

Abstract
Background:
The occurrence of primitive neuroectodermal tumors (PNET) in patients with neurofibromatosis type 1 (NF1) has only been reported in two other cases in English-Language literature. Owing to the rarity of intraspinal PNET and the extremely high gene mutation variability in NF1, there is currently no conclusive evidence to suggest that PNET is associated with NF1. Here, we report a case of intradural PNET in a patient with NF1.
Case Description:
A 27-year-old male underwent a C1-C3 laminectomy for resection of an intramedullary mass. Histopathology and immunohistopathology analysis was performed. Microscopic examination and immunohistochemical staining indicated the mass was a primitive neuroectodermal tumor. Within 1 month after tumor resection, the patient developed leptomeningeal carcinomatosis. The patient was not a candidate for radiation therapy but underwent palliative systemic chemotherapy. He subsequently developed neutropenia and died 3 months after tumor resection.
Conclusion:
To our knowledge, this is the first reported intraspinal PNET associated with NF1. Genetic analysis of CNS PNETs suggests a possible correlation, but larger case series are needed to support this theory.
Keywords: Intramedullary primitive neuroectodermal tumors, neurofibromatosis type 1, primitive neuroectodermal tumor
INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal dominant neurocutaneous disorder attributed to mutations in the tumor suppressor gene, NF1, located on chromosome 17q11.2.[41,42] Although it is characterized by almost 100% penetrance, variable expression produces a wide range of clinical manifestations of the disorder.[41,42] As such, two or more of the recognized diagnostic criteria for NF1 are required for diagnosis [Table 1].
Table 1.

Although NF1 is the most common neurocutaneous disorder and is associated with numerous tumors - such as astrocytomas, optic nerve gliomas, and neurofibromas - the occurrence of a primitive neuroectodermal tumor in a patient with NF1 has only been described in two cases in the English language literature.[4,43] Primitive neuroectodermal tumors (PNET) are divided into CNS primitive neuroectodermal tumors (cPNET) and peripheral primitive neuroectodermal tumors (pPNET). cPNETs refer to a group of embryonal tumors and include CNS neuroblastoma, CNS ganglioblastoma, medulloepithelioma, and ependymoblastoma.[29,40] Peripheral primitive neuroectodermal tumors most commonly occur in soft tissue and bone outside of the nervous system and are classified with Ewing Sarcoma in the Ewing family of tumors due to their shared histological resemblance and the high prevalence of t(11;22)(q24;q12) chromosomal translocation.[3,15]
Although there has been an increased number of reported cases of intraspinal PNET over the past decade, it remains a relatively rare entity.[10] Of the 82 cases of intraspinal PNET described in the English language literature to date, only 25 have been reported as primary intramedullary lesions. Here we report a case of intramedullary spinal cord PNET in a patient with NF1.
CASE REPORT
History and physical examination
A 27-year-old man with no significant past medical history was referred to our institution from an outside facility with mild myelopathy and gait instability. The patient reported a 2-month history of intermittent neck pain and 2 weeks of right upper extremity weakness and numbness accompanied by progressive gait instability. Physical exam revealed one café au lait spot on his front chest, axillary freckling, decreased vibratory sensation along right upper extremity, positive Romberg test, dysmetria on finger to nose with the right upper extremity only, and decreased right upper and lower extremity strength (4/5 biceps, triceps, hip flexion, and plantar flexion).
An MRI of the brain and neck revealed a cervical intramedullary mass extending from the medulla to the C4 level and detailed the presence of several other masses including: 1) a dumbbell-shaped enhancing mass expanding the left C5-C6 neural foramen 2) a right paraspinous soft tissue neck mass hyperintense on T2 with heterogenous enhancement 3) a dumbbell-shaped hetergeneously enhancing mass expanding the left T10-T11 neural foramen and 4) an ill-defined paraspinal paraortic enhancing soft tissue mass extending from T9-T11 [Figure 1].
Figure 1.
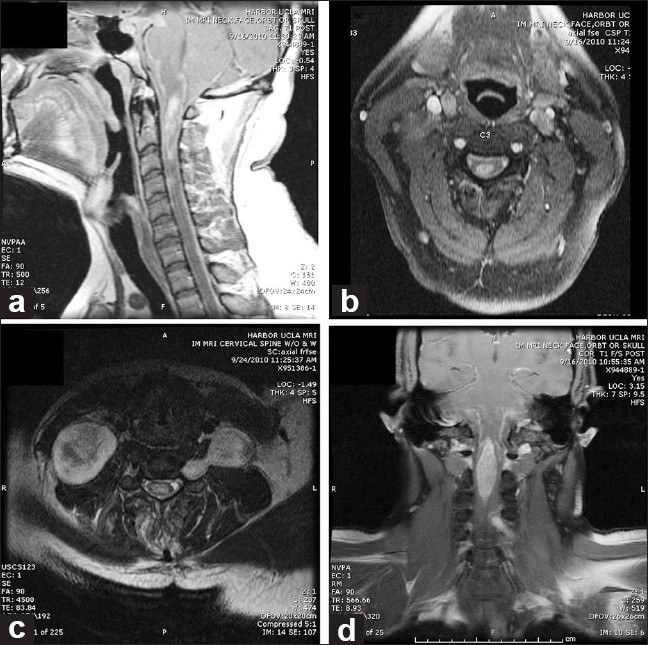
MRI of the neck with gadolineum. a) Sagittal T1 image illustrating an intramedullary enhancing mass from the cervicomedullary junction to C4. Leptomeningeal enhancement is also present. b) Axial T1 image illustrating the intramedullary mass and leptomeningeal enhancement. c) Axial image of cervical spine illustrating a dumbbell mass extending through C5-C6 neural foramen and paraspinal mass. d) Coronal T1 image illustrating intramedullary mass
The presence of multiple parapsinal masses with characteristics of nerve sheath tumors and the presence of axillary freckling resulted in a clinical diagnosis of neurofibromatosis type 1. The patient indicated he had no known family history of NF1 and had not been previously diagnosed with NF1 or any prior malignancy.
Procedure
The patient was recommended to undergo excisional biopsy of the intramedullary spine lesion as it was the likely symptomatic lesion. The peripheral nerve lesions – likely neurofibromas – were recommended to be observed. The patient agreed to the above, understanding the risks and benefits and subsequently underwent a wide C1 to C3 laminectomy microscopic resection of the intramedullary mass. Intraoperatively the mass was found to be a large, exophytic lesion extremely adherent to surrounding spinal cord tissue. Approximately 80% of the tumor was resected, with further resection halted when cardiovascular instability was encountered.
Pathological examination
Microscopic examination of the tumor revealed a highly cellular, poorly differentiated neoplasm with focal areas of necrosis. The cells exhibited characteristic scanty cytoplasm, intense basophilic and pleomorphic nuclei, and atypical mitosis [Figure 2]. Immunohistochemical staining was positive for GFAP, CD99 and synaptophysin [Figure 3].
Figure 2.
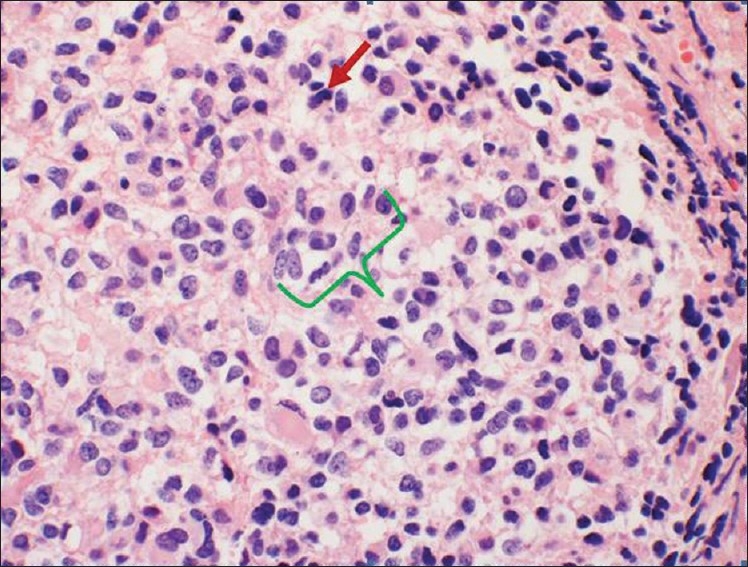
HandE staining illustrating small round blue cells with high mitotic rate, atypical mitosis (red arrow), and pleomorphic nuclei (green bracket) characteristic of a primitive neuroectodermal tumor
Figure 3.
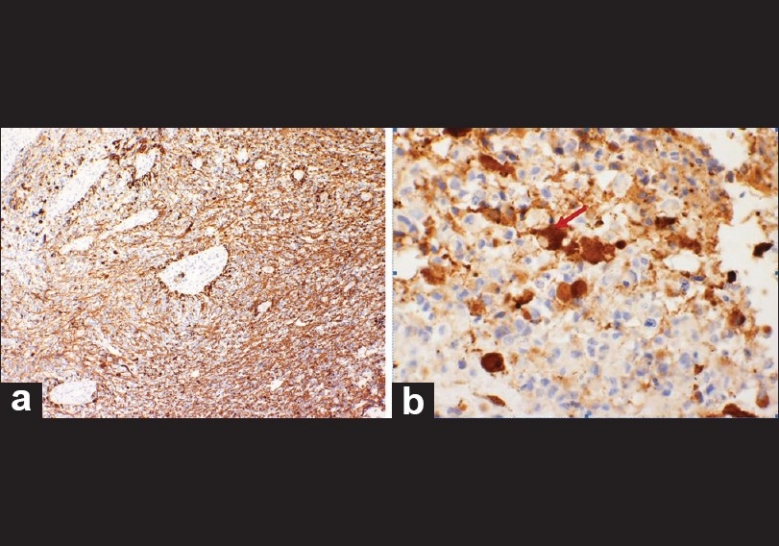
Immunohistomchemical staining. a) GFAP positive b) Synapthophysin positive, illustrating positivity around a binucleated cell (red arrow)
Postoperative course and treatment
During the immediate postoperative period, the patient's neurological exam remained at baseline. A postoperative MRI of the spine illustrated a small amount residual intramedullary tumor, largely localized in the superior region of the tumor. The patient was referred for radiation and chemotherapy. However, about 1 month post-operatively the patient developed a sudden loss of vision. An MRI of the orbits revealed abnormal enhancement of the optic nerve sheaths bilaterally, a nodular enhancement posterior to the optic chiasm, and diffuse basilar meningeal nodularity and enhancement consistent with leptomeningeal carcinomatosis. As such, the patient was not a candidate for radiation therapy. The patient underwent palliative chemotherapy, which was complicated by neutropenia. During that hospitalilzation, the patient experienced cardiopulmonary collapse, was unable to be resuscitated and subsequently expired. An autopsy was not performed.
DISCUSSION
Neurofibromatosis type 1 is the most common neurocutaneous disorder in the United States with prevalence estimates ranging from 1 per 2000 to 1 per 5000 people.[41] It has been well-documented that people with neurofibromatosis type 1 are at increased risk for certain malignancies including optic gliomas, malignant peripheral nerve sheath tumors, non-lymphocytic leukemias, rhabdomyosarcomas and pheochromocytomas.[24,41,43] In larger studies examining the association between brain tumors and NF1, other gliomas such as cerebellar astrocytomas, ependymomas, third ventricle astrocytomas, cerebral astrocytomas, brain stem gliomas, and spinal cord tumors were not only found to be increased in patients with NF1, but the frequency of malignant change in adults with NF1 was also found to be higher when compared to controls.[24] Although not confirmed by larger cohort studies of NF1, cases of Wilm's tumor, medulloblastoma, and neuroblastoma have also been reported.[24,30] The lack of more substantial data to demonstrate the association between these tumors and NF1 may partly be due to the relative rarity of these tumors.
Primary intraspinal PNET is a rare entity, with 82 cases reported in the literature to date. Interestingly, this tumor occurs more frequently in the cauda equina and rarely occurs as a primary intramedullary tumor.[1,6,8,9,11,14,18,26,27,30,31,33,38,43,45] Before a diagnosis of primary intrapsinal PNET can be made, the presence of any intracranial neoplasm must be eliminated to ensure the spinal cord lesion is not a drop metastasis. In our patient, no preoperative evidence of an intracranial neoplasm was found, confirming primary intrapsinal PNET. Without immunohistochemical staining or cytogenetic analysis it is difficult to differentiate intraspinal cPNET from pPNET, as histologically they are identical, consisting of small round, undifferentiated neuroepithelial cells with a high mitotic rate.[21] One role of immunohistochemistry is to determine whether an intrapsinal PNET originates peripherally or from the CNS. Peripheral PNET strongly expresses the glycoprotein p30/32 (CD99) encoded by the MIC2 gene while cPNET does not.[15,21] Although cytogenetic analysis demonstrating a translocation of (11;22)(q24;q12) is the definitive way to differentiate between pPNET and cPNET, MIC2/CD99 positivity is generally considered an appropriate method to distinguish cPNET from pPNET.[10,13,15,17,21,28] This distinction would be less important if cPNET and pPNET could not present in the same location. However, even though theoretically an intramedullary PNET should be central in origin, several cases of primary intramedullary pPNET have been confirmed [Table 1].[2,6,27] Our case represents the 25th case of primary intramedullary PNET reported in the literature and the 5th case of confirmed intramedullary peripheral PNET.
The two reported occurrences of PNET in NF1 include a 37-year-old male with a lumbar intraspinal pPNET and a 1-month-old male child with pPNET originating from within a plexiform neurofibroma.[4,43]
In the first case, the man presented with back pain and right lower extremity weakness. On imaging, an epidural lesion from L3-L5 that extended through the foramina into a paravertebral mass was noted. The mass was partially resected during an L3-L5 laminectomy and patient showed a slight clinical improvement postoperatively. Immunohistochemistry of the mass was positive for CD99, vimentin, enolase, neurofilaments, synaptophysin and was negative for GFAP, S100, EMA, LCA and chromograine. The patient was treated with lumbosacral radiation of 30 Gy (limited due to radiation side effects), was unable to receive chemotherapy, and died 6 months later.[43]
In the second case, an infant presented at birth with 3 café au lait spots and a large plexiform neurofibroma covering his anterior abdominal wall, lower back, buttock, and upper part of the thighs including the scrotum. In 2 month's time a new mass appeared over his left flank and right groin over the existing neurofibroma. The child underwent chemotherapy but the tumor metastasized and the child died at 12 months of age. Histological examination of the skin and tumor tissue demonstrated a mixed picture- with some areas having a characteristic appearance of plexiform neurofibroma and some areas consisting of small cells with a high nucleocytoplasmic ratio. Immunohistochemical staining was positive for S100 but negative for NSE. Cytogenetic studies revealed a hyportriploidy and structural abnormalities of chromosomes 13 and 14 but no abnormality of chromosome 17.[4] Although both of these cases are examples of the occurrence of peripheral PNET in patients with NF1, the site of origin of these lesions and that of the case reported here are dramatically different. Even after the intraspinal PNET metastasized in the first case, there was no intramedullary component. As such, our case is the first documented case of intramedullary PNET in a patient with NF1.
Including the present case, most documented cases of intramedullary pPNET were in male patients with an age range of 17-38 years at time of diagnosis. The demographics of these cases resonate with a recent review that reiterated that the majority of intraspinal pPNET occurs in young adult males.[2,10,16] However, as Table 2 illustrates, pPNET was only present in half of the cases of intramedullary PNET in which a distinction between pPNET and cPNET could be made.
Table 2.
Cases of intramedullary primitive neuroectodermal tumors
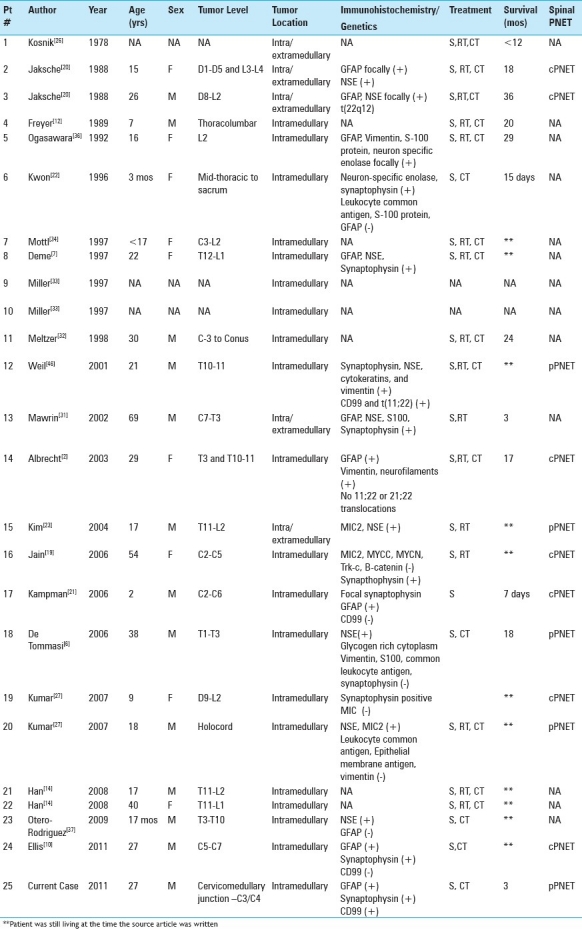
As alluded to above, the genetic distinction between these two entities is relatively clear, with pPNET demonstrating the chromosomal translocation t(11;22)(q24;q12) and cPNET being associated with isochromosome 17q.[3,15,16,21,39] However, few studies have examined the possible relationship between PNET and NF1 on the basis of their shared chromosome.[30,35,44]
The NF1 gene is expressed in various cells throughout the body including neurons, glial cells, and cells derived from the neural crest.[25] Under normal conditions, the protein product of the NF1 gene (neurofibromin) controls cell division by inhibiting the Ras oncogene.[25,35] In NF1, mutations produce either defective neurofibromin or insufficient quantities of neurofibromin- resulting in uncontrolled cell division.[25] Additionally, three exons in the NF1 gene are known to undergo alternative splicing- producing slightly different forms of neurofibromin. Although alternative splicing is a normal phenomenon, it may be one contributor to the varied clinical manifestations of NF1.[25]
Several studies have examined the presence of two specific splicing variants of the NF1 gene. Both Scheurlen et al., and Nishi et al., found a NF1-GRD splicing variant in primitive neuroectodermal tumors and suggest that this splicing variant might interfere with differentiation of neuroectodermal tissues- contributing to tumor formation.[35,44] Nishi et al., found the NF1-GRD splicing variant to be increased in undifferentiated primitive neuroectodermal tumors (under which they included neuroblastoma, medulloblastoma, and retinoblastoma). Scheurlen et al., found the NF1-GRD splicing variant to only be increased in PNET and immature teratoma when they scanned numerous pediatric brain tumors for the variant. The prevalence of this splicing variant in primitive neuroectodermal tumors points to an association between NF1 and PNET.
However, the studies examining the possible association between PNET and NF1 did not categorize PNET into cPNET and pPNET and did not conduct cytogentic analysis to determine if the chromosomal translocation t(11;22)(q24;q12) was present in the same tumors in which the NF1-GRD splicing variant was prevalent. The chromosomal translocation t(11;22)(q24;q12) present in pPNET produces a fusion protein that can act as a transcriptional activator, leading to aberrant cell growth.[5] Although this mechanism is distinct from the suspected role of NF1 and tumor formation, further cytogenetic analysis and detailed classification of spinal cord tumors in patients with NF1 may shed some light on the possible link between PNET and NF1.
CONCLUSIONS
Patients with NF1 are at increased risk for developing many different malignancies, including those not traditionally associated with NF1. Although intraspinal PNET is a very rare condition and has not been reported in patients with NF1, based on several genetic studies, an association is still plausible. Further cytogenetic analysis of patients presenting with intraspinal PNET should be conducted to better understand the genetic makeup and pathophysiology of the neoplasm.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2011/2/1/155/86835
Contributor Information
Celene B. Mulholland, Email: cmulholland@mednet.ucla.edu.
Garni Barkhoudarian, Email: gbarkhoudarian@mednet.ucla.edu.
Marcia E. Cornford, Email: mcornford@dhs.lacounty.gov.
Duncan Q. McBride, Email: dmcbride@mednet.ucla.edu.
REFERENCES
- 1.Akyüz M, Demiral AN, Gürer IE, Uçar T, Tuncer R. Primary primitive neuro-ectodermal tumor of cauda equina with intracranial seeding. Acta Neurochir (Wien) 2004;146:525–8. doi: 10.1007/s00701-003-0212-1. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht CF, Weiss E, Schulz-Schaeffer WJ, Albrecht T, Fauser S, Wickboldt J, et al. Primary intraspinal primitive neuroectodermal tumor: Report of two cases and review of the literature. J Neurooncol. 2003;61:113–20. doi: 10.1023/a:1022118317876. [DOI] [PubMed] [Google Scholar]
- 3.Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for ewing's sarcoma and peripheral primitive neuroectodermal tumors.Evidence for a common histogenesis of ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67:1886–93. doi: 10.1002/1097-0142(19910401)67:7<1886::aid-cncr2820670712>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 4.Chan GC, Nicholls JM, Lee AC, Chan LC, Lau YL. Malignant peripheral neuroectodermal tumor in an infant with neurofibromatosis type 1. Med Pediatr Oncol. 1996;26:215–9. doi: 10.1002/(SICI)1096-911X(199603)26:3<215::AID-MPO12>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 5.De Alava E, Gerald WL. Molecular biology of the Ewing's sarcoma/primitive neuroectodermal tumor family. J Clin Oncol. 2000;18:204. doi: 10.1200/JCO.2000.18.1.204. [DOI] [PubMed] [Google Scholar]
- 6.De Tommasi A, De Tommasi C, Occhiogrosso G, Cimmino A, Parisi M, Sanguedolce F, et al. Primary intramedullary primitive neuroectodermal tumor (PNET) – Case report and review of the literature. Eur J Neurol. 2006;13:240–3. doi: 10.1111/j.1468-1331.2006.01183.x. [DOI] [PubMed] [Google Scholar]
- 7.Deme S, Ang L, Skaf G, Rowed D. Primary intramedullary primitive neuroectodermal tumor of the spinal cord: Case report and review of the literature. Neurosurgery. 1997;41:1417. doi: 10.1097/00006123-199712000-00040. [DOI] [PubMed] [Google Scholar]
- 8.Dhatt S, Dhillon MS, Tripathy SK, Goyal T, Jagadeesh V. Peripheral primitive neuroectodermal tumor causing cauda equina syndrome with destruction of L5 vertebra. Indian J Orthop. 2010;44:339–41. doi: 10.4103/0019-5413.65153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorfmüller G, Würtz FG, Umschaden HW, Kleinert R, Ambros PF. Intraspinal primitive neuroectodermal tumour: Report of two cases and review of the literature. Acta Neurochir (Wien) 1999;141:1169–75. doi: 10.1007/s007010050414. [DOI] [PubMed] [Google Scholar]
- 10.Ellis JA, Rothrock RJ, Moise G, McCormick PC, Tanji K, Canoll P, et al. Primitive neuroectodermal tumors of the spine: A comprehensive review with illustrative clinical cases. Neurosurg Focus. 2011;30:E1. doi: 10.3171/2010.10.FOCUS10217. [DOI] [PubMed] [Google Scholar]
- 11.Fabre E, Guillevin R, Chretien F, Le Guerinel C, Duffau H. Peripheral primitive neuroectodermal tumor of the cauda equina in an elderly patient. J Neurosurg Spine. 2006;5:68–71. doi: 10.3171/spi.2006.5.1.68. [DOI] [PubMed] [Google Scholar]
- 12.Freyer DR, Hutchinson J, McKeever PE. Primary primitive neuroectodermal tumor of the spinal cord associated with neural tube defect. Pediatr Neurosurg. 1989;15:181–7. doi: 10.1159/000120466. [DOI] [PubMed] [Google Scholar]
- 13.Furuno Y, Nishimura S, Kamiyama H, Numagami Y, Saito A, Kaimori M, et al. Intracranial peripheral-type primitive neuroectodermal tumor case report. Neurol Med Chir (Tokyo) 2008;48:72–6. doi: 10.2176/nmc.48.72. [DOI] [PubMed] [Google Scholar]
- 14.Han IH, Kuh SU, Chin DK, Kim KS, Jin BH, Cho YE. Surgical treatment of primary spinal tumors in the conus medullaris. J Korean Neurosurg Soc. 2008;44:72–7. doi: 10.3340/jkns.2008.44.2.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harimaya K, Oda Y, Matsuda S, Tanaka K, Chuman H, Iwamoto Y. Primitive neuroectodermal tumor and extraskeletal Ewing sarcoma arising primarily around the spinal column: Report of four cases and a review of the literature. Spine (Phila Pa 1976) 2003;28:E408–12. doi: 10.1097/01.BRS.0000085099.47800.DF. [DOI] [PubMed] [Google Scholar]
- 16.Hrabálek L, Kalita O, Svebisova H, Ehrmann J, Hajduch M, Trojanec R, et al. Dumbbell-shaped peripheral primitive neuroectodermal tumor of the spine: Case report and review of the literature. J Neurooncol. 2009;92:211–7. doi: 10.1007/s11060-008-9744-9. [DOI] [PubMed] [Google Scholar]
- 17.Ishii N, Hiraga H, Sawamura Y, Shinohe Y, Nagashima K. Alternative EWS-FLI1 fusion gene and MIC2 expression in peripheral and central primitive neuroectodermal tumors. Neuropathology. 2001;21:40–4. doi: 10.1046/j.1440-1789.2001.00367.x. [DOI] [PubMed] [Google Scholar]
- 18.Isotalo PA, Agbi C, Davidson B, Girard A, Verma S, Robertson SJ. Primary primitive neuroectodermal tumor of the cauda equina. Hum Pathol. 2000;31:999–1001. doi: 10.1053/hupa.2000.16532. [DOI] [PubMed] [Google Scholar]
- 19.Jain A, Jalali R, Nadkarni TD, Sharma S. Primary intramedullary primitive neuroectodermal tumor of the cervical spinal cord. J Neurosurg Spine. 2006;4:497–502. doi: 10.3171/spi.2006.4.6.497. [DOI] [PubMed] [Google Scholar]
- 20.Jaksche H, Wöckel W, Wernert N. Primary spinal medulloblastomas? Neurosurg Rev. 1988;11:259–65. doi: 10.1007/BF01741419. [DOI] [PubMed] [Google Scholar]
- 21.Kampman WA, Kros JM, De Jong TH, Lequin MH. Primitive neuroectodermal tumours (PNETs) located in the spinal canal: The relevance of classification as central or peripheral PNET. J Neurooncol. 2006;77:65–72. doi: 10.1007/s11060-005-9006-z. [DOI] [PubMed] [Google Scholar]
- 22.Kwon OK, Wang KC, Kim CJ, Kim IO, Chi JG, Cho BK. Primary intramedullary spinal cord primitive neuroectodermal tumor with intracranial seeding in an infant. Childs Nerv Syst. 1996;12:633–6. doi: 10.1007/BF00261661. [DOI] [PubMed] [Google Scholar]
- 23.Kim Y, Jin B, Kim T, Cho Y. Primary intraspinal primitive neuroectodermal tumor at conus medullaris. Yonsei Med J. 2004;45:533–8. doi: 10.3349/ymj.2004.45.3.533. [DOI] [PubMed] [Google Scholar]
- 24.Korf BR. Malignancy in Neurofibromatosis Type 1. Oncologist. 2000;5:477–85. doi: 10.1634/theoncologist.5-6-477. [DOI] [PubMed] [Google Scholar]
- 25.Korf BR, Rubenstein AE. Neurofibromatosis: A Handbook for Patients, Families, and Health Care Professionals. In: Hiscock TY, editor. 2nd ed. New York: Thieme; 2005. [Google Scholar]
- 26.Kosnik EJ, Boesel CP, Bay J, Sayers MP. Primitive neuroectodermal tumors of the central nervous system in children. J Neurosurg. 1978;48:741–6. doi: 10.3171/jns.1978.48.5.0741. [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, Reddy SJ, Wani AA, Pal L. Primary spinal primitive neuroectodermal tumor: Case series and review of the literature. Pediatr Neurosurg. 2007;43:1–6. doi: 10.1159/000097517. [DOI] [PubMed] [Google Scholar]
- 28.Llombart-Bosch A, Navarro S. Immunohistochemical detection of EWS and FLI-1 proteinss in Ewing sarcoma and primitive neuroectodermal tumors: Comparative analysis with CD99 (MIC-2) expression. Appl Immunohistochem Mol Morphol. 2001;9:255–60. doi: 10.1097/00129039-200109000-00010. [DOI] [PubMed] [Google Scholar]
- 29.Louis D, Ohgaki H, Wiestler O, Cavenee W. Lyon, France: International Agency for Research on Cancer; 2007. WHO classification of tumours of the central nervous system. [Google Scholar]
- 30.Martinez-Lage J, Salcedo C, Corral M, Porta M. Medulloblastomas in neurofibromatosis type 1. Case report and literature review. Neurocirugia (Astur) 2002;13:128–31. doi: 10.1016/s1130-1473(02)70634-9. [DOI] [PubMed] [Google Scholar]
- 31.Mawrin C, Synowitz HJ, Kirches E, Kutz E, Dietzmann K, Weis S. Primary primitive neuroectodermal tumor of the spinal cord: Case report and review of the literature. Clin Neurol Neurosurg. 2002;104:36–40. doi: 10.1016/s0303-8467(01)00171-8. [DOI] [PubMed] [Google Scholar]
- 32.Meltzer CC, Townsend DW, Kottapally S, Jadali F. FDG imaging of spinal cord primitive neuroectodermal tumor. J Nucl Med. 1998;39:1207–9. [PubMed] [Google Scholar]
- 33.Miller D, Rorke L, Weinberg J, Allen J, Epstein F. Histopathologic diagnoses of intramedullary spinal cord tumors in children. J Neuropathol Exp Neurol. 1997;56:607. Mottl H, Koutecky J. Treatment of spinal cord tumors in children Med Pediatr Oncol 1997;29:293-5. [Google Scholar]
- 34.Nishi T, Saya H. Neurofibromatosis type 1 (NF1) gene: Implication in neuroectodermal differentiation and genesis of brain tumors. Cancer Metastasis Rev. 1991;10:301–10. doi: 10.1007/BF00554792. [DOI] [PubMed] [Google Scholar]
- 35.Ogasawara H, Kiya K, Kurisu K, Muttaqin Z, Uozumi T, Sugiyama K, et al. Intracranial metastasis from a spinal cord primitive neuroectodermal tumor: Case report. Surg Neurol. 1992;37:307–12. doi: 10.1016/0090-3019(92)90158-j. [DOI] [PubMed] [Google Scholar]
- 36.Otero-Rodríguez A, Hinojosa J, Esparza J, Muñoz MJ, Iglesias S, Rodríguez-Gil Y, et al. Purely intramedullary spinal cord primitive neuroectodermal tumor: Case report and review of the literature. Neurocirugia (Astur) 2009;20:381–7. doi: 10.1016/s1130-1473(09)70159-9. [DOI] [PubMed] [Google Scholar]
- 37.Ozdemir N, Usta G, Minoglu M, Erbay AM, Bezircioglu H, Tunakan M. Primary primitive neuroectodermal tumor of the lumbar extradural space. J Neurosurg Pediatr. 2008;2:215–21. doi: 10.3171/PED/2008/2/9/215. [DOI] [PubMed] [Google Scholar]
- 38.Perry R, Gonzales I, Finlay J, Zacharoulis S. Primary peripheral primitive neuroectodermal tumors of the spinal cord: Report of two cases and review of the literature. J Neurooncol. 2007;81:259–64. doi: 10.1007/s11060-006-9178-1. [DOI] [PubMed] [Google Scholar]
- 39.Pytel P, Lukas RV. Update on diagnostic practice: Tumors of the nervous system. Arch Pathol Lab Med. 2009;133:1062–77. doi: 10.5858/133.7.1062. [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33–40. doi: 10.1093/oxfordjournals.aje.a010118. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds RM, Browning GG, Nawroz I, Campbell IW. Von Recklinghausen's neurofibromatosis: Neurofibromatosis type 1. Lancet. 2003;361:1552–4. doi: 10.1016/s0140-6736(03)13166-2. [DOI] [PubMed] [Google Scholar]
- 42.Sarmiento U, Bujanda D, Galán R, Vera J, Morales J. Lumbar region intra-spinal primitive neuroectodermal tumour (PNET) combined with neurofibromatosis type 1. Clin Transl Oncol. 2005;7:464–7. doi: 10.1007/BF02716598. [DOI] [PubMed] [Google Scholar]
- 43.Scheurlen WG, Senf L. Analysis of the gap-related domain of the neurofibromatosis type 1 (NF1) gene in childhood brain tumors. Int J Cancer. 1995;64:234–8. doi: 10.1002/ijc.2910640404. [DOI] [PubMed] [Google Scholar]
- 44.Virani M, Jain S. Primary intraspinal primitive neuroectodermal tumor (PNET): A rare occurrence. Neurol India. 2002;50:75. [PubMed] [Google Scholar]
- 45.Weil RJ, Zhuang Z, Pack S, Kumar S, Helman L, Fuller BG, et al. Intramedullary Ewing sarcoma of the spinal cord: Consequences of molecular diagnostics. J Neurosurg Spine. 2001;95:270–5. doi: 10.3171/spi.2001.95.2.0270. [DOI] [PubMed] [Google Scholar]