropterin. FEBS Lett. 2002;523:234–238. doi: 10.1016/s0014-5793(02)02997-6. [DOI] [PubMed] [Google Scholar]
- 40.Ploom T, Thony B, Yim J, Lee S, Nar H, Leimbacher W, Richardson J, Huber R, Auerbach G. Crystallographic and kinetic investigations on the mechanism of 6-pyruvoyl tetrahydropterin synthase. J. Mol. Biol. 1999;286:851–860. doi: 10.1006/jmbi.1998.2511. [DOI] [PubMed] [Google Scholar]
- 41.Van Lanen SG, Reader JS, Swairjo MA, de Crecy-Lagard V. From cyclohydrolase to oxidoreductase: Discovery of nitrile reductase activity in a common fold. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4264–4269. doi: 10.1073/pnas.0408056102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suhadolnik RJ. Nucleoside antibiotics. New York: Wiley-Interscinece; 1970. Pyrrolopyrimidine nucleosides; pp. 298–353. [Google Scholar]
- 43.Nishioka H, Sawa T, Nakamura H, Iinuma H, Ikeda D, Sawa R, Naganawa H, Hiyashi C, Hamada M, Takeuchi T, Iitaka Y, Umezawa K. Isolation and structure determination of novel phosphatidylinositol kinase inhibitors, echiguanines A and B, from Streptomyces sp. J. Nat. Prod. 1991;54:1321–1325. doi: 10.1021/np50077a014. [DOI] [PubMed] [Google Scholar]
- 44.Isaac BG, Ayer SW, Letendre LJ, Stonard RJ. Herbicidal nucleosides from microbial sources. J. Antibiot. 1991;44:729–732. doi: 10.7164/antibiotics.44.729. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka N, Wu T, Okabe H, Yamashita A, Shimazu A, Nishimura TJ. Cadeguomycin, a novel nucleoside analog antibiotic. I. The producing organism, producation and isolation of cadeguomycin. J. Antibiot. 1982;35:272–278. doi: 10.7164/antibiotics.35.272. [DOI] [PubMed] [Google Scholar]
- 46.McCormick JRD, Morton GO. Identity of cosynthetic factor 1 of Streptomyces aureofaciens and fragment F0 from coenzyme F420 of Methanobacterium species. J. Am. Chem. Soc. 1982;104:4014–4015. [Google Scholar]
- 47.Kuo MT, Yurek DA, Coats JH, Li GP. Isolation and identification of 7,8-didemethyl-8-hydroxy-5-deazaflavin, an unusual cosynthetic factor in streptomycetes, from Streptomyces lincolnensis. J. Antibiot. 1989;42:475–478. doi: 10.7164/antibiotics.42.475. [DOI] [PubMed] [Google Scholar]
- 48.Bereswill S, Fassbinder F, Volzing C, Covacci A, Haas R, Kist M. Hemolytic properties and riboflavin synthesis of Helicobacter pylori: cloning and functional characterization of the ribA gene encoding GTP-cyclohydrolase II that confers hemolytic activity to Escherichia coli. Med. Microbiol. Immunol. (Berlin) 1998;186:177–187. doi: 10.1007/s004300050062. [DOI] [PubMed] [Google Scholar]
- 49.Fassbinder F, Kist M, Bereswill S. Structural and functional analysis of the riboflavin synthesis genes encoding GTP cyclohydrolase II (ribA), DHBP synthase (ribBA), riboflavin synthase (ribC), and riboflavin deaminase/reductase (ribD) from Helicobacter pylori strain P1. FEMS Microbiol. Lett. 2000;191:191–197. doi: 10.1111/j.1574-6968.2000.tb09339.x. [DOI] [PubMed] [Google Scholar]
- 50.Worst DJ, Gerrits MM, Vandenbroucke-Grauls CM, Kusters JG. Helicobacter pylori ribBA-mediated riboflavin production is involved in iron acquisition. J. Bacteriol. 1998;180:1473–1479. doi: 10.1128/jb.180.6.1473-1479.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeLano WL. The PyMol molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]