

Abstract
We have examined the structural and functional effects of site-directed methionine oxidation in Dictyostelium (Dicty) myosin II using mutagenesis, in vitro oxidation, and site-directed spin-labeling for electron paramagnetic resonance (EPR). Protein oxidation by reactive oxygen and nitrogen species is critical for normal cellular function, but oxidative stress has been implicated in disease progression and biological aging. Our goal is to bridge understanding of protein oxidation and muscle dysfunction with molecular-level insights into actomyosin interaction. In order to focus on methionine oxidation and to facilitate site-directed spectroscopy, we started with a Cys-lite version of Dicty myosin II. For Dicty myosin containing native methionines, peroxide-treatment decreased actin-activated myosin ATPase activity, consistent with the decline in actomyosin function previously observed in biologically aged or peroxide-treated muscle. Methionine-to-leucine mutations, used to protect specific sites from oxidation, identified a single methionine that is functionally sensitive to oxidation: M394, near the myosin cardiomyopathy loop in the actin-binding interface. Previously characterized myosin labeling sites for spectroscopy in the force-producing region and actin-binding cleft were examined; spin label mobility and distance measurements in the actin-binding cleft were sensitive to oxidation, but particularly in the presence of actin. Overall secondary structure and thermal stability were unaffected by oxidation. We conclude that the oxidation-induced structural change in myosin includes a redistribution of existing structural states of the actin-binding cleft. These results will be applicable to the many biological and therapeutic contexts in which a detailed understanding of protein oxidation, function and structure relationships are sought.
We cannot survive more than minutes without oxygen—nor have we escaped vulnerability to oxidative stress, the natural consequence of aerobic respiration. Disease progression and biological aging are familiar contexts in which the role of “oxidative damage” -- to DNA, lipids, and proteins -- has been recognized, even popularized through the promotion of antioxidant supplements for longevity and disease resistance. Under healthy conditions, too, molecules within cells must sensitively detect and respond to cellular redox state to maintain homeostasis. While irreversible oxidative modifications are potentially damaging, they might also serve as signals for protein repair, degradation and transcriptional regulation (1, 2). Reversible protein modifications, particularly those involving cysteine and methionine, have been implicated as protein sensors of the cellular environment (3–6). Yet critical questions remain unanswered: What are the molecular structural principles that govern how protein molecules sense, respond, and are eventually damaged by oxidation? Are these principles broadly applicable?
Redox modulation of muscle function
Muscle contraction produces reactive oxygen and nitrogen species (RONS) that are countered by an elaborate system of antioxidant enzymes and molecules (7). Muscle is highly adaptive tissue that must dynamically respond to metabolic, mechanical and functional demands such as exercise, hormonal fluctuations and development; redox signaling meditates many of these adaptations (8). Disruption of the RONS/antioxidant equilibrium in cardiac muscle contributes to adverse outcomes after myocardial infarction (9) and in models of heart failure (10). In skeletal muscle, the physiological function of oxidation is complex and biphasic, with lower levels of RONS potentiating function and higher levels inhibiting (11–13). However, elevated production of RONS can lead to the accumulation of oxidatively modified proteins, contributing to a decline of function in biologically aged or diseased muscle (11, 14–16). Impaired force with aging is due not only to muscle atrophy, but also to structural and functional defects involving both contractile and regulatory proteins (16–19). Knowing the site-specific functional and structural impact of oxidative modifications will aid in developing targeted therapies for oxidative stress-related muscle pathologies.
Methionine oxidation
Knowledge of the site and the chemistry of oxidative modifications to proteins, in combination with functional measurements, reveals that amino acid sequences encode not only protein structure and function, but also sensitivity to chemical modification (20). While cysteine oxidation and reduction is a well-established component of cell signaling, methionine oxidation and specific reduction by methionine sulfoxide reductase is emerging as a novel regulatory mechanism with diverse and far-reaching functional implications (1, 3, 5, 20–22). For example, where methionine is part of a hydrophobic kinase recognition sequence, methionine oxidation can prevent phosphorylation by disrupting the kinase binding site, coupling oxidative environment and phosphorylation (23). Methionine oxidation regulates immune response by attenuating calcineurin function in signal transduction pathways (24, 25). Methionine and cysteine redox sensors have been identified for proteins involved in calcium regulation (26–33), but few such sensors have been characterized for contractile proteins (7). In the current study, we have begun to elucidate the principles that govern how the muscle contractile protein myosin is structurally poised to respond to methionine oxidation.
Muscle myosin oxidation
We have previously shown that treating skinned muscle fibers with peroxide (H2O2) resulted in functional and structural changes in myosin with a pattern similar to changes for myosin extracted from aged rats (16, 34, 35). Myosin purified from oxidized muscle fibers perturbs structural transitions within the actomyosin complex that are crucial for force generation. Proteomics analysis revealed that myosin functional and structural changes are associated with oxidation of multiple methionine residues in the myosin catalytic domain and essential light chain (35). It is not clear which methionines, one or many, are responsible for the observed functional and structural changes in skeletal myosin. In the present study, we have directly tested the hypothesis that site-specific methionine oxidation causes functional decline in actomyosin interaction and have identified aspects of myosin internal dynamics and structure that are most sensitive to oxidation.
Approach
In order to characterize the functional and structural impact of site-specific methionine oxidation in myosin, we have used Met to Leu mutagenesis to control which myosin methionines are susceptible to oxidation within a Cys-lite Dictyostelium (Dicty) myosin II (S1dC) background suitable for site-directed spectroscopy. Our approach is possible because S1dC mutants can be expressed in milligram quantities; this is not feasible for skeletal muscle S1. A direct comparison of structural transitions at equivalent sites in the force-generating region of S1dC and skeletal S1 showed that these motors occupy identical structural states, but with altered coupling to biochemical state (36). Thus S1dC serves as a malleable platform for understanding the chemical and structural principles that determine how a myosin motor responds to oxidation at selected residues. Our goal was not to mimic physiological conditions of oxidation, but rather to achieve significant functional perturbation while obtaining complete oxidation of the remaining accessible methionines and thus obtaining a chemically well-defined and homogeneous preparation. We varied the concentration of hydrogen peroxide to achieve this goal in a brief (30-min) treatment of purified myosin. We then examined the impact of methionine oxidation on (a) functional sensitivity of myosin ATPase and actin-activated ATPase and (b) structural changes in myosin, measured by site-directed spin labeling and electron paramagnetic resonance (EPR), focusing on sites previously shown to be highly sensitive to key structural transitions in the actomyosin ATPase cycle (37).
MATERIALS AND METHODS
Protein Preparations, Spin-Labeling and Oxidation
Cysteine mutations for spin labeling and methionine substitution mutations were introduced into a Dicty myosin II gene truncated at residue 762 (S1dC), containing only a single (non-reactive) cysteine at position 655 (37). Mutations were C49T, C312S, C442S, C470V, C599T, and C678S. These proteins were expressed and purified from Dictyostelium orf+ cells. Spin labeling was carried out overnight on ice using 100 μM myosin and 800 μM IPSL [3-(2-Iodoacetamido)-2,2,5,5-tetramethyl-1-pyrolidinyloxy] (Toronto Research Chemicals, North York, Ontario), IASL [4-(2-Iodoacetamido)-2,2,6,6-tetramethyl-1-piperidinyloxy] (Sigma-Aldrich, USA), or MSL [N-(1-Oxyl-2,2,6,6-tetramethyl-4-piperidinyl)maleimide] (Toronto Research Chemicals). Spin-labeled sample was reacted with 0 to 500 mM hydrogen peroxide (purchased as a stabilized 30% solution from Sigma-Aldrich) for thirty minutes on ice. Unreacted label and peroxide were removed by buffer exchange using two sequential Zeba desalting columns (Thermo Fisher Scientific, Rockford, Illinois). Unless otherwise indicated, experiments were carried out at 4°C in EPR buffer (50 mM KCl, 3 mM MgCl2, and 10 mM MOPS, pH 7.5). F-actin was prepared from rabbit skeletal muscle (38, 39) and dissolved in F-actin (FA) buffer: 3 mM MgCl2, 0.2 mM ATP and 10 mM Tris (pH 7.5).
Activity Measurements
ATPase activity was measured as the release of inorganic phosphate (40, 41) at 25° C. High-salt Ca/K ATPase activity was measured in a solution containing 0.0125 mg/mL myosin, 10 mM CaCl2, 600 mM KCl, 5 mM ATP, and 50 mM MOPS (pH 7.5). Actin-activated ATPase activity was measured as the increase in activity due to the addition of rabbit skeletal actin (usually 10 μM) to a solution containing 1 μM myosin, 3.5 mM MgCl2, 10 mM KCl, and 10 mM MOPS (pH 7.5).
ESI (electrospray ionization) mass spectrometry (ESI-MS)
Myosin samples for mass spectrometry were exchanged into 10 mM NH4HCO3 (pH 7.9) using two sequential 0.5 mL Zeba desalting columns. Myosin samples at 2 mg/mL were diluted 1:10 into a solution containing 50:50 acetonitrile:water and 0.1% formic acid. Additional formic acid (10%) was added as needed (1–2 μL) to reach pH < 3. Myosin mass was determined using a QSTAR quadrupole-TOF mass spectrometer (ABI) with an electrospray ionization source. Prior to infusion of the intact myosin solution, a solution of 50:50 acetonitrile:water and 0.1% formic acid (load solution) was infused at 50 μL per minute using an integrated syringe pump. After a stable baseline total ion current (TIC) was established for the load solution, the protein solution (20 μM myosin, pH 3) was introduced into the solvent stream using a 10 μL injection loop installed in the integrated loop injector. After the TIC returned to baseline intensity, four more consecutive loop injections of the protein solution were made for a total of five injections per sample. Data were acquired continuously during load buffer infusion and protein infusions over the range 500 – 2000 m/z. ESI spectra were analyzed with BioAnalyst QS (ABI) software v 1.1.5. The Bayesian Protein Reconstruction tool was used to generate peak lists from the spectra, using a signal-to-noise threshold of 20 for intact myosin. Individual reconstruction error ranged from 0.1 – 0.5 Da.
Circular Dichroism
CD spectra were recorded in the far-UV region (200–260 nm) using a JASCO J-815 spectrophotometer with an automated temperature controller. A path length of 10 mm was used, with spectra recorded at 1 nm intervals for 0.25 mg/mL Dicty myosin in phosphate buffer (50 mM KCl, 1 mM MgCl2, pH 7.0) at 25°C. Baseline scans were obtained using the same acquisition parameters with buffer alone, which were subtracted from the respective CD data scans of Dicty myosin. The raw CD signal θλ (millidegrees of ellipticity) was converted to mean residue molar ellipticity θ:
| Eq. 1 |
where MRE = 110, C = protein concentration, and ℓ = path length. Far- UV CD spectra were analyzed using CDPro Analysis Software (42). Thermal denaturation of Dicty myosin was monitored at 222 nm while temperature was increased from 10 to 70°C in 1°C steps, with a 1 min incubation at each step. At the end of each thermal denaturation experiment, the sample was rapidly cooled to 25°C to determine the extent of refolding. Thermal denaturation curves were fit using Origin 8.1 (OriginLab Corp, Northampton, MA).
CW EPR (Continuous wave EPR): Mobility
EPR on spin-labeled Dicty myosin was carried out as described previously (37). EPR samples contained 100 μM Dicty myosin in EPR buffer. The ADP.V state was formed by addition of 5 mM ADP, followed by 5 mM Na3VO4. The actomyosin state was formed by addition of 200 μM F-actin. EPR was performed on deoxygenated samples at 4°C using a Bruker E500 spectrometer (Billerica, MA) at X-band (9.5 GHz), with modulation frequency 100 kHz and modulation amplitude 2 G. Mobility was measured in gas-permeable Teflon tubes (0.6 mm I.D., 20 μL sample volume) sealed with critoseal, placed into the quartz temperature control dewar inside an SHQ cavity (ER4122 ST). Scan width was 120 G and the microwave power of 2 mW produced approximately 40% saturation, with no significant line broadening. Plotted spectra (Fig. 6, Fig. 8) were normalized to the double integral. Order parameters were measured from EPR spectra by simulation and fitting with the software NLSL (Nonlinear Least-Squares Analysis of Slow-Motional EPR Spectra) (43–45). Spectra were simulated for a two-component system, each with its own cone angle (θc), correlation time (τr) and mole fraction (xi).
Fig. 6.

Effect of oxidation on structural dynamics of actin-binding cleft. CW EPR spectra for MSL-F270C in the apo (A) and strongly actin-bound (B) states.
Fig. 8.

CW-EPR spectra for peroxide treated (500 mM, red) and untreated (black) Dicty myosin II spin-labeled with IASL at positionsT688C (SH1 helix)
Pulsed EPR: Spin-Spin Distance Measurements
EPR samples for spin-spin distance measurements were the same as in other EPR experiments (described above), except that the buffer also contained 10% glycerol (v/v), and the 100 μL samples were flash-frozen using liquid nitrogen in a 5 mm OD quartz NMR tube (Wilmad glass, Buena NJ) and stored at −80°C until use. Pulsed EPR experiments were performed with a Bruker E580 spectrometer (Billerica, MA) at X-band (9.5 GHz) with a Bruker dielectric ring resonator (MD-5) using a 4-pulse DEER (double electron-electron resonance) protocol (46). The π/2 pulse width was 16 ns, and the ELDOR pulse width was 40–44 ns. The static field was set to the low-field resonance of the nitroxide signal. Temperature was controlled at 65° K during acquisition, which lasted 4–12 h.
Spin-spin distances were determined by fitting the experimental EPR data with simulations assuming a distance distribution function consisting of a sum of Gaussians:
| Eq. 2 |
| Eq. 3 |
The 3n-1 variable parameters in the fit were xi (mole fraction), ri (center distance), and σi (standard deviation), where the full width at half maximum is given by . The number of components n in the best fit was defined as the one for which n + 1 produced no further improvement, as defined by the residual plot and the residual sum of squares. The distribution function (Eq. 3) was convoluted with the Pake pattern (47) to simulate the weighted sum of the dipolar broadening function over the distance distribution (48), and this was used to simulate the EPR data, essentially as described previously for CW EPR (49, 50) and for DEER (51). CW EPR spectra were fitted using a Monte Carlo search procedure with laboratory-developed software (WACY, Edmund Howard). DEER background correction, distance determination by Tikhonov regularization, and fits based on Gaussian distance distributions were performed using methods provided in the software DEERAnalysis2006.1 (51, 52). Distances extracted by Tikhonov regularization were consistent with fits based on Gaussian distance distributions. We report the results based on Gaussian distance distributions because they are more useful for discussing models based on discrete conformational states that are common to different biochemical states.
RESULTS
Methionines in Dictyostelium (Dicty) myosin II subfragment 1 (S1)
A version of Dicty myosin II truncated to contain only the myosin motor domain and devoid of reactive cysteines (“S1dC Cys-lite”(37)) was used as a model system to examine the functional and structural consequences of myosin methionine oxidation. Using Dicty myosin as such a model is justified by its high level of structural and functional homology with muscle myosin catalytic domain (55, 56). Dicty myosin contains nine methionines, four of which are strictly conserved among class II myosins: Met 91 in the 25 kDa domain, Met 166 on a loop behind the nucleotide binding pocket, Met 486 at the bend in the relay helix, and Met 642 in the lower 50 kDa domain (Fig. 1A). Met 394, located at the C-terminus of an α-helix that transitions into the cardiomyopathy loop in the actin-binding interface, is Cys in cardiac and skeletal myosin and Leu in smooth muscle myosin. Met 91, Met 166 and Met 331 are expected to be mostly inaccessible to solvent based on the crystal structure shown in Fig. 1B.
Fig. 1.

A. Location of native methionines in Dictyostelium myosin II (1FMV). Lower 50 kDa domain (blue); upper 50 kDa domain (green), relay helix (red); nucleotide binding pocket (orange); methionines (yellow spheres). Conserved Met residues are shown bold. B. Accessible surface area calculated from the crystal structure 1FMV and reported as a ratio of accessible side-chain surface area to the value calculated for a random coil (53). VMD (Visual Molecular Dynamics) was used to create renderings of molecular structures (54).
Functional sensitivity of Dicty myosin II to global methionine oxidation
The physical properties and biochemical activity of Dicty myosin II containing all native methionines and a single spin-labeled (therefore, non-reactive) cysteine were characterized as a function of oxidation by hydrogen peroxide (used as an oxidative agent). Our goal was to produce a sample in which there is substantial functional perturbation and the accessible methionines were oxidized completely in a brief (30 min) peroxide treatment, producing a homogeneous sample for functional and structural analysis. In an initial series of experiments, the concentration of hydrogen peroxide was varied to achieve this goal as evaluated by mass spectrometry (Fig. 2, Fig. 3). It is clear from these data that peroxide concentration on the order of 500 mM is optimal. The change in myosin molecular weight upon treatment of wild-type myosin with 500 mM hydrogen peroxide is 46 Da, equivalent to the addition of 2.9 oxygens (Fig. 2A), suggesting that at least three methionines are susceptible to oxidation. The mass spectrum for peroxide-treated Dicty myosin II is substantially broadened relative to the untreated myosin, indicating the presence of multiple unresolved oxidized states. Treatment with hydrogen peroxide induces a substantial decrease in both the maximum Mg2+ actin-activated ATPase rate in the presence of saturating actin (Vmax) and the actin concentration required for half-maximal activation (KATPase) (Fig. 2B), consistent with the decline in actomyosin function observed previously for biologically aged or peroxide-treated skeletal myosin (16, 35). The Mg2+ ATPase activity decreases exponentially with increasing peroxide concentration and is more pronounced in the presence of 10 μM F-actin than under basal conditions (0 μM F-actin) (Fig. 2C), indicating that actomyosin interaction is altered with oxidation. High salt Ca2+/K+ myosin ATPase, a non-physiological measure of myosin ATP hydrolysis rate in the absence of actin, also decreases exponentially with increasing peroxide concentration (Fig. 2D).
Fig. 2.

A. ESI-MS of intact Dicty myosin. Deconvoluted mass spectra (normalized to maximum intensity) for 0 mM (black) and 500 mM (gray) peroxide. Peak full width at half maximum is 30 Da for 0 mM and 67 Da for 500 mM. B. Functional assays for the same samples. For 0 mM peroxide, Vmax= 2.3 ± 0.5 and KATPase = 16± 7. For 500 mM peroxide, Vmax= 0.65 ± 0.04 and KATPase = 3.0± 0.8. C. Peroxide dependence of actin-activated ATPase activity in the presence of 10 μM actin (●) and no actin (▲). D. High salt myosin ATPase activity.
Fig. 3.

A. ESI-MS of intact Dicty myosin. Change in mass with increasing peroxide concentration is reported with respect to the unoxidized, spin-labeled sample. B. Mg2+ actin-activated myosin ATPase for 10 μM F-actin for wildtype (WT) myosin (T688C spin-labeled) and M-to-L mutants. C. M-to-L mutants are as listed.
Functional sensitivity of Dicty myosin II to site-specific methionine oxidation
In order to determine the consequences of methionine oxidation site-specifically, we made methionine substitutions in a Dicty myosin II construct devoid of native reactive cysteines. All constructs used for functional measurements contained the T688C mutation in order to introduce a cysteine labeling site for spectroscopy experiments; spin-labeling was carried out prior to peroxide treatment under conditions that assured complete labeling. Leucine was used to substitute for methionine at sites we desired to protect from oxidation (mutants are described in Fig. 3C). Leucine is typically considered the most conservative substitute for methionine, and has been shown to produce substantially less structural perturbation than methionine oxidation (28, 57). Of the M to L mutants tested, only L6 had slightly compromised actin-activated ATPase activity before oxidation (Fig. 3A). Attempts to express mutants with M to L mutations at 486 and 642 were unsuccessful, possibly because these mutations produce myosins that are unstable or interfere with Dictyostelium function.
Substituting leucine for methionine at all positions except M468 and M642 (L7) protects myosin from the functional decline induced by peroxide-treatment (Fig. 3 L7). This suggests neither of these two conserved methionines M486, nor M642, are responsible for oxidation-induced functional decline in myosin. In fact, when M394 is re-introduced (L6), functional sensitivity to oxidation is restored (Fig. 3 L6), indicating that M394 is a functionally sensitive target of oxidation. It is quite surprising that none of the conserved myosin methionines (M91, M166, M486, M642) were functionally sensitive to oxidation. Of the non-conserved methionines, M394 was a strong candidate for the site of oxidatively sensitive function because of its proximity to the cardiomyopathy loop in the actin-binding interface. The M394L point mutation is sufficient to rescue the oxidation-induced functional decline in myosin (Fig. 3 L_Cleft).
The decline in myosin actin-activation parallels an increase in myosin molecular weight (Fig. 3). The mass spectrum of unoxidized myosin appears as a broad peak with full width at half maximum equal to 30 Da (Fig. 2A), consistent with width predicted for the isotopic envelope of a 89 K protein (58). Full resolution of singly oxidized states is not possible because of the intrinsic peak broadening due to myosin’s isotopic envelope. For 500 mM peroxide, myosin containing native Met, the peak maximum increases 46 Da, indicating that, on average, each myosin has an additional 2.9 oxygens (peak width), suggesting at least three of myosin’s nine methionines are susceptible to oxidation, but possibly more. The shift in mass is slightly less when leucine replaced methionine at 394 (L_Cleft); each myosin has an additional 2.4 oxygens after full oxidation, indicating that M394 is at least partially susceptible to oxidation. The mass of L7 shifts 34 Da, suggesting that the two remaining methionines (M486 and M642) are both highly susceptible to oxidation. The mass of L6 shifts, on average, 30 Da indicating that on average only two of the three methionines present (M394, M486 and M642) are oxidized. The increase in average mass should therefore be interpreted with the understanding that peroxide-treatment results in a distribution of oxidized states. We cannot rule out the possibility that some methionines are oxidized before they are treated, not can we rule out the unlikely possibility of oxidation of residues other than methionines.
Structural consequences of methionine oxidation in Dicty myosin II
Circular dichroism (CD) spectroscopy and site-directed spin labeling in combination with electron paramagnetic resonance (EPR) were used to examine myosin’s structural sensitivity to methionine oxidation. Global effects on myosin secondary structure and structural stability were examined using far-UV CD and thermal denaturation. EPR experiments focused on regions that undergo conformational changes related to myosin ATPase activity and actomyosin interaction using previously characterized labeling sites in the actin-binding interface, actin-binding cleft and force-generating region of myosin (Fig. 4) (36, 37).
Fig. 4.
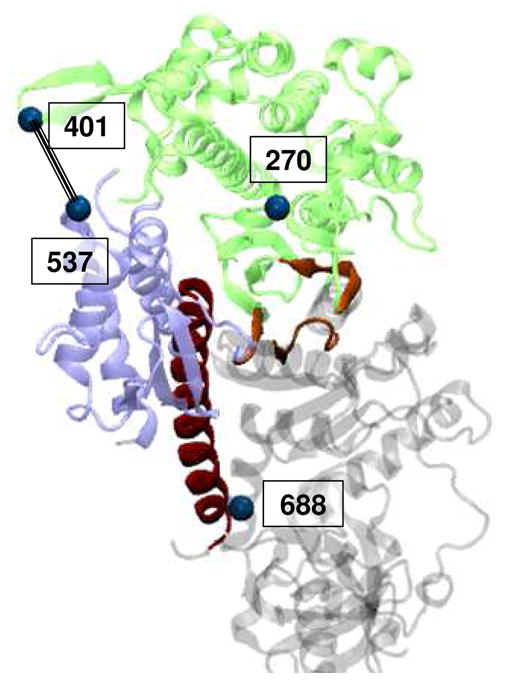
Location of Dicty myosin II spin-labeling sites. Myosin domains colored as in Fig 1.
Secondary structure and thermal stability of native and oxidatively modified Dicty myosin II
CD spectroscopy was used to assess changes in myosin secondary structure as a result of oxidative modification. No spectral differences were detected in the far-UV range (200–260 nm) at any peroxide concentration, suggesting that oxidation does not affect the relative amounts of α helix, β sheet, and random coil (Fig. 5A). Thermal denaturation was monitored at 222 nm by CD spectroscopy. All myosins exhibited similar transition midpoints (Tm) of ~41°C (Fig. 5D), consistent with reported values for myosin motor domains (59). All samples exhibited highly cooperative unfolding, demonstrated by the sharpness of the unfolding transition, indicating that each myosin forms a well-folded structure (Fig. 5C).
Fig. 5.

CD spectroscopy and thermal denaturation of peroxide-treated Dicty myosin containing native Met. A. Far UV spectra of myosin with no spin-label (black) and treatment with 0 (blue), 100 (green), 200 (purple), 350 (magenta), and 500 (red) mM H2O2, respectively. B. Ratio of molar ellipticity at 222 nm and 208 nm as a function of [H2O2]. C. Thermal denaturation curves of myosin. Samples same as part A. D. Transition midpoints derived from thermal denaturation curves as a function of [H2O2].
Oxidation-induced changes in myosin’s actin-binding cleft
Labeling sites within the myosin actin-binding cleft and the actomyosin interface were used to probe the structural dynamics of myosin alone and in complex with actin. F270C is located in the corner of the actin binding cleft, near switch I of the nucleotide binding site (Fig. 4). When F270C is labeled with maleimide spin label (MSL), the spin label motion, characterized by a rotational correlation time and cone angle of allowed motion, is sensitive to actin-binding. In the apo and strongly actin-bound biochemical states, two motional components whose relative populations shift in response to actin binding are resolved (Fig. 6). Global fitting of spectra reveals that the faster component (Fig. 6, innermost arrow) moves isotropically with a rotational correlation time of 3.7 × 10−9 s−1. The slower component (Fig. 6, outermost arrow) has a rotational correlation time of 7.9 × 10−9 s−1 and is restricted to a cone angle of 40°. In the apo state, the faster component dominates the spectrum with a mole fraction (xfast) equal to 0.9 and when myosin is strongly bound to actin, the slow component dominates (xfast = 0.4). Oxidation with 500 mM peroxide shifts the equilibrium toward the slower component for both the actomyosin complex (xfast = 0.2) and to a lesser extent, for apo myosin (xfast = 0.8).
Distances across the actin-binding cleft from N537C on the lower 50 kDa domain to G401C in the upper 50 kDa domain were measured by double electron-electron resonance (DEER) after spin labeling both sites with IPSL spin label. DEER is a pulsed EPR technique well-suited for measuring distances in the 2–6 nm range (51). N537C and G401C are located within the actomyosin interface (Fig. 4) and are sensitive to conformational changes in the actin-binding cleft at key points in force generation. Notably, the actin-binding cleft populates two distinct conformations when strongly bound to actin, the shorter distance corresponding to an ‘open’ cleft and longer distance corresponding to a ‘closed’ cleft (37).
While the effects of methionine oxidation are subtle in the apo DEER decay, they are pronounced in the decay for the strongly actin-bound complex (Fig. 7). By visual inspection, the decay for the oxidized actomyosin sample (red) is initially more rapid than for the unoxidized sample (black) (Fig. 7A); this indicates a shift toward shorter distances. The resulting distance distributions show how oxidation enhanced the stability of the conformation that corresponds to the shorter distance from 40% to 55% (Table 1). The DEER decay for the oxidized sample had slightly more distinct oscillations, indicating more narrow distance distributions. Oxidation restricted small scale conformational sampling in both the open and closed cleft conformations, and significantly stabilized the open cleft conformation.
Fig. 7.

Effect of oxidation on structural dynamics of actin-binding cleft near the actomyosin interface. DEER decay (A) and distance and distribution (B) for spin labels at G401C and N537C in the apo and strongly actin-bound states.
Table 1.
Summary of DEER-derived distance distributions for IPSL-537 and IPSL-401.
| DEER Sample | x1 | r1 | w1 | x2 | r2 | w2 |
|---|---|---|---|---|---|---|
| 537/401 apo, (−) H2O2 | 0.781 | 2.13 | 1.72 | 0.219 | 4.02 | 2.03 |
| 537/401 apo, (+)H2O2 | 0.815 | 2.20 | 1.20 | 0.186 | 2.96 | 2.30 |
| 537/401 actin-bound, (−)H2O2 | 0.396 | 1.84 | 2.01 | 0.604 | 3.81 | 1.21 |
| 537/401 actin-bound, (+)H2O2 | 0.545 | 2.34 | 1.58 | 0.455 | 3.77 | 1.04 |
Simulated spectra were fit to experimental spectra as described in Experimental Procedures. Fitting parameters for i=1, 2, or 3 Gaussian distance distributions are defined as follows: ri is the center distance in nm, wi is the full width half-maximum in nm, and xi is the mole fraction. Uncertainties were estimated from SD of repeated experiments (n = 3–5). For distances 1.6 to 3.0 nm, fractional uncertainties in r and w were approximately 12%.
No oxidation-induced changes in myosin dynamics in the force-generating region
Spin label mobility was measured at the T688C site in the myosin SH1 helix, a well-characterized labeling site that is sensitive to nucleotide binding and hydrolysis (36). Oxidation does not alter spin label mobility for any biochemical states tested, including the strongly actin-bound state (Fig. 8).
DISCUSSION
Functional consequences of methionine oxidation in myosin
Oxidation of Dicty myosin containing all nine native methionines is associated with a decrease in myosin ATPase, and a decline in actomyosin interaction (Fig. 2B,C, D). At least three of myosin’s nine methionines are susceptible to oxidation under the conditions tested (Fig. 2A). Methionine to leucine substitutions, in combination with oxidation, made it possible to determine which sites are functionally sensitive to oxidation. Leucine substitutions, even at seven of the nine myosin methionines, caused only slight functional change in myosin, a result consistent with what other groups have reported for Leu to Met substitutions (28). We identified at least three myosin methionines that are at least partially susceptible to oxidation by peroxide: M486, M642 and M394. The M394L point mutation is sufficient to prevent oxidation-induced functional decline in myosin ATPase activity and actomyosin interaction (Fig. 3).
Prochniewicz et al. found that peroxide treatment of permeabilized muscle fibers is associated with myosin methionine oxidation and a decline in actomyosin interaction (35). We find that M394 in Dicty myosin II, equivalent to C402 in skeletal myosin, is most functionally sensitive to oxidation. Cysteine oxidation was not detected in myosin purified from peroxide-treated muscle fibers, but was detected in myosin purified from aged rats (16, 34, 35). At least 9 of 22 methionines in the skeletal myosin catalytic domain are susceptible to oxidation (4 of these 9 methionines are oxidized before peroxide treatment). Of the three oxidizable methionines in S1dC (M394, M486 and M642), only M486 corresponds to an oxidizable Met in skeletal myosin (M496). M486 is located at the bend in the relay helix, a major path of communication between the nucleotide-binding site and the force-generating domain. Although we did not detect oxidation-induced structural changes at the nearby SH1 helix, a detailed examination of relay helix dynamics structural is warranted.
Skeletal myosin contains nearly double the number of methionines as Dicty myosin S1, so it is likely that some of the peroxide-induced loss of function in skeletal myosin (35) is due to methionines that are not present in S1dC. A next step might include introducing novel methionines into Dicty myosin at locations equivalent to skeletal myosin to determine the functional and structural impact of methionines that are unique to skeletal myosin.
Cysteine 400 in cardiac myosin, equivalent to M394 in Dicty myosin, is one of at least two cysteines in the myosin heavy chain susceptible to glutathionylation and associated with a decline in myosin ATPase (60). Glutathionylation is a reversible oxidative modification that could act to modulate actomyosin function during episodes of oxidative stress (61, 62). During episodes of oxidative stress, when the ratio of oxidized glutathione (glutathione disulfide) to glutathione is high, susceptible Cys are likely to be glutathionylated, while normal cellular conditions promote reduced Cys. It is reasonable to suggest that M394 in Dicty myosin II and C400 in cardiac myosin have evolved as redox sensors that act to modulate actomyosin interaction in response to oxidative stress. It remains unknown whether oxidation at M394 is reversible by methionine sulfoxide reductase, as reversibility would strengthen the argument that M394 is a redox sensor.
Structural consequences of methionine oxidation in myosin
Overall myosin secondary structure and thermal stability were unaffected by oxidation (Fig. 5). Changes in myosin structural dynamics due to oxidation were examined for two major locations: the actin-binding cleft and the force-generating region (Fig. 4). Spectroscopic methods and spin-labeling sites sensitive to key conformational changes have been previously established for these regions (36, 37). IASL spin label mobility at T688C in the SH1 helix in the force-generating domain was insensitive to oxidation in every biochemical state tested, including when actin was present (Fig. 8). It is surprising that no sensitivity toward oxidation is observed for the nearby SH1 helix since we have shown that M486, situated in the bend of the relay helix, is susceptible to oxidation. It remains to be known whether relay helix dynamics are sensitive to oxidation. For the actin-binding cleft, myosin structural dynamics was sensitive to oxidation, but mainly in the presence of actin. Both spin label mobility inside the cleft and distances measured across the cleft in the actin-binding interface resolved two distinct components that probably correspond to two different structural states (Fig. 6, Fig. 7). Oxidation shifted the equilibrium between states, indicating that the functional effects of oxidation are due not to the stabilization of a new structural state of myosin, but through a change in the distribution of existing structural states.
M394 is located in the actin-binding interface, so it is reasonable to expect that oxidation to methionine sulfoxide at this location is responsible for the changes observed in cleft structural dynamics observed in the presence of actin. M394 is the last residue of an α-helix bordering the cardiomyopathy loop (Fig. 9). Although methionine is well-suited for helical structure, methionine sulfoxide can destabilize helices (3). We hypothesize that oxidation of M394 destabilizes the end of the helix, effectively lengthening the cardiomyopathy loop, altering the interaction of the upper 50 kDa domain with actin, and therefore changing the distribution of cleft structural states in the presence of actin. It is likely that it is this change in actomyosin structural dynamics that underlies the functional decline in actomyosin interaction observed with peroxide treatment.
Fig. 9.
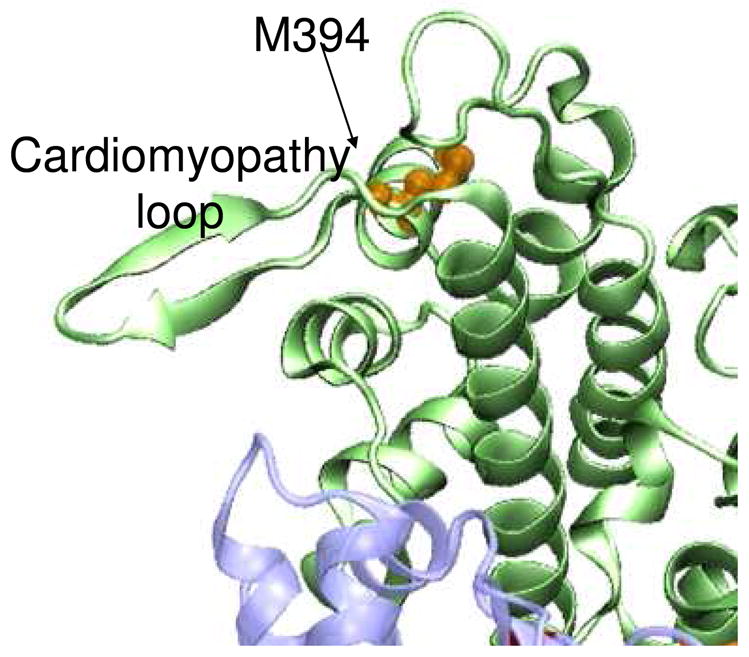
Location of M394 (orange) near the cardiomyopathy loop in the myosin actin-binding interface.
CONCLUSION
Site-directed mutagenesis was used to control Met and Cys susceptibility to oxidation by peroxide in order to determine the functional and structural impact of site-specific Met oxidation in myosin. At least three Mets, including two conserved (M486 and M642) and one nonconserved (M394), are susceptible to oxidation. Oxidation is associated with a steep decline in myosin ATPase and actin-activation; the M394L mutation fully blocks this functional sensitivity. Structural dynamics in the actin-binding cleft, both near the nucleotide pocket and in the actomyosin interface, are sensitive to oxidation in the presence of actin. The overall oxidation-induced structural impact includes reduced sampling of conformational space and a large redistribution of existing structural states of the actin-binding cleft. While this work on Dicty myosin does not provide direct insight into oxidative modifications in muscle, it contributes broadly to our understanding of how myosin senses and responds to oxidative stress. Knowledge of the functional and structural impact of site-specific methionine oxidation will be an important factor in the development of targeted therapies for degenerative muscle diseases.
Acknowledgments
This work was supported by grants to DDT from NIH (AR32961 and AG26160) and the University of Minnesota Biomedical Genomics Center.
This work was supported by NIH grants to DDT (AR32961, AG26160, and AR057220). JCK was supported by NIH training grant AG029796. RJM was supported by AG037303. We received excellent assistance with manuscript preparation from Octavian Cornea. The Biophysical Spectroscopy Facility and Minnesota Supercomputing Institute were essential for this work.
ABBREVIATIONS
- EPR
electron paramagnetic resonance
- DEER
double electron electron resonance
- SDSL
site-directed spin labeling
- ESI-MS
Electrospray ionization mass spectrometry
- S1
subfragment 1 of skeletal myosin
- Dicty
Dictyostelium discoideum
- S1dC
subfragment 1 of Dictyostelium myosin II heavy chain
- IASL
4-(2-Iodoacetamido)-2,2,6,6-tetramethyl-1-piperidinyloxy spin label
- IPSL
3-(2-Iodoacetamido)-2,2,5,5-tetramethyl-1-pyrolidinyloxy spin label
- MSL
N-(1-oxy-2,2,5,5-tetramethyl-4-piperidinyl)maleimide spin label
- ROS
reactive oxygen species
- RONS
reactive oxygen and nitrogen species
References
- 1.Hoshi T, Heinemann S. Regulation of cell function by methionine oxidation and reduction. J Physiol. 2001;531:1–11. doi: 10.1111/j.1469-7793.2001.0001j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50:279–289. doi: 10.1080/713803728. [DOI] [PubMed] [Google Scholar]
- 3.Bigelow DJ, Squier TC. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta. 2005;1703:121–134. doi: 10.1016/j.bbapap.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Stadtman ER. Protein oxidation and aging. Free Radic Res. 2006;40:1250–1258. doi: 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- 5.Emes MJ. Oxidation of methionine residues: the missing link between stress and signalling responses in plants. Biochem J. 2009;422:e1–2. doi: 10.1042/BJ20091063. [DOI] [PubMed] [Google Scholar]
- 6.Prentice HM, Moench IA, Rickaway ZT, Dougherty CJ, Webster KA, Weissbach H. MsrA protects cardiac myocytes against hypoxia/reoxygenation induced cell death. Biochem Biophys Res Commun. 2008;366:775–778. doi: 10.1016/j.bbrc.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 7.Jackson MJ. Redox regulation of skeletal muscle. IUBMB Life. 2008;60:497–501. doi: 10.1002/iub.72. [DOI] [PubMed] [Google Scholar]
- 8.Palomero J, Jackson MJ. Redox regulation in skeletal muscle during contractile activity and aging. J Anim Sci. 2010;88:1307–1313. doi: 10.2527/jas.2009-2436. [DOI] [PubMed] [Google Scholar]
- 9.Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S, Utsumi H, Takeshita A. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. 2000;87:392–398. doi: 10.1161/01.res.87.5.392. [DOI] [PubMed] [Google Scholar]
- 10.Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, Laufs U. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108:1567–1574. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- 11.Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve. 2007;35:411–429. doi: 10.1002/mus.20743. [DOI] [PubMed] [Google Scholar]
- 12.Reid MB, Durham WJ. Generation of reactive oxygen and nitrogen species in contracting skeletal muscle: potential impact on aging. Ann N Y Acad Sci. 2002;959:108–116. doi: 10.1111/j.1749-6632.2002.tb02087.x. [DOI] [PubMed] [Google Scholar]
- 13.Reid MB. Invited Review: redox modulation of skeletal muscle contraction: what we know and what we don’t. J Appl Physiol. 2001;90:724–731. doi: 10.1152/jappl.2001.90.2.724. [DOI] [PubMed] [Google Scholar]
- 14.Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol. 2007;102:1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- 15.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 16.Prochniewicz E, Thompson LV, Thomas DD. Age-related decline in actomyosin structure and function. Exp Gerontol. 2007;42:931–938. doi: 10.1016/j.exger.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delbono O. Molecular mechanisms and therapeutics of the deficit in specific force in ageing skeletal muscle. Biogerontology. 2002;3:265–270. doi: 10.1023/a:1020189627325. [DOI] [PubMed] [Google Scholar]
- 18.Lowe DA, Surek JT, Thomas DD, Thompson LV. Electron paramagnetic resonance reveals age-related myosin structural changes in rat skeletal muscle fibers. Am J Physiol Cell Physiol. 2001;280:C540–547. doi: 10.1152/ajpcell.2001.280.3.C540. [DOI] [PubMed] [Google Scholar]
- 19.Lowe DA, Warren GL, Snow LM, Thompson LV, Thomas DD. Muscle activity and aging affect myosin structural distribution and force generation in rat fibers. J Appl Physiol. 2004;96:498–506. doi: 10.1152/japplphysiol.00842.2003. [DOI] [PubMed] [Google Scholar]
- 20.Ghesquiere B, Jonckheere V, Colaert N, Van Durme J, Timmerman E, Goethals M, Schymkowitz J, Rousseau F, Vandekerckhove J, Gevaert K. Redox proteomics of protein-bound methionine oxidation. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Bigelow DJ, Squier TC. Thioredoxin-dependent redox regulation of cellular signaling and stress response through reversible oxidation of methionines. Mol Biosyst. 7:2101–2109. doi: 10.1039/c1mb05081h. [DOI] [PubMed] [Google Scholar]
- 23.Hardin SC, Larue CT, Oh MH, Jain V, Huber SC. Coupling oxidative signals to protein phosphorylation via methionine oxidation in Arabidopsis. Biochem J. 2009;422:305–312. doi: 10.1042/BJ20090764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agbas A, Moskovitz J. The Role of Methionine Oxidation/Reduction in the Regulation of Immune Response. Curr Signal Transduct Ther. 2009;4:46–50. doi: 10.2174/157436209787048748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carruthers NJ, Stemmer PM. Methionine oxidation in the calmodulin-binding domain of calcineurin disrupts calmodulin binding and calcineurin activation. Biochemistry. 2008;47:3085–3095. doi: 10.1021/bi702044x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balog EM, Norton LE, Bloomquist RA, Cornea RL, Black DJ, Louis CF, Thomas DD, Fruen BR. Calmodulin oxidation and methionine to glutamine substitutions reveal methionine residues critical for functional interaction with ryanodine receptor-1. J Biol Chem. 2003;278:15615–15621. doi: 10.1074/jbc.M209180200. [DOI] [PubMed] [Google Scholar]
- 27.Balog EM, Norton LE, Thomas DD, Fruen BR. Role of calmodulin methionine residues in mediating productive association with cardiac ryanodine receptors. Am J Physiol Heart Circ Physiol. 2006;290:H794–799. doi: 10.1152/ajpheart.00706.2005. [DOI] [PubMed] [Google Scholar]
- 28.Balog EM, Lockamy EL, Thomas DD, Ferrington DA. Site-Specific Methionine Oxidation Initiates Calmodulin Degradation by the 20S Proteasome. Biochemistry. 2009;48:3005–3016. doi: 10.1021/bi802117k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boschek CB, Jones TE, Smallwood HS, Squier TC, Bigelow DJ. Loss of the calmodulin-dependent inhibition of the RyR1 calcium release channel upon oxidation of methionines in calmodulin. Biochemistry. 2008;47:131–142. doi: 10.1021/bi701352w. [DOI] [PubMed] [Google Scholar]
- 31.Anbanandam A, Bieber Urbauer RJ, Bartlett RK, Smallwood HS, Squier TC, Urbauer JL. Mediating molecular recognition by methionine oxidation: conformational switching by oxidation of methionine in the carboxyl-terminal domain of calmodulin. Biochemistry. 2005;44:9486–9496. doi: 10.1021/bi0504963. [DOI] [PubMed] [Google Scholar]
- 32.Bartlett RK, Bieber Urbauer RJ, Anbanandam A, Smallwood HS, Urbauer JL, Squier TC. Oxidation of Met144 and Met145 in calmodulin blocks calmodulin dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 2003;42:3231–3238. doi: 10.1021/bi026956z. [DOI] [PubMed] [Google Scholar]
- 33.Sun H, Gao J, Ferrington DA, Biesiada H, Williams TD, Squier TC. Repair of oxidized calmodulin by methionine sulfoxide reductase restores ability to activate the plasma membrane Ca-ATPase. Biochemistry. 1999;38:105–112. doi: 10.1021/bi981295k. [DOI] [PubMed] [Google Scholar]
- 34.Prochniewicz E, Thomas DD, Thompson LV. Age-related decline in actomyosin function. J Gerontol A Biol Sci Med Sci. 2005;60:425–431. doi: 10.1093/gerona/60.4.425. [DOI] [PubMed] [Google Scholar]
- 35.Prochniewicz E, Lowe DA, Spakowicz DJ, Higgins L, O’Conor K, Thompson LV, Ferrington DA, Thomas DD. Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am J Physiol Cell Physiol. 2008;294:C613–626. doi: 10.1152/ajpcell.00232.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agafonov RV, Nesmelov YE, Titus MA, Thomas DD. Muscle and nonmuscle myosins probed by a spin label at equivalent sites in the force-generating domain. Proc Natl Acad Sci U S A. 2008;105:13397–13402. doi: 10.1073/pnas.0801342105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein JC, Burr AR, Svensson B, Kennedy DJ, Allingham J, Titus MA, Rayment I, Thomas DD. Actin-binding cleft closure in myosin II probed by site-directed spin labeling and pulsed EPR. Proc Natl Acad Sci U S A. 2008;105:12867–12872. doi: 10.1073/pnas.0802286105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prochniewicz E, Zhang Q, Howard EC, Thomas DD. Microsecond rotational dynamics of actin: spectroscopic detection and theoretical simulation. J Mol Biol. 1996;255:446–457. doi: 10.1006/jmbi.1996.0037. [DOI] [PubMed] [Google Scholar]
- 39.Prochniewicz E, Walseth TF, Thomas DD. Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry. 2004;43:10642–10652. doi: 10.1021/bi049914e. [DOI] [PubMed] [Google Scholar]
- 40.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 41.Fiske CH, Subbarow Y. The colorimetric Determination of Phosphorus. The Journal of Biological Chemistry. 1925;26:375–400. [Google Scholar]
- 42.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 43.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson A. 1996;120:155–189. [Google Scholar]
- 44.Nesmelov YE, Karim CB, Song L, Fajer PG, Thomas DD. Rotational dynamics of phospholamban determined by multifrequency electron paramagnetic resonance. Biophys J. 2007;93:2805–2812. doi: 10.1529/biophysj.107.108910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nesmelov YE, Agafonov RV, Burr AR, Weber RT, Thomas DD. Structure and dynamics of the force-generating domain of myosin probed by multifrequency electron paramagnetic resonance. Biophys J. 2008;95:247–256. doi: 10.1529/biophysj.107.124305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 47.Pake GE. Nuclear Resonance Absorption in Hydrated Crystals: Fine Structure of the Proton Line. Journal of Chemical Physics. 1948;16:327–336. [Google Scholar]
- 48.Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci U S A. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinhoff HJ, Radzwill N, Thevis W, Lenz V, Brandenburg D, Antson A, Dodson G, Wollmer A. Determination of interspin distances between spin labels attached to insulin: comparison of electron paramagnetic resonance data with the X-ray structure. Biophys J. 1997;73:3287–3298. doi: 10.1016/S0006-3495(97)78353-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 51.Jeschke G. Distance measurements in the nanometer range by pulse EPR. Chemphyschem. 2002;3:927–932. doi: 10.1002/1439-7641(20021115)3:11<927::AID-CPHC927>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 52.Jeschke G, Koch A, Jonas U, Godt A. Direct conversion of EPR dipolar time evolution data to distance distributions. J Magn Reson. 2002;155:72–82. doi: 10.1006/jmre.2001.2498. [DOI] [PubMed] [Google Scholar]
- 53.Fraczkiewicz R, Braun W. Exact and Efficient Analytical Calculation of the Accessible Surface Areas and Their Gradients for Macromolecules. J Comp Chem. 1998;19:319–333. [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Schroder RR, Manstein DJ, Jahn W, Holden H, Rayment I, Holmes KC, Spudich JA. Three-dimensional atomic model of F-actin decorated with Dictyostelium myosin S1. Nature. 1993;364:171–174. doi: 10.1038/364171a0. [DOI] [PubMed] [Google Scholar]
- 56.Bauer CB, Holden HM, Thoden JB, Smith R, Rayment I. X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J Biol Chem. 2000;275:38494–38499. doi: 10.1074/jbc.M005585200. [DOI] [PubMed] [Google Scholar]
- 57.Kim YH, Berry AH, Spencer DS, Stites WE. Comparing the effect on protein stability of methionine oxidation versus mutagenesis: steps toward engineering oxidative resistance in proteins. Protein Eng. 2001;14:343–347. doi: 10.1093/protein/14.5.343. [DOI] [PubMed] [Google Scholar]
- 58.Yergey J, Heller D, Hansen G, Cotter RJ, Fenselau C. Isotopic distributions in mass spectra of large molecules. Analytical Chemistry. 1983;55:353–356. [Google Scholar]
- 59.Zolkiewski M, Redowicz MJ, Korn ED, Ginsburg A. Thermal unfolding of Acanthamoeba myosin II and skeletal muscle myosin. Biophys Chem. 1996;59:365–371. doi: 10.1016/0301-4622(95)00129-8. [DOI] [PubMed] [Google Scholar]
- 60.Passarelli C, Petrini S, Pastore A, Bonetto V, Sale P, Gaeta LM, Tozzi G, Bertini E, Canepari M, Rossi R, Piemonte F. Myosin as a potential redox-sensor: an in vitro study. J Muscle Res Cell Motil. 2008;29:119–126. doi: 10.1007/s10974-008-9145-x. [DOI] [PubMed] [Google Scholar]
- 61.Rasmussen HH, Hamilton EJ, Liu CC, Figtree GA. Reversible oxidative modification: implications for cardiovascular physiology and pathophysiology. Trends Cardiovasc Med. 2010;20:85–90. doi: 10.1016/j.tcm.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 62.Passarelli C, Di Venere A, Piroddi N, Pastore A, Scellini B, Tesi C, Petrini S, Sale P, Bertini E, Poggesi C, Piemonte F. Susceptibility of isolated myofibrils to in vitro glutathionylation: Potential relevance to muscle functions. Cytoskeleton (Hoboken) 2010;67:81–89. doi: 10.1002/cm.20425. [DOI] [PubMed] [Google Scholar]