

The results of a phase II study of bevacizumab and everolimus in patients with metastatic colorectal cancer are presented. Modest activity was observed.
Keywords: Phase II, Bevacizumab, Everolimus, Refractory colorectal cancer
Abstract
Purpose.
For patients with metastatic colorectal cancer (mCRC), no standard therapy exists after progression on 5-fluorouracil, oxaliplatin, irinotecan, bevacizumab, and cetuximab or panitumumab. Preclinical data demonstrated that combined vascular endothelial growth factor and mammalian target of rapamycin inhibition has greater antiangiogenic and antitumor activity than either monotherapy. A phase I study of bevacizumab plus everolimus demonstrated that the combination is safe; activity was seen in several patients with refractory mCRC.
Methods.
Fifty patients with refractory mCRC were enrolled and received bevacizumab at 10 mg/kg every 2 weeks and everolimus at 10 mg orally daily.
Results.
Of the 50 patients enrolled, the median age was 56 years and the median number of prior regimens was four. Forty-seven patients (96%) had prior bevacizumab exposure and 42 patients (84%) had documented progression on prior bevacizumab-based therapy. Forty-nine patients were evaluable for response; eight patients had minor responses (16%) and an additional 15 patients (30%) had stable disease (SD). No complete or partial responses were seen. The median progression-free survival interval was 2.3 months; however, 26% of patients achieved prolonged SD for ≥6 months, and three patients (6%) were on study for >1 year. The median overall survival duration was 8.1 months. The most common grade 1–2 toxicities were mucositis (68%) and hyperlipidemia (64%). Clinically significant grade ≥3 toxicities included hypertension (14%), fistula/abscess/perforation (8%), mucositis (6%), and hemorrhage (2%).
Conclusions.
Bevacizumab plus everolimus is generally tolerable but may have risks related to mucosal damage and/or wound healing. Bevacizumab plus everolimus appears to have modest activity in refractory mCRC in patients.
Introduction
Colorectal cancer continues to be the second leading cause of cancer deaths for men and women in the U.S. [1]. Bevacizumab, the monoclonal antibody against vascular endothelial growth factor (VEGF), when administered in combination with 5-fluorouracil (FU)-based cytotoxic chemotherapy, has emerged as a standard first-line treatment for metastatic colorectal cancer (mCRC) [2, 3]. Recent data suggested that continued use of bevacizumab following progression may be associated with a longer median overall survival (OS) time [4]. However, eventually all patients progress on these bevacizumab-based treatments, which suggests that acquired resistance to VEGF inhibition may contribute to disease progression. For patients with mCRC, treatment options are absent after progression on 5-FU, oxaliplatin, irinotecan, bevacizumab, and cetuximab or panitumumab (if clinically appropriate). Everolimus is an inhibitor of the mammalian target of rapamycin (mTOR) pathway and is approved for use in patients with metastatic renal cell carcinoma, for which trials have shown a longer OS time than with best supportive care in the refractory setting [5]. The mTOR pathway receives upstream signals from the phosphoinositide 3-kinase (PI3K)/AKT pathway, and these pathways are key regulators of tumor and endothelial cell proliferative and survival functions, including compensatory responses to hypoxia [6]. In endothelial cells, the mTOR and PI3K/AKT pathways mediate many of the functions associated with VEGF stimulation [7, 8]. Therefore, combined blockade of these two pathways may overcome several possible mechanisms of tumor- and endothelial cell–mediated resistance to monotherapy. Phase I data by our group demonstrated that the combination of bevacizumab and everolimus is safe, and prolonged stable disease (SD) (16–112 weeks) and minor responses were observed in several patients with mCRC previously refractory to bevacizumab-based therapy [9]. We sought to further evaluate this combination in a phase II trial in refractory mCRC patients to better define the activity and tolerability of this regimen. In addition, this study included blood and tumor-related biomarkers analyses, which are reported separately.
Materials and Methods
Patient Eligibility
Patients were required to have histologically proven adenocarcinoma of the colon or rectum and must have progressed on, or could not tolerate, all of the following standard of care treatments for mCRC: fluoropyrimidines, oxaliplatin, irinotecan, and the epidermal growth factor receptor inhibitor cetuximab and/or panitumumab (if wild-type KRAS). Additional inclusion criteria included the following: a Karnofsky performance status score ≥70%; adequate hematologic, hepatic, and renal function; and adequately controlled hypertension <150/100 mmHg. All patients were required to have measurable disease defined by the Response Evaluation Criteria in Solid Tumors (RECIST) [10]. Key exclusion criteria included treatment for cancer <28 days prior to the study, known central nervous system metastases, poorly controlled cardiovascular disease, and a history of an abdominal fistula, perforation, abscess, or major bleeding event within 6 months.
This study (ClinicalTrials.gov identifier, NCT00597506) was approved by the Duke University Institutional Review Board and followed the Helsinki Guidelines and Good Clinical Practice. All patients provided informed written consent prior to any study-related procedure. Subjects were accrued at Duke University Medical Center and at Community Memorial Healthcenter in South Hill, Virginia through the Duke Oncology Network.
Safety Evaluations
Toxicity and safety clinical assessments, including a physical examination and hematology and biochemistry analyses, were performed weekly during the first cycle and on day 1 and day 15 of each subsequent cycle. Monthly safety assessments included evaluation of the urine protein-to-creatinine ratio, fasting lipid profile, creatinine phosphokinase for patients on statins, and pregnancy β-human chorionic gonadotropin for women of child-bearing potential. An electrocardiogram, the prothrombin time and partial thromboplastin time, and the thyroid-stimulating hormone level were assessed every two cycles; cardiac ejection fractions were obtained every 6 months. Guidelines for supportive care and toxicity management, including dose modifications, were included in the protocol.
Tumor response as assessed by serum carcinoembryonic antigen and radiographic imaging by computed tomography or magnetic resonance imaging were completed at baseline and every two cycles. Toxicities were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0) [11].
Study Treatment Schedule
Bevacizumab (Avastin®; Genentech/Hoffmann-La Roche, Basel, Switzerland) was administered as a biweekly i.v. infusion of 10 mg/kg on day 1 and day 15 of each 28-day cycle. Everolimus (Afinitor®; Novartis Pharmaceuticals Corporation, East Hanover, NJ) was orally administered daily at 10 mg. Treatment was discontinued for disease progression, unacceptable toxicity, or physician and/or patient decision. Patients whose therapy was discontinued for any reason were followed for survival every 3 months.
Statistical Methods
This was an open-label, nonrandomized, phase II trial to assess the safety, tolerability, and efficacy of bevacizumab and everolimus in patients with refractory mCRC. The primary endpoints were overall response (complete and partial response) as defined by the RECIST (version 1.0) and progression-free survival (PFS). Complete response was defined as the disappearance of all target and nontarget lesions, and partial response was defined as a ≥30% decrease in the sum of the diameters of target lesions. Secondary endpoints included OS, safety, and tolerability.
For sample size determination, the trial was to be considered positive if either the response rate was ≥10% or the 8-week PFS rate was ≥67%. In refractory mCRC patients, a response rate of 10% is considered promising. This study of 50 patients had at least 82% power to simultaneously detect a response rate of 10%, compared with 1%, and a 6-week longer median PFS interval, compared with a historical median PFS duration of 8 weeks (which corresponds to an improvement from 50% to 67% for the 8-week PFS rate) given a type I error of 7.4%. If three or more responses (6%) or ≥31 instances (62%) of an 8-week PFS time were observed among the 50 evaluable patients, this treatment regimen would be considered worthy of further investigation in this disease. Power calculations were based on the assumption that the response rate and PFS rate were uncorrelated. If they were positively correlated, the overall power would be slightly lower and the type I error would be slightly higher. Survival duration was calculated using the Kaplan–Meier method; 90% and 95% confidence intervals (CIs) for the 8-week PFS rate were calculated, and 95% CIs for the median PFS and OS times were also calculated.
The “general” PFS duration was defined as the interval between the start of treatment and the date of disease progression or death. Patients who were alive were censored at their last follow-up. The “on treatment” PFS interval was defined as the start of study treatment to the date of disease progression or death, whichever occurred earlier, with censoring of patients at the time of loss to follow-up or start of a new line of treatment (for patients who discontinued study treatment for reasons other than disease progression).
Results
The characteristics of the 50 enrolled patients are shown in Table 1. The patient population was highly refractory, having progressed on a median of four prior treatments (range, two to eight). Forty-two patients (84%) had progressed on prior bevacizumab-based therapy. Five patients (10%) had received bevacizumab but had not progressed on bevacizumab, and three patients (6%) had never received bevacizumab. Forty-nine patients were evaluable for progression and all 50 were evaluable for toxicity. Twenty-three patients (46%) had SD as their best response, and eight of these 23 patients (16% overall) achieved a minor response, defined as a radiographic decrease in tumor volume by 10%–20% (Fig. 1). Twenty-one patients (42%) had progressive disease as their best response on treatment; 17 patients (34%) progressed radiographically whereas four (8%) progressed clinically. Five patients (10%) discontinued study treatment because of toxicity in the absence of progression or response, prior to their first restaging. No objective partial or complete responses were seen.
Table 1.
Patient characteristics

Figure 1.

Waterfall plot of best radiographic response. Data from 37 of 50 patients are presented. Not pictured are: three patients with new radiographic lesions, four patients with clinical progression, five patients who came off treatment for toxicity prior to first restaging, and one nonevaluable patient who died from a nontreatment-related illness.
The 8-week PFS rate was 0.58 using the general definition (95% CI, 0.43–0.70; 90% CI, 0.46–0.68) and 0.59 using the on treatment definition (95% CI, 0.44–0.71; 90% CI, 0.47–0.70). The median PFS interval, using the general and on treatment definitions, was 2.3 months (95% CI, 1.9–3.7 months), as seen in Figure 2. However, the median OS time was 8.1 months (95% CI, 5.5–11.5 months) (Fig. 3). Thirteen patients (26%) achieved SD on treatment for a period lasting >6 months, eight of whom had seen and progressed through two or more bevacizumab-containing regimens. Three patients (6%) were treated in this study for >1 year.
Figure 2.

Kaplan–Meier curve of general progression-free survival.
Figure 3.

Kaplan–Meier curve of overall survival.
Grade ≥3 treatment-related adverse events (AEs) are listed in Table 2. The most common grade ≥3 AEs were laboratory only; the only grade 4 AE was asymptomatic hypokalemia. Clinically significant grade 3 AEs included mucositis, abscess, fistula, perforation, and gastrointestinal hemorrhage. The incidence of grade 1–2 mucositis (including oropharyngitis, proctitis, and vaginitis) was 68% (n = 34), and the incidence of grade 1–2 hyperlipidemia was 64% (n = 32). Everolimus was dose reduced in 15 patients (30%), and either bevacizumab or everolimus were temporarily held for toxicity in 34% (n = 17) and 62% (n = 31) of patients, respectively. There were no treatment-related deaths.
Table 2.
Grade ≥3 treatment-related adverse events
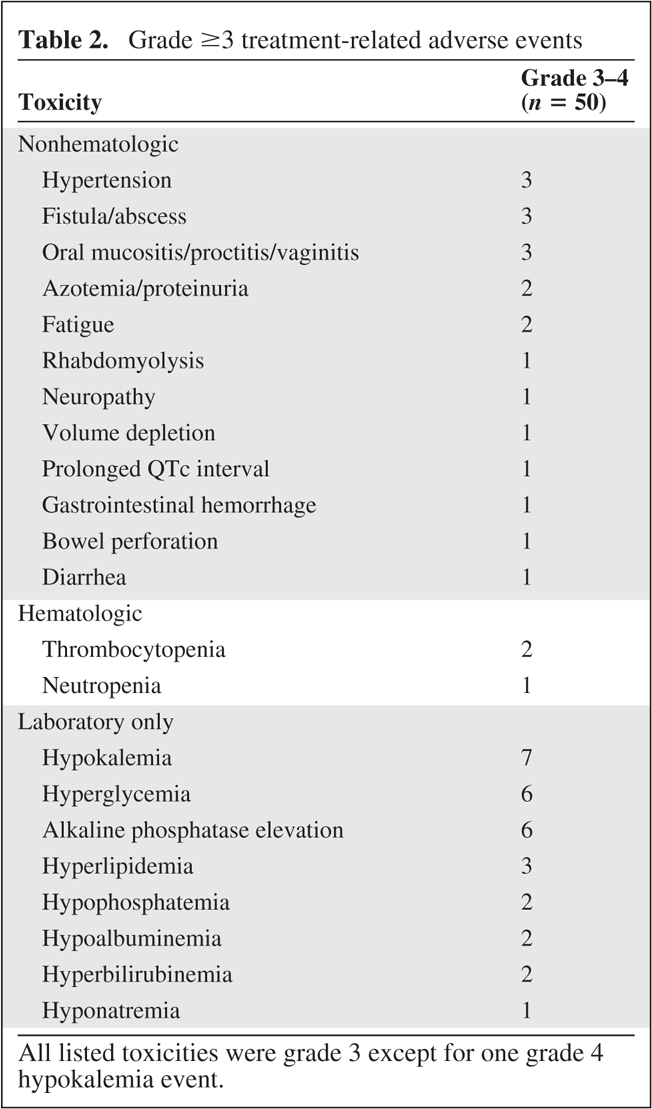
All listed toxicities were grade 3 except for one grade 4 hypokalemia event.
Discussion
Targeting molecular pathways of tumor growth has recently become a major focus of anticancer treatments, and rational combinations of targeted agents have the potential for better efficacy through mediating sensitivity and/or resistance to therapy. The mTOR inhibitors temsirolimus and everolimus are approved by the U.S. Food and Drug Administration for the treatment of patients with refractory renal cell cancer [5, 12]. For other cancers, mTOR inhibition has shown single-agent activity; phase II and phase III trials in several cancer types are currently ongoing. Many mechanisms of resistance to VEGF inhibitors have been described, with recent emphasis on hypoxia responses [13, 14]. In cell culture models, mTOR inhibition by sirolimus specifically abrogated hypoxia-mediated proliferation and angiogenesis [15]. Rapamycins have also been shown, in cell culture, to inhibit hypoxia-inducible factor 1α, a transcription factor that regulates expression of VEGF, suggesting that combined VEGF and mTOR inhibition could have greater antiangiogenic and antitumor activity than either monotherapy [16–18].
In this trial, it is noteworthy that bevacizumab and everolimus were tolerable at full doses of each agent, with only one treatment-related grade 4 toxicity of clinically asymptomatic hypokalemia. This regimen also appears to be well tolerated based on preliminary data from several other ongoing studies in patients with other tumor types [19–21]. However, a study evaluating the combination of another mTOR inhibitor, temsirolimus, with bevacizumab in renal cell carcinoma patients found that full doses of both agents could not be administered as a result of unacceptable toxicities [22]. The reasons for these apparent differences may be related to specific toxicities among the different mTOR agents. In the present study, mucositis was the most common toxicity, reported by 64% of patients at grade 1–2 and by 12% of patients at grade 3, which was readily managed with standard supportive care and appropriate dose holding and/or reductions.
In mCRC patients, neither bevacizumab nor everolimus has shown clinically significant single-agent activity [2, 23]. Although there were no objective responses, treatment with bevacizumab plus everolimus led to prolonged SD of >6 months in a subset of patients, and three patients were treated on study for >1 year. Even without tumor regression, SD can be clinically meaningful, provided it is prolonged and not associated with significant toxicity. In our study, 11 of the 13 patients achieving prolonged SD had progressed on at least one prior bevacizumab-containing regimen, and eight had been treated with bevacizumab in at least two and as many as four lines of treatment. This finding suggests that, for some patients, the combination of bevacizumab plus everolimus may help overcome resistance to bevacizumab.
In summary, in this heavily pretreated CRC population, bevacizumab plus everolimus is tolerable but has limited clinical activity, primarily seen as prolonged SD.
Acknowledgments
We gratefully acknowledge the invaluable contributions of the patients, their families, and the Duke University GI Oncology clinical trials team, with special recognition to Anthony Amara for data management and Anuradha Bulusu for statistical assistance.
Johanna C. Bendell is currently at the Sarah Cannon Research Institute, Nashville, Tennessee, USA. Karen E. Bullock is currently with the U.S. Navy, Portsmouth, Virginia, USA.
This study was funded by Genentech/Roche and Novartis.
Author Contributions
Conception/Design: Herbert I. Hurwitz, Johanna C. Bendell, Karen E. Bullock
Financial support: Herbert I. Hurwitz
Administrative support: Herbert I. Hurwitz
Provision of study material or patients: Herbert I. Hurwitz, Johanna C. Ben- dell, Hope E. Uronis, Michael A. Morse, S. David Hsu, S. Yousuf Zafar, Wanda Honeycutt, Linda Sutton, Gerard C. Blobe
Collection and/or assembly of data: Ivy Altomare
Data analysis and interpretation: Herbert I. Hurwitz, Ivy Altomare, Herbert Pang
Manuscript writing: Herbert I. Hurwitz, Ivy Altomare, Herbert Pang
Final approval of manuscript: Herbert I. Hurwitz, Ivy Altomare, Johanna C. Bendell, Karen E. Bullock, Hope E. Uronis, Michael A. Morse, S. David Hsu, S. Yousuf Zafar, Herbert Pang, Wanda Honeycutt, Linda Sutton, Gerard C. Blobe
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539–1544. doi: 10.1200/JCO.2006.09.6305. [DOI] [PubMed] [Google Scholar]
- 3.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 4.Grothey A, Sugrue MM, Purdie DM, et al. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: Results from a large observational cohort study (BRiTE) J Clin Oncol. 2008;26:5326–5334. doi: 10.1200/JCO.2008.16.3212. [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 6.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Bjornsti MA, Houghton PJ. The TOR pathway: A target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 8.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Bullock KE, Petros WP, Younis I, et al. A phase I study of bevacizumab (B) in combination with everolimus (E) and erlotinib (E) in advanced cancer (BEE) Cancer Chemother Pharmacol. 2011;67:465–474. doi: 10.1007/s00280-010-1507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 11.National Institutes of Health, Cancer Therapy Evaluation Program. Bethesda, MD: 2003. Jun 10, Common Terminology Criteria for Adverse Events (CTCAE) Version 3.0. [Google Scholar]
- 12.Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 13.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–26. doi: 10.1016/j.cytogfr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Humar R, Kiefer FN, Berns H, et al. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16:771–780. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 16.George DJ, Kaelin WG., Jr The von Hippel-Lindau protein, vascular endothelial growth factor, and kidney cancer. N Engl J Med. 2003;349:419–421. doi: 10.1056/NEJMp030061. [DOI] [PubMed] [Google Scholar]
- 17.Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 19.Whorf RC, Hainsworth JD, Spigel DR, et al. Phase II study of bevacizumab and everolimus (RAD001) in the treatment of advanced renal cell carcinoma (RCC) J Clin Oncol. 2010;28:2129–2136. doi: 10.1200/JCO.2009.26.3152. [DOI] [PubMed] [Google Scholar]
- 20.Yao JC, Phan AT, Fogleman D, et al. Randomized run-in study of bevacizumab (B) and everolimus (E) in low- to intermediate-grade neuroendocrine tumors (LGNETs) using perfusion CT as functional biomarker. J Clin Oncol. 2010;28(15 suppl):4002. [Google Scholar]
- 21.Peyton JD, Spigel DR, Burris HA, et al. Phase II trial of bevacizumab and everolimus in the treatment of patients with metastatic melanoma: Preliminary results. Cancer. 2010;116:4122–4129. doi: 10.1002/cncr.25320. [DOI] [PubMed] [Google Scholar]
- 22.Escudier BJ, Negrier S, Gravis G, et al. Can the combination of temsirolimus and bevacizumab improve the treatment of metastatic renal cell carcinoma (mRCC)? Results of the randomized TORAVA phase II trial. J Clin Oncol. 2010;28(15 suppl):4516. [Google Scholar]
- 23.Fuchs CS, Tabernero JM, Hwang J, et al. Multicenter phase II study of RAD001 in patients with chemotherapy-refractory metastatic colorectal cancer (mCRC). 2009 Gastrointestinal Cancers Symposium; p. 446. [Google Scholar]