

Abstract
Systemic sclerosis (SSc) is an autoimmune disease characterized by vascular alterations and immunological disturbances and fibrosis, the order of which remains to be fully determined. Clinically, patients show clear signs of hypoxia in skin and internal organs. The low oxygen tension is potentially caused by a yet to be indentified circuitry involving the three features that typify SSc. In addition, once present, the hypoxia creates a vicious circle of ongoing pathology. In this paper, we provide an overview of the evidence that points towards the mechanisms causing hypoxia in SSc. In addition, data that suggest how hypoxia itself may orchestrate worsening of symptoms is presented. Altogether, it is clear that hypoxia is an important hallmark in SSc patients. By providing an overview of the mechanisms at play and the possible therapeutic avenues that have emerged, we hope to stimulate researchers to provide novel clues into the conundrum in SSc patients.
1. Introduction
Systemic sclerosis (SSc) is typified by vascular alterations and immunological disturbances and fibrosis of the skin and internal organs, which culminates in severe disabilities and not seldom premature death. Although the abovementioned pathways are all clearly involved, their sequel and relative contributions are still a matter of debate. About 90% of the patients diagnosed with SSc experienced Raynaud's phenomenon long before the appearance of other clinical symptoms that drives the patient to visit a physician [1]. The diagnosis is often made when patients suffer from a full-blown SSc with rarefaction of the small capillaries as identified by capillaroscopy, digital ulcers, and progressive fibrosis of the skin [2, 3]. Both Raynaud's phenomenon and the rarefaction of capillaries suggest the presence of hypoxia during certain stages of disease. In line with these thoughts, several studies demonstrated a lowered oxygen pressure in SSc skin [4–6]. Data showing a lower pO2 only in the lesional SSc skin and a correlation between skin thickness and pO2 suggests a direct connection between fibrosis and tissue hypoxia. Lastly, evidence of increased oxidative stress, thought to be caused by ischemia-reperfusion events, is convincing [7, 8]. The precise contribution of the clinically clear hypoxia to the cellular derogatory mechanism is exposed bit by bit. Next to this aberrant regulation of oxygen system, a role for the dysregulated immune system is illustrated by the fact that 75–90% of patients have ANA positivity, mostly explained by the presence of anticentromere or antitopoisomerase antibodies (reviewed in [9]). Recent literature shows the potential interaction between the vascular alterations and the immunopathology found in SSc. The rarefaction of the small vessels clearly lowers oxygen pressure in the skin, and there are signs of increased oxidative stress, which both are able to alarm and activate the plethora of different cell types of the immune system (Figure 1).
Figure 1.
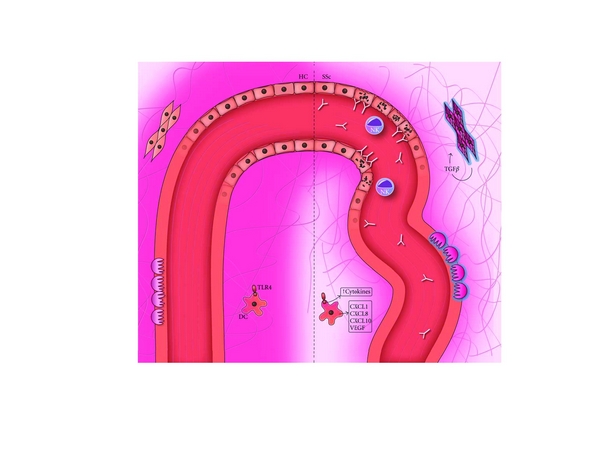
Hypoxia in the pathogenesis of systemic sclerosis. The left side illustrates the normal situation, with a healthy blood vessel delivering oxygen to the surrounding tissue. The right side represents the situation in SSc, where the diseased vessel and overwhelming deposition of collagen fibers prevent the oxygen from reaching the periphery. In the vessel, endothelial apoptosis as a result of antibody-dependent cytotoxicity is visible. Surrounding the ECs, activated pericytes are responsible for the deposition of collagen. The resulting hypoxia leads to a higher cyto- and chemokine production by DCs, in part triggered by TLR stimulation, and to a continuing loop of TGFβ production, collagen synthesis, and myofibroblast differentiation of fibroblasts. This in turn leads to a hindered dispersion of oxygen, keeping the vicious circle going on.
1.1. HIF-1α
As a result of the hypoxia in skin and other affected tissue in SSc, activation of important hypoxia pathways is plausible. The identification of hypoxia-inducible factor 1 (HIF-1) unlighted the cellular mechanism of oxygen homeostasis. As a transcription factor of genes important in the metabolism of the cell but also angiogenesis, apoptosis, proliferation, and matrix production, this factor seems to play a more then central role in the cellular response to changing oxygen pressures (reviewed in [10]). HIF-1α is the tightly regulated form of HIF-1, which is quickly hydroxylated and degraded in normoxic conditions by prolyl hydroxylases [11]. But in case of hypoxia, the levels of HIF-1α increase dramatically [12]. Counterintuitive however, HIF-1α was found to be decreased in the epidermis of SSc patients compared to healthy controls [6]. As nicely suggested in the review by Beyer et al. [13], this could be caused by a negative feedback loop causing an increase in prolyl hydroxylases resulting in a faster degradation of HIF-1α. The persistent upregulation of VEGF (further discussed below) in SSc illustrates the activation of pathways sensitive to oxygen pressure and thus the low expression of HIF-1α must be compensated by other factors like HIF-2α and HIF-3α. The presence of these factors in SSc is currently unknown, and, moreover, the function of these factors is not mutually exclusive with HIF-1α [14]. Furthermore, a French cohort SSc patients showed an increased presence of a genetic variant of HIF-1α, implying a role in SSc pathogenesis [15]. How a defective HIF-1α regulation is precisely involved in the development of SSc will be discussed in detail later in this paper.
2. Vasculopathy
Of the three pathogenic features in SSc, vasculopathy is thought to be the first one to occur [16]. The vascular defects can be made visible with nailfold capillaroscopy, showing giant capillaries, loss of architectural arrangement, hyperpermeability, and dropout of capillaries [2]. The lower density and quality of vessels leads to a reduced blood flow and consequential tissue hypoxia.
The absence or aberrant function of dermal endothelial cells (ECs) is one mechanism often addressed to explain the onset of vasculopathy. ECs can be distinguished on immunohistochemistry by the expression of CD31, von Willebrand factor (vWF), and vascular endothelial cadherin (VE-cadherin). Dermis of early diffuse SSc patients (disease duration <2 years in 27/30 patients) shows a population of CD31+ endothelial cells with no expression of vWF and VE-cadherin [17]. Another group observed apoptotic vWF positive cells in skin section from early diffuse SSc patients, indicating apoptosis of EC. In the UCD 200/206 chicken SSc model, this apoptosis of ECs was already present before the appearance of perivascular infiltrates or fibrosis [16] and was also identified in the lungs, esophagus, and kidneys [18]. The presence of immunoglobulins (Ig) on these ECs [16] points towards a role of antiendothelial cell antibodies (AECA), which are known to be present in the serum of SSc patients [19]. The induction of EC apoptosis is thought to be the result of antibody-dependent cellular cytotoxicity [20, 21], and Sgonc et al. have suggested that natural killer (NK) cells form a likely candidate to initiate this process [22].
Next to the increased loss of ECs due to apoptosis, there might also be a problem in endothelial repair in SSc. SSc patients show a decreased amount of bone-marrow (BM) resident endothelial progenitors, while the hematopoiesis of other cell lines seems to be undisturbed [23]. The decreased amount of BM resident endothelial precursors could be a consequence of the above-mentioned AECA, which can induce apoptosis of the precursors upon exposure to AECA-positive serum [23]. Another mechanism to be considered is a higher efflux of these progenitor cells to the periphery, possibly as a result of elevated VEGF levels (as discussed in the next paragraph) [24]. Of interest in this light are the articles showing an increased amount of circulating endothelial progenitors cells (CEP) [25, 26], although there are also groups reporting a conflicting lower number of CEP [27]. These latter are more in line with a hampered recruitment of the BM progenitor cells to the periphery. There was no difference between patient and healthy control CEP when looking at VE-cadherin, CD31, and vascular endothelial growth factor receptor 2 (VEGFR-2) [27, 27]. Nevertheless, the capacity to differentiate in vitro to EC is diminished in SSc progenitors, as shown by a tempered induction of vWF after prolonged culture [27]. On the other hand, healthy control (HC) and SSc CEP show an equal ability to form tubules when cultured on a Matrigel matrix in vitro [28]. Thus, in principal, SSc CEP should be able to form coherent and functional vessels.
In normal circumstances, hypoxia induces the expression of angiogenic factors to stimulate angiogenesis. One of the key angiogenic growth factors is vascular endothelial growth factor (VEGF), which is produced by, among others, fibroblasts in response to HIF-1α [6]. Despite the low presence of HIF-1α in SSc, the expression of VEGF is increased in serum and skin of SSc patients [6, 29]. The ongoing induction of VEGF must then be caused by another mechanism, like the effect of the inflammatory cytokines interleukin 1 (IL-1) and platelet-derived growth factor (PDGF). These are highly present in SSc [30, 31] and can indeed induce the expression of VEGF in fibroblasts [6].
The effects of VEGF are regulated by its receptors, VEGF receptor 1 and 2 (VEGFR-1 and VEGFR-2). From these, VEGFR-2 is the effecting molecule and VEGFR-1s main function is to regulate the phosphorylation and activation of the other [32]. In scleroderma skin, both receptors are found to be higher present with an intenser staining for VEGFR-2 than for VEGFR-1 [6]. Exposure of EC to hypoxia in vitro showed a downregulation of VEGFR-2 and upregulation of VEGFR-1 in HC and SSc samples; the latter showed a lower expression in SSc EC [28]. This differential regulation can make the cells more responsive for VEGF. Correct timing of VEGF upregulation results in recruitment of EPC and formation of new vessels, which improves the oxygen supply to the tissue. In contrast, an ongoing VEGF stimulus caused by a higher VEGF and a defective regulation due to a relative decrease in VEGFR-1 elicits the formation of aberrant vessels [33], resembling the anomalous vessels seen on nailfold capillaroscopy in SSc [2].
In conclusion, in SSc, the vascular reaction to hypoxia is dominated by the growth factor VEGF. On the other hand, there is the altered quantity and phenotype of the ECs and their progenitors, which could be a causal factor in the appearance of hypoxia in SSc. The fact that this altered phenotype is not present in the EC of patients who were treated with intensive immunotherapy and subsequent stem cell transplantation is a promising thought [17]. This could imply reversibility of the vascular defects and might open novel avenues for therapeutic intervention.
3. The Dysfunctional Immune System
It is by now generally accepted that hypoxia can induce inflammation and that inflammation itself causes hypoxia in tissues. The decrease in oxygen tension during inflammation is a logic result of the increasing metabolic demand of cells, thrombosis, and compression of the vessels due to edema (interstitial hypertension). The effect of hypoxia, causing a proinflammatory environment throughout the body, is nicely illustrated by individuals that spend time at extreme height and display increased levels of IL-6 and CRP [34]. Furthermore, we know from transplantation medicine that the expression of Toll-like receptors (TLRs) and pro-inflammatory cytokines correlates with the amount of ischemia [35, 35, 36].
The main TLR-expressing cells are the cells of the innate immune system, especially the antigen-presenting cells. In response to the hypoxic environment during inflammation, these cells need to increase their expression of HIF-1α for a proper function. This was elegantly demonstrated in phagocytes from HIF knockout mice that could not efficiently remove bacteria and get persistent ulcers [37, 38]. Differentiation and maturation of dendritic cells (DCs) is inhibited under hypoxic conditions [39] but they showed an increased production of CXCL1, VEGF, CXCL8, and CXCL10 [40, 41], all of which increased in SSc [29, 42, 43]. In contrast with these latter observations, these hypoxic DCs showed a lowered CCL2 and CCL18 production [40, 41], two pivotal chemokines that are repeatedly found to be increased in the circulation of SSc patients [43, 44]. Interestingly, other research showed an increased DC maturation and costimulatory capacity after TLR ligation during hypoxia [45, 46], suggesting that these cells are already in an activated state. Taking into account the endogenous TLR4 ligands found in SSc serum [47], the increased cytokine production in response to TLR ligands [48], and the increase in circulating cytokines mainly produced by these antigen presenting cells, it is tempting to speculate about a role for the hypoxic environment in this condition and this justifies further research to unravel this conundrum.
The effects of lowered oxygen pressure on the adaptive immune system are also quite evident. Hypoxia increases the expression of IL-10, TGF-b, galectin-1, and Foxp3, all supporting the development of regulatory T cells (Tregs) in hypoxic conditions. This is in line with several studies that reported increased numbers of Tregs in the circulation of SSc patients [49–51]. HIF-1α expression in T lymphocytes enhances the apoptotic rate and decreases T-cell function. In line with this, conditional deletion of HIF-1α in T and B cells is associated with the appearance of autoimmune responses in mice [52]. More intriguingly, the HIF-1α deficient T lymphocytes produced more pro-inflammatory cytokines in response to TCR triggering than HIF-1α expressing control cells [53, 54]. Taken into account the low HIF-1α expression in SSc patients, even in low oxygen conditions, it is tempting to suggest that the function of T lymphocytes in SSc is diverted by their impaired response to the hypoxic condition. Altogether, a hypoxic environment as present in SSc patients has clear effects on the immune system which might be augmented by an inherent defect to adept to low oxygen concentrations by SSc cells. Research focused on the effect of hypoxia on immune cells from SSc patients however is scarce and needs more attention in future investigations. Next to that, the underlying circuitry that explains this possible altered response to hypoxia by SSc cells remains elusive and warrants further investigation.
4. Fibrosis
Fibrosis is regarded to be the end stage of SSc and is often thought to cause the majority of the clinical symptoms. It is no wonder that most of the research of the last decades in this field focused on fibroblast biology and hypoxia as a causative factor for the extensive extracellular matrix (ECM) deposition.
A lower supply of oxygen to the tissue can be caused by vessel depletion or impaired diffusion. Where vasculopathy can be the cause of the former, fibrosis can lead to the latter. An important effector cell in this perivascular fibrosis is the pericyte. Pericytes can be found together with ECs around capillaries and show characteristics resembling vascular smooth muscle cells (SMCs) [55, 56]. In the perivascular area in SSc skin, pericytes with a myofibroblast phenotype have been observed, expressing alpha smooth muscle actin (α-SMA) and ED-A splice variant fibronectin (ED-A FN). These cells also show expression of the cell-surface glycoprotein Thy1 [57], which is essential in the differentiation of fibroblasts to myofibroblasts [58]. The upregulation of α-SMA in pericytes is associated with collagen production [59]. Furthermore, a reduced endothelial growth was observed when combining pericytes and EC in culture [56]. Taking together, these myofibroblast-like cells can lead to both perivascular fibrosis and endothelial dysfunction and thus cause a reduced supply of oxygen to the tissue.
The excessive production of ECM components by fibroblasts in reaction to transforming growth factor beta (TGFβ) and connective tissue growth factor (CTGF) is a paradigm in scleroderma [60]. Lately, there seems to be a paradigm shift noting that this excessive ECM production is not only due to an intrinsic defect in the fibroblasts, but also in part caused by a normal reaction of these fibroblasts to the pathological environment present in SSc patients. Indeed, in hypoxic circumstances, fibroblasts from SSc patients and HC react in the same profibrotic way. Exposure of fibroblasts to a low oxygen environment leads to HIF-1α-dependent upregulation of both TGFβ and CTGF [61–63]. Stabilization of HIF-1α, as occurs during hypoxia, also elevates the sensitivity of cells towards TGFβ, leading to a quicker TGFβ-dependent upregulation of CTGF. Herein could lay the explanation for the heightened serum CTGF in SSc [62]. Moreover, during hypoxia, the fibroblasts show an upregulation of genes involved in ECM synthesis and regulation, including fibronectin, thrombospondin, proα2(I) collagen (COL1A), and lysyl hydroxylase 2 (LH2) [64, 65]. This upregulation is in part regulated by HIF-1α, but totally dependent on TGFβ [64]. Last but not least, the hypoxic damage and oxidative stress could induce tissue damage and the subsequent release of associated molecular patterns (DAMPs), which could in turn stimulate TLRs on the fibroblasts and thus lead to activation [66].
Epithelial-to-mesenchymal transition (EMT) is a novel concept in fibrosis of lung and kidney, two severe complications of SSc. It describes a process in which epithelial cells develop mesenchymal characteristics, such as an increased expression of α-SMA and vimentin and a decrease in expression of E-cadherin. TGFβ is the prototypic stimulus of EMT [67]. EMT can also be induced by a lower oxygen pressure, as is shown by hypoxic culture of alveolar and kidney tubulus epithelial cells. The role of HIF-1α and TGFβ in this transition seems to be dependent on the origin of the epithelial cells. In alveolar cells, both molecules are required for EMT, while in kidney epithelium the process seems to be independent of these two proteins [4, 61]. Recently, EMT has been observed in the skin of a murine SSc model, suggesting that this process may be involved in both dermal and pulmonal fibrosis [68].
In contrast to the long-thought intrinsic defect in fibroblasts, data show that these cells respond in a normal way to abnormal circumstances. Taking the above mentioned together, there is evidence that in SSc hypoxia can lead to differentiation and activation of fibroblasts but might have similar effects on primary immune cells all contributing to an extensive ECM deposition.
5. Conclusion
Most human cells are able to adapt to a broad range of oxygen tension varying between 40–100 mmHg in the circulation and 4–20 mmHg in tissue. In contrast to these physiological differences, in certain condition pathological hypoxia develops. In SSc patients pathological hypoxia is obvious only by looking at their hands and seeing the digital ulcers patients suffer from. The response to this loss of oxygen has to be well coordinated and tightly regulated by factors like HIF-1α. The hypoxia caused by a combination of activated immune cells (increasing metabolic demands), rarefaction of blood vessels, and perivascular fibrosis becomes detrimental without this regulation. In SSc, there is no sign of increasing HIF-1α activity and therefore the low oxygen tension can go on activating fibroblasts (etc.), aggravating immune responses, and impairing endothelial cells and thereby further increasing the hypoxic environment. Moreover, the ongoing hypoxia will increase TGFβ levels and will therefore increase the production of extracellular matrix, further increasing fibrosis and thus the distance to the closest blood vessel [64, 69, 69]. Interestingly, another detrimental effect of hypoxia is shown in cancer research by the profibrotic and proinflammatory effect of the increased reactive oxygen species and autophagy caused by low oxygen tension [70]. Eventually, it seems inescapable that the system will choke in its own vicious circle. As shown by the effect of stem cell therapy [17], there are options to interfere in this ongoing process. For example, one would be able to interfere in the impaired response to hypoxia more specifically by the use of propyl-hydroxylase inhibitors (reviewed in [71]). In addition, the pharmacological increase of bilirubin by the use of atazanavir could directly provide more anti-inflammatory agents thereby bypassing the dysfunctional hypoxic pathway [72]. These therapeutic interventions could be given systemically but, alternatively, could also be specifically targeted to certain cells of the immune cells by exploiting recent knowledge on liposomes or even nanoparticles. In conclusion, the research field on therapeutic interventions in detrimental hypoxia is growing which will hopefully lead to the broadening of our therapeutic armamentarium to treat this disease.
Acknowledgment
The autors would like to thank T. Oort for his help with the graphic design of the figure.
References
- 1.Jimenez SA, Derk CT. Following the Molecular Pathways toward an Understanding of the Pathogenesis of Systemic Sclerosis. Annals of Internal Medicine. 2004;140(1):37–50. [PubMed] [Google Scholar]
- 2.Cutolo M, Grassi W, Cerinic MM. Raynaud’s phenomenon and the role of capillaroscopy. Arthritis and Rheumatism. 2003;48(11):3023–3030. doi: 10.1002/art.11310. [DOI] [PubMed] [Google Scholar]
- 3.Maricq HR, LeRoy EC. Correlation of capillary morphology and flow during cold exposure in Raynaud syndrome. Bibliotheca Anatomica. 1979;(18):382–384. [PubMed] [Google Scholar]
- 4.Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. Journal of Clinical Investigation. 2007;117(12):3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silverstein JL, Steen VD, Medsger TA, Falanga V. Cutaneous hypoxia in patients with systemic sclerosis (scleroderma) Archives of Dermatology. 1988;124(9):1379–1382. [PubMed] [Google Scholar]
- 6.Distler O, Distler JHW, Scheid A, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circulation Research. 2004;95(1):109–116. doi: 10.1161/01.RES.0000134644.89917.96. [DOI] [PubMed] [Google Scholar]
- 7.Stein CM, Tanner SB, Awad JA, Roberts LJ, Morrow JD. Evidence of free radical-mediated injury (isoprostane overproduction) in scleroderma. Arthritis and Rheumatism. 1996;39(7):1146–1150. doi: 10.1002/art.1780390711. [DOI] [PubMed] [Google Scholar]
- 8.Lau CS, Bridges AB, Muir A, Scott N, Bancroft A, Belch JJF. Further evidence of increased polymorphonuclear cell activity in patients with Raynaud’s phenomenon. British Journal of Rheumatology. 1992;31(6):375–380. doi: 10.1093/rheumatology/31.6.375. [DOI] [PubMed] [Google Scholar]
- 9.Ho KT, Reveille JD. The clinical relevance of autoantibodies in scleroderma. Arthritis Research and Therapy. 2003;5(2):80–93. doi: 10.1186/ar628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brihimi-Horn MC, Pouysségur J. HIF at a glance. Journal of Cell Science. 2009;122(8):1055–1057. doi: 10.1242/jcs.035022. [DOI] [PubMed] [Google Scholar]
- 11.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 12.Ivan M, Haberberger T, Gervasi DC, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13459–13464. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beyer C, Schett G, Gay S, Distler O, Distler JH. Hypoxia. Hypoxia in the pathogenesis of systemic sclerosis. Arthritis Research & Therapy. 2009;11(2):p. 220. doi: 10.1186/ar2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? Journal of Clinical Investigation. 2007;117(4):862–865. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wipff J, Dieude P, Avouac J, et al. Association of hypoxia-inducible factor 1A (HIF1A) gene polymorphisms with systemic sclerosis in a French European Caucasian population. Scandinavian Journal of Rheumatology. 2009;38(4):291–294. doi: 10.1080/03009740802629432. [DOI] [PubMed] [Google Scholar]
- 16.Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. Journal of Clinical Investigation. 1996;98(3):785–792. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming JN, Nash RA, McLeod DO, et al. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001452. Article ID e1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen VA, Sgonc R, Dietrich H, Wick G. Endothelial in jury in internal organs of University of California at Davis line 200 (UCD 200) chickens, an animal model for systemic sclerosis (Scleroderma) Journal of Autoimmunity. 2000;14(2):143–149. doi: 10.1006/jaut.1999.0355. [DOI] [PubMed] [Google Scholar]
- 19.Rosenbaum J, Pottinger BE, Woo P, et al. Measurement and characterisation of circulating anti-endothelial cell IgG in connective tissue diseases. Clinical and Experimental Immunology. 1988;72(3):450–456. [PMC free article] [PubMed] [Google Scholar]
- 20.Holt CM, Lindsey N, Moult J, et al. Antibody-dependent cellular cytotoxicity of vascular endothelium: characterization and pathogenic associations in systemic sclerosis. Clinical and Experimental Immunology. 1989;78(3):359–365. [PMC free article] [PubMed] [Google Scholar]
- 21.Penning CA, Cunningham J, French MAH. Antibody-dependent cellular cytotoxicity of human vascular endothelium in systemic sclerosis. Clinical and Experimental Immunology. 1984;57(3):548–556. [PMC free article] [PubMed] [Google Scholar]
- 22.Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis and Rheumatism. 2000;43(11):2550–2562. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Del PN, Quirici N, Scavullo C, et al. Antiendothelial cell antibodies induce apoptosis of bone marrow endothelial progenitors in systemic sclerosis. Journal of Rheumatology. 2010;37(10):2053–2063. doi: 10.3899/jrheum.091346. [DOI] [PubMed] [Google Scholar]
- 24.Gill M, Dias S, Hattori K, et al. Vascular trauma induces rapid but transient mobilization of VEGFR2+ AC133+ endothelial precursor cells. Circulation Research. 2001;88(2):167–174. doi: 10.1161/01.res.88.2.167. [DOI] [PubMed] [Google Scholar]
- 25.Avouac J, Juin F, Wipff J, et al. Circulating endothelial progenitor cells in systemic sclerosis: association with disease severity. Annals of the Rheumatic Diseases. 2008;67(10):1455–1460. doi: 10.1136/ard.2007.082131. [DOI] [PubMed] [Google Scholar]
- 26.Del PN, Colombo G, Fracchiolla N, et al. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis and Rheumatism. 2004;50(4):1296–1304. doi: 10.1002/art.20116. [DOI] [PubMed] [Google Scholar]
- 27.Kuwana M, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y. Defective vasculogenesis in systemic sclerosis. Lancet. 2004;364(9434):603–610. doi: 10.1016/S0140-6736(04)16853-0. [DOI] [PubMed] [Google Scholar]
- 28.Avouac J, Wipff J, Goldman O, et al. Angiogenesis in systemic sclerosis: impaired expression of vascular endothelial growth factor receptor 1 in endothelial progenitor-derived cells under hypoxic conditions. Arthritis and Rheumatism. 2008;58(11):3550–3561. doi: 10.1002/art.23968. [DOI] [PubMed] [Google Scholar]
- 29.Distler O, Del RA, Giacomelli R, et al. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Research. 2002;4(6):p. R11. doi: 10.1186/ar596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umehara H, Kumagai S, Murakami M, et al. Enhanced production of interleukin-1 and tumor necrosis factor α by cultured peripheral blood monocytes from patients with scleroderma. Arthritis and Rheumatism. 1990;33(6):893–897. doi: 10.1002/art.1780330619. [DOI] [PubMed] [Google Scholar]
- 31.Gay S, Jones RE, Huang GQ, Gay RE. Immunohistologic demonstration of platelet-derived growth factor (PDGF) and sis-oncogene expression in scleroderma. Journal of Investigative Dermatology. 1989;92(2):301–303. doi: 10.1111/1523-1747.ep12276895. [DOI] [PubMed] [Google Scholar]
- 32.Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. American Journal of Pathology. 2004;164(5):1531–1535. doi: 10.1016/S0002-9440(10)63711-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dor Y, Djonov V, Abramovitch R, et al. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO Journal. 2002;21(8):1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartmann G, Tschöp M, Fischer R, et al. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine. 2000;12(3):246–252. doi: 10.1006/cyto.1999.0533. [DOI] [PubMed] [Google Scholar]
- 35.Krüger B, Krick S, Dhillon N, et al. Donor toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(9):3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrade CF, Kaneda H, Der S, et al. Toll-like receptor and cytokine gene expression in the early phase of human lung transplantation. Journal of Heart and Lung Transplantation. 2006;25(11):1317–1323. doi: 10.1016/j.healun.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 37.Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyssonnaux C, Datta V, Cramer T, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. Journal of Clinical Investigation. 2005;115(7):1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mancino A, Schioppa T, Larghi P, et al. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008;112(9):3723–3734. doi: 10.1182/blood-2008-02-142091. [DOI] [PubMed] [Google Scholar]
- 40.Bosco MC, Puppo M, Santangelo C, et al. Hypoxia modifies the transcriptome of primary human monocytes: modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. Journal of Immunology. 2006;177(3):1941–1955. doi: 10.4049/jimmunol.177.3.1941. [DOI] [PubMed] [Google Scholar]
- 41.Elia AR, Cappello P, Puppo M, et al. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. Journal of Leukocyte Biology. 2008;84(6):1472–1482. doi: 10.1189/jlb.0208082. [DOI] [PubMed] [Google Scholar]
- 42.Furuse S, Fujii H, Kaburagi Y, et al. Serum concentrations of the CXC chemokines interleukin 8 and growth-regulated oncogene-α are elevated in patients with systemic sclerosis. Journal of Rheumatology. 2003;30(7):1524–1528. [PubMed] [Google Scholar]
- 43.Antonelli A, Ferri C, Fallahi P, et al. CXCL10 (α) and CCL2 (β) chemokines in systemic sclerosis—a longitudinal study. Rheumatology. 2008;47(1):45–49. doi: 10.1093/rheumatology/kem313. [DOI] [PubMed] [Google Scholar]
- 44.van Lieshout AWT, Vonk MC, Bredie SJH, et al. Enhanced interleukin-10 production by dendritic cells upon stimulation with Toll-like receptor 4 agonists in systemic sclerosis that is possibly implicated in CCL18 secretion. Scandinavian Journal of Rheumatology. 2009;38(4):282–290. doi: 10.1080/03009740802572467. [DOI] [PubMed] [Google Scholar]
- 45.Jantsch J, Chakravortty D, Turza N, et al. Hypoxia and hypoxia-inducible factor-la modulate lipopolysaccharide-induced dendritic cell activation and function. Journal of Immunology. 2008;180(7):4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 46.Spirig R, Djafarzadeh S, Regueira T, et al. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1α and dendritic cell maturation under normoxic conditions. PLoS ONE. 2010;5(6) doi: 10.1371/journal.pone.0010983. Article ID e10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roelofs MF, Joosten LAB, Abdollahi-Roodsaz S, et al. The expression of Toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of Toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis and Rheumatism. 2005;52(8):2313–2322. doi: 10.1002/art.21278. [DOI] [PubMed] [Google Scholar]
- 48.van Bon L, Popa C, Huijbens R, et al. Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Annals of the Rheumatic Diseases. 2010;69(8):1539–1547. doi: 10.1136/ard.2009.128207. [DOI] [PubMed] [Google Scholar]
- 49.Radstake TRDJ, van Bon L, Broen J, et al. Increased frequency and compromised function of T regulatory cells in systemic sclerosis (SSc) is related to a diminished CD69 and TGFβ expression. PLoS ONE. 2009;4(6) doi: 10.1371/journal.pone.0005981. Article ID e5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giovannetti A, Rosato E, Renzi C, et al. Analyses of T cell phenotype and function reveal an altered T cell homeostasis in systemic sclerosis. Correlations with disease severity and phenotypes. Clinical Immunology. 2010;137(1):122–133. doi: 10.1016/j.clim.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Slobodin G, Ahmad MS, Rosner I, et al. Regulatory T cells (CD4+CD25brightFoxP3+) expansion in systemic sclerosis correlates with disease activity and severity. Cellular Immunology. 2010;261(2):77–80. doi: 10.1016/j.cellimm.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 52.Kojima H, Gu H, Nomura S, et al. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1α-deficient chimeric mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(4):2170–2174. doi: 10.1073/pnas.052706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lukashev D, Klebanov B, Kojima H, et al. Cutting edge: hypoxia-inducible factor 1α and its activation-inducible short isoform I.1 negatively regulate functions of CD4 + and CD8+ T lymphocytes. Journal of Immunology. 2006;177(8):4962–4965. doi: 10.4049/jimmunol.177.8.4962. [DOI] [PubMed] [Google Scholar]
- 54.Thiel M, Chouker A, Ohta A, et al. Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biology. 2005;3(6):p. e174. doi: 10.1371/journal.pbio.0030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circulation Research. 2005;97(6):512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 56.Hirschi KK, D’Amore PA. Pericytes in the microvasculature. Cardiovascular Research. 1996;32(4):687–698. [PubMed] [Google Scholar]
- 57.Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Research & Therapy. 2005;7(5):R1113–R1123. doi: 10.1186/ar1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koumas L, Smith TJ, Feldon S, Blumberg N, Phipps RP. Thy-1 expression in human fibroblast subsets defines myofibroblastic or lipofibroblastic phenotypes. American Journal of Pathology. 2003;163(4):1291–1300. doi: 10.1016/S0002-9440(10)63488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. American Journal of Pathology. 2008;173(6):1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LeRoy EC. Increased collagen synthesis by scleroderma skin fibroblasts in vitro. A possible defect in the regulation or activation of the scleroderma fibroblast. Journal of Clinical Investigation. 1974;54(4):880–889. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou G, Dada LA, Wu M, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. American Journal of Physiology, Lung Cellular and Molecular Physiology. 2009;297(6):L1120–L1130. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong KH, Yoo SA, Kang SS, Choi JJ, Kim WU, Cho CS. Hypoxia induces expression of connective tissue growth factor in scleroderma skin fibroblasts. Clinical and Experimental Immunology. 2006;146(2):362–370. doi: 10.1111/j.1365-2249.2006.03199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of Ctgf is directly mediated by Hif-1. American Journal of Physiology, Renal Physiology. 2004;287(6):F1223–F1232. doi: 10.1152/ajprenal.00245.2004. [DOI] [PubMed] [Google Scholar]
- 64.Distler JHW, Jüngel A, Pileckyte M, et al. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis and Rheumatism. 2007;56(12):4203–4215. doi: 10.1002/art.23074. [DOI] [PubMed] [Google Scholar]
- 65.Brinckmann J, Kim S, Wu J, et al. Interleukin 4 and prolonged hypoxia induce a higher gene expression of lysyl hydroxylase 2 and an altered cross-link pattern: important pathogenetic steps in early and late stage of systemic scleroderma? Matrix Biology. 2005;24(7):459–468. doi: 10.1016/j.matbio.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Wei J, Bhattacharyya S, Tourtellotte WG, Varga J. Fibrosis in systemic sclerosis: emerging concepts and implications for targeted therapy. Autoimmunity Reviews. 2010;10:267–275. doi: 10.1016/j.autrev.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nature Reviews Molecular Cell Biology. 2006;7(2):131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 68.Xu H, Zaidi M, Struve J, et al. Abnormal fibrillin-1 expression and chronic oxidative stress mediate endothelial mesenchymal transition in a murine model of systemic sclerosis. American Journal of Physiology - Cell Physiology. 2011;300(3):C550–C556. doi: 10.1152/ajpcell.00123.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Falanga V, Tiegs SL, Alstadt SP. Transforming growth factor-beta: selective increase in glycosaminoglycan synthesis by cultures of fibroblasts from patients with progressive systemic sclerosis. Journal of Investigative Dermatology. 1987;89(1):100–104. doi: 10.1111/1523-1747.ep12580445. [DOI] [PubMed] [Google Scholar]
- 70.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, et al. Understanding the "lethal" drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biology and Therapy. 2010;10(6):537–542. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyata T, Takizawa S, Van Ypersele De Strihou C. Hypoxia. 1. Intracellular sensors for oxygen and oxidative stress: novel therapeutic targets. American Journal of Physiology, Cell Physiology. 2011;300(2):C226–C231. doi: 10.1152/ajpcell.00430.2010. [DOI] [PubMed] [Google Scholar]
- 72.Dekker D, Dorresteijn MJ, Pijnenburg M, et al. The bilirubin-increasing drug atazanavir improves endothelial function in patients with type 2 diabetes mellitus. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(2):458–463. doi: 10.1161/ATVBAHA.110.211789. [DOI] [PubMed] [Google Scholar]