

Abstract
The Steroidogenic factor 1 (SF-1, also known as NR5A1) is a transcription factor belonging to the nuclear receptor superfamily. Whereas most of the members of this family have been extensively characterized, the therapeutic potential and pharmacology of SF-1 still remains elusive. Described here is the identification and characterization of selective inhibitory chemical probes of SF-1 by a rational ultra-high-throughput screening (uHTS) strategy. A set of 64,908 compounds from the National Institute of Health’s Molecular Libraries Small Molecule Repository (MLSMR) was screened in a transactivation cell-based assay employing a chimeric SF-1 construct. Two analogous isoquinolinones, SID7969543 and SID7970631, were identified as potent submicromolar inhibitors, yielding IC50 values of 760 nM and 260 nM. The compounds retained their potency in a more physiologic functional assay employing the full-length SF-1 protein and its native response element, yielding IC50 values of 30 and 16 nM, respectively. The selectivity of these isoquinolinones was confirmed via transactivation-based functional assays for RORA, VP-16 and LRH-1. Their cytotoxicity, solubility, permeability and metabolic stability were also measured. These isoquinolinones represent valuable chemical probes to investigate the therapeutic potential of SF-1.
Introduction
Nuclear receptors (NRs) are transcription factors that regulate the expression of downstream genes through the binding of lipophilic ligands such as hormones, vitamins, lipids and/or small molecules (Giguere, 1999). They are involved in diverse biological processes, such as embryogenesis, homeostasis, reproduction, cell growth and death (Mangelsdorf et al., 1995). With numerous NR-targeting drugs marketed or in development, NRs have proven to be successful therapeutic targets for a wide range of diseases (Moore et al., 2006). Whereas natural or synthetic ligands have been reported for numerous members of the NR superfamily, the pharmacology of so-called “orphan” nuclear receptors -for which no natural ligand has been reported- as well as those recently “adopted” remains poorly characterized (Giguere, 1999). We are currently investigating the therapeutic potential of such unexplored nuclear receptors, among them the Steroidogenic Factor 1 (SF-1, also known as NR5A1).
SF-1 plays a central role in sex determination and the formation of steroidogenic tissues during development, and is involved in endocrine function throughout life (Luo et al., 1995a; Parker et al., 2002; Val et al., 2003). SF-1 is expressed in the pituitary, testes, ovaries, and adrenal gland where it regulates the expression of several genes involved in steroidogenesis (Val et al., 2003). SF-1-deficient mice exhibit male-to-female sex reversal (Luo et al., 1994), an impaired development of adrenals and gonads (Luo et al., 1995b; Sadovsky et al., 1995), defective pituitary gonadotroph, and an agenesis of the ventromedial hypothalamic nucleus (Ikeda et al., 1995; Shinoda et al., 1995). Although SF-1 has been shown to be rarely associated with clinical disorders of sexual differentiation (Parker et al., 2002), it has been reported to have a potential role in obesity (Majdic et al., 2002). More recently it has been observed that an increased concentration of SF-1 causes adrenocortical cell proliferation and cancer (Doghman et al., 2007).
Small-molecule pharmacologic probes of SF-1 activity represent valuable investigational tools to better understand target involvement in both physiological and pathophysiological contexts (Lazo et al., 2007). Presented here is the use of cell-based functional assays in a rational high-throughput screening approach that led to the identification of two efficacious and selective isoquinolinone inhibitors of SF-1 activity.
Materials and Methods
Materials
Compounds SID7969543 and SID7970631 were purchased from Life Chemicals (Kiev, Ukraine). Compound AC-45594 (Del Tredici et al., 2007) was acquired from Sigma-Aldrich (Milwaukee, WI).
Vector construction
pGal4DBD_SF-1LBD and pGal4DBD_RORALBD were generated by cloning PCR fragments encoding either human SF-1 (aa 198–462) or mouse RORA (aa 266–523) LBD in frame with the DBD of the yeast transcriptional factor Gal4 encoded by the pFA-CMV vector (Stratagene, La Jolla, CA). SF-1 (aa 198–462) was amplified from an Invitrogen EST clone (San Diego, CA; clone# 5163875). BamHI and XbaI sites introduced by the primers GATCGGATCCCCGGAGCCTTATGCCAGCCC (forward) and GATCTCTAGATCAAGTCTGCTTGGCTTGCAGCATTTCGATGAG (reverse) were used for subcloning the amplicon into pFA-CMV. RORA (aa 266–523) was generated by PCR primers GCCGCCCCCGGGCCGAACTAGAACACCTTGCCC (forward) and TATATAAAGCTTTCCTTACCCATCGATTTGCATGG (reverse) from a mouse liver cDNA library from Clontech (Mountain View, CA) and subcloned through XmaI and HindIII restriction sites into pFA-CMV.
Cell culture and transient transfection conditions
Chinese Hamster Ovary (CHO) cells of the K1 subtype (ATCC, Manassas, VA) were grown in T-175 flasks (Corning, Lowell, MA) at 37°C, 5% CO2, 95% relative humidity in F12 media (Gibco, Carlsbad, CA) supplemented with 10% v/v fetal bovine serum (Gemini Bio-products, West Sacramento, CA) and 1% v/v penicillin-streptomycin-neomycin mix (Gibco, Carlsbad, CA). Cells were routinely cultured by splitting them from 1:4 to 1:8.
The day before transfection, cells were rinsed with PBS and trypsinized with a 0.25% trypsin-EDTA solution (Gibco, Carlsbad, CA), then 6 million CHO-K1 cells were seeded in T-175 flasks containing 20 mL of F12 media supplemented as mentioned above. Cells were allowed to incubate overnight at 37°C, 5% CO2 and 95% relative humidity (RH). On the following day, CHO-K1 cells were transiently co-transfected with either 250 ng of pGal4DBD_SF-1LBD plasmid or 125 ng of pGal4DBD_RORALBD in combination with 9 µg of pG5luc (Promega Corporation, Madison, WI) and 8.75 µg of empty pcDNA3.1 (Invitrogen, Carlsbad, CA), in 1.2 mL of F12 media containing 54 µL of TransIT®-CHO reagent and 9 µL of TransIT-CHO® Mojo reagent, according to the manufacturer's protocol (Mirus Bioproducts, Madison, WI).
For uHTS assays, cells transfected with the pG5-luc and empty pcDNA3.1 plasmids alone, designated “−NR” cells (as opposed to “+NR” cells, which are co-transfected with the Gal4-LBD encoding plasmid) were used as positive control for inhibition. Flasks were then placed back in the incubator at 37°C, 5%CO2 and 95% relative humidity. Four hours after transfection, cells were trypsinized and suspended to a concentration of 800,000 cells per milliliter in supplemented F12 media.
1536-well format SF-1 and RORA uHTS assays
The assay began by dispensing 5 µL of transfected cell suspension to each well (i.e. 4,000 cells/well) of a white solid-bottom 1536-well plate (Greiner, North Carolina, USA) using a Bottle Valve liquid dispenser (GNF/Kalypsys, San Diego, CA). Cells from flasks designated −NR were seeded in the first two columns of the 1536-well plate (Low Control) and the remaining 46 columns were filled with +NR cells. One hour after seeding, +NR cells were treated with 50 nL/well of test compounds or DMSO alone (High Control) using a 1536-well head PinTool unit (GNF/Kalypsys, San Diego, CA). Plates were then incubated at 37°C, 5%CO2 and 95% relative humidity. Twenty hours later, plates were equilibrated to room temperature for 20 minutes and a luciferase assay was performed by adding 5 µL per well of SteadyLite HTS™ reagent (PerkinElmer, Waltham, MA). After a 15 minutes incubation time, light emission was measured for 30 seconds with the ViewLux™ reader (PerkinElmer, Turku, Finland). Activity of each compound was calculated on a per-plate basis using the following equation:
where High Control represents wells containing +NR cells treated with DMSO (n=24) and Low Control represents wells containing −NR cells treated with DMSO (n=24).
Primary high-throughput screening and hit selection
A library of 64,908 compounds provided by the Molecular Library Screening Center Network (MLSCN) in fifty-two 1536-well plates was tested in the SF-1 assay as described above. The final nominal test concentration was 10 µM, in a final DMSO concentration of 1%.
The uHTS campaign was executed on the automated GNF/Kalypsys robotic platform of the Scripps Research Institute Molecular Screening Center (Jupiter, FL). Raw data from each primary campaign were uploaded and analyzed in our institutional HTS database (MDL Information Systems, San Ramon, CA). The percent inhibition of each tested compound was calculated on a per-plate basis as described in the previous section. A mathematical algorithm was used to determine active compounds (Hodder et al., 2003). Two values were calculated: (1) the average percent inhibition of all compounds tested, and (2) three times their standard deviation. The sum of these two values was used as a cutoff parameter, i.e. any compound that exhibited greater percent inhibition than the cutoff parameter was declared active. Z’ value was calculated as previously described (Zhang et al., 1999). Detailed information regarding this screen can be found in the MLSCN PubChem website (pubchem.ncbi.nlm.nih.gov, Bioassay AID 525).
HTS dose-response experiments
Compounds found active during primary screens were selected from the NIH’s Molecular Libraries Small Molecule Repository (MLSMR, San Francisco, CA) and 10-point, 1:3 serial dilutions starting from a nominal 10 mM solution were prepared using an automated liquid handler (Beckman-Coulter, Fullerton, CA). Titration experiments were performed as mentioned above, by transferring 50 nL of the compound solutions in the titration plate into 3 different assay plates. For each compound, triplicate percentage inhibitions were plotted against compound concentration. A four parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using Assay Explorer software (MDL® Information Systems). IC50 values were generated from fitted curves by solving for X-intercept at the 50% inhibition level of Y-intercept. For compounds where no curve was fitted by the algorithm, an IC50 was determined manually. Detailed information regarding the SF-1 titration assay and the RORA counterscreen can be found at the MLSCN PubChem website (Bioassays AID 600 and 599, respectively).
VP16 promiscuity assay
This assay used the same protocol as the uHTS titration assays described in the previous section, apart that 125 ng of pGal4DBD_VP16LBD plasmid (a kind gift from Dr. Michael Conkright, Dept. of Cancer Biology, Scripps Florida) was used for the transfection step (Amelio et al., 2007).
SFRE assays
Prior to the assay, an 11 point, 1:3 serial dilution of compound starting at 0.4 mM (40X of final assay concentration) was prepared in PBS containing 5% DMSO using a liquid handling robot (PlateMate® Plus, Matrix Technologies, Hudson, NH). Human Embryonic Kidney (HEK) cells of the 293T subtype (ATCC, Manassas, VA) were grown in DMEM (Gibco, Carlsbad, CA) supplemented with 10% v/v fetal bovine serum and 1% v/v penicillin-streptomycin-glutamine mix (Gibco, Carlsbad, CA). Cells were co-transfected in white 384-well plate (5,000 cells per well in 40uL) using a mix of 25 ng of pCMV-SF-1 or pCMV-LRH-1 (Open Biosystems, Huntsville, AL), 25 ng of p5×SFRE (a kind gift of Dr. Donald McDonnell, Duke University Medical Center, Durnham, NC) and a 3:1 lipid to DNA ratio of FuGENE® 6 transfection reagent (Roche, Indianapolis, IN). Plates were then incubated at 37°C, 5% CO2, and 95% RH. Twenty-four hours later, cells were treated with 1 µL of the intermediate 40× serial dilution (n=6), giving a final DMSO concentration of 0.125%. Cells were then returned to standard incubation conditions for twenty four hours. Plates were then allowed to equilibrate for 15 minutes at room temperature and a luciferase assay was performed by adding 25 µL/well of BriteLite™ (PerkinElmer, Waltham, MA). After a 2 minutes incubation time, plates were read using the EnVision™ Multilabel Plate Reader (PerkinElmer, Shelton, CT).
Cell viability assay
CHO-K1 cells were plated at 500 cells per well in 1536-well plates in 5 µL of media (F12 supplemented with 10% FBS and 1% Pen/Strep/Neo). Compounds (50nL of 100× DMSO solution per well) were prepared as 10-point, 1:3 serial dilutions starting at 10 mM, then added to the cells using the pin tool. Plates were then incubated 20 hours at 37°C, 5%CO2 and 95% RH. After incubation, five microliters of CellTiter-Glo® (Promega, Madison, WI) were added to each well, and plates were allowed to incubate for 15 minutes at room temperature. Luminescence was recorded for 30 seconds using the ViewLux™ reader (PerkinElmer, Turku, Finland). Viability was expressed as a percentage relative to wells containing media only (0%) and wells containing cells treated with DMSO only (100%).
Hepatic microsomal stability
Microsome stability was evaluated by incubating 1 µM compound with 2 mg/ml hepatic microsomes from either human, monkey, rat, dog, or mouse in 100 mM potassium phosphate buffer, pH 7.4. The reactions were held at 37° C with continuous shaking. The reaction was initiated by adding NADPH (1mM final concentration). The final incubation volume was 300 µl and 40 µl aliquots were removed at 0, 1, 3, 5, 8, and 10 minutes for rapidly metabolized compounds, or at 0, 5, 10, 20, 40, and 60 minutes for more stable compounds. The removed aliquot was added to 160 µl acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction is evaluated with parallel incubations without NADPH. At the end of the assay, the samples were centrifuged through a 0.45 micron filter plate (Millipore, Billerica, MA) and analyzed by LC-MS/MS. The data was log transformed and results are reported in units of half-life.
Solubility
In a glass test tube, 1–2 mg of probe compound was added to 1 mL of either pH 7.4 or pH 3.5 potassium phosphate buffer. The samples were allowed to invert for 24 hours at room temperature. The samples were centrifuged and the supernatant was analyzed by HPLC against a known reference.
Parallel Artificial Membrane Permeability Assay (PAMPA)
An assessment of permeability was done using a commercial PAMPA kit (BD Biosciences, Franklin Lakes, NJ). Compounds were evaluated over a range of concentrations by addition of 300 µl of PBS containing the compound to the bottom donor plate. Compounds were from DMSO stocks and the final DMSO concentration in the donor wells was one percent. 200 µl of blank PBS was added to the top receiver plate. The plates were allowed to incubate at room temperature. After 5 hours, aliquots were taken from the donor and receiver plates and the concentration of drug was determined. Compound permeability was calculated using the equation
where Pe is expressed in units of cm/s, CA(t) is drug concentration in the acceptor at time t, VD is donor well volume, VA is acceptor well volume, A is the area of the filter (0.3 cm2), t is time in seconds, and .
Data representation
All data representations have been done with GraphPad Prism version 4.03 (GraphPad Software, San Diego, CA). Curve fitting and IC50 determination were performed using the variable slope sigmoidal dose-response analysis tool of GraphPad Prism.
Cheminformatics
In silico analyses were performed using tools and screening data available in the Pubchem website (http://pubchem.ncbi.nlm.nih.gov/). For determining whether compounds were luciferase assay artefact, data existing in PubChem was used (Bioassay AID 411).
Results
SF-1 uHTS assay development and validation
The SF-1 uHTS assay was designed to specifically monitor the SF-1 activation state via co-transfection of CHO-K1 cells with a plasmid encoding a Gal4-SF-1 chimerical transcription factor, pGal4DBD-SF-1LBD, and a second plasmid driving luciferase expression under control of 5 multimerized Gal4 binding sites (Fig. 1). Experiments were executed to validate this functional response (Fig 2A). Co-transfected cells emitted a strong luminescent signal when compared to cells transfected with the luciferase reporter plasmid alone, suggesting that the chimerical pGal4DBD-SF-1LBD proteins were functional and the endogenous co-activators required for activation are present in CHO-K1 cells. Additionally, specific amino-acid substitutions in SF-1 LBD (M455A/L456A) known to disrupt co-activator interaction for most nuclear receptors (Li et al., 2003) and specifically to suppress SF-1 transcriptional activity (Hammer et al., 1999) reduced the luciferase signal by 90%, confirming that the elevated basal transcriptional activity was due to a functional LBD and not to the Gal4 DBD alone.
Figure 1. SF-1 uHTS assay principle.

Transiently transfected CHO-K1 cells express a chimeric SF-1 nuclear receptor in which the original DNA binding domain (DBD) has been replaced with the Gal4 DBD. Upon interaction with endogenous ligands (L) and/or coactivators (Co-Act) present in CHO-K1 cells, the construct triggers the transcription of a co-transfected luciferase-encoding plasmid via its multimerized Gal4 binding sites. After cell lysis, D-luciferin substrate is used to determine luciferase expression levels by measuring its conversion to light-emitting oxiluciferin. The presence of an SF-1 inhibitor is detected by a decrease of the measured luminescence.
Figure 2. A. Gal4DBD_SF-1LBD activity is LBD-dependent.


CHO-K1 cells were transiently transfected with the non-fused Gal4 expressing vector pFA-CMV (Vec), or with the Gal4DBD_SF-1LBD expressing plasmid in its wild-type (WT) or mutated (Mut) form. Transfected cells were incubated and assayed for luciferase activity. Luminescent signal is expressed as a percentage of the maximal signal given by WT Gal4DBD_SF-1LBD. Bars represent the mean of three replicates plus or minus their standard deviation. B. Cell seeding density optimization. Both −SF-1 and +SF-1 CHO-K1 cells were seeded in a 1536-well plate at densities ranging from 1,000 to 5,000 cells per well (n=128 wells for each condition). Z’ (circles) and S/B (squares) calculated based on RLUs values measured for −NR and +NR are shown for each tested cell density. The star indicates the selected optimal cell density.
The SF-1 uHTS assay was implemented in 1536-well plates in a total volume of 10 µL per well. Experimental conditions were optimized to give the best balance between assay performance (determined by Z’-factor), reagent consumption, and suitability of the protocol on the robotic screening platform. A cell seeding density of 4,000/well (Fig. 2B) and 20 hour incubation time with test compound yielded the best assay results (please see the materials & methods section for all final conditions). During assay optimization efforts, no inhibitors of SF-1 activity were available, and therefore a pharmacologic positive control could not be used in the uHTS campaign. Instead cells transiently transfected with the luciferase reporter plasmid alone were used as the positive control, representing 100% inhibition.
High-throughput screening for SF-1 inhibitors
The SF-1 assay was used to screen a collection of 64,908 compounds made publicly available by the National Institutes of Health (NIH) through the Molecular Libraries Screening Center Network (MLSCN) initiative (Austin et al., 2004). As run, the steady state throughput of the uHTS campaign was ~11,520 compounds tested per hour. After having tested each member of the library at a final nominal concentration of 10 µM (Fig. 3), three hundred and fifty nine compounds exhibited a percent inhibition greater than that calculated by a nominal cutoff algorithm (47.96%, see Material and Methods for details). To further confirm their activity, the 359 primary hits were selected and retested in dose-response experiments.
Figure 3. MLSCN compound library screening and hit selection.

The SF-1 cell-based assay was screened against 64,908 compounds (black dots). The average Z’ value during the screen was 0.72 ± 0.06. The dotted-line represents the activity cutoff, which was calculated at 47.96% inhibition. Compounds with inhibition results above the cutoff are located above the dotted-line.
Selection and characterization of the isoquinolinones
As a first step in triage, non-promiscuous SF-1 inhibitors were identified by titrating all 359 primary hits in parallel in the SF-1 assay and in a second cell-based dose-response assay targeting another nuclear receptor, the Retinoic Acid Receptor-related orphan receptor A (RORA). The majority of compounds originating from the SF-1 uHTS effort gave comparable IC50 values in both the SF-1 and RORA assays (Fig 4A). Among these were the compound doxorubicin (SID855944), known to be cytotoxic in cancer-derived mammalian cell lines. It yielded an IC50 value of 443 ± 25 nM (n=3) in the SF-1 assay and of 392 ± 34 nM (n=3) in the RORA assay. Compounds with similar properties, e.g. vinblastine sulfate (SID855758), vincristine sulfate (SID855866), and daunorubicin hydrochloride (SID855543) yielded comparable results. Moreover, analogs of 3-phenoxy-methyl-benzoic acid (SID4263122, SID4255902, SID4261716, SID4243980), previously identified as luciferase detection format artefacts, gave almost identical potencies in both SF-1 and RORA assays (IC50 values in the range of 500 nM were typical). These observations suggested that other compounds from the SF-1 uHTS campaign with comparable IC50 values in the SF-1 and RORA assays were most likely promiscuous inhibitors and/or cytotoxic compounds and therefore were not considered further. Compounds selected for further follow-up passed the two following criteria: (1) the IC50 in the SF-1 assay was at least 10 fold lower than the one determined in the RORA assay, and (2) the calculated SF-1 IC50 was lower than 1 µM. Two isoquinolinone derivatives, annotated as SID7969543 and SID7970631, passed these criteria (Fig. 4B).
Figure 4. Selection of the SF-1 isoquinolinone inhibitors.

(A) Comparison of the titration results in the SF-1 and RORA assays. Graphed are the IC50 values of 359 primary hits, titrated in parallel in the SF-1 and RORA assays. Each compound is located according to its IC50 determined in the SF-1 (X-axis) or the RORA (Y-axis) assay. The isoquinolinones SID7969543 and SID7970631 are designated by an arrowhead. (B) Structures of SID7969543 and SID7970631. The isoquinolinone scaffold is indicated in bold.
Fresh powders of SID7969543 and SID7970631 were retested in the SF-1 and RORA assays (Fig. 5). An SF-1 inverse agonist recently described in the literature, AC-45594 (Del Tredici et al., 2007), was used as a positive control in the SF-1 experiment. Consistent with the screening results SID7969543 and SID7970631 dose-dependently inhibited luciferase expression in the SF-1 assay, giving IC50 values of 760 ± 102 nM (n=3) and 255 ± 63 nM (n=3), respectively (Table 1).
Figure 5. Dose-response results of isoquinolinones.

SID7969543 (A) and SID7970631 (B) were assessed in the SF-1 assay (circles), the RORA assay (squares) and a viability assay (triangles). Additionally (C), SID7969543 (triangles) and SID7970631 (squares) were also assessed in the SFRE/SF-1 assay. Error bars represent the standard error of three separate experiments.
Table 1.
Activity profile of SF-1 inhibitors
 |
 |
|||||
|---|---|---|---|---|---|---|
| SID7969543 | SID7970631 | AC-45594 | ||||
| % Inh. a (% cytotoxicity) |
IC50b (CC50) |
% Inh. a (% cytotoxicity) |
IC50b (CC50) |
% Inh. | IC50 | |
| Gal4-fusion assays | ||||||
| SF-1 | 84 ± 2 | 760 ± 102 | 80 ± 2 | 255 ± 63 | 59 ± 3 | 7,384 ± 368 |
| ROR-a | 16 ± 4 | >33,333 | 20 ± 13 | >33,333 | NT | NT |
| VP16 | 18 ± 2 | >33,333 | −48 ± 21 | >33,333 | NT | NT |
| SFRE promoter assays with full-length proteins | ||||||
| SF-1 | 73 ± 5 | 36 ± 15 | 77 ± 9 | 13 ± 9 | NT | NT |
| LRH-1 | 0 ±3 | NA | −9 ± 9 | NT | NT | NT |
| Cytotoxicity Assay | ||||||
| Cytotoxicity | −1 ± 6 | >99,000 | 19 ± 1 | >33,333 | NT | NT |
Values represent mean percentage of inhibition measured at 10 µM plus or minus standard deviation (n=3)
Values represent mean IC50 (or CC50) plus or minus standard deviation where applicable (n=3), reported in nanomolar.
NA: not applicable, since compound did not reach 50% inhibition
NT: not tested
SAR of the isoquinolinone scaffold
An in silico structure-activity relationship (SAR) study was performed by retrieving primary HTS results of compounds containing the isoquinolinone scaffold (Table 2). Compound SID7971227 differs from SID7970631 only by the absence of the branched methyl on the phenoxy group and was found to be less active (30.6% inhibition vs. 83.1% at 10 µM). As observed in SID7970257, SID7970995, SID7969723, SID7970701 and SID7970398, compounds with substitutions of the SID7970631’s dioxolane moiety exhibited reduced activity in the SF-1 uHTS assay.
Table 2.
In Silico Similarity Search Results of Scaffolds Related to SID7969543 and SID7970631.
 | |||
|---|---|---|---|
| Compound | R1 | R2 | SF-1 % Inh.a |
| SID7970631 | –CH3 |  |
83.1 |
| SID7969543 | –CH3 |  |
81.8 |
| SID7970257 | –CH3 |  |
17.6 |
| SID7969723 | –CH3 |  |
8.4 |
| SID7970398 | –CH3 | 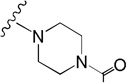 |
−0.2 |
| SID7971227 | –H |  |
30.6 |
| SID7970995 | –H |  |
10.8 |
| SID7970701 | –H |  |
−12.3 |
Inhibition measured at 10 µM, n=1,
N.B. Compounds showing % Inhibition greater than the calculated hit-cutoff (47.96%) were considered active.
The Liver Receptor Homolog 1 (LRH-1, NR5A2) is considered the closest NR related to SF-1 (Fayard et al., 2004). Its LBD shares 65% identity with SF-1 and its active site is thought to accommodate similar ligands (Whitby et al., 2006). In order to further investigate their selectivity on a closely related target, both isoquinolinones were assessed in an assay similar to the SFRE/SF-1 assay described below, apart that full-length LRH-1 was transiently expressed in place of SF-1. Used as a positive control for this assay, SHP (short heterodimer partner, NR0B2) repressed LRH-1 activity (Lee and Moore, 2002) (data not shown). In contrast, neither compound SID7969543 nor SID7970631 inhibited LRH-1-triggered SFRE activation (Table 1).
SFRE/SF-1activity confirmation assay
To ensure SID7969543 and SID7970631 potency was not unique to the chimeric construct functional assay, the isoquinolinones were further investigated in a more relevant biological system (Figure 5). Specifically, both compounds were assayed for functional activity against HEK 293T cells co-transfected with a plasmid encoding full length SF-1 and a second vector allowing expression of the luciferase gene under control of five tandem repeats of the natural SF-1 response element (SFRE). Both compounds inhibited SF-1-triggered luciferase expression with IC50 values of 30 ± 15 nM (n=3) and 16 ± 7 nM, respectively, a rank order consistent with the chimeric SF-1 construct experiments (Table 1).
Transactivation promiscuity and cytotoxicity assays
Similar in format to the SF-1 uHTS and RORA assays, a VP16 assay was also performed to confirm that the isoquinolinones do not promiscuously inhibit transactivation reporter systems. Gal4-VP16 is a synthetic fusion protein that links the yeast Gal4 protein DBD and the herpes simplex virus protein VP16 (also called Vmw65) together, thus acting as a potent transcriptional activator (Sadowski et al., 1988). In the VP16 assay both isoquinolinones exhibited an inhibition profile similar to that observed in the RORa assay (i.e. IC50 values greater than 33,333 µM) suggesting their SF-1 inhibition cannot be attributed to transactivation assay artefact (Table 1). A cell viability assay was also performed to determine the cytotoxicity of both compounds (Fig. 5 and Table 1). Although cytotoxicity was observed at higher test concentrations the isoquinolinones did not exhibit cytotoxicity at a concentration near their respective IC50 values.
Solubility, Permeability and Microsome Studies
In the perspective of investigating potential in vivo stability, physico-chemical properties of both isoquinolinones were evaluated, including solubility, permeability and microsomal stability. As reported Table 3, both compounds demonstrated excellent permeability and solubility, adding confidence to the results obtained from the various cell-based assays performed in this study. Short half-life values were observed in the hepatic-microsomal stability experiments for both compounds in the five species evaluated.
Table 3.
Physico-chemical properties of selected SF-1 inhibitors
| Compounds | ||
|---|---|---|
| SID7969543 | SID7970631 | |
| Solubility (µmoles/L) | ||
| at pH3.5 | 183 | 263 |
| at pH7.4 | 150 | 140 |
| PAMPA Permeabilitya (logP) | −4.74 | −4.67 |
| Microsome stabilityb (t½ min) | ||
| Dog | 1.12 | ≤1 |
| Monkey | ≤1 | ≤1 |
| Mouse | ≤1 | ≤1 |
| Human | 1.24 | ≤1 |
| Rat | ≤1 | 1.41 |
Compounds were evaluated at 10 µM. Antipyrine and propanolol, used as references of highly permeable compounds, both exhibited a logP of −4.81 in the same assay. In contrast, ranitidine, a poorly permeable compound, exhibited a logP value of −6.67.
The following compounds were also tested in the same assays: highly stable reference compounds tolbutamide and dapsone exhibited t½ ≥120 minutes and ranging from 13.04 to ≥120 minutes in the same microsome panel, respectively. Sunitinib, a moderately stable compound, gave t½ ranging from 13.21 to 54.57 minutes. The poorly stable compound verapamil exhibited t½ ranging from 2.34 to 7.37 minutes in the same panel.
Discussion
Development of functional assays for SF-1 activity
It is well known that transcription factors of the NR superfamily share two important conserved structural features, namely a DNA-binding domain (DBD) and a ligand-binding domain (LBD) (Steinmetz et al., 2001). Transactivation reporter gene functional assays have been extensively used to study NRs and characterize their natural or synthetic ligands (Jausons-Loffreda et al., 1994). Accordingly, for the research presented here several cell-based transactivation assays were implemented to discover novel inhibitors of SF-1 activity as well as probe for selectivity of the most potent compounds. Combined with appropriate assays to identify cytotoxic and promiscuous transactivation assay inhibitors, this chemical biology approach facilitates the rapid characterization of physiologically relevant, cell-penetrant chemical probes.
Choice of the RORA functional assay to triage SF-1 uHTS assay results
There were several reasons for choosing RORA as a preliminary selectivity assay for compounds found active in the SF-1 uHTS assay. First, SF-1 and RORA are both transcriptionally active in cell-based functional assays (Carlberg et al., 1994; Mellon and Bair, 1998) and they both bind DNA as monomers whereas most of NRs do so only in a homo- or heterodimerized state (Giguere, 1999). Secondly, their ligand binding domains may be responsive to small-molecule ligands (Kallen et al., 2004; Li et al., 2005) and their phylogeny is distant enough to make them susceptible to different ligands (Moore et al., 2006). An ancillary benefit is that assay protocols and reagents used for both assays were nearly identical; the major difference is the transient transfection procedure for each receptor’s particular ligand binding domain.
Interpreting results of the functional and cytotoxicity assays
For the two isoquinolinones presented here, the correlation observed between the RORA inhibition cell viability assay results suggest that the inhibition measured in the RORA assay is attributed to the general toxicity of the isoquinolinones at higher concentrations rather than via specific RORA inhibition. The pharmacology of doxorubicin in the SF-1 and RORA assays serves as a useful example. Exhibiting behavior typical of a non-specific inhibitor, i.e. identical potency in both SF-1 and RORA activity assays, doxorubicin’s selectivity index (defined as the ratio RORA IC50/SF-1 IC50) was unity. In contrast, SID7969543 and SID7970631 gave much higher IC50 values in the RORA assay (>33,333 nM) compared to those measured in the SF-1 assay. The selectivity indexes of >44 and >131 for SID7969543 and SID7970631, respectively, suggest that the compounds are indeed selective to SF-1.
The SF-1 uHTS assay was a useful tool to discover the isoquinolinones presented here, and the SFRE/SF-1 assay, employing full length SF-1 protein and a luciferase reporter under control of the natural promoter (SFRE), confirmed their efficacy in a more physiologically relevant context. Their potency in both the SF-1 uHTS (performed in a CHO cell line background) and SFRE/SF-1 (performed in an HEK cell line background) assays support the hypothesis that abrogation of SF-1 activity by SID7969543 and SID7970631 is not cell-line specific. The increased potency in the SFRE assay is attributed to the difference in cell backgrounds: when HEK cells were transfected with the chimeric SF-1 construct, the potency of the isoquinolinones increased (data not shown). Lastly, the results of the LRH-1 selectivity and VP16 promiscuity assays manifest that they are selective inhibitors of SF-1 and also not responsible for general transactivation assay artefact.
Possible mechanisms of action of the isoquinolinone inhibitors
As mentioned above, SF-1 demonstrates constitutive transcriptional activity, and therefore alternative hypotheses can be formed on the isoquinolinones’ mechanism of action. Since crystallographic studies show lipids can occupy the canonical lipid-binding pocket of SF-1(Krylova et al., 2005; Li et al., 2005; Wang et al., 2005), one possible mechanism of action is that they behave as antagonists, competing with endogenous lipids located in this pocket. Another mechanism of action could be that they behave as inverse agonists, originally described by Klein and co-workers (Klein et al., 1996). Since there is no direct evidence that lipids occupy the binding site in living cells, it may be hypothesized that the isoquinolinones behave in this way, or they may bind somewhere outside of the ligand-binding pocket, perhaps in a non-competitive fashion. Although their exact mode of action remains to be elucidated in future studies, the advantage of the SF-1 assays described here is that they provide a rapid method of identifying ligands that affect SF-1 function and potentially facilitate the discovery of novel NR pathway targets.
The results of permeability & solubility studies show that the isoquinolinones presented here are practical tools to probe SF-1 activity in cell-based and biochemical experimentation. The short half-life observed in microsomal studies is attributed to the presence of three features on these compounds, namely (1) the ethyl ester, (2) the amide bound, and (3) the 1,4-dioxane or 1,3-dioxolane ring. The addition of NADPH accelerated the metabolism of SID7969543 and SID7970631 by hepatic microsomes (data not shown). However, the compounds were also rapidly metabolized in the absence of NADPH, presumably by hepatic esterases and/or amidases. There is no evidence to suggest that the compounds are metabolized during the course of the functional assay. However, it can be hypothesized that such metabolism would result in reducing the actual isoquinolinone concentration inside the cell and may lead to an underestimate of the efficacy of the inhibitors. Results of an ongoing medicinal chemistry effort aimed at improving isoquinolinone potency, selectivity and microsomal stability will be the subject of future reports.
Conclusion
Using a chemical biology approach, the goal of the presented research was to identify compounds that selectively inhibit SF-1 functional activity. To this end robust cell-based assays were implemented that employ a luciferase reporter gene transcriptional readout. The SF-1 uHTS assay was successfully executed against a publicly available 65,000 member compound library from the NIH’s Molecular Library Screening Center Network (MLSCN). Two isoquinolinone analogs, SID7969543 and SID7970631, demonstrated submicromolar efficacy and selectivity to SF-1 via a panel of nuclear receptor functional assays. Moreover, their inhibition was confirmed in a physiologically relevant assay employing a human cell-line with a full-length, non-chimerical SF-1 protein and a natural SF-1 response element. The isoquinolinones presented here are readily soluble and cell-permeable. They constitute excellent chemical probes for further elucidation of SF-1 pharmacology and represent a novel chemical scaffold for future studies on general NR modulation.
Acknowledgments
Pierre Baillargeon and Louis Scampavia (Lead Identification Dept., Scripps Florida) are acknowledged respectively for managing the compounds used for the various stages of this project and for the LC-MS analysis of compound powders. The authors would like to thank Dr. Holly Ingraham (Dept. of Physiology, School of Medicine, UCSF) for valuable insights concerning assay development for SF-1.
ABBREVIATIONS
- NR
nuclear receptor
- SF-1
steroidogenic factor 1
- SID7969543
ethyl 2-[2-[2-(2,3-dihydro-1,4-benzodioxin-7-ylamino)-2-oxoethyl]-1-oxoisoquinolin-5-yl]oxypropanoate
- SID7970631
ethyl 2-[2-[2-(1,3-benzodioxol-5-ylmethylamino)-2-oxoethyl]-1-oxoisoquinolin-5-yl]oxypropanoate
- SID4243980
3-[(4-methoxyphenoxy)methyl]benzoic acid
- SID4255902
3-[(4-ethoxyphenoxy)methyl]benzoic acid
- SID4261716
3-{[2-chloro-4-(hydroxymethyl)phenoxy]methyl}benzoic acid
- SID4263122
3-[(2-Chlorophenoxy)methyl]benzoic acid
- AC-45594
4 (heptyloxy)phenol
- aa
amino acid(s)
- LBD
ligand-binding domain
- DBD
DNA-binding domain
- RORA
RAR-related orphan receptor A
- CMV
cytomegalovirus
- VP16
Herpes simplex virus transcriptional activator protein Vmw65
- CHO
Chinese hamster ovary
- PBS
phosphate-buffered saline
- RH
relative humidity
- uHTS
ultra-high-throughput screening
- −NR
not cotransfected nuclear receptor
- +NR
with cotransfected nuclear receptor
- DMSO
dimethyl sulfoxide
- HTS
high-throughput screening
- HEK
human embryonic kidney
- SFRE
steroidogenic factor 1 response element
- MLSCN
Molecular Libraries Screening Center Network
- LRH-1
liver receptor homolog 1.
Footnotes
The SF-1 assay was initially developed with the NIH grant CA099875 (Dr. S. Thacher, Principal Investigator). The NIH grant MH077624-01 (Dr. Xiaolin Li, Principal Investigator) supported the transfer of the assay to the MLSCN. The efforts of M.C., P.C., P.G., and F.M. were supported by the National Institutes of Health (NIH) Molecular Library Screening Center Network (MLSCN) grant U54MH074404 (Hugh Rosen, Principal Investigator).
References
- 1.Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, Orth AP, Busby J, Hogenesch JB, Conkright MD. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(51):20314–20319. doi: 10.1073/pnas.0707999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306(5699):1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 3.Carlberg C, Hooft van Huijsduijnen R, Staple JK, DeLamarter JF, Becker-Andre M. RZRs, a new family of retinoid-related orphan receptors that function as both monomers and homodimers. Mol Endocrinol. 1994;8(6):757–770. doi: 10.1210/mend.8.6.7935491. [DOI] [PubMed] [Google Scholar]
- 4.Del Tredici AL, Andersen CB, Currier EA, Ohrmund SR, Fairbain LC, Lund BW, Olsson R, Piu F. Identification of the first synthetic Steroidogenic Factor 1 inverse agonists: Pharmacological modulation of steroidogenic enzymes. Mol Pharmacol. 2007 doi: 10.1124/mol.107.040089. [DOI] [PubMed] [Google Scholar]
- 5.Doghman M, Karpova T, Rodrigues GA, Arhatte M, De Moura J, Cavalli LR, Virolle V, Barbry P, Zambetti GP, Figueiredo BC, Heckert LL, Lalli E. Increased Steroidogenic Factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0120. [DOI] [PubMed] [Google Scholar]
- 6.Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14(5):250–260. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Giguere V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20(5):689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 8.Hammer GD, Krylova I, Zhang Y, Darimont BD, Simpson K, Weigel NL, Ingraham HA. Phosphorylation of the nuclear receptor SF-1 modulates cofactor recruitment: integration of hormone signaling in reproduction and stress. Mol Cell. 1999;3(4):521–526. doi: 10.1016/s1097-2765(00)80480-3. [DOI] [PubMed] [Google Scholar]
- 9.Hodder P, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, Culberson C, Bleicher L, Cosford ND, Bayly C, Suto C, Varney M, Strulovici B. Identification of metabotropic glutamate receptor antagonists using an automated high-throughput screening system. Anal Biochem. 2003;313(2):246–254. doi: 10.1016/s0003-2697(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol Endocrinol. 1995;9(4):478–486. doi: 10.1210/mend.9.4.7659091. [DOI] [PubMed] [Google Scholar]
- 11.Jausons-Loffreda N, Balaguer P, Roux S, Fuentes M, Pons M, Nicolas JC, Gelmini S, Pazzagli M. Chimeric receptors as a tool for luminescent measurement of biological activities of steroid hormones. J Biolumin Chemilumin. 1994;9(3):217–221. doi: 10.1002/bio.1170090316. [DOI] [PubMed] [Google Scholar]
- 12.Kallen J, Schlaeppi JM, Bitsch F, Delhon I, Fournier B. Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. The Journal of biological chemistry. 2004;279(14):14033–14038. doi: 10.1074/jbc.M400302200. [DOI] [PubMed] [Google Scholar]
- 13.Klein ES, Pino ME, Johnson AT, Davies PJ, Nagpal S, Thacher SM, Krasinski G, Chandraratna RA. Identification and functional separation of retinoic acid receptor neutral antagonists and inverse agonists. The Journal of biological chemistry. 1996;271(37):22692–22696. doi: 10.1074/jbc.271.37.22692. [DOI] [PubMed] [Google Scholar]
- 14.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120(3):343–355. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Lazo JS, Brady LS, Dingledine R. Building a pharmacological lexicon: small molecule discovery in academia. Mol Pharmacol. 2007;72(1):1–7. doi: 10.1124/mol.107.035113. [DOI] [PubMed] [Google Scholar]
- 16.Lee YK, Moore DD. Dual mechanisms for repression of the monomeric orphan receptor liver receptor homologous protein-1 by the orphan small heterodimer partner. The Journal of biological chemistry. 2002;277(4):2463–2467. doi: 10.1074/jbc.M105161200. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Molecular cell. 2005;17(4):491–502. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Lambert MH, Xu HE. Activation of nuclear receptors: a perspective from structural genomics. Structure. 2003;11(7):741–746. doi: 10.1016/s0969-2126(03)00133-3. [DOI] [PubMed] [Google Scholar]
- 19.Luo X, Ikeda Y, Lala DS, Baity LA, Meade JC, Parker KL. A cell-specific nuclear receptor plays essential roles in adrenal and gonadal development. Endocr Res. 1995a;21(1–2):517–524. doi: 10.3109/07435809509030469. [DOI] [PubMed] [Google Scholar]
- 20.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 21.Luo X, Ikeda Y, Schlosser DA, Parker KL. Steroidogenic factor 1 is the essential transcript of the mouse Ftz-F1 gene. Mol Endocrinol. 1995b;9(9):1233–1239. doi: 10.1210/mend.9.9.7491115. [DOI] [PubMed] [Google Scholar]
- 22.Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143(2):607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- 23.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mellon SH, Bair SR. 25-Hydroxycholesterol is not a ligand for the orphan nuclear receptor steroidogenic factor-1 (SF-1) Endocrinology. 1998;139(6):3026–3029. doi: 10.1210/endo.139.6.6129. [DOI] [PubMed] [Google Scholar]
- 25.Moore JT, Collins JL, Pearce KH. The nuclear receptor superfamily and drug discovery. ChemMedChem. 2006;1(5):504–523. doi: 10.1002/cmdc.200600006. [DOI] [PubMed] [Google Scholar]
- 26.Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. 2002;57:19–36. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]
- 27.Sadovsky Y, Crawford PA, Woodson KG, Polish JA, Clements MA, Tourtellotte LM, Simburger K, Milbrandt J. Mice deficient in the orphan receptor steroidogenic factor 1 lack adrenal glands and gonads but express P450 side-chain-cleavage enzyme in the placenta and have normal embryonic serum levels of corticosteroids. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(24):10939–10943. doi: 10.1073/pnas.92.24.10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335(6190):563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 29.Shinoda K, Lei H, Yoshii H, Nomura M, Nagano M, Shiba H, Sasaki H, Osawa Y, Ninomiya Y, Niwa O, et al. Developmental defects of the ventromedial hypothalamic nucleus and pituitary gonadotroph in the Ftz-F1 disrupted mice. Dev Dyn. 1995;204(1):22–29. doi: 10.1002/aja.1002040104. [DOI] [PubMed] [Google Scholar]
- 30.Steinmetz AC, Renaud JP, Moras D. Binding of ligands and activation of transcription by nuclear receptors. Annu Rev Biophys Biomol Struct. 2001;30:329–359. doi: 10.1146/annurev.biophys.30.1.329. [DOI] [PubMed] [Google Scholar]
- 31.Val P, Lefrancois-Martinez AM, Veyssiere G, Martinez A. SF-1 a key player in the development and differentiation of steroidogenic tissues. Nucl Recept. 2003;1(1):8. doi: 10.1186/1478-1336-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, Shelloe R, Mehra U, Eng K, Nguyen H, Settachatgul C, Powell B, Milburn MV, West BL. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(21):7505–7510. doi: 10.1073/pnas.0409482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, Willson TM. Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. J Med Chem. 2006;49(23):6652–6655. doi: 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]
- 34.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]