

Abstract
Structure-function relationships in the respiratory system are often a result of the emergence self-organized patterns or behaviors that are characteristic of certain respiratory diseases. Proper description of such self-organized behavior requires network models that include nonlinear interactions among different parts of the system. This review focuses on 2 models that exhibit self-organized behavior: a network model of the lung parenchyma during the progression of emphysema that is driven by mechanical force-induced breakdown, and an integrative model of bronchoconstriction in asthma that describes interactions among airways within the bronchial tree. Both models suggest that the transition from normal to pathologic states is a nonlinear process that includes a tipping point beyond which interactions among the system components are reinforced by positive feedback, further promoting the progression of pathologic changes. In emphysema, the progressive destruction of tissue is irreversible, while in asthma, it is possible to recover from a severe bronchoconstriction. These concepts may have implications for pulmonary medicine. Specifically, we suggest that structure–function relationships emerging from network behavior across multiple scales should be taken into account when the efficacy of novel treatments or drug therapy is evaluated. Multiscale, computational, network models will play a major role in this endeavor.
Keywords: self-organization, emergence, complex behavior, tipping point, network model
I. INTRODUCTION
The most complex phenomenon known to us is life. Indeed, the evolution of life and the evolution of biological complexity are stunning, and many attempts have been aimed at understanding the origin of life and biological complexity both at the experimental and theoretical levels. Despite dramatic advances in biological and medical research, many phenomena cannot simply be explained using the traditional reductionist approach, which seeks linear correlations between parameters. The reason for this limitation is that those phenomena are emergent, meaning that the interactions among molecules, different metabolic or signaling pathways, cells, different regions of an organ, or, in general, various components of a system can lead to unexpected novel and coherent structures or behaviors that cannot be predicted by considering the components in isolation.
It has been argued that emergence is a result of self-organized behavior in complex nonlinear systems under the influence of nonequilibrium conditions.1 Nonequilibrium conditions force the nonlinear system to undergo transitions or bifurcations and, as a result, the emergent structure or behavior that develops depends on these nonequilibrium conditions as well as the history of the transitions. Reconstructing and understanding such structures or behaviors is not possible using simple extrapolation of the fundamental properties of the components of the system; new approaches are required that are as fundamental as the properties themselves.2 The science of complex systems offers time series analyses, computational tools, and mathematical theories that can be applied to emergent properties.
This review discusses two important areas of emergent behavior related to lung pathophysiology that have not yielded resolution via the traditional reductionist approach. The first is the progressive nature of emphysema in terms of tissue destruction and the second is the self-organization of airway constriction related to asthma. In both cases, the underlying microscopic structure undergoes spatially correlated changes in such a manner that new functional behavior emerges at the macroscopic scale that significantly influences health. These structure–function relations are emergent and need to be understood from the point of view of the overall system, which currently can only be done through computational modeling. As we show in this short review, computational bioengineering and physics can contribute to significant new insights with important clinical implications to these diseases.
II. PULMONARY EMPHYSEMA
A. characterization of Emphysema
Pulmonary emphysema is defined by pathologic criteria as the destruction of lung parenchyma distal to the terminal bronchioles without associated inflammation or fibrosis.3,4 The physiologic manifestations of the disease include impaired gas exchange due to loss of alveolar surface area, airflow limitation, increased lung compliance, and increased work of breathing. The main risk factors for emphysema are exposure to cigarette smoke, environmental irritants, genetic factors, and indoor pollutants.5 The major mechanisms classically thought to be responsible for and/or involved in the development and progression of emphysema include the protease–antiprotease imbalance, inflammation, oxidative stress, apoptosis, and matrix remodeling.5 However, it is likely that these are not entirely distinct processes and that each contributes to the development of this complex disease.6 Compelling evidence points to inflammation that can trigger a cascade of responses that culminate in the tissue destruction that is characteristic of this disease.7
Most of the mechanisms that have been proposed to be involved in the pathogenesis and/or progression of emphysema come from animal research.8,9 The usual animal models include knock out7 or transgenic mice10 or mice that have been treated with some agent such as cigarette smoke11,12 or elastase.13,14 In most studies, the presence of emphysema is confirmed from histologic slides using measurement methods such as the mean linear intercept.15–24 When the mean linear intercept is found to increase compared to control, the molecule that was knocked out or occurred in excess is concluded to be an important contributor to emphysema.21,25–28 Interestingly, many studies have reported or introduced a new animal model of emphysema. Indeed, at present, there are at least 20 different known animal models of emphysema.7,10,12,25,27,29–36 Nearly every year a new and important molecule or receptor is identified, implying that there may be 20 or more different molecules that are essential for the development or progression of this disease. These studies at the cellular and molecular levels are important, and undoubtedly, many of these molecules and their associated mechanisms could play a role in the pathogenesis or progression of this disease. Yet one outstanding question remains: How does the lack or excess of a single molecule lead to airspace enlargement? Since the very essence of confirming the presence of emphysema is airspace enlargement, all of these mechanisms must generate tissue elimination. Therefore, we must ask whether these mechanisms converge somewhere along the path to airspace enlargement or whether each follows a different pathway.
If a common mechanism was involved in the last stage of airspace enlargement, then such a mechanism could not possibly be related to a single molecule. Instead, such a mechanism must act on all molecules involved; in other words, there must be some interaction among the molecules. Here we argue that this mechanism is simply a mechanical force whose role in the progressive nature of emphysema has been overlooked in the biological and medical research community. To put it simply, the lung is a mechanical device that has evolved to serve the gas exchange needs of an organism. For proper gas exchange with sufficient reserve capacity, a large surface area with a small diffusion distance is necessary.37 The 3-dimensional (3D) structure of the thin-walled alveolar septa forms a fractal with a huge surface area38 that is unstable at low inflating pressures. To prevent lung collapse, the alveolar wall network at the end of expiration is under a pre-existing mechanical stress, also called prestress, generated by the pleural pressure around the lung. In addition, breathing superimposes cyclic forces with intermittent deep inspirations. These forces are in fact capable of physically rupturing an entire alveolar wall after the sufficient enzymatic damage and repair that occur in emphysema.39
It has been suggested that mechanical factors should contribute to disease progression40 as measured by a decline in forced expiratory volume in 1 s (FEV1)41 or by changes in the properties of low attenuation areas (LAA) in computed tomography (CT) images.42 In fact, mechanical force-based tissue destruction serves as an organizing principle that can help explain how the various, apparently different, molecular mechanisms ultimately generate progressive airspace enlargement. Furthermore, since the lung parenchyma is a network of delicate septal walls, its macroscopic elasticity (characterized by lung compliance) is an emergent property arising from the combined effects of septal wall elasticity, surface tension, and, importantly, the network topology of the parenchyma. Thus, mechanical force-based elimination of septal walls weakens the network and reduces its bulk modulus, and hence, this is also the driving force behind any structure–function relation associated with disease progression.
B. the Progressive nature of Emphysema
Based on the previous discussion, we can formulate the following overall hypothesis about the progressive nature of emphysema: Independent of the initial pathogenesis, the irreversible and relentless progression of emphysema is associated with the physical failure of the alveolar septal walls due to the effects of mechanical forces during breathing on the enzyme-injured lung.
This hypothesis contains several important elements. First, it states that what drives progression is independent of the nature of pathogenesis. Thus, if this is true, the ultimate nature of progression is due to mechanical force-induced alveolar septal-wall rupture, independent of how the disease was triggered which would then be a main factor in all of the aforementioned animal models of emphysema. This, however, does not imply that mechanical forces cannot be involved in the pathogenesis. For example, it has been recently shown that mechanical forces on elastin fibers in the alveolar wall accelerate the cleavage of elastin through an increase in the unbinding rate of elastase and at the same time unfold cryptic binding sites along the fibers.43 Furthermore, we can speculate that changes in the mechanical properties of the alveolar walls due to smoke-related elastase injury also induce alterations in cellular mechanotransduction, which in turn can trigger further secretion of enzymes. This has not been proven, but the possibility cannot be excluded until it is actually confirmed experimentally.
The second factor is that the progression is irreversible. Clinicians know well that emphysema ultimately leads to death. The postulated role of mechanical forces is fully consistent with irreversibility. Once an alveolar wall ruptures due to mechanical forces, the separation of the wall tissue into fragments is fully irreversible. If cells could be guided to rebuild such walls or if some tissue-engineering method existed to replace the failed wall, then reversibility might occur. Unfortunately, both biology and tissue engineering are far from achieving this. Even if cells could be made to rebuild alveolar walls, there is no guarantee that the regeneration would be as functional as the original tissue, since the cells would have to do their regenerative work under different conditions (e.g., altered parenchymal architecture, biochemical milieu, and prestress on the tissue).
The third element of this hypothesis states that progression is relentless. This is something that is not easily understood in terms of the biology or biochemistry of the disease. It has been proposed that the almost unnoticed residual, or perhaps even viral inflammation, keeps the “cigarette” burning44 even after smoking has ceased. However, the same question arises: How does inflammation eliminate alveolar septal walls? Mechanical force-based destruction offers again a simple and attractive scenario. As mentioned, previous experiments have shown that, when an alveolar wall is enzymatically weakened, mechanical forces can break the alveolar wall.39 Since the parenchyma is under tensile prestress, when an alveolar wall actually ruptures, the stress the wall carried just before failure is redistributed among the neighboring walls. Consequently, some neighboring areas experience an increased prestress, which unfolds new binding sites and increases the unbinding and cleaving rate.43 These mechanisms in turn result in a higher probability of the mechanical failure of the wall. Thus, a single rupture can lead to a cascade of ruptures near the first rupture and can serve as a positive feedback for further breakdown.45 Notice that a cascade of mechanical failures generates spatial interactions among more distant regions of the elastic network of the parenchyma, which in turn leads to emergent structure and behavior (as discussed below). Furthermore, such a cascade is exactly what persistent or relentless progression means, and this represents a tipping point beyond which the structure and the corresponding function cannot return to the normal condition that existed before the first failure. Of course, when a wall ruptures, the neighboring wall does not necessarily rupture immediately. A new mechanical equilibrium is formed, and the neighboring walls first experience more prestress. The new equilibrium may remain stable for a longer time period. However, sudden exacerbations interrupt these quiet periods. While the exacerbation might be triggered by viral or bacterial infections, the many cycles of breathing generate fatigue and/or the forceful coughing during exacerbation eventually lead to failure of septal walls, which in turn results in a sharp decline in function. Thus, if local fatigue and/or mechanotransduction play a role, the process can be slow yet irreversible and relentless as well as independent of the initial cause of enzyme-induced weakening of the tissue.
What is the evidence for the mechanical force-induced progression? The possibility that mechanical forces contribute to the progression of emphysema was put forth as early as 1971 by West,46 who showed that the topographical distribution of emphysema scores closely resemble the regional distribution of mechanical stresses in the lung due to deformation under its own weight. This insight emerged from extensive computational work. West then argued that mechanical forces contribute to tissue pathophysiology. This idea was revisited 30 years by Kononov et al., who used imaging of the alveolar walls coupled with simultaneous measurement of the mechanical properties of tissue strips during uniaxial stretching.39 Importantly, it was found that the alveolar walls from elastase-treated rats can break under the influence of mechanical forces akin to those likely to occur in vivo. Follow-up studies comparing lung function in smokers with upper-zone emphysema with that in a1-antitrypsin deficient patients with lower-zone emphysema clearly showed a faster rate of functional decline in those with upper-zone emphysema.47 More recent experimental evidence supporting this hypothesis has emerged from clinical observations among patients with advanced emphysema undergoing lung volume reduction surgery (LVRS). LVRS involves surgical resection and resizing of the hyperexpanded lung to the chest wall.48 This therapy produced an immediate 30–50% increase in recoil pressure.49 Since the remaining lung of these patients is stretched to fill the thoracic cavity, mechanical forces on the connective tissue must also increase. Many patients who undergo LVRS experience a deterioration in lung function over time that is accelerated compared to their rate of lung function decline prior to surgery49,50 and that is far greater than would be anticipated for nonsmokers. Also, a statistically significant correlation between the magnitude of short-term incremental improvement and the long-term rate of deterioration in FEV1 following LVRS has been reported.48 Thus, the increased mechanical forces on the fiber network that follow LVRS and dictate immediate physiological benefit may also be responsible for an accelerated rate of mechanical damage of the alveolar walls and subsequent deterioration of lung function. The notion that mechanical forces contribute to tissue destruction is also consistent with detailed analysis of CT images of the lung.42 Specifically, in this study a network model was introduced which was able to mimic the early progression of the disease as seen on CT images when airspace enlargement was based on mechanical force-induced rupture. The network model was further refined in several other studies that provided consistent agreement with lung mechanics in normal51 and emphysematous tissue52 as well as imaging of alveolar-wall stretching under a microscope.53
c. mechanical Forces and the Emergence of Structure–Function relations
As discussed in the previous section, the role of mechanical forces in the progression of emphysema is consistent with this hypothesis and can explain the irreversible and progressive nature of the disease. While this does not exclude other mechanisms, it can serve as an organizing principle since ultimately the mechanical failure of the alveolar wall is due to mechanical forces and not enzymatic dissolution. Perhaps most important is the fact that many reports present data both in patients and animal models of emphysema that are consistent with this hypothesis. In this section, we will describe how mechanical failure is responsible for both changes in structure and function using a simple elastic network model.
Let us consider a simple, small hexagonal network model of the lung parenchyma54 similar to that first proposed by Mead et al.55 The network is composed of identical perfectly elastic and linear springs (Fig. 1A). The borders of the network are fixed in space, whereas all the internal nodes are free to move when the equilibrium is perturbed. Additionally, all springs are prestressed which means that the initial length of the springs at which they do not generate recoil force is smaller than their current length. This condition is maintained via the fixed borders. Due to symmetry, each spring inside the network carries an identical force. Next, we mimic airspace enlargement by assuming that spring 1a becomes weak due to enzymatic digestion and that its threshold for failure decreases below the unit force it carries. Consequently, spring 1a ruptures, the force this spring carried just before rupture is redistributed among the neighbors, and a new equilibrium is established. This new configuration of the network is shown in Fig. 1B, where the color of each spring is proportional to the force it carries. Figure 1C compares the forces on springs 1, 2, and 3 before and after the failure of spring 1a. It can be seen that the force on spring 1, and due to symmetry on spring 1b, increases. The same happens with spring 2. However, the force on spring 3 decreases, since this spring is now in the horizontal direction forming a single spring with spring 2a. It is easy to understand how this new force distribution comes about. Before breaking, the total force in the horizontal direction is 3 units (Fig. 1C, gray bars) carried by 3 springs: 1, 1a, and 1b. After breaking, the force is carried only by 2 springs (1 and 1b); therefore, the force these 2 springs carry must increase. Notably, the force is not conserved here before and after breaking, since the boundary conditions are given as fixed nodes and not fixed force.
FIGURE 1.
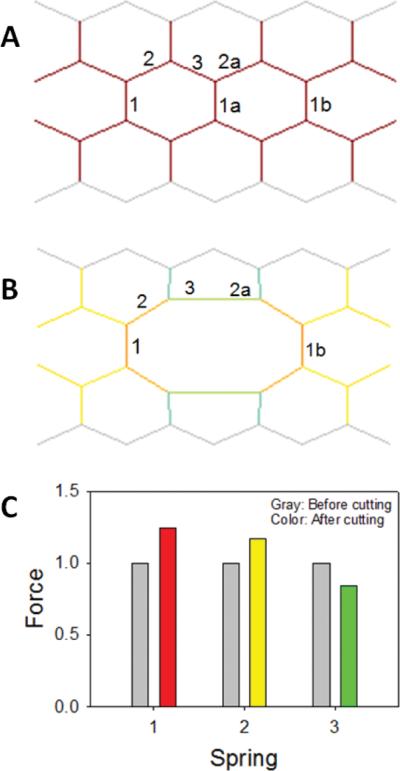
A small hexagonal network (A and B) and the distribution of normalized mechanical forces (C) on the network elements before (A) and after (B) mechanical failure. Springs are labeled with numbers. On (B) spring 1a is missing and the network was solved for its equilibrium configuration using simulated annealing. The color inside the network is proportional to force carried by the springs. See text for further explanation. Modified with permission from B. Suki.54
We can now see how the failure of a single element predisposes the neighboring elements to failure: elastic equilibrium following rupture requires that its neighbors carry a larger force than before rupture. At the same time, the failure of a single element also induces airspace enlargement. Furthermore, the elastic stiffness and consequently the recoil force of the network decreases since there are fewer springs in the network. This can be seen simply as follows. If we stretch the network by a small amount in the vertical direction, then the network before failure resists this stretch altogether by 11 springs. After failure there are only 10 springs remain to resist the stretch, and hence the network after failure is weaker with an elastic modulus that is expected to be about 10% smaller. For a more accurate analysis, the changes in angle between neighboring springs would have to be taken into account. When solved numerically using a uniform stretch in 2 dimensions (2D) (mimicking uniform volumetric stretch in 3D), the stiffness of the network indeed decreases following the failure of spring 1a.51 The inverse of this stiffness is the compliance which is a measure of the functional property of the network because it specifies the ability of the network to do work when stretched beyond its equilibrium configuration. The compliance of the network in Fig. 1B is larger by about 16% than that of the intact network in Fig. 1A. These findings may be summarized as follows. The rupture of a single spring in the network induces local airspace enlargement, which in turn also increases the global compliance of the network. Thus, mechanical failure simultaneously drives structural and functional changes. Hence, structure–function relations of the emphysematous lung emerge as a consequence of topological changes in the elastic network of septal walls due to mechanical forces acting on the enzymatically weakened parenchyma.
While mechanical failure alters the stress distribution in any network, in biological tissues it also sets up the network for further breakdown through various biochemical and biological mechanisms. One possibility is that increased stress leads to unfolding of binding sites and augments cleavage.43 Recently, it has been also shown that elastin fragments are chemotactic in that they recruit new inflammatory cells that can produce and secrete further enzymes driving the progression of the disease.56 Furthermore, the small molecular weight fragments of the proteoglycan hyaluronan have proinflammatory properties.57 These biochemical changes in the local extracellular matrix (ECM) region followed by rupture initiate a positive feedback loop which, in turn, generates slow avalanches of breakdown that eventually gives rise to progressive emphysema. This feedback loop likely starts in a small microscopic region as a seed. However, as successive ruptures open up larger and larger defect holes in a self-organized manner, the effects of rupture percolate to the macroscopic scale.58 The result can be seen in CT images as LAA. The distribution of LAA has been shown to follow a power law,42 which invariably signifies a complex system with emergent properties.59 Thus, mechanical force-induced network breakdown leads to an emergent structure that can be detected in clinical settings. Indeed, the analysis of the power law with regard to LAA distribution has been useful for the selection of patients for LVRS.60
With regard to early signs of the disease, it is instructive to further analyze the network. In addition to inducing airspace enlargement, we should also notice that the airspaces around the large hole contract (Fig. 1B). This is a result of the requirement for mechanical equilibrium. Thus, a homogeneous network becomes highly heterogeneous following a single rupture as the central airspace increases in size while the surrounding airspaces decrease in size. Therefore the first sign of the progression of emphysema is expected to be the appearance of heterogeneity on histological slides. This is exactly what was found in early emphysema in the pallid mouse,52 a model of α1-antitrypsin deficiency.61 Specifically, the standard deviation of alveolar size showed the largest difference between normal and the pallid mouse.52 The important finding here is that this increase in heterogeneity occurred at the age of 7 weeks. Previously, based on mean linear intercept measurements, the pallid mouse was thought to develop emphysema only in adulthood by about 10 months of age.61,62 However, analysis of the heterogeneity indicates that there must be early but subtle biochemical alterations in the tissue that allow a few ruptures to create topological heterogeneity. By looking at the mean linear intercept only, such subtle signals are easily missed.63 To maximize our ability to capture early signs of such heterogeneity, Parameswaran et al. recently introduced a new index, the area weighted mean alveolar diameter D2, and showed that it is significantly more sensitive to changes in heterogeneity than the mean linear intercept.63 In the case of the network model in Fig. 1, we should note that the mean area of the airspaces in Fig. 1B is the same as in Fig. 1A, since the boundaries of the network were fixed. Thus, the mean size does not change at all, and the only relevant signal is the heterogeneity of the network structure. This notion has also been corroborated in a 3D analysis of the alveolar structure in normal and elastase-induced emphysema in mice.64
Given this analysis, the next question is to what extent structural heterogeneity influences organ-level function. In a recent study, mean respiratory compliance Cmean was correlated to biochemical and structural parameters of the mouse lung before and after elastase-induced emphysema.65 Interestingly, Cmean did not correlate with bulk measures of soluble type I collagen, type III collagen, or elastin. There was, however, a strong association between Cmean and the mean equivalent diameter of airspaces (Deq), and an even stronger relation between Cmean and D2 with r2 values of 0.675 (p<0.01) and 0.933 (p<0.001), respectively (Fig. 2A). Since D2 includes higher-order moments of the distribution of equivalent airspace diameters, it is highly sensitive to heterogeneities. Thus, the correlations in Fig. 2A provide evidence that it is not the mean airspace size but its heterogeneity that primarily determines function in agreement with the network analysis. Further insight can be gained by examining how function emerges from the underlying structure, when heterogeneity is generated in various ways. To do this, a 2D rectangular network of linear elastic springs was used to predict the structure–function relations in Fig. 2A.65 A uniform negative pressure was applied at the boundaries of the network to mimic pleural pressure, which generated tensile forces on the springs. Emphysema progression was then simulated in 3 different ways at subsequent iterations: (1) springs were uniformly weakened, (2) springs were randomly eliminated, and (3) springs carrying high force were eliminated based on previous findings.39 The 2D compliance C of the network was calculated, and the iterations were repeated until C increased by 2, the same increase seen in Cmean in the experimental data (Fig. 2A). Following proper normalization, the model simulations can be compared to the data from the same study.65 It can be seen from Fig. 2B that each of these mechanisms produces an approximately linear relation between the normalized C and the normalized D2 of the network during simulated disease progression, but the slopes of these relations are very different. The data corresponding to the force-based destruction approximates best the experimental results suggesting that producing a few large defect holes has a much stronger effect on function than uniformly weakening the alveolar walls.
FIGURE 2.
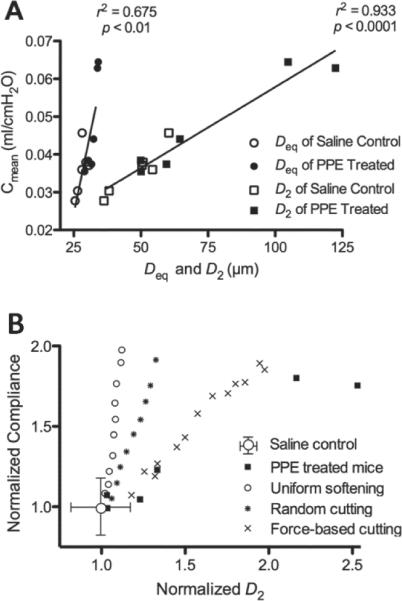
Structure–function relations obtained in saline control and porcine pancreatic elastase-treated mice. (A) Respiratory compliance, Cmean, as a function of the mean equivalent diameter (Deq) or the area weighted mean equivalent diameter (D2). (B) Comparison of structure-function relation from the network model and experiments. Experimental data were normalized with the mean value of Cmean and D2 of control mice. Each black square corresponds to data of a single mouse. The symbols corresponding to the various model cases represent model outputs normalized by the output of the base-line model. The data and simulations are from Hamakawa et al.65 Reprinted with permission of the American Thoracic Society. Copyright© American Thoracic Society.
D. Future Recommendations
Before concluding this section, we should point out possible areas of future research that could help unravel this debilitating disease. It is possible that extensive research will, in the end, identify a single molecule responsible for the pathogenesis of emphysema. However, it is not likely that a single molecule will be able to account for its progressive nature. First, recall that the parenchyma is a network and that its macroscopic compliance characterizing its functional behavior emerges from network effects during the progressive breakdown of the structure. The most important feature of emergence is that it is not the properties of the individual components, but their nonlinear interaction that leads to unexpected structure or behavior. Mechanical failure, a prerequisite for airspace enlargement, is a process that occurs at multiple-length scales.
At the lowest scale, the structure and the bio-mechanical properties of single collagen and elastin fibers are complex,66 and the physical process of how a fiber composed of thousands of interlinked fibrils and molecules fails under mechanical tension is not well understood. While enzymatic digestion weakens tissues and hence the fibers,43 it is not known how inflammation and remodeling alter the failure stress of single fibers. How does a septal wall consisting of a network of collagen and elastin fibers rupture? The failure of a simple elastin network embedded in and cross linked by proteoglycans already shows emergent network behavior.67 It is thus expected that the rupture of the alveolar wall as a composite material will exhibit even greater complexity.
Since collagen is the major load-bearing element at high strain, understanding the failure process both at the level of a fiber and the alveolar wall is important because inventing a way to protect the collagen from rupture may yield a treatment that slows down the progression of the disease. Aerosolized administration of hyaluronan creates a coating around elastin and this has been shown to reduce the severity of smoke-induced emphysema.24 Perhaps similarly, inducing a coating or cross-linking, or steering the cellular remodeling process appropriately, might lead to such protection of collagen. With regard to the network behavior of the parenchyma at a larger scale, a topological change induced by septal-wall rupture alters the elastic interactions among the neighboring network elements by increasing or decreasing the prestress.
It is not known how far such interactions reach in the network. Yet this is of crucial importance since it determines the size of the region in which a change in biological response is expected. The nature of this biological response is also not well characterized. For example, how do cells respond to increased stress in a biochemically altered ECM? While stretch alters binding-site availability and unbinding reaction rates in normal tissue,43 we do not know whether these phenomena depend on stretch in the biologically remodeled ECM. Are there any tissue-engineering possibilities? If so, how should the size and the physical and biological properties of the engineered tissue be optimized, and what should be the target location of tissue replacement? Some hypothetical solutions have been proposed based on network modeling.68 At an even larger scale, the ongoing failure process alters the properties of CT images,42 but it is not well understood how the pattern of destruction in the 3D geometry of the parenchyma influences function. Insight from 3D network modeling would be useful because linking regional compliance to destruction pattern might provide a way to statistically predict regions of deterioration or suitability for LVRS. Perhaps combining such modeling with clinical CT imaging could also be used to predict the risk of exacerbation with impact on treatment strategy. In summary, we believe that the tools of complexity combined with computational modeling and molecular and system level analyses can significantly contribute to resolving many of the outstanding issues which in turn may lead to the amelioration or perhaps the treatment of emphysema in the future.
III. BRONCHOCONSTRICTION IN ASTHMA
A. Characterization of Bronchoconstriction
The most severe manifestations of airway diseases are airway narrowing and closure. In asthma, which has a high prevalence and affects millions of people worldwide,69,70 acute bronchoconstriction leads to a rapid and sometimes life-threatening decrease in airway lumen and subsequently an increase in airway resistance and the work of breathing. Additionally, bronchoconstriction impairs gas exchange and leads to gas trapping and hyperinflation.71
Bronchoconstriction in asthma has been studied extensively and compared with the behavior of normal airways, including investigations of the behavior of the whole lung, tissue strips, cells, and molecular processes. These studies have lead to major advances in our understanding of both isolated mechanisms and global lung behavior. The primary, although mostly implicit, paradigm of these studies was that whole-organ behavior is determined by the sum of behaviors of the isolated components, which may be acceptable for certain specific questions. However, during the last decade it has become clear that there are nontrivial behaviors of the airway tree that emerge from interdependencies among the individual components that have previously only been studied in isolation.72
An increasing number of imaging studies suggest that the average behavior of the individual components of the lung does not predict the lung's overall behavior during bronchoconstriction. Examples of this disconnect come from magnetic resonance (MR) images of asymptomatic asthmatics,73 after methacholine- and exercise-induced challenges,74 and from dynamic positron emission tomography (PET) scans during bronchoconstriction in sheep75 and in humans,72,76,77 which have demonstrated the presence of large regions with very low ventilation. The large size and shape of these ventilation defects (VDefs) led initially to the speculation that VDefs may be caused by severe obstruction of large airways.74 However, experimental evidence78 and theoretical models79 suggest that VDefs are primarily caused by heterogeneous constriction of very small airways (<< 2 mm diameter) that cannot be directly measured by CT imaging. In addition to the heterogeneity in ventilation, there is evidence from imaging studies showing that VDefs affect the regional distribution of pulmonary perfusion.76
B. Mechanisms of Bronchoconstriction
Ventilation heterogeneity, including patchy patterns of VDefs, cannot be explained by linear correlations between 2 or more parameters of individual components of the lungs nor by the average behavior of independent airways, even if a random component is included (e.g., Venegas et al.72 and Anafi et al.80). Also, how is it possible that the most severe constriction of very small airways is not randomly scattered but clustered in VDefs?
To answer these questions, we have to consider that each airway is part of the airway tree and embedded in the lung parenchyma. The diameter of an airway embedded in the lung parenchyma can be determined by the balance of forces from parenchyma recoil, tissue deformation, and transmural pressures81 so that it represents a local static behavior.82 Static behavior refers here to a well-defined class of models in systems theory that have no memory or state parameters. Anafi and Wilson added two important mechanisms to this local static model: (1) the effects of the airway diameter on the airflow through the airway and, thus, on the local tidal expansion of the parenchyma and the embedded airway, and (2) the physiological effect of tidal expansions on the tension of ASM.83 Adding dynamics as a local mechanism showed that interactions among the different components, that affect the airway diameter during bronchoconstriction result, under pressure-controlled conditions, in instability below a critical diameter and, subsequently, in airway closure. Additionally, the reopening of closed airways requires pressures that are substantially higher than the critical pressure for airway closure.
An integrative model of bronchoconstriction was the first computational model that combined Anafi et al.'s local dynamic model of a single airway with a network structure of the airway tree.72 In this integrative model of airway behavior, the constriction of each individual airway is determined by its interactions with other airways and the regional lung inflation rather than by an average behavior as in lumped parameter models of bronchoconstriction. The model includes the dynamic changes of pressures, airflows, and local tidal expansions of a symmetric airway tree with 12 generations. The local dynamic behavior of each airway and its constriction is influenced by the interactions of pressure differences between inside and outside the airway wall, parenchymal tethering forces, and airway smooth-muscle forces modulated by tidal breathing.
C. Emergence of Self-Organized Heterogeneity in Bronchoconstriction
At low partial activation of airway smooth muscle, airway constriction is very homogeneous in the integrative model, with virtually no asymmetry. However, as activation gradually increases and crosses a threshold, or tipping point, a highly heterogeneous airway behavior emerges that leads to a patchy distribution of ventilation including VDefs.72 The characteristics of the emergent behavior in the model are consistent with VDefs in PET imaging studies72,76,77 and are similar to emergent behaviors in other natural phenomena and computational models.84,85 However, patchiness in ventilation during bronchoconstriction is one of the very few currently known examples, where self-organized patterns emerge in a system with a hierarchical structure, which is in this case the anatomical structure of the airway tree.
The emergent behavior in a virtually symmetric airway tree with a small random perturbation in airway wall thickness of 1% coefficient of variation and with uniform smooth muscle activation is remarkable. It suggests that the intrinsic heterogeneity in the pulmonary structure and its functional parameters is not necessary to explain the heterogeneous airway behavior during bronchoconstriction, although it may contribute to it. In other words, the interdependence among airways and other components may be the dominating mechanism for the severe heterogeneity observed in asthma.
It is also remarkable that interactions among the airways suddenly change at the tipping point, which triggers the emergence of self-organized heterogeneity and the clustering of constriction in peripheral airways that results in VDefs. Changes in airway diameter are also much faster after the transition at the tipping point.
The essential mechanism that leads to the emergence of self-organized heterogeneity in bronchoconstriction and VDefs involves a positive feedback loop, also called a vicious cycle, including airflow distribution, airway-wall mechanics, and airway smooth-muscle behavior (Fig. 3). For a simple explanation it is sufficient to consider a single airway bifurcation. Two identical daughter branches would receive the same airflow and have the same constriction. The homogeneous airflow distribution may even exist beyond the tipping point if the unstable symmetry is not perturbed. Assuming that each daughter airway is surrounded by lung tissue that the airway supplies with air, any small perturbation that slightly lowers airflow in one branch during inspiration also reduces the regional tidal expansion of the tissue surrounding that airway. The lower tidal stretch of the airway wall and, thus, of the ASM increases ASM constriction with a subsequent reduction in airway lumen, which in turn increases airway resistance. The increase in resistance in one daughter airway further decreases the airflow so that the positive feedback eventually results in severe constriction of this daughter airway. This severe constriction results in airflow redistribution to the other airway, if the global tidal volume is maintained, so that it increases the local tidal stretch and leads to dilation of this branch. This is reminiscent of the redistribution of stress in the parenchymal network following mechanical failure (Fig. 1). The interaction between the constricting and dilating daughter airways is a manifestation of parallel airway interdependence (Fig. 4A). In fact, the dilation of 1 daughter branch itself increases the asymmetry in airway resistance so that it further reduces the airflow in the other branch. Within the airway tree, the airflow and pressure distributions eventually determine the proximity of interactions among the airways and the pattern of constriction and dilation.86 A similar combination of airway constriction and dilation was found in a CT imaging study in response to stimulation of ASM by cold air or methacholine.87
FIGURE 3.
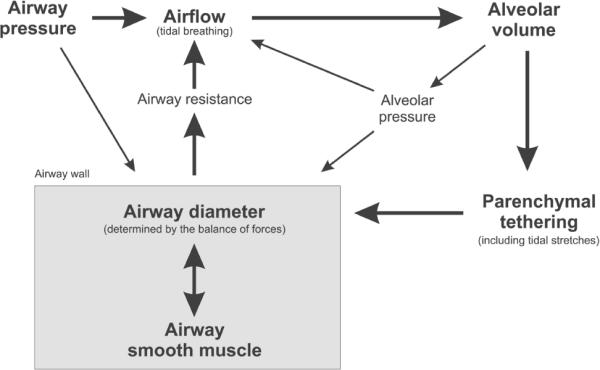
Schematic of local dynamic interactions that affect the airway diameter and the positive feedback loop that can result in airway closure during bronchoconstriction. The main components of the positive feedback (bold) dominate the airway behavior but minor components can, nevertheless, have a substantial influence on sensitive domains of the complex system.
FIGURE 4.
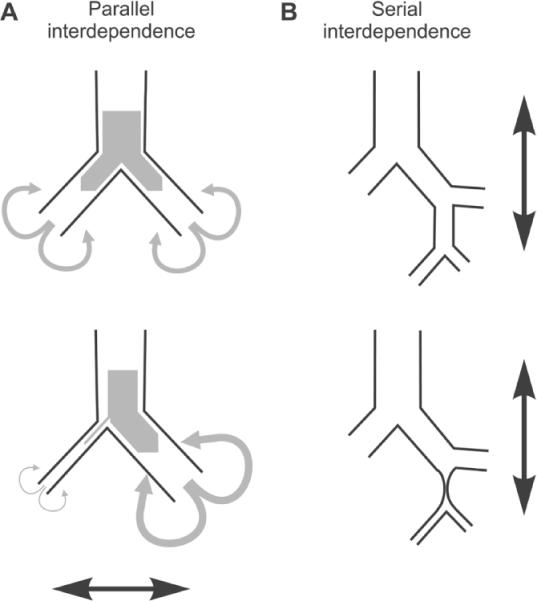
Typical Interdependencies among airways during bronchoconstriction. (A) Parallel interdependence results in asymmetric constriction of daughter airways at bifurcation of the bronchial tree including both constriction and dilation relative to the homogeneous constriction before the heterogeneity emerges. (B) Serial interdependence leads to more severe constriction in distal and, thus, smaller airways. Airways that are distal to a severe constriction or occlusion are not exposed to tidal stretches by airflow and have lower peak airway pressures so that the ASM constriction increases until it reaches the static equilibrium.
Does parallel interdependence also explain why the most severe bronchoconstriction occurs in the very small airways? Since parallel interdependence among airways is not limited to the small airways, it cannot, of course, explain the lack of severe constriction in large airways. Anatomical reasons have also been ruled out by an animal study that showed that the local stimulation of a single large airway at generation 3–5 can cause complete closure although the global stimulus after inhalation of an aerosol using 10 times the concentration of the local stimulation did not cause any closure of a large airway.88 Hence, unless the different responses were caused by differences in the concentration after aerosol deposition compared to the local stimulus, this finding shows that the constriction of an airway is not only a function of its ASM stimulation. In fact, the integrative model of bronchoconstriction shows the same behavior as in the animal study: local stimulation of a large airway leads to its complete closure, while uniform maximum stimulation does not.86
Why does global stimulation not cause any large airway closure although local does? Also, why are larger airways after uniform global stimulation on average less constricted than smaller airways?86 During inspiration, there is a pressure gradient within the airway tree from the large central airway to the smaller peripheral airways and terminal units. This gradient in intraluminal pressure affects local transmural pressures at the airway walls, local tidal stretches of ASM and, subsequently, the constriction of airways. The inspiratory pressure gradient increases during bronchoconstriction as airway lumen decreases and results in a serial interdependence among airways (Fig. 4B) in addition to the parallel interdependence. The gradient of transmural pressures throughout the bronchial tree can explain the higher level of constriction in the small peripheral airways compared to the larger central airways.
It is very important to understand that the airway resistance is distributed throughout the network of airways and that the heterogeneity in constriction causes heterogeneity in airway resistance that affects not only the airflows but also the distribution of pressures within the network. The distribution of pressure is an additional parameter that can affect airway diameter directly and indirectly by its contribution to the tidal stretches that affect ASM. The effects of the closure of a single large airway on its subtended distal airways constitute an excellent example of serial interdependence. The obstruction of the airflow and the dramatic reduction of peak pressures distal to the closure result in a shift of airway diameter to the static equilibrium of the forces at the airway wall, which is substantially smaller than that of a dynamic equilibrium in ventilated airways.86 This effect of tidal stretches on bronchoconstriction was also demonstrated in whole-organ studies89 and in tissue strips,90 although additional factors may be involved in the constriction of isolated airways.91
In complex systems it is possible that very small external stimuli cause dramatic changes in the system. Airway behavior during bronchoconstriction exhibits such characteristic behavior through its high sensitivity to certain changes in conditions that affect the parallel and serial interdependencies among airways. For example, pressure-controlled ventilation or similar conditions result in a catastrophic drop of the tidal volume when the airflows and pressures are insufficient for the stability of the airways.92 The positive feedback between bronchoconstriction and changes in tidal volume during pressure-controlled ventilation leads to a very high sensitivity for the pressure settings when the ventilation mode is changed from volume-controlled to pressure-controlled. Pressures that are only a few percent lower than the critical inspiratory pressure, that prevents changes in constriction, result in a substantial increase in constriction, while slightly higher pressures lead to a reduction or complete elimination of VDefs.92 Interestingly, ventilation heterogeneity emerges even under pressure-controlled ventilation although it does not lead to the clustering of severe airway constriction in VDefs, and the heterogeneity in airway constriction seems to be higher in distal airways. This observation suggests that parallel interdependence does not require volume-controlled ventilation, although the large differences in airway constriction that are associated with VDefs did not occur during pressure-controlled ventilation.
The complex behavior of the airways can be highly sensitive to changes in parameters that affect the balance of forces at the airway wall. This means that the tipping point for the emergence of VDefs, their size, and the specific characteristics of the airway constriction are affected by those parameters. For example, changes in tidal volume cause dramatic changes in the size of VDefs and show a history dependence of airway behavior.72 In a study where tidal volume was first decreased in multiple steps and then increased again, the VDefs were present at a higher tidal volume after the maneuver than before the maneuver. Additionally, the location of the VDefs after the maneuver was different from where it had first appeared. Another example is the effect of end-expiratory lung volume on bronchoconstriction,86 which showed that increased lung volume might reduce the size of ventilation defects. The effect is most likely caused by increased tethering forces of lung parenchyma affecting the equilibrium of forces at the airway wall. The characteristic behavior of this effect, including the volume dependence of a tipping point, is consistent with experimental findings.86,93
The combination of local dynamic interactions at the airway wall and interdependencies among the airways in the integrative model allows exploration of the complex behavior of the lungs and testing of sophisticated hypotheses. However, the value of computational modeling depends ultimately on the consistency between model predictions and experimental results, and the model's ability to explain experimental findings that are difficult to understand without the model. The integrative model of bronchoconstriction can explain at least 6 characteristic behaviors of bronchoconstriction that were found in human or animal studies, which suggests that it is a unified model of these behaviors: (1) emergence of VDefs,72 (2) combination of constriction and dilation,86,87 (3) differences between local and global stimulation of ASM,86,88 (4) effects of lung volume on VDefs,86,93 (5) apparent time delay between peripheral and central airway dilation during recovery,86,94 and (6) increase in the size of VDefs but not in large airway constriction when the concentration of an inhaled stimulus is at a high level and is further increased.86,95 None of the listed behaviors were expected to be linked to the other behaviors, illustrating a remarkable strength of the model.
D. Future Recommendations
Complex systems, such as the lungs during bronchoconstriction, are sensitive to numerous factors that are not independent from each other. For example, multiscale modeling including smaller length scales than in the models we discussed in this review may be able to explain how changes in molecular and intracellular mechanisms affect the emergent behavior of bronchoconstriction.96 Interactions between heterogeneous airway constriction in an airway tree and environmental stimuli that are distributed by airflow can lead to spatial and temporal heterogeneity in bronchoconstriction.97 Beyond bronchoconstriction, there is also evidence from computational modeling that interdependence among airways during mechanical ventilation in ARDS can lead to emergent behaviors and history dependence.98
The sensitivity of bronchoconstriction to changes in different parameters and combinations of changes is completely unknown. To identify parameters that are both effective in reducing or preventing severe bronchoconstriction and suitable as therapeutic targets is a challenge. However, computational modeling provides the necessary tools for studying the complex behavior of lungs. In fact, interdependencies among airways cannot be studied directly in experiments since it is impossible to control or measure the numerous interactions within the complex system. Eventually, additional factors such as gravitational gradients in lung inflation need to be incorporated in models including both spatially distributed parameters, as realized by Politi et al.,96 and dynamic interdependencies among airways72 to study how additional factors affect the emergent behavior of airways.
IV. CONCLUSIONS
Studies of self-organized and complex behavior in respiratory physiology are difficult but essential for the understanding of normal function as well as complex disease processes. The examples of complex models reviewed here suggest that self-organized behavior can emerge in networks from interactions among its elements. In contrast, the behavior of completely independent elements with an imprinted heterogeneity does not lead to emergent behavior. Emergence may include irreversible changes in the structure of a network, as in the case of emphysema, but it is not required, as the example of bronchoconstriction demonstrates. The network structure also implies that at least 1 parameter must be spatially distributed among the elements to allow for interactions and self-organized patterns in contrast to lumped parameter models that focus on the average behavior of the whole system.
A better understanding of complex disease processes may have important implications for new therapeutic concepts. Efficient targets are those which prevent the system from reaching the tipping points beyond which the healthy state transitions to a diseased state which in turn can trigger mechanisms of positive feedback such as the stress redistribution-induced ruptures in emphysema or the serial and parallel interdependencies during bronchoconstriction. Potential targets include mechanisms, properties, signaling pathways, or single-molecule inhibitors that affect the interactions within the system. However, since the result of the influence of a single target on the system's functional behavior depends on other properties of the network, unexpected side effects can arise. Hence, we suggest that emergent multiscale structure–function relations should be taken into account when the efficacy of novel treatments or drug therapies is evaluated. Advances will crucially depend on the development of more sophisticated theoretical and computational network models, and on experimental studies that are linked to these models. Based on multiscale modeling similar to those considered here, it should eventually be possible to set up recommendations for mechanical ventilation of patients, delivery of inhaled therapies, breathing during asthma attacks, or the type and strength of exercise that would impact quality of life. The ability to shift tipping points could open new opportunities to stop the progression of diseases or to trigger the body's own defense mechanisms for recovery. Such integrative approaches may be more successful in diseases where traditional approaches have yielded only marginal advances.
ACKNOWLEDGMENT
This study was supported by National Institutes of Health grants HL090757 and HL087281.
REFERENCES
- 1.Nicolis G, Prigogine I. Exploring complexity: an introduction. W.H. Freeman; New York: 1989. [Google Scholar]
- 2.Anderson PW. More is different. Science. 1972;177(4047):393–6. doi: 10.1126/science.177.4047.393. [DOI] [PubMed] [Google Scholar]
- 3.Snider GL. Chronic obstructive pulmonary disease: a definition and implications of structural determinants of airflow obstruction for epidemiology. Am Rev Respir Dis. 1989;140(3 Pt 2):S3–8. doi: 10.1164/ajrccm/140.3_Pt_2.S3. [DOI] [PubMed] [Google Scholar]
- 4.Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, November 1986. Am Rev Respir Dis. 1987;136(1):225–44. doi: 10.1164/ajrccm/136.1.225. [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343(4):269–80. doi: 10.1056/NEJM200007273430407. [DOI] [PubMed] [Google Scholar]
- 6.Churg A, Wright JL. Proteases and emphysema. Curr Opin Pulm Med. 2005;11(2):153–9. doi: 10.1097/01.mcp.0000149592.51761.e3. [DOI] [PubMed] [Google Scholar]
- 7.Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med. 2002;166(6):849–54. doi: 10.1164/rccm.200202-097OC. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro SD. Animal models for chronic obstructive pulmonary disease: age of klotho and marlboro mice. Am J Respir Cell Mol Biol. 2000;22(1):4–7. doi: 10.1165/ajrcmb.22.1.f173. [DOI] [PubMed] [Google Scholar]
- 9.Snider GL, Lucey EC, Stone PJ. Animal models of emphysema. Am Rev Respir Dis. 1986;133(1):149–69. doi: 10.1164/arrd.1986.133.1.149. [DOI] [PubMed] [Google Scholar]
- 10.D'Armiento J, Dalal SS, Okada Y, Berg RA, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell. 1992;71(6):955–61. doi: 10.1016/0092-8674(92)90391-o. [DOI] [PubMed] [Google Scholar]
- 11.Wright JL, Churg A. Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. Am J Physiol. 1995;268(1 Pt 1):L17–20. doi: 10.1152/ajplung.1995.268.1.L17. [DOI] [PubMed] [Google Scholar]
- 12.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–4. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 13.Lucey EC, Goldstein RH, Stone PJ, Snider GL. Remodeling of alveolar walls after elastase treatment of hamsters. Results of elastin and collagen mRNA in situ hybridization. Am J Respir Crit Care Med. 1998;158(2):555–64. doi: 10.1164/ajrccm.158.2.9705021. [DOI] [PubMed] [Google Scholar]
- 14.Ito S, Ingenito EP, Arold SP, Parameswaran H, Tgavalekos NT, Lutchen KR, Suki B. Tissue heterogeneity in the mouse lung: effects of elastase treatment. J Appl Physiol. 2004;97(1):204–12. doi: 10.1152/japplphysiol.01246.2003. [DOI] [PubMed] [Google Scholar]
- 15.Vlahovic G, Russell ML, Mercer RR, Crapo JD. Cellular and connective tissue changes in alveolar septal walls in emphysema. Am J Respir Crit Care Med. 1999;160(6):2086–92. doi: 10.1164/ajrccm.160.6.9706031. [DOI] [PubMed] [Google Scholar]
- 16.Mercer RR, Crapo JD. Structural changes in elastic fibers after pancreatic elastase administration in hamsters. J Appl Physiol. 1992;72(4):1473–9. doi: 10.1152/jappl.1992.72.4.1473. [DOI] [PubMed] [Google Scholar]
- 17.Martorana PA, van Even P, Gardi C, Lungarella G. A 16-month study of the development of genetic emphysema in tight-skin mice. Am Rev Respir Dis. 1989;139(1):226–32. doi: 10.1164/ajrccm/139.1.226. [DOI] [PubMed] [Google Scholar]
- 18.Fisk DE, Kuhn C. Emphysema-like changes in the lungs of the blotchy mouse. Am Rev Respir Dis. 1976;113(6):787–97. doi: 10.1164/arrd.1976.113.6.787. [DOI] [PubMed] [Google Scholar]
- 19.Ranga V, Grahn D, Journey TM. Morphologic and phenotypic analysis of an outcross line of blotchy mouse. Exp Lung Res. 1983;4(4):269–79. doi: 10.3109/01902148309055014. [DOI] [PubMed] [Google Scholar]
- 20.Lucey EC, Keane J, Kuang PP, Snider GL, Goldstein RH. Severity of elastase-induced emphysema is decreased in tumor necrosis factor-alpha and interleukin-1beta receptor-deficient mice. Lab Invest. 2002;82(1):79–85. doi: 10.1038/labinvest.3780397. [DOI] [PubMed] [Google Scholar]
- 21.Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170(5):492–8. doi: 10.1164/rccm.200404-511OC. [DOI] [PubMed] [Google Scholar]
- 22.Foronjy RF, Okada Y, Cole R, D'Armiento J. Progressive adult-onset emphysema in transgenic mice expressing human MMP-1 in the lung. Am J Physiol Lung Cell Mol Physiol. 2003;284(5):L727–37. doi: 10.1152/ajplung.00349.2002. [DOI] [PubMed] [Google Scholar]
- 23.D'Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26(2):204–13. doi: 10.1183/09031936.05.00095204. [DOI] [PubMed] [Google Scholar]
- 24.Cantor JO, Cerreta JM, Ochoa M, Ma S, Chow T, Grunig G, Turino GM. Aerosolized hyaluronan limits airspace enlargement in a mouse model of cigarette smoke-induced pulmonary emphysema. Exp Lung Res. 2005;31(4):417–30. doi: 10.1080/01902140590918669. [DOI] [PubMed] [Google Scholar]
- 25.Foronjy RF, Mirochnitchenko O, Propokenko O, Lemaitre V, Jia Y, Inouye M, Okada Y, D'Armiento JM. Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am J Respir Crit Care Med. 2006;173(6):623–31. doi: 10.1164/rccm.200506-850OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takubo Y, Guerassimov A, Ghezzo H, Triantafillopoulos A, Bates JH, Hoidal JR, Cosio MG. Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med. 2002;166(12 Pt 1):1596–603. doi: 10.1164/rccm.2202001. [DOI] [PubMed] [Google Scholar]
- 27.Tang K, Rossiter HB, Wagner PD, Breen EC. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol. 2004;97(4):1559–66. doi: 10.1152/japplphysiol.00221.2004. discussion 49. [DOI] [PubMed] [Google Scholar]
- 28.van Kuppevelt TH, van de Lest CH, Versteeg EM, Dekhuijzen PN, Veerkamp JH. Induction of emphysematous lesions in rat lung by beta-D-xyloside, an inhibitor of proteoglycan synthesis. Am J Respir Cell Mol Biol. 1997;16(1):75–84. doi: 10.1165/ajrcmb.16.1.8998082. [DOI] [PubMed] [Google Scholar]
- 29.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106(11):1311–9. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodera T, McGaha TL, Phelps R, Paul WE, Bona CA. Disrupting the IL-4 gene rescues mice homozygous for the tight-skin mutation from embryonic death and diminishes TGF-beta production by fibroblasts. Proc Natl Acad Sci U S A. 2002;99(6):3800–5. doi: 10.1073/pnas.052709999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32(4):311–8. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 32.Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature. 2003;422(6928):169–73. doi: 10.1038/nature01413. [DOI] [PubMed] [Google Scholar]
- 33.Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–8. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes PJ. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–9. doi: 10.1165/ajrcmb.26.5.4685. [DOI] [PubMed] [Google Scholar]
- 35.Snider GL, Lucey EC, Faris B, Jung-Legg Y, Stone PJ, Franzblau C. Cadmium-chloride-induced air-space enlargement with interstitial pulmonary fibrosis is not associated with destruction of lung elastin. Implications for the pathogenesis of human emphysema. Am Rev Respir Dis. 1988;137(4):918–23. doi: 10.1164/ajrccm/137.4.918. [DOI] [PubMed] [Google Scholar]
- 36.Zheng T, Kang MJ, Crothers K, Zhu Z, Liu W, Lee CG, Rabach LA, Chapman HA, Homer RJ, Aldous D, Desanctis G, Underwood S, Graupe M, Flavell RA, Schmidt JA, Elias JA. Role of cathepsin S-dependent epithelial cell apoptosis in IFN-{gamma}-induced alveolar remodeling and pulmonary emphysema. J Immunol. 2005;174(12):8106–15. doi: 10.4049/jimmunol.174.12.8106. [DOI] [PubMed] [Google Scholar]
- 37.Weibel ER. What makes a good lung? Swiss Med Wkly. 2009;139(27–28):375–86. doi: 10.4414/smw.2009.12270. [DOI] [PubMed] [Google Scholar]
- 38.Sapoval B, Filoche M, Weibel ER. Smaller is better—but not too small: a physical scale for the design of the mammalian pulmonary acinus. Proc Natl Acad Sci U S A. 2002;99(16):10411–6. doi: 10.1073/pnas.122352499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kononov S, Brewer K, Sakai H, Cavalcante FS, Sabayanagam CR, Ingenito EP, Suki B. Roles of mechanical forces and collagen failure in the development of elastase-induced emphysema. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1920–6. doi: 10.1164/ajrccm.164.10.2101083. [DOI] [PubMed] [Google Scholar]
- 40.Stehbens WE. Proteinase imbalance versus biomechanical stress in pulmonary emphysema. Exp Mol Pathol. 2000;69(1):46–62. doi: 10.1006/exmp.2000.2307. [DOI] [PubMed] [Google Scholar]
- 41.Croxton TL, Weinmann GG, Senior RM, Wise RA, Crapo JD, Buist AS. Clinical research in chronic obstructive pulmonary disease: needs and opportunities. Am J Respir Crit Care Med. 2003;167(8):1142–9. doi: 10.1164/rccm.200207-756WS. [DOI] [PubMed] [Google Scholar]
- 42.Mishima M, Hirai T, Itoh H, Nakano Y, Sakai H, Muro S, Nishimura K, Oku Y, Chin K, Ohi M, Nakamura T, Bates JH, Alencar AM, Suki B. Complexity of terminal airspace geometry assessed by lung computed tomography in normal subjects and patients with chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 1999;96(16):8829–34. doi: 10.1073/pnas.96.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jesudason R, Sato S, Parameswaran H, Araujo AD, Majumdar A, Allen PG, Bartolak-Suki E, Suki B. Mechanical forces regulate elastase activity and binding site availability in lung elastin. Biophys J. 2010;99(9):3076–83. doi: 10.1016/j.bpj.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC, Rogers RM, Hayashi S, Hogg JC. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med. 2001;164(3):469–73. doi: 10.1164/ajrccm.164.3.2007149. [DOI] [PubMed] [Google Scholar]
- 45.Suki B, Lutchen KR, Ingenito EP. On the progressive nature of emphysema: roles of proteases, inflammation, and mechanical forces. Am J Respir Crit Care Med. 2003;168(5):516–21. doi: 10.1164/rccm.200208-908PP. [DOI] [PubMed] [Google Scholar]
- 46.West JB. Distribution of mechanical stress in the lung, a possible factor in localisation of pulmonary disease. Lancet. 1971;1(7704):839–41. doi: 10.1016/s0140-6736(71)91501-7. [DOI] [PubMed] [Google Scholar]
- 47.Hughes JA, Hutchison DC, Bellamy D, Dowd DE, Ryan KC, Hugh-Jones P. Annual decline of lung function in pulmonary emphysema: influence of radiological distribution. Thorax. 1982;37(1):32–7. doi: 10.1136/thx.37.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brenner M, McKenna RJ, Jr, Gelb AF, Fischel RJ, Wilson AF. Rate of FEV1 change following lung volume reduction surgery. Chest. 1998;113(3):652–9. doi: 10.1378/chest.113.3.652. [DOI] [PubMed] [Google Scholar]
- 49.Gelb AF, McKenna RJ, Jr, Brenner M, Schein MJ, Zamel N, Fischel R. Lung function 4 years after lung volume reduction surgery for emphysema. Chest. 1999;116(6):1608–15. doi: 10.1378/chest.116.6.1608. [DOI] [PubMed] [Google Scholar]
- 50.Gelb AF, McKenna RJ, Jr, Brenner M, Epstein JD, Zamel N. Lung function 5 yr after lung volume reduction surgery for emphysema. Am J Respir Crit Care Med. 2001;163(7):1562–6. doi: 10.1164/ajrccm.163.7.2009048. [DOI] [PubMed] [Google Scholar]
- 51.Cavalcante FS, Ito S, Brewer KK, Sakai H, Alencar AM, Almeida MP, Andrade JS, Jr, Majumdar A, Ingenito EP, Suki B. Mechanical interactions between collagen and proteoglycans: implications for the stability of lung tissue. J Appl Physiol. 2005;98:672–9. doi: 10.1152/japplphysiol.00619.2004. [DOI] [PubMed] [Google Scholar]
- 52.Ito S, Bartolak-Suki E, Shipley JM, Parameswaran H, Majumdar A, Suki B. Early emphysema in the tight skin and pallid mice: roles of microfibril-associated glycoproteins, collagen, and mechanical forces. Am J Respir Cell Mol Biol. 2006;34(6):688–94. doi: 10.1165/rcmb.2006-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brewer KK, Sakai H, Alencar AM, Majumdar A, Arold SP, Lutchen KR, Ingenito EP, Suki B. Lung and alveolar wall elastic and hysteretic behavior in rats: effects of in vivo elastase treatment. J Appl Physiol. 2003;95(5):1926–36. doi: 10.1152/japplphysiol.00102.2003. [DOI] [PubMed] [Google Scholar]
- 54.Suki B. Structure-function relations in the emphysematous lung. Kokyū. 2009;28(3):315–29. [Google Scholar]
- 55.Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol. 1970;28(5):596–608. doi: 10.1152/jappl.1970.28.5.596. [DOI] [PubMed] [Google Scholar]
- 56.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. The Journal of clinical investigation. 2006;116(3):753–9. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cantor JO, Nadkarni PP. Hyaluronan: the Jekyll and Hyde molecule. Inflamm Allergy Drug Targets. 2006;5(4):257–60. doi: 10.2174/187152806779010936. [DOI] [PubMed] [Google Scholar]
- 58.Bates JH, Davis GS, Majumdar A, Butnor KJ, Suki B. Linking parenchymal disease progression to changes in lung mechanical function by percolation. Am J Respir Crit Care Med. 2007;176(6):617–23. doi: 10.1164/rccm.200611-1739OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suki B. Fluctuations and power laws in pulmonary physiology. Am J Respir Crit Care Med. 2002;166(2):133–7. doi: 10.1164/rccm.200202-152pp. [DOI] [PubMed] [Google Scholar]
- 60.Coxson HO, Whittall KP, Nakano Y, Rogers RM, Sciurba FC, Keenan RJ, Hogg JC. Selection of patients for lung volume reduction surgery using a power law analysis of the computed tomographic scan. Thorax. 2003;58(6):510–4. doi: 10.1136/thorax.58.6.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martorana PA, Brand T, Gardi C, van Even P, de Santi MM, Calzoni P, Marcolongo P, Lungarella G. The pallid mouse. A model of genetic alpha 1-anti-trypsin deficiency. Lab Invest. 1993;68(2):233–41. [PubMed] [Google Scholar]
- 62.Keil M, Lungarella G, Cavarra E, van Even P, Martorana PA. A scanning electron microscopic investigation of genetic emphysema in tight-skin, pallid, and beige mice, three different C57 BL/6J mutants. Lab Invest. 1996;74(2):353–62. [PubMed] [Google Scholar]
- 63.Parameswaran H, Majumdar A, Ito S, Alencar AM, Suki B. Quantitative characterization of airspace enlargement in emphysema. J Appl Physiol. 2006;100(1):186–93. doi: 10.1152/japplphysiol.00424.2005. [DOI] [PubMed] [Google Scholar]
- 64.Parameswaran H, Bartolak-Suki E, Hamakawa H, Majumdar A, Allen PG, Suki B. Three-dimensional measurement of alveolar airspace volumes in normal and emphysematous lungs using micro-CT. J Appl Physiol. 2009;107(2):583–92. doi: 10.1152/japplphysiol.91227.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamakawa H, Bartolák-Suki E, Parameswaran H, Majumdar A, Lutchen K, Suki B. Structure-function relations in an elastase-induced mouse model of emphysema. Am J Respir Cell Mol Biol. 2011;45(3):517–24. doi: 10.1165/rcmb.2010-0473OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Suki B, Ito S, Stamenovic D, Lutchen KR, Ingenito EP. Biomechanics of the lung parenchyma: critical roles of collagen and mechanical forces. J Appl Physiol. 2005;98(5):1892–9. doi: 10.1152/japplphysiol.01087.2004. [DOI] [PubMed] [Google Scholar]
- 67.Ritter MC, Jesudason R, Majumdar A, Stamenovic D, Buczek-Thomas JA, Stone PJ, Nugent MA, Suki B. A zipper network model of the failure mechanics of extracellular matrices. Proc Natl Acad Sci U S A. 2009;106(4):1081–6. doi: 10.1073/pnas.0808414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suki B, Majumdar A, Nugent MA, Bates JH. In silico modeling of interstitial lung mechanics: implications for disease development and repair. Drug Discov Today Dis Models. 2007;4(3):139–45. doi: 10.1016/j.ddmod.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.National Asthma Education and Prevention Program . Expert panel report 2: guidelines for the diagnosis and management of asthma. National Institutes of Health; 1997. [Google Scholar]
- 70.Global strategy for asthma management and prevention (GINA) 2007. p. 7. NIH Publication No. 02-3659. Jan 1995 (updated 2007) [DOI] [PubMed] [Google Scholar]
- 71.Bosse Y, Riesenfeld EP, Pare PD, Irvin CG. It's not all smooth muscle: non-smooth-muscle elements in control of resistance to airflow. Annu Rev Physiol. 2010;72:437–62. doi: 10.1146/annurev-physiol-021909-135851. [DOI] [PubMed] [Google Scholar]
- 72.Venegas JG, Winkler T, Musch G, Vidal Melo MF, Layfield D, Tgavalekos N, Fischman AJ, Callahan RJ, Bellani G, Harris RS. Self-organized patchiness in asthma as a prelude to catastrophic shifts. Nature. 2005;434(7034):777–82. doi: 10.1038/nature03490. [DOI] [PubMed] [Google Scholar]
- 73.Altes TA, Powers PL, Knight-Scott J, Rakes G, Platts-Mills TA, de Lange EE, Alford BA, Mugler JP, 3rd, Brookeman JR. Hyperpolarized 3He MR lung ventilation imaging in asthmatics: preliminary findings. J Magn Reson Imaging. 2001;13(3):378–84. doi: 10.1002/jmri.1054. [DOI] [PubMed] [Google Scholar]
- 74.Samee S, Altes T, Powers P, de Lange EE, Knight-Scott J, Rakes G, Mugler JP, 3rd, Ciambotti JM, Alford BA, Brookeman JR, Platts-Mills TA. Imaging the lungs in asthmatic patients by using hyperpolarized helium-3 magnetic resonance: assessment of response to methacholine and exercise challenge. J Allergy Clin Immunol. 2003;111(6):1205–11. doi: 10.1067/mai.2003.1544. [DOI] [PubMed] [Google Scholar]
- 75.Vidal Melo MF, Harris RS, Layfield JD, Venegas JG. Topographic basis of bimodal ventilation-perfusion distributions during bronchoconstriction in sheep. Am J Respir Crit Care Med. 2005;171:714–21. doi: 10.1164/rccm.200409-1296OC. [DOI] [PubMed] [Google Scholar]
- 76.Harris RS, Winkler T, Tgavalekos N, Musch G, Melo MF, Schroeder T, Chang Y, Venegas JG. Regional pulmonary perfusion, inflation, and ventilation defects in bronchoconstricted patients with asthma. Am J Respir Crit Care Med. 2006;174(3):245–53. doi: 10.1164/rccm.200510-1634OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harris RS, Winkler T, Musch G, Vidal Melo MF, Schroeder T, Tgavalekos N, Venegas JG. The prone position results in smaller ventilation defects during bronchoconstriction in asthma. J Appl Physiol. 2009;107(1):266–74. doi: 10.1152/japplphysiol.91386.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beigelman-Aubry C, Capderou A, Grenier PA, Straus C, Becquemin MH, Similowski T, Zelter M. Mild intermittent asthma: CT assessment of bronchial cross-sectional area and lung attenuation at controlled lung volume. Radiology. 2002;223(1):181–7. doi: 10.1148/radiol.2231010779. [DOI] [PubMed] [Google Scholar]
- 79.Lutchen KR, Jensen A, Atileh H, Kaczka DW, Israel E, Suki B, Ingenito EP. Airway constriction pattern is a central component of asthma severity: the role of deep inspirations. Am J Respir Crit Care Med. 2001;164(2):207–15. doi: 10.1164/ajrccm.164.2.2008119. [DOI] [PubMed] [Google Scholar]
- 80.Anafi RC, Beck KC, Wilson TA. Impedance, gas mixing, and bimodal ventilation in constricted lungs. J Appl Physiol. 2003;94(3):1003–11. doi: 10.1152/japplphysiol.00569.2002. [DOI] [PubMed] [Google Scholar]
- 81.Lai-Fook SJ, Hyatt RE, Rodarte JR. Effect of parenchymal shear modulus and lung volume on bronchial pressure-diameter behavior. J Appl Physiol. 1978;44(6):859–68. doi: 10.1152/jappl.1978.44.6.859. [DOI] [PubMed] [Google Scholar]
- 82.Winkler T. In silico modeling of airway mechanics. Drug Discovery Today: Disease Models. 2007;4(3):125–9. doi: 10.1016/j.ddmod.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anafi RC, Wilson TA. Airway stability and heterogeneity in the constricted lung. J Appl Physiol. 2001;91(3):1185–92. doi: 10.1152/jappl.2001.91.3.1185. [DOI] [PubMed] [Google Scholar]
- 84.Kondo S, Miura T. Reaction-diffusion model as a framework for understanding biological pattern formation. Science. 2010;329(5999):1616–20. doi: 10.1126/science.1179047. [DOI] [PubMed] [Google Scholar]
- 85.Rietkerk M, Dekker SC, de Ruiter PC, van de Koppel J. Self-organized patchiness and catastrophic shifts in ecosystems. Science. 2004;305(5692):1926–9. doi: 10.1126/science.1101867. [DOI] [PubMed] [Google Scholar]
- 86.Winkler T, Venegas JG. Complex airway behavior and paradoxical responses to bronchoprovocation. J Appl Physiol. 2007;103(2):655–63. doi: 10.1152/japplphysiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 87.Kotaru C, Coreno A, Skowronski M, Muswick G, Gilkeson RC, McFadden ER., Jr. Morphometric changes after thermal and methacholine bronchoprovocations. J Appl Physiol. 2005;98(3):1028–36. doi: 10.1152/japplphysiol.01186.2003. [DOI] [PubMed] [Google Scholar]
- 88.Brown RH, Mitzner W. The myth of maximal airway responsiveness in vivo. J Appl Physiol. 1998;85(6):2012–7. doi: 10.1152/jappl.1998.85.6.2012. [DOI] [PubMed] [Google Scholar]
- 89.Shen X, Wu MF, Tepper RS, Gunst SJ. Mechanisms for the mechanical response of airway smooth muscle to length oscillation. J Appl Physiol. 1997;83(3):731–8. doi: 10.1152/jappl.1997.83.3.731. [DOI] [PubMed] [Google Scholar]
- 90.Fredberg JJ, Inouye D, Miller B, Nathan M, Jafari S, Raboudi SH, Butler JP, Shore SA. Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am J Respir Crit Care Med. 1997;156(6):1752–9. doi: 10.1164/ajrccm.156.6.9611016. [DOI] [PubMed] [Google Scholar]
- 91.Laprad AS, Szabo TL, Suki B, Lutchen KR. Tidal stretches do not modulate responsiveness of intact airways in vitro. J Appl Physiol. 2010;109(2):295–304. doi: 10.1152/japplphysiol.00107.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wongviriyawong C, Winkler T, Harris RS, Venegas JG. Dynamics of tidal volume and ventilation heterogeneity under pressure-controlled ventilation during bronchoconstriction: a simulation study. J Appl Physiol. 2010;109(4):1211–8. doi: 10.1152/japplphysiol.01401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ding DJ, Martin JG, Macklem PT. Effects of lung volume on maximal methacholine-induced bronchoconstriction in normal humans. J Appl Physiol. 1987;62(3):1324–30. doi: 10.1152/jappl.1987.62.3.1324. [DOI] [PubMed] [Google Scholar]
- 94.Bayat S, Porra L, Suhonen H, Nemoz C, Suortti P, Sovijarvi AR. Differences in the time course of proximal and distal airway response to inhaled histamine studied by synchrotron radiation CT. J Appl Physiol. 2006;100(6):1964–73. doi: 10.1152/japplphysiol.00594.2005. [DOI] [PubMed] [Google Scholar]
- 95.Bayat S, Porra L, Suhonen H, Suortti P, Sovijarvi AR. Paradoxical conducting airway responses and heterogeneous regional ventilation after histamine inhalation in rabbit studied by synchrotron radiation CT. J Appl Physiol. 2009;106(6):1949–58. doi: 10.1152/japplphysiol.90550.2008. [DOI] [PubMed] [Google Scholar]
- 96.Politi AZ, Donovan GM, Tawhai MH, Sanderson MJ, Lauzon AM, Bates JH, Sneyd J. A multiscale, spatially distributed model of asthmatic airway hyper-responsiveness. J Theor Biol. 2010;266(4):614–24. doi: 10.1016/j.jtbi.2010.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Amin SD, Majumdar A, Frey U, Suki B. Modeling the dynamics of airway constriction: effects of agonist transport and binding. J Appl Physiol. 2010;109(2):553–63. doi: 10.1152/japplphysiol.01111.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma B, Bates JH. Modeling the complex dynamics of derecruitment in the lung. Ann Biomed Eng. 2010;38(11):3466–77. doi: 10.1007/s10439-010-0095-2. [DOI] [PubMed] [Google Scholar]