

Abstract
Objective
To examine the relation of cerebral amyloid angiopathy (CAA) to cognitive domains in older community-dwelling persons with and without dementia.
Methods
Subjects were 404 persons in the Religious Orders Study, a cohort study of aging, who underwent annual clinical evaluations, including 19 neuropsychological tests from which 5 cognitive domain and global summary scores were derived, and brain autopsy at time-of-death (mean age-at-death 86). Using amyloid-β immunostaining, CAA severity was graded in 5 regions (midfrontal, inferior temporal, angular, calcarine, and hippocampal cortices), as 0 = none, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe. Because severity was related across regions (all rs > 0.63), and almost all persons had some CAA, we averaged regional CAA scores and created class variable predictors for no-to-minimal (<0.5), mild-to-moderate (0.5-2.5) and moderate-to-very severe CAA (>2.5).
Results
CAA was very common (84.9%; 94 had no-to-minimal, 233 mild-to-moderate, and 76 moderate-to-very severe disease) and was related to AD pathology (rs = 0.68). In linear regression analyses controlling for age, sex, education, AD pathology, infarcts, and Lewy bodies, moderate-to-very severe CAA was associated with lower perceptual speed (p = 0.012) and episodic memory (p = 0.047), but not semantic memory, working memory, visuospatial skills, or a composite of all cognitive measures. No associations of mild-to-moderate CAA with cognition were found. Dementia did not modify these findings.
Interpretation
CAA pathology is very common in older community-dwelling persons and is associated with AD pathology. Moderate-to-very severe CAA, but not mild-to-moderate CAA, is associated with lower performance in specific cognitive domains, most notably perceptual speed, separately from the effect of AD pathology.
Cerebral amyloid angiopathy (CAA) is common in older persons,1,2 and in a small number, particularly when severe, has been related to serious adverse neurological conditions, most commonly hemorrhage.3 CAA has also been implicated in dementia,4 infarcts,5 and more recently brain microbleeds.6 A general role for CAA in cognitive impairment in aging is controversial. Indeed, CAA has been related to one of the main causes of dementia, Alzheimer's disease (AD) pathology,7–9 yet little data are available on the relation of CAA to cognitive impairment, the principal clinical manifestation of AD.3,10–12 Further, we are not aware of any previously published study of the relation of CAA to different cognitive domains. A better understanding of the relation of common neuropathology such as CAA to a common and disabling condition of aging, cognitive impairment, may impact future research in aging.
In this study, we tested the hypothesis that CAA pathology is associated with specific cognitive domains in older community-dwelling women and men, with and without dementia. We used data from the Religious Orders Study, an ongoing epidemiologic clinical-pathologic study of aging and dementia. In more than 400 autopsied persons, we describe characteristics of CAA data, including frequency and distribution of pathology, inter-relations of severity scores across brain regions, and associations with relevant variables, including AD pathology. We then examine associations of CAA severity with 5 different cognitive domains and global cognition proximate-to-death, in analyses controlling for AD pathology and other covariates.
Patients and Methods
Subjects
Subjects were older Catholic priests, nuns, and brothers enrolled in the Religious Orders Study, a longitudinal clinical-pathologic study of aging and dementia, from more than 40 groups across the United States. The study was approved by the Institutional Review Board of Rush University Medical Center. All subjects agreed to annual clinical evaluations and brain donation at time of death, and signed an informed consent and anatomical gift act donating their brain to Rush researchers.
There were 1,135 persons enrolled in the Religious Orders Study between January 1994 and December 2009 who had a baseline clinical evaluation. The study follow-up rate is 94%, with up to 15 years of annual follow-up. Over the course of the study, 500 persons died, 468 of whom underwent a brain autopsy (94% autopsy rate). Analyses for this study were conducted on the first consecutive 404 persons on whom neuropathological data were available.
Clinical Evaluations
Clinical evaluations followed procedures recommended by the Consortium to Establish a Registry for Alzheimer's Disease,13 and have been previously described in detail elsewhere.14 Each subject underwent a baseline uniform structured clinical evaluation, which included a medical history, neuropsychological testing (see below), and a neurological examination. Annual follow-up clinical evaluations were identical to the baseline evaluation in all essential details, and were performed by examiners blinded to previously collected data. Data were collected on laptop computers with forms programmed in a Pascal-based entry program.
Cognitive function was evaluated at baseline and each follow-up evaluation using a standardized battery of neuropsychological tests that were selected to assess a broad range of abilities commonly affected in aging, as reported previously.14–16 All neuropsychological data were reviewed by a neuropsychologist blinded to previously collected data. The Mini-Mental State Examination17 was used for descriptive purposes. Nineteen neuropsychological tests were used to form composite measures of 5 separate cognitive domains and global cognition. Episodic memory was based on 7 tests, semantic memory on 4 tests, working memory on 4 tests, perceptual speed on 2 tests, and visuospatial ability on 2 tests, as previously described.14 The global cognition measure was based on all 19 neuropsychological tests.14 Valid measures required scores on at least half the components of the tests. Composite measures were derived by converting the raw scores of individual tests to z scores, using the mean and standard deviation from the baseline evaluation of all participants, and averaging the z scores. Composite measures of cognition allow for decreased floor and ceiling artifacts and other sources of measurement error, and these measures have been used in numerous studies in this and other cohorts.16,18,19
Identification of dementia was based on the recommendation of the Joint Working Group of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association,20 and determined by clinicians experienced in diseases of aging, after review of all available clinical data from that year. At time of death, a board-certified neurologist, blinded to pathologic data, reviewed all clinical information to render a classification of dementia status proximate-to-death.
Autopsy and Neuropathologic Measures
Brain autopsies were performed blinded to clinical data and using standard techniques, as previously described.21,22 The mean postmortem interval was 8.2 hours. After obtaining a total brain weight, brains were placed in a Plexiglas jig and hemispheres were cut coronally into 1-cm slabs. Slabs not designated for freezing were fixed in 4% paraformaldehyde. A uniform examination for cerebral infarcts was conducted by examiners blinded to all clinical data. The age, size, and location of all cerebral infarcts visible to the naked eye were recorded, as previously reported.22 For these analyses, we dichotomized chronic macroscopic infarcts as present (1 or more infarcts) or absent, as previously reported.22
CAA pathology was assessed in 5 brain regions. The four neocortical regions were midfrontal (Brodmann area {BA} 46/9), inferior temporal (BA20), angular gyrus (BA39), and calcarine cortices (BA17), and 1 mesial temporal region, the hippocampus (BA24). Tissue was dissected from 1-cm-thick paraformaldehyde-fixed slabs from 2 or more blocks from adjacent slabs from each region, then paraffin-embedded, cut into 20-μm sections, and mounted on glass slides. The presence of CAA was assessed in each region using immunohistochemical labeling with anti-amyloid-β (Clone 6F/3D, M 0872; DAKO; 1:100). Positive controls were included in each run. CAA pathology was measured in 5 brain regions using a 5 point scale (0 through 4), similar to other studies,5,8 with 0 = none (no immunohistostaining for CAA), 1 = mild (scattered positivity in either leptomeningeal or cortical blood vessels), 2 = moderate (strong, circumferential positivity in some but not all leptomeningeal or cortical blood vessels), 3 = severe (widespread, strong, circumferential positivity in leptomeningeal and cortical blood vessels), and 4 = very severe (same as 3, but with additional changes of positivity emanating from vessels into surrounding neuropil), as illustrated in the Figure.
Figure.
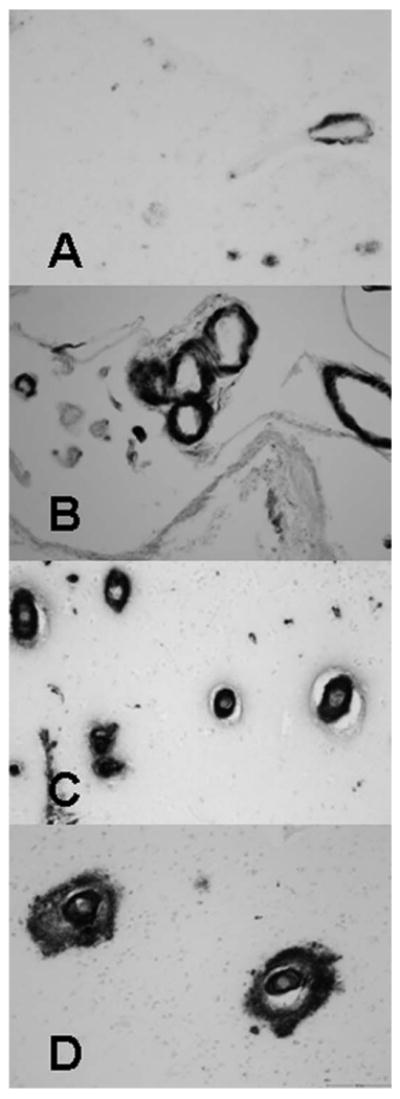
Immunostaining of CAA showing increasing severity of CAA. All panels are shown in original magnification ×200. (A) Mild severity (grade 1) showing incomplete circumferential staining of leptomeningeal blood vessels in a case with sparse positivity. (B) Moderate severity (grade 2) showing strong circumferential staining of leptomeningeal blood vessels. (C) Severe (grade 3) with multiple blood vessels with strong circumferential staining in a case with widespread positivity. (D) Very severe (grade 4) with amyloid positivity emanating from the blood vessels into surrounding neuropil, in a case with widespread circumferential staining. CAA = cerebral amyloid angiopathy.
All brains were examined for pathological markers of AD. A modified Bielschowsky silver stain was used to visualize pathological markers of AD in separate cortical regions, as previously described.22 Briefly fixed tissue from the midfrontal, middle, or superior temporal, inferior parietal, and entorhinal cortex were dissected, paraffin embedded, and cut into 6-μm sections, which were then mounted on glass slides. After silver staining, a board-certified neuropathologist or trained technician counted neuritic plaques, diffuse plaques, and neurofibrillary tangles in the area with the greatest density of each marker, using a graticule in a 1-mm2 area (×100 magnification) in each region. Neuritic plaque, diffuse plaque, and neurofibrillary tangle counts were then standardized separately in each of the 4 cortical regions, by dividing the raw score by the standard deviation of the mean raw count of that marker from that region from the entire deceased cohort, and averaged across the 4 regions to create summary scores of the 3 markers for each subject. A summary measure of global AD pathology was created, by averaging the summary scores of neuritic plaques, diffuse plaques, and neurofibrillary tangles for each subject (average score = 0.6, range 0–2.7, SD = 0.5).21
Because neocortical Lewy bodies are also a significant cause of cognitive impairment in older persons, 6-μm sections of cortex and substantia nigra were also evaluated using α-synuclein immunohistochemistry (Zymed, 1:100) to delineate the presence of Lewy body pathology.23 We required Lewy bodies in the midfrontal, middle temporal, or inferior parietal cortex for a diagnosis of neocortical Lewy body disease similar to established guidelines.24
Statistical Analysis
We used Spearman rank correlation coefficients to examine the inter-relations of the 5 brain region CAA severity scores. Because severity was related across regions (rs range from 0.36 to 0.91, and all p < 0.0001), we averaged the 5 regional scores to create an overall CAA severity score. Because almost all persons had some CAA (84.9%), mostly mild-to-moderate disease (Table 1), and in order to make analyses easier to interpret, the overall CAA severity score was converted into a 3-level class variable predictor. We used an overall severity score of > 2.5 to separate persons with mild-to-moderate from moderate-to-very severe CAA, with the reference group consisting of persons with no-to-minimal CAA, defined by a score of <0.5. Similarly, to create the neocortical measure of CAA we averaged the 4 neocortical regional scores and converted the mean score to a 3-level class variable using the same mean cutoff points. The cutoff levels chosen for the CAA variables were based on the distribution of CAA and ease of interpretation of CAA severity, without consideration of cognitive outcomes and before any analyses were conducted. We also performed intra-rater and inter-rater reliability studies for the CAA scoring system. The original rater repeated 12 cases selected to represent the 3 strata of CAA. A second rater also rated these 12 cases. The intra-rater variability was 5.4% of the variation between cases and the variation between raters was 5.7% of the variation between cases.
Table 1. Clinical and Pathologic Characteristics of Cohort.
| Dementia (n = 181) | No Dementia (n = 223) | Total (n = 404) | |
|---|---|---|---|
| Age at death, yr | 88.9 (6.5) | 84.5 (6.8) | 86.5 (7.0) |
| Male sex, n (%) | 65 (35.9%) | 98 (44.0%) | 163 (40.4%) |
| Education, yr | 17.6 (3.3) | 18.2 (3.7) | 17.9 (3.5) |
| MMSE score proximate-to-death | 14.0 (8.6) | 27.3 (3.1) | 21.4 (9.0) |
| AD pathology score | 0.8 (0.6) | 0.4 (0.4) | 0.6 (0.5) |
| Cerebral infarct present, n (%) | 84 (46.4%) | 62 (27.8%) | 146 (36.1%) |
| Neocortical Lewy bodies present, n (%) | 33 (18.2%) | 6 (2.7%) | 39 (9.7%) |
| CAA present, n (%)a | 170 (93.9%) | 173 (77.8%) | 343 (84.9%) |
| No-to-minimal CAA, n (%)b | 20 (11.1%) | 74 (33.2%) | 94 (23.3%) |
| Mild-to-moderate CAA, n (%)b | 114 (63.3%) | 119 (53.4%) | 233 (57.8%) |
| Moderate-to-very severe CAA, n (%)b | 46 (25.6%) | 30 (13.4%) | 76 (18.9%) |
Values are mean (SD) unless otherwise indicated.
CAA severity score of 1 or more in any of the 5 brain regions.
CAA pathology was measured in 5 brain regions using a 5-point scale (0–4), with 0 = none (no immunohistostaining for CAA), 1 = mild (scattered positivity in either leptomeningeal or cortical blood vessels), 2 = moderate (strong, circumferential positivity in some but not all leptomeningeal or cortical blood vessels), 3 = severe (widespread, strong, circumferential positivity in leptomeningeal and cortical blood vessels), and 4 = very severe (same as 3, but with additional changes of positivity emanating from vessels into surrounding neuropil).
AD = Alzheimer disease; CAA = cerebral amyloid angiopathy; MMSE = mini-mental state examination; SD = standard deviation.
We examined clinical differences proximate-to-death among those with and without dementia, using chi square tests for categorical variables and nonpaired t tests for continuous variables. We then examined the association of CAA with demographic and neuropathologic variables using Spearman correlations.
We used regression analyses to examine the effect of CAA severity on cognitive domains. All analyses were adjusted for age-at-death, sex, and education. Models also adjusted for 3 additional terms of common neuropathology known to be associated with cognitive impairment, including AD pathology, macroscopic cerebral infarcts, and neocortical Lewy bodies. AD pathology is related to CAA and cognition and represents an important possible confounder that needs to be considered in analyses and interpretation of the role of CAA and cognition. Though macroscopic cerebral infarcts were not related to CAA (see Results), given their potential role in CAA and known association with cognition, we also included a term controlling for infarcts. Because neocortical Lewy bodies were associated with CAA in univariate analyses, and are related to cognitive impairment, this was also included as a covariate in analyses.
In primary analyses using adjusted linear regressions, we examined the association of CAA with cognition proximate-to-death, considering each of the 5 cognitive domain and global cognition scores separately. To examine for potential regional effects of CAA on cognition, we also replaced the CAA variables with neocortical CAA and, separately, hippocampal CAA.
Finally, because we were interested in whether the associations differed in persons with and without dementia, we conducted additional analyses with an interaction term of CAA with dementia.
Analyses were carried out using SAS/STAT software version 9.2 (SAS Institute Inc, Cary, NC) on a SunUltraSparc (Sun Microsystems Inc, Santa Clara, CA) workstation. Model assumptions were evaluated graphically and analytically and were judged to be adequately met.25
Results
Clinical and Neuropathologic Characteristics
Of the 404 persons included in the analyses, 45% had dementia and 55% did not (see Table 1). The mean time interval between the last clinical evaluation and death was 6.7 months. Compared to those without dementia, subjects with dementia were older, more likely to have a lower MMSE score, and more pathology, including AD pathology, cerebral infarcts, neocortical Lewy bodies, and CAA (all p < 0.01).
Summary of CAA Data
CAA was present in almost all persons with dementia and the majority of those without dementia (see Table 1). Severity scores across region were related (rs range from 0.36 to 0.91, and all p < 0.0001). Examining severity by brain region, we found that moderate-to-very severe CAA was present most commonly in the calcarine cortex, and least commonly in the hippocampus (Table 2). Using the class variable, about a quarter of the cohort had no-to-minimal CAA (severity score < 0.5), over half had mild-to-moderate CAA (severity score 0.5–2.5), and about one-fifth of the total group had moderate -to-very severe CAA (severity score > 2.5) (see Table 1). The overall CAA severity score was associated with age (rs = 0.25, p < 0.001) and female sex (rs = 0.16, p = 0.002) but not education (p = 0.560). CAA was strongly associated with AD pathology (rs = 0.68, p < 0.001), was also associated with neocortical Lewy bodies (rs = 0.127, p = 0.011), but not cerebral infarct (p = 0.112).
Table 2. Number (%) of Persons with Moderate-to-Very Severe CAA by Region.
| Dementia | No Dementia | Total | |
|---|---|---|---|
| Hippocampus | 24 (16.0%) | 17 (9.5%) | 41 (12.5%) |
| Mid-frontal cortex | 52 (29.1%) | 33 (14.9%) | 85 (21.2%) |
| Inferior temporal cortex | 49 (30.2%) | 30 (15.4%) | 79 (22.1%) |
| Angular gyrus cortex | 46 (26.3%) | 37 (17.0%) | 83 (21.2%) |
| Calcarine cortex | 70 (39.6%) | 52 (23.9%) | 122 (31.0%) |
CAA – cerebral amyloid angiopathy.
CAA and Cognition
We examined associations of CAA severity with cognitive domains and global cognition, separate from of AD pathology, using a series of adjusted linear regression models. All models adjusted for age-at-death, sex, and education. To minimize potential confounding by other common neuropathologies all regression analyses also had terms for AD pathology, cerebral infarct, and neocortical Lewy bodies.
We found that moderate-to-very severe CAA was associated with lower levels of perceptual speed and episodic memory, but not semantic memory, working memory, or visuospatial skills, or global cognition (Table 3). Specifically, compared to subjects with no-to-minimal CAA, those with moderate-to-very severe CAA had a 0.50-unit (SE = 0.20, p = 0.012) lower score on perceptual speed. This effect was equivalent to approximately 10 years of age (estimate = −0.05, SE = 0.01, p < 0.001), and more than the effect observed for 1 unit of AD pathology (estimate = −0.37, SE = 0.12, p = 0.002; average AD pathology in person with dementia = 0.8 unit). Compared to subjects with no-to-minimal CAA, those with moderate-to-very severe CAA had a 0.46-unit (SE = 0.23, p = 0.047) lower score on episodic memory, approximately equivalent to 12 years of age (estimate = −0.03, SE = 0.01, p < 0.001) and half the effect of 1 unit of AD pathology (estimate = −1.06, SE = 0.14, p < 0.001). No associations were found of mild-to-moderate CAA with any of the cognitive domains (all p > 0.05) or global cognition (p = 0.939).
Table 3. Relation of Moderate-to-Very Severe CAA to Five Cognitive Domains and Global Cognition.
| Cognitive Score | Estimatea (SE), p |
|---|---|
| Perceptual speed | −0.50 (0.20), 0.012 |
| Episodic memory | −0.46 (0.23), 0.047 |
| Semantic memory | −0.21 (0.22), 0.340 |
| Working memory | 0.03 (0.16), 0.865 |
| Visuospatial abilities | −0.27 (0.17), 0.102 |
| Global cognition | −0.35 (0.19), 0.066 |
Model terms include CAA class variable predictors for moderate-to-very severe CAA (shown), mild-to-moderate CAA, and terms adjusting for age-at-death, sex, education, AD pathology, cerebral infarcts, and Lewy bodies.
β-Coefficient.
AD = Alzheimer disease; CAA = cerebral amyloid angiopathy; SE = standard error.
To further examine the role of CAA in cognition separately from AD pathology, we conducted analyses to determine whether the relationship between CAA and cognitive domains differed among those with differing levels of AD pathology. Using regression analyses as previously described, we added interaction terms for CAA and AD pathology, and no interactions were found (all p > 0.109), suggesting the relationship between CAA and cognitive domains is not modified by AD pathology.
Because CAA severity in neocortical and limbic regions may be differentially related to cognitive impairment, we also examined whether neocortical CAA severity or, separately, hippocampal CAA severity, were associated with perceptual speed or episodic memory. The neocortical CAA severity score (average of CAA severity scores in the 4 neocortical regions) was 1.5 (SD = 1.3). In analyses adjusted for clinical (age-at-death, sex, and education) and pathologic variables (AD pathology, Lewy bodies, and infarcts), moderate-to-very severe neocortical CAA was associated with a lower level of perceptual speed (estimate = −0.41, SE = 0.19, p = 0.033), but not episodic memory (p = 0.116). There were no associations of mild-to-moderate neocortical CAA with perceptual speed (p = 0.209) or episodic memory (p = 0.375). We conducted similar analyses with the hippocampal CAA severity score (score = 0.8, SD = 1.3), and did not find associations of hippocampal CAA with perceptual speed (p = 0.300) or episodic memory (p = 0.592).
CAA, Cognition, and Dementia
Because the effect of CAA on cognition may vary by dementia status, we conducted additional linear regression analyses with terms for dementia. Controlling for dementia did not change the observed association of moderate-to-very severe CAA with perceptual speed (estimate = −0.46, SE = 0.16, p = 0.005) or episodic memory (estimate = −0.40, SE = 0.17, p = 0.018). In a separate analysis, there was no evidence for interactions of CAA with dementia (p = 0.126 for perceptual speed and p = 0.138 for episodic memory), suggesting that the associations of CAA with perceptual speed and episodic memory do not differ among those with and without dementia.
Discussion
In this clinical-pathologic study of more than 400 community-dwelling, older persons, we found that CAA was very common in persons with and without dementia and has a strong association with AD pathology. After accounting for AD pathology, we found that moderate-to-very severe CAA was specifically associated with lower levels of perceptual speed and to a lesser extent with episodic memory. The effects of CAA on perceptual speed and episodic memory did not differ among those with or without dementia, or in those with differing levels of AD pathology.
Similar to AD pathology, CAA is very common in the aging brains, yet reports vary on the exact frequency of CAA pathology in older persons. High frequencies have generally been found in the larger published clinic-based autopsy series (of about 100 persons), with CAA found in 83% of persons with pathologically-confirmed AD,2 and 64% to 68% of those with and without dementia.11,26 Previous population-based autopsy studies have report a relatively lower frequency of CAA. Two studies, of which 1 was in men only and the other was in persons with an average age of 97 years, reported a frequency of CAA of just under 50%.10,27 Another study that assessed for the presence of severe CAA specifically, reported its presence in 21% of older persons.28 In our study, which included community-dwelling persons with and without dementia, we found that CAA pathology is very common (85%), and present in almost all those with dementia (94%) and in most of those without (77%); however, only about 1/5 had moderate-to-very severe disease, in keeping with a previous report.28 Cohort and study design differences may account for discrepancies in the reported frequency of CAA pathology. In particular, differences in assessment of CAA, such as methodology used to detect CAA (Congo red stain27 vs immunohistochemistry), staging,10 and determination of cutoff values for identification CAA (severe stage28 vs other) may account for some of the differences. We examined multiple sections of brain with specific antibodies to beta-amyloid. Amyloid found in any meningeal or cortical vessel was considered evidence of CAA. Similar to other studies, we found that CAA is typically of mild-to-moderate severity and most severe in the calcarine cortex, with increased severity in persons with dementia (and consequently AD pathology) compared to those without.1,28,29
There is little data on the association of CAA with cognitive domains.2 Indeed, we are aware of only 3 studies that directly examined the relation of CAA pathology to cognition in older persons with and without dementia.10–12 In a small clinical-pathologic study, extent of CAA was associated with the Clinical Dementia Rating scale, but this association was no longer present when controlling for AD pathology, suggesting that CAA does not have a separate effect on cognition.11 In a population-based study of about 200 persons, CAA was reported to be associated with cognition, but information regarding specific cognitive measures and covariates were not provided.12 In a third study, also population-based and of a similar size but in men only, authors found that the association of CAA with cognition was present only in a subgroup of men with dementia.10 Findings were unchanged when controlling for AD pathology and other variables. Our study extends the understanding of the relation of CAA to cognitive domains in aging. We find that CAA is associated most strongly with perceptual speed and to a lesser extent with episodic memory, even after accounting for AD pathology. Discrepant findings in previous studies may be related to the lack of consideration of AD pathology as a covariate, and the differential effects of CAA on different cognitive domains.
Because AD pathology is associated with lower cognitive function and CAA is associated with AD pathology,30 AD pathology represents an important possible confounder. Several findings increase confidence that our findings, particularly the association of CAA with perceptual speed, are separate from that of AD pathology. First we controlled for AD pathology using a construct that enabled us to control for all levels of pathology, from minimal to severe. Second, the effect of AD pathology is strongest for episodic memory whereas our main finding was that CAA was related to lowered perceptual speed with a lesser effect on episodic memory. This differs from what is seen with AD pathology, which preferentially effects episodic memory. This suggests that the relation between CAA and cognitive domains is not solely a proxy for more severe AD pathology. Third, in some specific forms of CAA, such as the relatively uncommon hereditary forms, cognitive effects of CAA appear to be independent of AD pathology.31 Finally, we found that the magnitude of the association between moderate-to-very severe CAA and cognitive systems is clinically meaningful. For example, the association of CAA with the lowering of perceptual speed is comparable to that of 0.5 unit of AD pathology (on average persons with dementia have about 0.8 units of AD pathology) and to about 10 years of age. Overall, these factors support a separate and important association between CAA and specific cognitive domains. Further study will be important to confirm the specificity of these relationships between CAA and different cognitive domains.32
There are several potential mechanisms whereby CAA may lead to impaired perceptual speed and episodic memory, including diffuse brain microbleeds, microinfarcts, and white matter hypoxia. Brain microbleeds, which are common and increasingly recognized with advances in neuroimaging, have been related to both CAA and cognition.33,34 CAA may also be related to microinfarcts35 and white matter changes and degeneration.36 White matter hyperintensities on MRI have been associated with decreased perfusion in persons with CAA,37 and an extensive body of literature supports the link of white matter abnormalities with impaired cognition.38–40 Indeed, these and other tissue related injury may be an important factor in the development of cognitive impairment in persons with CAA. Other neurobiologic mechanisms linking CAA to cognition may include inflammation,41,42 oxidative stress, tissue microstructural changes,43 and others.44 Further studies are needed to help elucidate the role of these and other factors in the relation of CAA with cognition.
Some literature suggests that executive function, such as perceptual speed, declines with advancing age.45 It is interesting to note that in our study, CAA affects perceptual speed in persons both with and without dementia. Thus, CAA may be a factor in some of the common cognitive changes noted in older persons without dementia.
There are limitations to this study. First, we did not have data on the tissue effects of the pathology (eg, microhemorrhage, scarring, and inflammation). Persons with CAA and tissue damage may be particularly susceptible to cognitive impairment. Second, although homogeneity of the cohort may control for a variety of factors and provides internal validity to the study, subjects in this study have a unique lifestyle and findings may not be generalizable to the population. Results will need to be replicated in more diverse cohorts representative of the general population.
Confidence in these findings is strengthened by several factors. We used neuropathologic data collected blinded to clinical data, and assessed CAA in multiple regions according to a previously published method. Next, we accounted for important potential confounders in the relation of CAA to cognition, including AD pathology. Further, we summarized detailed neuropsychological data proximate-to-death to yield quantitative data on the different cognitive domains. Finally, the study has a very high follow-up and autopsy rate, which contributes to internal validity. Overall, these data support a separate role for CAA in the lowering of function in specific cognitive domains in older persons with and without dementia.
Acknowledgments
This research was supported by National Institute on Aging grants (K23 AG23675 to Z.A.; P30 AG10161 to D.A.B.; and R01 AG15819 to D.A.B.).
We thank the nuns, priests, and brothers from the following groups who participated in the Religious Orders Study: archdiocesan priests of Chicago, IL, Dubuque, IA, and Milwaukee, WI; Benedictine monks of Lisle, IL, Collegeville, MN, and St. Meinrad, IN; Benedictine Sisters of Erie, Erie, PA; Benedictine Sisters of the Sacred Heart, Lisle, IL; Capuchins, Appleton, WI; Christian Brothers, Chicago, IL and Memphis, TN; Diocesan priests of Gary, Gary, IN; Dominicans, River Forest, IL; Felician Sisters, Chicago, IL; Franciscan Handmaids of Mary, New York, NY; Franciscans, Chicago, IL; Holy Spirit Missionary Sisters, Techny, IL; Maryknolls, Los Altos, CA and Ossining, NY; Norbertines, DePere, WI; Oblate Sisters of Providence, Baltimore, MD; Passionists, Chicago, IL; Presentation Sisters, B.V.M., Dubuque, IA; Servites, Chicago, IL; Sinsinawa Dominican Sisters, Chicago, IL and Sinsinawa, WI; Sisters of Charity, B.V.M., Chicago, IL and Dubuque, IA; Sisters of the Holy Family, New Orleans, LA; Sisters of the Holy Family of Nazareth, Des Plaines, IL; Sisters of Mercy of the Americas, Chicago, IL, Aurora, IL and Erie, PA; Sisters of St. Benedict, St. Cloud, MN and St. Joseph, MN; Sisters of St. Casimir, Chicago, IL; Sisters of St. Francis of Mary Immaculate, Joliet, IL; Sisters of St. Joseph of LaGrange, LaGrange Park, IL; Society of Divine Word, Techny, IL; Trappists, Gethsemani, KY and Peosta, IA; and Wheaton Franciscan Sisters, Wheaton, IL. We also thank Traci Colvin, MPH, and George Hoganson for Religious Orders Study Coordination; Woojeong Bang, MS, for analytic programming; Greg Klein for data management; and the staff and faculty of the Rush Alzheimer's Disease Center.
Footnotes
Potential Conflicts of Interest: Nothing to report.
References
- 1.Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke. 1983;14:924–928. doi: 10.1161/01.str.14.6.924. [DOI] [PubMed] [Google Scholar]
- 2.Ellis RJ, Olichney JM, Thal LJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- 3.Itoh Y, Yamada M, Hayakawa M, et al. Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci. 1993;116:135–141. doi: 10.1016/0022-510x(93)90317-r. [DOI] [PubMed] [Google Scholar]
- 4.Matthews FE, Brayne C, Lowe J, et al. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. 2009;6:e1000180. doi: 10.1371/journal.pmed.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olichney JM, Hansen LA, Hofstetter CR, et al. Cerebral infarction in Alzheimer's disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol. 1995;52:702–708. doi: 10.1001/archneur.1995.00540310076019. [DOI] [PubMed] [Google Scholar]
- 6.Nakata-Kudo Y, Mizuno T, Yamada K, et al. Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr Cogn Disord. 2006;22:8–14. doi: 10.1159/000092958. [DOI] [PubMed] [Google Scholar]
- 7.Mandybur TI. The incidence of cerebral amyloid angiopathy in Alzheimer's disease. Neurology. 1975;25:120–126. doi: 10.1212/wnl.25.2.120. [DOI] [PubMed] [Google Scholar]
- 8.Attems J, Jellinger KA. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol. 2004;107:83–90. doi: 10.1007/s00401-003-0796-9. [DOI] [PubMed] [Google Scholar]
- 9.Weller RO, Subash M, Preston SD, et al. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. 2008;18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfeifer LA, White LR, Ross GW, et al. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–1634. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 11.Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62:1287–1301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- 12.Fernando MS, Ince PG MRC Cognitive Function and Ageing Neuropathology Study Group. Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci. 2004;226:13–17. doi: 10.1016/j.jns.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer's disease. Neurology. 1989;39:1159–1165. doi: 10.1212/wnl.39.9.1159. [DOI] [PubMed] [Google Scholar]
- 14.Bennett DA, Wilson RS, Schneider JA, et al. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- 15.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging. 2002;17:179–193. [PubMed] [Google Scholar]
- 16.Wilson RS, Mendes De Leon CF, Barnes LL, et al. Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA. 2002;287:742–748. doi: 10.1001/jama.287.6.742. [DOI] [PubMed] [Google Scholar]
- 17.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 18.Wilson RS, Barnes LL, Krueger KR, et al. Early and late life cognitive activity and cognitive systems in old age. J Int Neuropsychol Soc. 2005;11:400–407. [PubMed] [Google Scholar]
- 19.Arvanitakis Z, Bennett DA, Wilson RS, Barnes LL. Diabetes and cognitive systems in older black and white persons. Alzheimer Dis Assoc Disord. 2010;24:37–42. doi: 10.1097/WAD.0b013e3181a6bed5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 21.Bennett DA, Wilson RS, Schneider JA, et al. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology. 2003;60:246–252. doi: 10.1212/01.wnl.0000042478.08543.f7. [DOI] [PubMed] [Google Scholar]
- 22.Schneider JA, Wilson RS, Cochran EJ, et al. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology. 2003;60:1082–1088. doi: 10.1212/01.wnl.0000055863.87435.b2. [DOI] [PubMed] [Google Scholar]
- 23.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer's disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. Erratum in: Neurology 2005;65:1992. [DOI] [PubMed] [Google Scholar]
- 25.Collett D. Modeling survival data in medical research. 2nd. Boca Raton, FL: Chapman & Hall; 2003. [Google Scholar]
- 26.Attems J, Quass M, Jellinger KA, Lintner F. Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J Neurol Sci. 2007 Jun 15;257:49–55. doi: 10.1016/j.jns.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Tanskanen M, Lindsberg PJ, Tienari PJ, et al. Cerebral amyloid angiopathy in a 95+ cohort: complement activation and apolipoprotein E (ApoE) genotype. Neuropathol Appl Neurobiol. 2005;31:589–599. doi: 10.1111/j.1365-2990.2005.00652.x. [DOI] [PubMed] [Google Scholar]
- 28.Neuropathology Group. Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 29.Yamada M, Tsukagoshi H, Otomo E, Hayakawa M. Cerebral amyloid angiopathy in the aged. J Neurol. 1987;234:371–376. doi: 10.1007/BF00314080. [DOI] [PubMed] [Google Scholar]
- 30.Attems J, Jellinger KA, Lintner F. Alzheimer's disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol. 2005;110:222–231. doi: 10.1007/s00401-005-1064-y. [DOI] [PubMed] [Google Scholar]
- 31.Natté R, Maat-Schieman ML, Haan J, et al. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol. 2001;50:765–772. doi: 10.1002/ana.10040. [DOI] [PubMed] [Google Scholar]
- 32.Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009;40:2601–2606. doi: 10.1161/STROKEAHA.108.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeerakathil T, Wolf PA, Beiser A, et al. Cerebral microbleeds: prevalence and associations with cardiovascular risk factors in the Framingham Study. Stroke. 2004;35:1831–1835. doi: 10.1161/01.STR.0000131809.35202.1b. [DOI] [PubMed] [Google Scholar]
- 34.Greenberg SM, Vernooij MW, Cordonnier C, et al. Microbleed Study Group. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soontornniyomkij V, Lynch MD, Mermash S, et al. Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol. 2010;20:459–467. doi: 10.1111/j.1750-3639.2009.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salat DH, Smith EE, Tuch DS, et al. White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. Stroke. 2006;37:1759–1764. doi: 10.1161/01.STR.0000227328.86353.a7. [DOI] [PubMed] [Google Scholar]
- 37.Holland CM, Smith EE, Csapo I, et al. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. Stroke. 2008;39:1127–1133. doi: 10.1161/STROKEAHA.107.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charlton RA, McIntyre DJ, Howe FA, et al. The relationship between white matter brain metabolites and cognition in normal aging: the GENIE study. Brain Res. 2007;1164:108–116. doi: 10.1016/j.brainres.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 39.Smith EE, Egorova S, Blacker D, et al. Magnetic resonance imaging white matter hyperintensities and brain volume in the prediction of mild cognitive impairment and dementia. Arch Neurol. 2008;65:94–100. doi: 10.1001/archneurol.2007.23. [DOI] [PubMed] [Google Scholar]
- 40.Tiehuis AM, Vincken KL, Mali WP, et al. Automated and visual scoring methods of cerebral white matter hyperintensities: relation with age and cognitive function. Cerebrovasc Dis. 2008;25:59–66. doi: 10.1159/000111500. [DOI] [PubMed] [Google Scholar]
- 41.Xu F, Grande AM, Robinson JK, et al. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid beta-protein precursor transgenic mice. Neuroscience. 2007;146:98–107. doi: 10.1016/j.neuroscience.2007.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68:1411–1416. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- 43.Viswanathan A, Patel P, Rahman R, et al. Tissue microstructural changes are independently associated with cognitive impairment in cerebral amyloid angiopathy. Stroke. 2008;39:1988–1992. doi: 10.1161/STROKEAHA.107.509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farris W, Schütz SG, Cirrito JR, et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol. 2007;171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Earles JL, Salthouse TA. Interrelations of age, health, and speed. J Gerontol B Psychol Sci Soc Sci. 1995;50:P33–P41. doi: 10.1093/geronb/50b.1.p33. [DOI] [PubMed] [Google Scholar]