

Abstract
Corticosteroid insensitivity (CI) is a major barrier to treating severe asthma. Despite intensive research, the molecular mechanism of CI remains uncertain. The aim of this study was to determine abnormality in corticosteroid action in severe asthma and to identify the molecular mechanism of the long-acting β2-adrenergic agonists (LABAs) formoterol and salmeterol on restoration of corticosteroid sensitivity in severe asthma in vitro. Peripheral blood mononuclear cells (PBMCs) were obtained from 16 subjects with severe corticosteroid-insensitive asthma, 6 subjects with mild corticosteroid-sensitive asthma, and 11 healthy volunteers. Corticosteroid (dexamethasone) sensitivity was determined on tumor necrosis factor-α (TNF-α)-induced interleukin (IL)-8 production. Glucocorticoid receptor (GR) phosphorylation and kinase phosphorylation were evaluated by immunoprecipitation-Western blotting analysis and kinase phosphorylation array in IL-2/IL-4-treated corticosteroid insensitive model in PBMCs. In vitro corticosteroid sensitivity on TNF-α-induced IL-8 production was significantly lower in patients with severe asthma than in healthy volunteers and patients with mild asthma. This CI seen in severe asthma was associated with reduced GR nuclear translocation and with hyperphosphorylation of GR, which were reversed by LABAs. In IL-2/IL-4-treated PBMCs, LABAs inhibited phosphorylation of Jun-NH2-terminal kinase and p38 mitogen-activated protein kinase-γ (p38MAPK-γ) as well as GR. In addition, cells with p38MAPK-γ knockdown by RNA interference did not develop CI in the presence of IL-2/IL-4. Furthermore, p38MAPK-γ protein expression was up-regulated in PBMCs from some patients with severe asthma. In conclusion, p38 MAPK-γ activation impairs corticosteroid action and p38 MAPK-γ inhibition by LABAs has potential for the treatment of severe asthma.
Introduction
Most patients with asthma, symptoms are now effectively controlled with inhaled corticosteroids. However, approximately 5% of patients with asthma do not respond well to corticosteroids or require high-dose inhaled or oral corticosteroids to control asthma symptoms, although side effects are still a problem. Thus, corticosteroid insensitivity (CI) presents considerable management problems, accounting for a disproportionate amount of healthcare spending in asthma (Leung and Szefler, 1998; Adcock and Ito, 2004).
The biological actions of corticosteroids are mediated by glucocorticoid receptors (GRs), which are normally located in cell cytoplasm. Corticosteroids cross the cell membrane and bind to GR, which then translocates into the nucleus, and its homodimers bind to DNA at glucocorticoid response elements in the promoter region of corticosteroid-responsive anti-inflammatory genes, such as secretory leukoprotease inhibitor (SLPI), mitogen-activated kinase phosphatase-1 (MKP-1), and glucocorticoid inducible leucine zipper (GILZ), increasing gene transcription. As well as this GR-glucocorticoid response element binding, GR may directly influence proinflammatory signaling by forming inhibitory interactions with proinflammatory DNA-binding transcription factors such as activator protein-1 and nuclear factor-κB (NF-κB), or by recruitment of corepressors such as histone deacetylase 2 (Ito et al., 2006a,b). GR nuclear translocation, therefore, is an essential and critical step for corticosteroid action. However, as we reported previously (Matthews et al., 2004), some patients with severe asthma showed defect of GR nuclear translocation.
Numerous studies demonstrated possible mechanisms of corticosteroid insensitivity, such as overexpression of transcription factors (Adcock et al., 1995), histone deacetylase reduction (Cosío et al., 2004; Hew et al., 2006) and increased decoy receptor (Leung et al., 1998). Post-translational modifications of GR, such as phosphorylation, acetylation, and ubiquitination, are also important components for the mechanism of corticosteroid resistance (Ito et al., 2006a,b). For example, Rogatsky et al. (1998) demonstrated that the ability of GR of transcriptional activation was reduced once Ser467 of rat GR (equivalent to Ser226 of human GR) was phosphorylated by c-Jun N-terminal kinases (JNK). Irusen et al. (2002) showed that an inhibitor of p38 mitogen-activate protein kinase (MAPK)-α and -β isoforms inhibited interleukin (IL)-2/IL-4-induced GR phosphorylation in whole-cell extracts though Rogatsky et al. (1998) showed that GR at Ser-246 was not phosphorylated by p38 MAPK. Thus, GR phosphorylation is reported to be associated with CI, but GR phosphorylation has not been detected in clinical samples.
p38 MAPK-γ is one of four isoforms of p38MAPKs (Mertens et al., 1996; Cuenda et al., 1997). This kinase is also called stress-activated protein kinase-3, extracellular signal-regulated kinase 6, or MAPK12 and is able to phosphorylate postsynaptic density 95/disc-large/zona occludens motif-containing proteins, such as stress-activated protein (SAP) 90 and SAP97. p38MAPK-γ is activated by environmental stress, such as oxidative stress and osmotic stress, or pro-inflammatory cytokines, and it phosphorylates several downstream targets. p38 MAPK-γ is expressed in T lymphocytes, macrophages, and skeletal muscle cells, but its function is not certain.
The combinations of a long-acting β2-agonist (LABA) with a low dose of inhaled corticosteroid have been reported to achieve better asthma control than either drug alone or a higher dose of inhaled corticosteroid alone (Reynolds et al., 2005; Miller-Larsson and Selroos, 2006). LABAs alone have been shown to induce GR nuclear translocation in smooth muscle cells and fibroblasts (Eickelberg et al., 1999) and enhance corticosteroid actions in vitro and in vivo (Pang and Knox, 2000; Roth et al., 2002; Usmani et al., 2005). In this way, LABAs may enhance the anti-inflammatory action of corticosteroids, but the molecular mechanism has not been fully elucidated. Here we show that p38 MAPK-γ causes corticosteroid insensitivity in severe asthma through hyperphosphorylation of GR. In addition, we found that the steroid sensitivity and the defective mechanism are reversed by LABAs.
Materials and Methods
Materials.
Formoterol [rac-(R,R)-N-[2-hydroxy-5-[1-hydroxy-2-[1-(4-methoxyphenyl)propan-2-ylamino]ethyl] phenyl]formamide] and salmeterol [(R,S)-2-(hydroxymethyl)-4-{1-hydroxy-2-[6-(4-phenylbutoxy) hexylamino]ethyl}phenol] were provided by AstraZeneca (Lund, Sweden) and GlaxoSmithKline (Greenford, UK), respectively. Dexamethasone [(8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihydroxy-17-(2-hydroxyacetyl)-10,13,16- trimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-3H-cyclopenta[a]phenanthren-3-one] and SB203580 [4-[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl]pyridine] were purchased from Sigma-Aldrich (Poole, UK).
Subjects.
Eleven healthy nonsmoking subjects (mean age ± S.E.M., 35.8 ± 1.6 years; three women; mean forced expiratory volume in 1 s (FEV1 ± S.E.M., 98.2 ± 2.2% of predicted), six patients with mild asthma (41.5 ± 3.3 years; three women; FEV1, 84.5 ± 4.6% of predicted), and 16 patients with severe asthma (age, 35.1 ± 2.6 years; 11 women; FEV1, 55.0 ± 3.4% predicted) were recruited (Table 1). This study was approved by the Ethics Committee of the Royal Brompton and Harefield Hospitals National Health Service Trust, and all subjects gave written informed consent. Fifty milliliters of blood were taken, and PBMCs were separated by Ficoll-Paque (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) gradients.
TABLE 1.
Characteristics of the subjects recruited
| Healthy Volunteers | Mild asthma | Severe asthma | |
|---|---|---|---|
| N | 11 | 6 | 16 |
| Sex, no. male/female | 8/3 | 3/3 | 5/11 |
| Age, years | 35.8 ± 1.6 | 41.5 ± 3.3 | 35.1 ± 2.6 |
| Asthma duration, years | N.A. | N.D. | 16.3 ± 2.6 |
| Atopy | 2/11 | 2/6 | 6/15 |
| FEV1, % pred. | 98.2 ± 2.2 | 84.5 ± 4.6 | 55.0 ± 3.4 |
| FEV1/FVC, % | 96.8 ± 3.1 | 79.9 ± 3.2 | 46.6 ± 12.6 |
| Oral steroid, mg | 0 | 0 | 16.8 ± 4.0 |
| Inhaled steroid, μg | 0 | 0 | 1860 ± 206 |
| Others | N.A. | Albuterol on demand | Albuterol on demand |
N.A., not applicable, N.D.: not determined; FVC, forced vital capacity.
Corticosteroid-Insensitive Model.
PBMCs from healthy volunteers were incubated with human recombinant IL-2 (2 ng/ml) and IL-4 (10 ng/ml) for 48 h.
FITC-Dexamethasone Incorporation.
PBMCs were incubated with FITC-conjugated dexamethasone (FITC-Dex; 10−6 M) for 30 min at 37°C. Nonspecific FITC diffusion was determined in the presence of 10−5 M nonconjugated Dex and subtracted from the total FITC fluorescence value. The nuclear fraction was prepared by hypotonic buffer (Active Motif, Rixensart, Belgium) for 10-min incubation, followed by pulse vortexing with 0.1% NP-40 containing PBS. FITC-dexamethasone in nuclei was extracted with 0.5% NP-40 containing PBS on ice for 20 min. The concentration of FITC-Dex was determined using a standard curve to different concentrations of FITC-Dex. FITC was detected at 488 nm in a fluorescent plate reader.
Detection of GR and Phosphorylated GR.
Whole-cell extracts were prepared with modified radioimmunoprecipitation assay buffer (Ito et al., 2000). For the detection of GR, whole-cell extracts were separated by Tris-glycine SDS/polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The GR level was normalized to β-actin expression. For the detection of phosphorylated GR, GR was immunopurified using anti-GR antibody conjugated agarose A/G (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and separated by SDS-polyacrylamide gel electrophoresis/Western blotting. Phosphorylation level was determined with anti-pan-phosphoserine antibody (Santa Cruz Biotechnology Inc.) and was normalized to GR expression. In addition, GR phosphorylated at Ser226 was determined using anti-phosphorylated (S226) GR antibody (New England Biolabs UK Ltd., Hitchin, Hertfordshire, UK). The band density was calculated by densitometry (UVP Bioimaging Systems, Cambridge, UK) using Labworks software (Ultra-Violet Products, Cambridge, UK).
Reverse Transcription-PCR.
Cells were harvested for total RNA isolation. Commercially available kits were used to extract total cellular RNA (RNeasy; QIAGEN, Crawley, UK) and to perform reverse transcription (Omniscript RT; QIAGEN). Gene transcript level of p38 MAPK-α, -γ, and -δ and the housekeeping gene GNB2L1 or GAPDH were quantified by real-time PCR using a TaqMan PCR kit (Applied Biosystems, Warrington, UK) on a Rotor-Gene 3000 PCR apparatus (Corbett Research, Mortlake, NSW, Australia).
ELISA.
Cells were treated with dexamethasone (10−12–10−6 M) for 30 min in the presence or absence of LABA and then stimulated overnight with either TNF-α (1 ng/ml) or a combination of anti-human CD3 (10 μg/ml) and CD28 antibodies (8 μg/ml) (BD Biosciences, Oxford, UK). IL-8 and IL-2 levels in supernatant were determined by sandwich ELISA (Duoset ELISA for human IL-8; R&D Systems Europe, Abingdon, UK) according to the manufacturer's instructions.
Kinase Profiling.
The phosphorylation of 19 different kinases was evaluated using the Human Phospho-MAPK Array Kit Proteome Profiler (R&D Systems Europe) according to the manufacturer's instructions. HSP27 (phosphorylated and total) and p38MAPK-γ (phosphorylated p38MAPK/stress-activated protein kinase and total) were detected by Western blotting. All antibodies were purchased from R&D Systems Europe.
Measurement of Phosphorylated and Total p38MAPK-γ in Cells.
Phosphorylated p38MAPK-γ and total p38MAPK-γ were detected in PBMCs obtained from healthy subjects using p38MAPK-γ (Thr183/Tyr185) phosphorylation and total cell-based ELISA (Duoset intracellular ELISA). In brief, cells were stimulated with human recombinant IL-2 (2 ng/ml) and IL-4 (10 ng/ml) for 48 h and then treated with formoterol, salmeterol, or salbutamol for 20 min. Cells were collected and lysed using lysis buffer according to the manufacturer's instructions.
RNA Interference.
Short interference RNA (siRNA) of the p38 MAPK-δ (MAPK13) and p38 MAPK-γ (MAPK 12) were purchased from Dharmacon Inc. (Colorado Springs, CO, USA) and transfected by nucleofection using AMAXANucreofector (Lonza GmbH, Cologne, Germany) according to the manufacturer's instructions (100 nM each). Cells were incubated for 24 h and then stimulated with IL-2/IL-4 for further 48 h. Nonspecific control duplex (scrambled oligonucleotide, 47% GC content) were also purchased from Dharmacon RNA Technologies (Lafayette, CO).
Statistical Analysis.
Results are expressed as means ± S.E.M. Analysis of variance was done by Kruskal-Wallis analysis; when significant, comparisons were made by Mann Whitney U test using the PC analysis package SPSS 10.0 (SPSS Inc., Chicago, IL) or Prism 4 (GraphPad Software, San Diego, CA). The differences between treatment groups in the in vitro data were analyzed by Welch's t test. The correlation between two parameters was determined by Spearman methods. A p value < 0.05 was considered statistically significant.
Results
PBMCs From Severe Asthma Were Corticosteroid-Insensitive Because of Defects of GR Nuclear Translocation.
As shown in Fig. 1A, PBMCs produced IL-8 when stimulated with TNF-α in patients with severe asthma (SA; 1430 ± 286 pg/ml), to a level similar to that seen in healthy volunteers (HV; 1650 ± 304 pg/ml), although the IL-8 production was significantly higher in patients with mild asthma (MA; 2160 ± 94.9 pg/ml) than that in HV. In contrast, when 50% inhibitory activity of dexamethasone (Dex-IC50) on TNF-α-induced IL-8 release was calculated as an index of corticosteroid sensitivity, the Dex-IC50 values in PBMCs from patients with SA (181 ± 28.7 nM) were significantly higher than those from HV (15.5 ± 4.2 nM; p < 0.01) and patients with MA (20.0 ± 3.8 nM; p < 0.01) (Fig. 1B).
Fig. 1.

TNF-α-induced IL-8 production (A), dexamethasone sensitivity to inhibit this response (IC50-Dex; B), and GR nuclear translocation by FITC-Dex incorporation assay (C) were determined in PBMCs obtained from HV and patients with MA or SA. NS, not significant.
In PBMCs, there were no significant differences in GRα mRNA expression (GRα/GNB2L1: SA, 70.4 ± 17.8; HV, 102.4 ± 34.4; MA, 89.3 ± 24.3), GRβ mRNA expression (GRβ/GNB2L1: SA, 0.00042 ± 0.00015; HV, 0.000002 ± 0.0000005; MA, 0.00043 ± 0.00029) or in GRα protein expression (GRα/β-actin: SA, 1681 ± 205; HV, 4914 ± 763; MA, 3050 ± 670).
As a marker of GR nuclear translocation after ligand binding, we determined the amount of FITC-Dex in nuclei. Because the anti-inflammatory efficacy of FITC-Dex was 10 times lower than that of unlabeled Dex (IC50 values, FITC-Dex, 5.6 × 10−8 M versus unlabeled Dex, 4.3 × 10−9 M on IL-1β-induced IL-8 production in A549 cells), we used a relatively high concentration (10−6 M) of FITC-Dex for the assay. As shown in Fig. 1C, GR nuclear translocation was significantly impaired in SA cells [FITC-Dex in nuclei, 4.8 ± 0.9 nM; p < 0.01 versus HV (11.0 ± 1.5 nM); p < 0.05 versus MA (9.7 ± 1.4 nM)]. In addition, there was a significant, negative correlation between the IC50-Dex value on TNF-α-induced IL-8 release and the amount of FITC-Dex in nuclei (Spearman r = −0.55; p = 0.0035), suggesting that less Dex efficacy was associated with defects of GR nuclear translocation.
Formoterol Reversed Corticosteroid Insensitivity in PBMCs from Patients with SA by Enhancing GR Nuclear Translocation.
Treatment with formoterol (FOR, 1 nM) decreased the Dex-IC50 value for TNF-α induced IL-8 release in PBMCs from patients with SA (Dex-IC50 with versus without FOR, 42.8 ± 21.0 versus 182.5 ± 28.2 nM, respectively; n = 6, p < 0.05; Fig. 2A) and a similar change but with a lower effect was found with salmeterol (SAL, 100 nM) (Dex-IC50 with versus without SAL, 83.7 ± 16.7 versus 180.7 ± 28.7 nM, respectively; n = 15, p < 0.05) (Table 2). In patients with SA, treatment with FOR 1 nM showed better improvement index than SAL (for SA: 4.3 for FOR versus 2.2 for SAL; Table 2), but the efficacy was not significantly different in FOR and SAL. Neither FOR nor SAL significantly changed corticosteroid sensitivity in HV (Dex-IC50, with FOR, 21.5 ± 4.8 nM; with SAL, 31.0 ± 8.3 nM; no treatment, 15.5 ± 4.2 nM; Table 2) or in patients with MA (Dex-IC50 with SAL, 14.7 ± 3.5 nM versus no treatment, 20.0 ± 3.8 nM; FOR was not tested in patients with MA). As well as reduced corticosteroid sensitivity to TNFα-induced IL-8 release, corticosteroid sensitivity to CD3/CD28-induced IL-2 release from PBMC was also decreased in SA (Dex-IC50, 77.6 ± 25.4 nM in SA, n = 8, versus 11.5 ± 3.3 nM in HV, n = 7, and 40.1 ± 16.4 nM in MA, n = 6) and reversed by SAL (100 nM) (Dex-IC50 with SAL, 30.4 ± 14.4 nM; p = 0.0065; FOR was not tested), suggesting that SAL increased Dex sensitivity. In addition, in SA, FOR (1 nM) enhanced GR nuclear translocation as evaluated by FITC-Dex (nuclear FITC-Dex with versus without FOR, 9.5 ± 1.5 versus 4.8 ± 0.9 nM; p < 0.05; Fig. 2B). Similar results were found with SAL (100 nM) (nuclear FITC-Dex, with versus without SAL, 14.2 ± 2.7 versus 4.8 ± 0.9 nM; p < 0.05; n = 16). The improvement in Dex-IC50 by FOR (increased ratio of Dex-IC50 without FOR versus Dex-IC50 with FOR) correlated well with the improvement of FITC-Dex accumulation in nuclei by FOR (decreased ratio of FITC-Dex in nuclei without FOR versus FITC-Dex in nuclei with FOR; Spearman r = −0.77, p = 0.042). In addition, the improvement in Dex-IC50 by FOR was also negatively correlated with FITC-Dex values in nuclei (Spearman r = −0.73, p = 0.0013; Fig. 2C), suggesting that PBMCs with defect of GR nuclear translocation are more sensitive to FOR treatment.
Fig. 2.

Effects of FOR (1 nM) on dexamethasone sensitivity to inhibit TNF-α-induced IL-8 production (IC50-Dex; A), and GR nuclear translocation determined by FITC-Dex incorporation assay (B) were evaluated in PBMCs obtained from patients with SA. C, correlation between FITC-Dex in nuclei and ratio of Dex-IC50 without versus with FOR, as an index of the improvement of corticosteroid sensitivity with FOR.
TABLE 2.
Effect of treatment with formoterol and salmeterol on corticosteroid sensitivity
The IC50 values of dexamethasone are shown. Values in square brackets are Dex-IC50 without treatment/Dex-IC50 with LABAs.
| No. Patients Studied | Nontreated | Formoterol (1 nM) | Salmeterol (100 nM) | |
|---|---|---|---|---|
| nM | nM | nM | ||
| Healthy | 7 | 15.5 ± 4.2 | 21.5 ± 4.8 | 31.0 ± 8.3 |
| [0.72] | [0.5] | |||
| Mild asthma | 6 | 20.0 ± 3.8 | N.D. | 14.7 ± 3.5 |
| [1.4] | ||||
| Severe asthma | 6 | 182.5 ± 28.2 | 42.8 ± 21.0 | N.D. |
| [4.3] | ||||
| 15 | 180.7 ± 28.27 | N.D. | 83.7 ± 16.7 | |
| [2.2] |
N.D., not determined.
GRs Were Highly Phosphorylated in PBMCs of Severe Asthma and Dephosphorylated by Formoterol.
As shown in Fig. 3, A and B, GR in cytoplasm of PBMCs was highly phosphorylated at serine residues in SA (ratio of phospho-GR/GR, 0.48 ± 0.065; Fig. 3B) compared with those of HV (0.22 ± 0.083; Fig. 3B) and MA (0.25 ± 0.050; not shown). FOR significantly inhibited phosphorylation of GR after 20-min incubation (ratio of phospho-GR/GR, 0.58 ± 0.093 in SA, 0.22 ± 0.037 with 10 nM FOR, 0.25 ± 0.050 with 1 M FOR; p < 0.05 versus nontreated SA; Fig. 3C).
Fig. 3.

GR phosphorylation in severe asthma and restoration by formoterol. Serine phosphorylation of immunoprecipitated GR in PBMCs of patients with SA and HV (A and B). Effects of FOR on GR phosphorylation in PBMCs obtained from patients with SA (C) and in IL-2/IL-4-treated PBMCs from HV (D and E). pGR, phosphorylated GR. *, p < 0.05 versus severe asthma PBMCs (C) or versus IL-2/IL-4-treated PBMCs without FOR (E) by Wilcoxon ranked-pair test. F, GR phosphorylation at Ser226 was also detected in PBMCs from HV (n = 4) and patients with SA (n = 6); #, p < 0.05 for SA versus HV.
IL-2/IL-4 treatment of PBMCs from HV induced corticosteroid insensitivity with impaired GR nuclear translocation in PBMCs (FITC-Dex in nuclei, with versus without IL-2/IL-4, 2.3 ± 1.1 versus 10.7 ± 1.3 nM; p < 0.05). IL-2/IL-4 treatment of PBMCs also induced phosphorylation of GR (ratio of phospho-GR/GR, with versus without IL-2/IL-4, 0.42 ± 0.047 versus 0.11 ± 0.024; p < 0.05; Fig. 3, D and E), and FOR significantly inhibited this effect (ratio of phospho-GR/GR, 0.17 ± 0.044 with 10 nM FOR; 0.17 ± 0.048 with 1 nM FOR; for both, p < 0.05 versus 0.42 ± 0.047 without FOR; Fig. 3, D and E). In addition, the GR phosphorylated at Ser 226 was determined in same samples. As shown in Fig. 3F, GR phosphorylation at Ser 226 was also significantly (p < 0.05) elevated in PBMCs from patients with SA.
p38 MAPK-γ Activation Caused Corticosteroid Insensitivity and Was Inhibited by Formoterol.
To determine the kinase activated by 48-h cell incubation with IL-2 and IL-4, which may be involved in the GR phosphorylation (directly or indirectly), kinase phosphorylation array analysis was performed in PBMCs from healthy volunteers. The 48-h exposure to IL-2/IL-4 up-regulated phosphorylation of all four isoforms of p38 MAPK (α, β, γ, and δ), RSK1 and -2, Akt2 (and pan-Akt), JNK2, JNK3 (and pan-JNK), MSK1, HSP27, glycogen synthase kinase-3α and -β, and p70S6 (Fig. 4, A and B). In this experiment, higher concentration of FOR (10 nM) was used to maximize the effect. FOR significantly inhibited only phosphorylation of pan-JNK and p38 MAPK-γ (Fig. 4, A and B). Phosphorylation of p38 MAPK-α and -β was up-regulated by IL-2/IL-4 but was not inhibited by FOR. Western blot analysis also showed that FM (1 and 10 nM) decreased p38MAPK-γ phosphorylation but not p38α phosphorylation (Fig. 4C). In contrast, although p38MAPK α/β inhibitor SB203580 inhibited phosphorylation of HSP27, a p38MAPK-α downstream molecule, it did not inhibit p38MAPK-γ phosphorylation (Fig. 4C). These results were also quantified and shown in Supplemental Fig. 1. Furthermore, inhibitory effects of β-adrenoceptor agonists on phosphorylation of p38MAPK-γ were evaluated by cell-based ELISA. As shown in Fig. 4D, FOR and SAL concentration-dependently inhibited phosphorylation of p38MAPK-γ, and the IC50 values were 0.97 and 26 nM, respectively, although salbutamol showed partial inhibition at 100 nM.
Fig. 4.

Kinase profiling in IL-2/IL-4-treated PBMCs from healthy volunteers. Representative image (A) and densitometric analysis (B) of kinase phosphorylation array. PBMCs were incubated with IL-2/IL-4 for 48 h and then incubated with FOR (10 nM) for 20 min. The density of each dot was calculated, and the percentage increase over nontreatment was determined. Open bars indicate treatment with IL-2/IL-4 alone, and closed bars indicate IL-2/IL-4 treatment with FOR. Significant induction or reduction by FOR are indicated by the bold arrows. Data were plotted as means ± S.E.M. of n = 3 independent experiments. C, Western blot of phosphorylation of p38MAPK-α and -γ and HSP27. Cells were treated as shown in A. SB, SB203580. D, cell-based ELISA of p38MAPK-γ phosphorylation. Formoterol, salmeterol, and salbutamol were treated 20 min after 48-h treatment of IL-2/IL-4, and inhibitory effects versus IL-2/IL-4 control were calculated and plotted.
siRNAs against p38 MAPK-γ and -δ were transfected to PBMCs from healthy volunteers to obtain knockdowns (KD). After 24 h, mRNA levels of p38MAPK-γ and -δ were reduced by more than 75%. KD cells (and normal cells) were treated with IL-2 and IL-4 for 48 h and stimulated with CD3/CD28 in the presence or absence of Dex to determine Dex-IC50 on IL-2 production. The expected IL2/IL-4-induced corticosteroid resistance to CD3/CD28-induced IL-2 release was not seen in cells with p38 MAPK-γ KD by RNA interference (Fig. 5A). By contrast, p38 MAPK-δ KD did not prevent corticosteroid insensitivity (Fig. 5A). Although FOR (1 nM) reversed IL-2/IL-4-dependent corticosteroid insensitivity, SB203580, a selective p38 MAPK-α and -β inhibitor, did not restore corticosteroid sensitivity in IL-2/IL-4-treated PBMCs (Fig. 5A). Improvement of Dex-IC50 on CD3/CD28-induced IL-2 release by FOR correlated well with the improvement by p38 MAPK-γ KD (r = 0.53, p = 0.0079; Fig. 5B) in the same subjects.
Fig. 5.

p38 MAPK-γ causes corticosteroid insensitivity. A, effects of p38 MAPK-γ or -δ KD by RNA interference on IL2/IL-4-induced dexamethasone resistance to CD3/CD28-induced IL-2 release sensitivity in PBMCs from healthy volunteers. FOR (1 nM) or SB-203580 (SB, 1 μM) were also preincubated for 20 min before CD3/CD28 treatment. B, Spearman correlation analysis between formoterol efficacy and p38 MAPK-γ KD efficacy in each sample from healthy volunteer. C, representative image of p38MAPK-γ knockdown in U937 cells. SC, scrambled oligonucleotides. E, effect of p38 MAPK-γ on anti-inflammatory action of dexamethasone in U937 cells on TNF-α-induced IL-8 (D) and GR phosphorylation by IL-2/IL-4. F, effects of FOR (1 or 10 nM) or SB-203580 (SB; 1 μM) on GR phosphorylation in U937 cells. G, p38 MAP-γ mRNA expression in PBMCs from patients with SA, HV, and patients with MA. H. The relationship between phosphorylated GR (p-GR) corrected to total GR expression and p38MAPK-γ protein expression corrected to α-tublin, evaluated by Western blot analysis in PBMCs from three healthy volunteers and six patients with SA (Supplemental Fig. 2).
U937 cells (a monocytic cell line) were transfected with siRNAs against p38 MAPK-γ or scrambled oligonucleotides as control for 24 h. As shown in Fig. 5C, p38MAPK-γ was clearly knocked down in this condition. Cells were then treated with IL-2/IL-4 for 48 h and stimulated with TNF-α (1 ng/ml) in the presence or absence of Dex to determine Dex-IC50 values. Dex inhibited TNFα-induced IL-8 production with an IC50 of 4.6 nM, but Dex potency was decreased in the presence of IL-2/IL-4 (Dex-IC50, 36 nM). However, p38 MAPK-γ KD shifted the dose-response curve of Dex leftward (Dex-IC50, 9.6 versus 36 nM with scrambled oligonucleotide treatment as a control; all cells were treated with IL-2/IL-4; p < 0.05), suggesting that Dex counteracts the development of IL-2/IL-4-induced corticosteroid insensitivity (Fig. 5D). In contrast, KD of p38 MAPK-δ did not affect IL-2/IL-4-induced corticosteroid insensitivity (Dex-IC50, 21 versus 36 nM with scrambled oligonucleotide treatment; not significant; data not shown). Furthermore, p38 MAPK-γ KD, but not p38 MAPK-δ KD, also inhibited GR phosphorylation by IL-2/IL-4 (Fig. 5E) where FOR, but not SB203580, inhibited GR phosphorylation in this U937 system (Fig. 5F). When mRNAs of p38MAPKs were determined in PBMCs, there was no significant difference between HV and patients with SA in mRNA encoding p38 MAPK-α, -β, and -δ (data not shown) or p38MAPK-γ (Fig. 5G). However, there was a good correlation between mRNA expression of p38 MAPK-γ and GR nuclear translocation (nuclear FITC-Dex) (p < 0.05, date not shown) in patients with SA, suggesting that higher p38MAPK-γ causes defect of GR function. More importantly, when p38MAPK-γ protein expression was analyzed and the level was normalized to α-tubulin expression (a housekeeping gene) in PBMCs from three HV and six patients with SA (Supplemental Fig. 2, there was significant correlation between GR phosphorylation and p38MAPK-γ protein expression (Fig. 5H). For further confirmation of role of p38MAPK-γ in corticosteroid insensitivity, p38MAPK-γ was also overexpressed in U937 cells, and Dex-IC50 was determined. The U937 cells with p38MAPK-γ overexpression showed significantly higher Dex-IC50 value on TNFα-induced IL-8 release after IL-2/IL-4 treatment (Supplemental Fig. 3).
Discussion
Severe asthma is characterized by corticosteroid insensitive inflammation. We showed here that IC50 value of Dex on TNF-α stimulated IL-8 release in PBMCs of patients with SA was higher by approximately 10-fold than those of HV or patients with MA, suggesting that PBMCs from patients with SA were also steroid-insensitive in vitro.
At a molecular level, the reduction in corticosteroid responsiveness observed in cells from patients with SA has been ascribed to a reduced number of GR, altered affinity of the ligand for GR, reduced ability of the GR to bind to DNA, increased expression of inflammatory transcription factors (such as activator protein-1) that compete for DNA binding, or reduction of histone deacetylase-2 (Adcock et al., 2006). In this study, there were no significant differences in GRα mRNA and protein expression. GRβ mRNA expression likely increased in patients with SA but the difference was not significant, possibly because of lack of power; however, several reports have shown that GRβ overexpression was not critical to corticosteroid insensitivity in patients with SA (Irusen et al., 2002; Torrego et al., 2004). Irusen et al. (2002) also demonstrated that GR affinity in nuclei was decreased in patients with SA, although we did not analyze the GR function in nuclei in present study (Irusen et al., 2002). In contrast, we found the defect of GR in cytoplasm.
An increase of activated GR in nuclei is critical for GR action. For the detection of ligand-bound GR in nuclei, we determined the amount of FITC-conjugated Dex in nuclei instead of using classic immunocytochemistry or Western blotting methods, which are not quantitative, but are time-consuming and require a large number of cells. Because the anti-inflammatory efficacy of FITC-conjugated Dex is 10-fold weaker than that of unlabeled Dex on IL-1β-induced IL-8 production in A549 cells, we used a relatively high concentration (10−6 M) of FITC-Dex for the assay. There was a good correlation between percentage of GR positive nuclei in immunocytochemistry assay and the absolute value of FITC-Dex in nuclei in PBMC of healthy volunteers (data not shown, r = 0.65, p < 0.05), suggesting that this FITC-Dex method is useful for quantification of GR nuclear translocation in a small number of cells. We demonstrated that GR nuclear translocation was significantly impaired in SA PBMCs (Fig. 1C), which is supported by previous report (Matthews et al., 2004). In addition, there was a significant, negative correlation between the IC50-Dex value on TNF-α-induced IL-8 release and the amount of FITC-Dex in nuclei, indicating that patients with less GR nuclear translocation are more corticosteroid insensitive (Spearman r = −0.55 p = 0.0035). LABAs are reported to be able to enhance corticosteroid sensitivity in several in vitro systems and clinical trials, even in our PBMC systems; we also confirmed that add-on-treatment with FOR (1 nM) and SAL (100 nM) decreased the Dex-IC50 value for TNF-α-induced IL-8 release in PBMCs from patients with SA (Fig. 2A), although the efficacy of SAL on restoration of corticosteroid sensitivity was weaker than that of FOR. Neither FOR nor SAL changed corticosteroid sensitivity in HV and patients with MA (Table 2). In addition, FOR (1 nM) enhanced GR nuclear translocation evaluated with FITC-Dex in patients with SA (Fig. 2B). The levels of corticosteroid sensitivity and restoration by FOR or SAL were not affected by current medication (oral steroid or inhaled steroid therapy; data not shown). The improvement in Dex-IC50 with FOR (ratio of Dex-IC50 with versus without FOR) correlated well with the improvement of FITC-Dex accumulation in nuclei by FOR (Spearman r = −0.77, p = 0.042), suggesting that FOR reversed corticosteroid insensitivity by enhancement of GR nuclear translocation. The improvement in Dex-IC50 with FOR was negatively correlated with basal FITC-Dex values in nuclei (Spearman r = −0.73; p = 0.0013; Fig. 2C), suggesting that patients with less GR nuclear translocation are more sensitive to FOR-dependent reversal of corticosteroid resistance.
GR are reported to be phosphoproteins, and phosphorylation of inactive GR may block subsequent hormone binding and affect GR subcellular localization and GR nuclear cytoplasmic trafficking through the nuclear pore complex (Ismaili and Garabedian, 2004). We demonstrated that GR in cytoplasm was highly phosphorylated at serine residues in patients with SA compared with those of HV and patients with MA (Fig. 3, A and B), and there was a good correlation between GR phosphorylation and FITC-Dex nuclear translocation (as an index of capability of GR on nuclear translocation). Phosphorylation of Ser226 is reported to cause defect of GR-mediated transcriptional activation (Rogatsky et al., 1998) or enhancement of nuclear export (Itoh et al., 2002). Very interestingly, the level of GR phosphorylation at Ser226 was higher in patients with SA compared with HV (Fig. 3F). Even more importantly, FOR significantly inhibited phosphorylation of GR (Fig. 3C). IL-2/IL-4 treatment, which is known to mimic corticosteroid insensitivity seen in SA (Kam et al., 1993; Larsson et al., 1997; Irusen et al., 2002), also induced phosphorylation of GR (Fig. 3, D and E) and significantly inhibited by FOR (Fig. 3, D and E).
Several kinases, such as MAPK, cyclin-dependent kinase, glycogen synthase kinase-3, and JNK are reported to phosphorylate GR, each of them having distinct specificities for potential phosphorylation sites (Ito et al., 2006). Kinase phosphorylation array analysis demonstrated that IL-2/IL-4 treatment up-regulated phosphorylation of several stress kinases, including p38 MAPK (α, β, γ, δ) and JNK (Fig. 4, A and B). Very interestingly FOR (10−8 M) significantly inhibited only phosphorylation of pan-JNK and p38 MAPK-γ (Fig. 4, A and B). GR Ser226 phosphorylation is reported to be catalyzed by JNK and to inactivate GR (Rogatsky et al., 1998). The leucine-rich sequences flanking Ser246 in rat GR (Ser226 in human GR) are also reported to be involved in nuclear export (Itoh et al., 2002), and phosphorylation of this site may increase GR nuclear export as the means of inactivating GR transcriptional enhancement, leading eventually to accumulation of phosphorylated GR in the cytoplasm. Thus, JNK will be a key kinase on GR phosphorylation in this finding, but the roles of p38 MAPK-γ on GR phosphorylation and corticosteroid effects have not previously been reported.
In U937 cells and PBMCs, where p38 MAPK-γ was knocked down by RNA interference, IL2/IL-4 exposure did not induce corticosteroid insensitivity (Fig. 5, A and D). Furthermore, p38 MAPK-γ KD, but not p38 MAPK-δ KD, inhibited GR phosphorylation by IL-2/IL-4 (Fig. 5E). Thus, p38 MAPK-γ seems to be a key kinase regulating corticosteroid sensitivity, probably by phosphorylation of cytoplasmic GR. In fact, PBMCs with higher levels of GR phosphorylation at Ser226 showed higher levels of mRNA expression of p38 MAPK-γ (Fig. 5H). The p38MAPK-γ protein expression was also higher in patients with SA than in healthy subjects (Supplemental Fig. 2). Further analysis with p38MAPK-γ overexpression in U937 cells (Supplemental Fig. 3) supported the finding that p38MAPK-γ overexpression in SA is likely to be one of molecular mechanisms of steroid insensitivity.
In our system, FOR converted phosphorylated GR and p38 MAPK-γ to nonphosphorylated forms within 20 min after IL-2/IL-4 treatment for 48 h, suggesting that FOR might dephosphorylate GR via dephosphorylation of p38 MAPK-γ rather than by inhibiting p38 MAPK-γ directly. In fact, FOR (or SAL) (100, 1000 nM) did not directly inhibit p38 MAPK-γ kinase activity [kinase profiler assay (Millipore, Billerica, MA; data not shown]. That is, FOR might enhance a specific phosphatase to dephosphorylate p38 MAPK-γ. The phosphatase that specifically dephosphorylates p38 MAPK-γ has not yet been identified. However, cAMP-PKA signals are reported to enhance activity of protein phosphatase (PP)2A (Feschenko et al., 2002). PP2C is also known as cAMP-coupled phosphatase (Yokoyama et al., 1995), and PP1 and PP5 are reported to be involved in GR localization (DeFranco et al., 1991; Dean et al., 2001; Hinds and Sánchez, 2008). Further studies will be required to clarify the molecular mechanism of LABA on dephosphorylation of p38MAPK-γ (and GR).
Thus, LABAs restored corticosteroid sensitivity defected by phosphorylation (Fig. 6). Corticosteroid itself is reported to increase β-adrenoceptor expression (Aksoy et al., 2002) and also LABAs are reported to enhance GR nuclear translocation(Eickelberg et al., 1999; Usmani et al., 2005). This should be a self-enforcing loop induced by the combination therapy of LABA and inhaled corticosteroid. Corticosteroid-insensitive severe diseases are heterogeneous, but our study demonstrated that at least a subpopulation of patients with SA is characterized by defective GR nuclear translocation and GR hyperphosphorylation, which were reversed by LABA via a p38 MAPK-γ-dependent mechanism (Fig. 6). Our studies provide new insights into the regulation of inflammation and raise the prospects of new classes of compounds to treat SA and other inflammatory diseases.
Fig. 6.
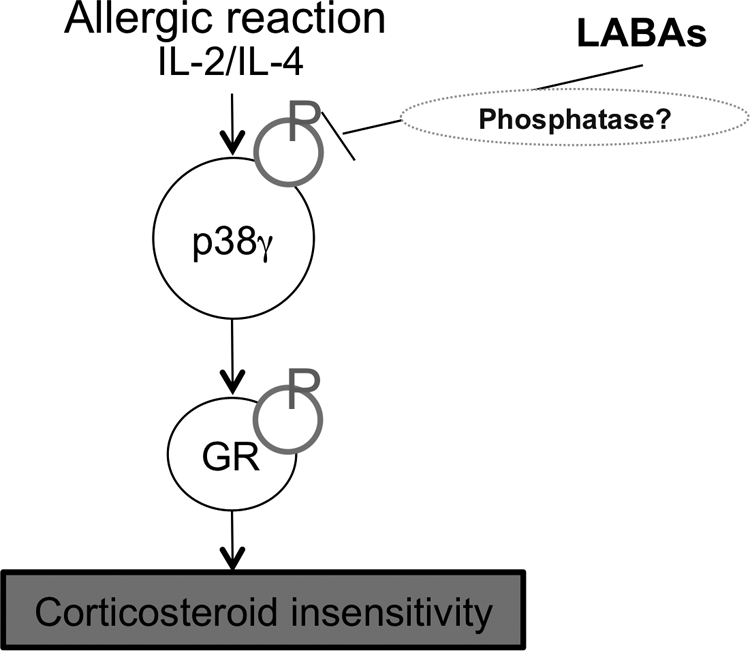
Precise mechanism of formoterol on restoration of corticosteroid sensitivity in severe asthma. Severe allergic inflammation associated with production of IL-2 and IL-4 induced corticosteroid insensitivity via GR phosphorylation by p38MAPK-γ activation. Formoterol inhibits p38MAPK-γ phosphorylation, possibly via phosphatase activation, and inhibits GR phosphorylation.
Supplementary Material
Acknowledgments
We are indebted to Dr. Sergei A. Kharitonov, Debby Campbell, and Sally Meah for assistance in providing clinical samples, Misako Ito and Dr. Masashi Deguchi for assistance in the in vitro analysis.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This study was supported by Asthma UK [Grant 04-56]; the UK Medical Research Council [Grant G0401662]; GlaxoSmithKline; and AstraZeneca (Lund).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/mol.111.071993.
- CI
- corticosteroid insensitivity
- GR
- glucocorticoid receptor
- NF-κB
- nuclear factor-κB
- JNK
- c-Jun-NH2-terminal kinase
- MAPK
- mitogen-activate protein kinase
- IL
- interleukin
- LABA
- long-acting β2-agonist
- SB203580
- 4-[4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl]pyridine
- FEV1
- forced expiratory volume in 1 s
- PBMC
- peripheral blood mononuclear cell
- FITC
- fluorescein isothiocyanate
- Dex
- dexamethasone
- PCR
- polymerase chain reaction
- ELISA
- enzyme-linked immunosorbent assay
- HSP
- heat shock protein
- KD
- knockdown
- FOR
- formoterol
- SAL
- salmeterol
- HV
- healthy volunteer(s)
- SA
- severe asthma
- MA
- mild glucocorticoid-sensitive asthma
- PP
- protein phosphatase.
Authorship Contributions
Participated in research design: Mercado, To, Adcock, and Ito.
Conducted experiments: Mercado, To, Kobayashi, and Ito.
Performed data analysis: Mercado, To, Kobayashi, and Ito.
Wrote or contributed to the writing of the manuscript: Barnes and Ito.
References
- Adcock IM, Caramori G, Ito K. (2006) New insights into the molecular mechanisms of corticosteroids actions. Curr Drug Targets 7:649–660 [DOI] [PubMed] [Google Scholar]
- Adcock IM, Ito K. (2004) Steroid resistance in asthma: a major problem requiring novel solutions or a non-issue? Curr Opin Pharmacol 4:257–262 [DOI] [PubMed] [Google Scholar]
- Adcock IM, Lane SJ, Brown CR, Lee TH, Barnes PJ. (1995) Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. J Exp Med 182:1951–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy MO, Mardini IA, Yang Y, Bin W, Zhou S, Kelsen SG. (2002) Glucocorticoid effects on the beta-adrenergic receptor-adenylyl cyclase system of human airway epithelium. J Allergy Clin Immunol 109:491–497 [DOI] [PubMed] [Google Scholar]
- Cosío BG, Mann B, Ito K, Jazrawi E, Barnes PJ, Chung KF, Adcock IM. (2004) Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med 170:141–147 [DOI] [PubMed] [Google Scholar]
- Cuenda A, Cohen P, Buée-Scherrer V, Goedert M. (1997) Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/P38). EMBO J 16:295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean DA, Urban G, Aragon IV, Swingle M, Miller B, Rusconi S, Bueno M, Dean NM, Honkanen RE. (2001) Serine/threonine protein phosphatase 5 (PP5) participates in the regulation of glucocorticoid receptor nucleocytoplasmic shuttling. BMC Cell Biol 2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFranco DB, Qi M, Borror KC, Garabedian MJ, Brautigan DL. (1991) Protein phosphatase types 1 and/or 2A regulate nucleocytoplasmic shuttling of glucocorticoid receptors. Mol Endocrinol 5:1215–1228 [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Roth M, Lörx R, Bruce V, Rüdiger J, Johnson M, Block LH. (1999) Ligand-independent activation of the glucocorticoid receptor by Beta2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 274:1005–1010 [DOI] [PubMed] [Google Scholar]
- Feschenko MS, Stevenson E, Nairn AC, Sweadner KJ. (2002) A novel CAMP-stimulated pathway in protein phosphatase 2A activation. J Pharmacol Exp Ther 302:111–118 [DOI] [PubMed] [Google Scholar]
- Hew M, Bhavsar P, Torrego A, Meah S, Khorasani N, Barnes PJ, Adcock I, Chung KF. (2006) Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am J Respir Crit Care Med 174:134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds TD, Jr, Sánchez ER. (2008) Protein phosphatase 5. Int J Biochem Cell Biol 40:2358–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. (2002) P38 mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol 109:649–657 [DOI] [PubMed] [Google Scholar]
- Ismaili N, Garabedian MJ. (2004) Modulation of glucocorticoid receptor function via phosphorylation. Ann NY Acad Sci 1024:86–101 [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM. (2000) Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 20:6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Chung KF, Adcock IM. (2006a) Update on glucocorticoid action and resistance. J Allergy Clin Immunol 117:522–543 [DOI] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. (2006b) Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med 203:7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. (2002) Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol 16:2382–2392 [DOI] [PubMed] [Google Scholar]
- Kam JC, Szefler SJ, Surs W, Sher ER, Leung DY. (1993) Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J Immunol 151:3460–3466 [PubMed] [Google Scholar]
- Larsson S, Brattsand R, Linden M. (1997) Interleukin-2 and -4 induce resistance of granulocyte-macrophage colony-stimulating factor to corticosteroids. Eur J Pharmacol 334:265–271 [DOI] [PubMed] [Google Scholar]
- Leung DY, de Castro M, Szefler SJ, Chrousos GP. (1998) Mechanisms of glucocorticoid-resistant asthma. Ann NY Acad Sci 840:735–746 [DOI] [PubMed] [Google Scholar]
- Leung DY, Szefler SJ. (1998) New insights into steroid resistant asthma. Pediatr Allergy Immunol 9:3–12 [DOI] [PubMed] [Google Scholar]
- Matthews JG, Ito K, Barnes PJ, Adcock IM. (2004) Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J Allergy Clin Immunol 113:1100–1108 [DOI] [PubMed] [Google Scholar]
- Mertens S, Craxton M, Goedert M. (1996) SAP kinase-3, a new member of the family of mammalian stress-activated protein kinases. FEBS Lett 383:273–276 [DOI] [PubMed] [Google Scholar]
- Miller-Larsson A, Selroos O. (2006) Advances in asthma and COPD treatment: combination therapy with inhaled corticosteroids and long-acting beta 2-agonists. Curr Pharm Des 12:3261–3279 [DOI] [PubMed] [Google Scholar]
- Pang L, Knox AJ. (2000) Synergistic inhibition by beta(2)-agonists and corticosteroids on tumor necrosis factor-alpha-induced interleukin-8 release from cultured human airway smooth-muscle cells. Am J Respir Cell Mol Biol 23:79–85 [DOI] [PubMed] [Google Scholar]
- Reynolds NA, Lyseng-Williamson KA, Wiseman LR. (2005) Inhaled salmeterol/fluticasone propionate: a review of its use in asthma. Drugs 65:1715–1734 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Logan SK, Garabedian MJ. (1998) Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA 95:2050–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Johnson PR, Rüdiger JJ, King GG, Ge Q, Burgess JK, Anderson G, Tamm M, Black JL. (2002) Interaction between glucocorticoids and beta2 agonists on bronchial airway smooth muscle cells through synchronised cellular signalling. Lancet 360:1293–1299 [DOI] [PubMed] [Google Scholar]
- Torrego A, Pujols L, Roca-Ferrer J, Mullol J, Xaubet A, Picado C. (2004) Glucocorticoid receptor isoforms alpha and beta in in vitro cytokine-induced glucocorticoid insensitivity. Am J Respir Crit Care Med 170:420–425 [DOI] [PubMed] [Google Scholar]
- Usmani OS, Ito K, Maneechotesuwan K, Ito M, Johnson M, Barnes PJ, Adcock IM. (2005) Glucocorticoid receptor nuclear translocation in airway cells after inhaled combination therapy. Am J Respir Crit Care Med 172:704–712 [DOI] [PubMed] [Google Scholar]
- Yokoyama N, Kobayashi T, Tamura S, Sugiya H. (1995) PP2C phosphatase activity is coupled to CAMP-mediated pathway in rat parotid acinar cells. Biochem Mol Biol Int 36:845–853 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.