

Abstract
The E. coli trp repressor (trpR) homodimer recognizes its palindromic DNA-binding site through a pair of flexible helix-turn-helix (HTH) motifs displayed on an intertwined helical core. Flexible N-terminal arms mediate association between dimers bound to tandem DNA sites. The 2.5 Å X-ray structure of trpR crystallized in 30% (v/v) isopropanol reveals a substantial conformational rearrangement of HTH motifs and N-terminal arms, with the protein appearing in the unusual form of an ordered 3D domain-swapped supramolecular array. Small angle X-ray scattering measurements show that the self-association properties of trpR in solution are fundamentally altered by isopropanol.
Three-dimensional domain swapping, defined as the noncovalent exchange of polypeptide segments between protein molecules, is a proposed mechanism for evolution of protein oligomers (Bennett et al., 1995; Schlunegger et al., 1997), and is hypothesized to participate in disease-related protein aggregation (Jaskolski, 2001; Lomas and Carrell, 2002; Zerovnik, 2002). By the strictest definition (Abona fide domain-swapping@), both swapped and unswapped folds either co-exist or can be interconverted by environmental influence. In most documented cases, a single reciprocal swap leads to symmetric dimerization of monomeric precursors, but in a few instances swapping among three or more chains yields cyclic or extended assemblies (Liu and Eisenberg, 2002; Liu et al., 2002; Newcomer, 2002). RNaseA has been shown to swap N-terminal helices, producing dimers, or C-terminal strands, producing dimers or cyclic trimers (Liu et al., 2002), and higher-order aggregates of RNaseA may reflect mixed swapping of both termini (Liu and Eisenberg, 2002; Liu et al., 2002). Domain swapping can lead to formation of infinite open-ended polymer chains, as observed in crystals of E. coli RecA (Story et al., 1992), T7 gene 4 helicase (Sawaya et al., 1999), a mutant form of plasminogen activator inhibitor-1 (Sharp et al., 1999), and M. thermoautotrophicum carbonic anhydrase (Strop et al., 2001).
We report here an unusual example of bona fide domain swapping that leads to assembly of an infinite crystalline 3D supramolecular array. The precursor is dimeric trpR, a sequence-specific DNA-binding protein activated by L-tryptophan. Given its highly intertwined fold (Fig. 1a), the trpR dimer itself is a probable example of quasi-domain swapping (Hakansson and Linse, 2002; Schlunegger et al., 1997), although no monomeric homolog has been identified. To our knowledge, this is the first example of an ordered, extended 3D network formed by domain-swapping.
Figure 1.
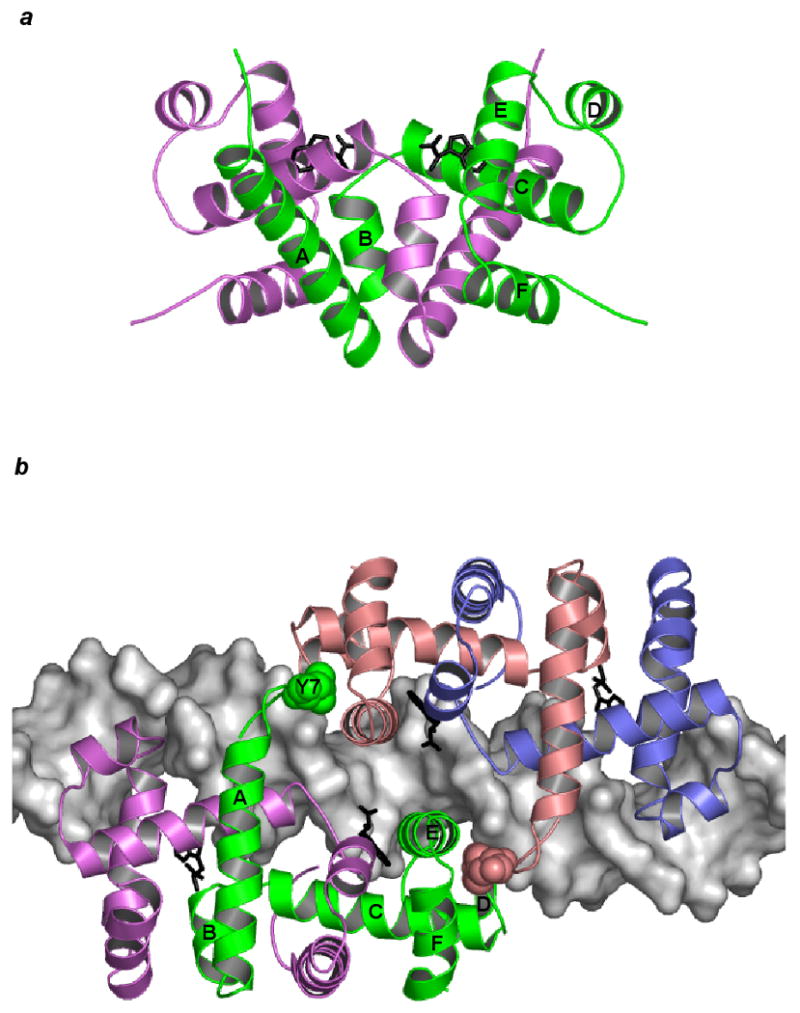
Dimeric trpR. (a) The trpR dimer is shown in schematic ribbon view. Two equivalent subunits are represented in green and purple (orthorhombic crystal form, PDB entry 2WRP (Lawson et al., 1988)). Two L-tryptophan corepressors (black) fill hydrophobic pockets within the extensive interface between the intertwined subunits. (b) Two trpR dimers are shown bound to DNA at tandem sites spaced 8 base-pairs apart (left dimer: green and purple subunits, right dimer: pink and blue subunits, tandem trpR-DNA complex, PDB entry 1TRR (Lawson and Carey, 1993)). For the subunits participating in tandem association between the two dimers (left dimer: green, right dimer: pink), flexible N-terminal arm residue Tyr 7 is displayed in space-fill and labeled on the green subunit. In both (a) and (b), helices A-F of the green subunit are labelled.
The smallest stable unit of trpR is the dimer (Joachimiak et al., 1983a), but higher-order association is well documented. When bound to tandem DNA sites, trpR dimers associate in a two-fold symmetric manner through a limited, predominantly hydrophobic interaction between flexible N-terminal arms and a concave surface adjacent to and involving the HTH (Fig. 1b) (Lawson and Carey, 1993). TrpR dimers also associate in the absence of DNA, yielding predominantly tetramers but also higher-order species (Fernando and Royer, 1992; Martin et al., 1994). Biochemical, mutational, and structural evidence support the hypothesis first put forward by Royer that dimer-dimer interactions in the absence of DNA involve N-terminal arm contacts similar to those made on DNA (Chae et al., 1999; Mackintosh et al., 1998; Vangala, 1998, A. Chin, B.B., and C.L.L., manuscript in preparation).
Domain swapping in trpR crystals was first identified by one of us (B.B.) for a mutant bearing a single substitution (Leu 75 replaced by Phe) that confers long-range effects on the protein's dynamics and structure (Jin et al., 1999; Tyler et al., 2002). The structure determination reported here, carried out as a wild-type control, shows that domain swapping is a general feature of the crystallization condition, and not a consequence of mutation. Domain-swapped trpR crystals are induced by addition of any of several alcohols (to 30-40% (v/v)) to aqueous buffered solutions of purified trpR. Interdimer subunit exchange of trpR is also induced by addition of alcohol, or by heating (Graddis et al., 1988; Hurlburt and Yanofsky, 1993).
Comparison of dimeric vs. domain-swapped crystal structures shows that alcohol increases the α-helix content of the flexible parts of trpR, the N-terminal arms and HTH motifs, with only minor effects on the core fold. The comparison also reveals decreased exposure of polar atoms to solvent, and increased salt-bridge formation. Small-angle X-ray scattering indicates that alcohol (16% (v/v) isopropanol) also perturbs dimer-dimer associations in solution. We suggest that domain-swapped array formation by trpR is associated with susceptibility of non-covalent interactions to alcohol at all structural levels.
Results
Domain-swapped trpR array
Crystals of apo-trpR are readily produced in the 30% (v/v) isopropanol condition of a popular sparse-matrix crystallization screen (Jancarik and Kim, 1991). Crystals with identical morphology form when isopropanol is substituted by other alcohols, including methanol, ethanol, 1-propanol, or 2-methyl-2,4-pentanediol (MPD). Unlike dimeric apo- and holo-trpR crystals produced in salt precipitants (Joachimiak et al., 1983b; Lawson et al., 1988; Zhang et al., 1987), solvent content is high (75 %), and diffraction is relatively poor. Native diffraction data were obtained to a resolution limit of 2.5 Å with synchrotron radiation (Table I), and the structure was solved by molecular replacement using a trpR dimer search model (107 residues/subunit). Initial maps featured an unexpected strong ∀-helical density bridging neighboring molecules, and lack of electron density at the expected position of helix D, the first helix of the HTH motif (Fig. 2a). The single polypeptide chain defining the crystal asymmetric unit was retraced through the spanning density, and a final model for domain-swapped trpR (ds-trpR) was obtained using standard modeling and refinement techniques (see Methods).
Table I. Crystal diffraction and final model statistics for ds-trpR.
| spacegroup cell constants |
P6122 a =b=85.3 Å, c = 114.0 Å, ∀=∃=90E, (=120E |
|
|---|---|---|
| dataset: | native | 5-Bromotryptophan derivative |
|
| ||
| synchrotron beamline | NSLS-X25 | NSLS-X12B |
| wavelength (Å) | 1.1 | 1.0 |
| resolution limits1 (Å) | 50-2.5 (2.6-2.5) | 50-3.2 (3.3-3.2) |
|
| ||
| measured reflections | 67359 (1340) | 39145 (3525) |
| unique reflections | 8304 (526) | 4374 (415) |
| completeness | .925 (.610) | .987 (.976) |
| linear Rmerge | .052 (.169) | .073 (.420) |
| model 2: | isotropic B | isotropic B + TLS |
|
| ||
| R-value | .274 (.353) | .253(.297) |
| free R-value | .315 (.359) | .287(.338) |
| Fo, Fc correlation coefficient | .902 | .913 |
| number of parameters | 3524 | 3544 |
| number of atoms | 881 | 881 |
| rms bonds (Å) | .016 | .018 |
| rms angles (E) | 1.58 | 1.53 |
The highest resolution shell of the diffraction data is defined in parentheses; on subsequent lines statistics for this shell are given in parentheses.
Model statistics vs. native diffraction data are given for two refined models that differ only in treatment of disorder. In both cases, overall anisotropic B and bulk solvent corrections were applied, and individual atom isotropic temperature factors were refined. For the model at right, TLS tensors for a single rigid body encompassing all model atoms (protein+solvent) were also refined (Winn et al., 2001).
Figure 2.
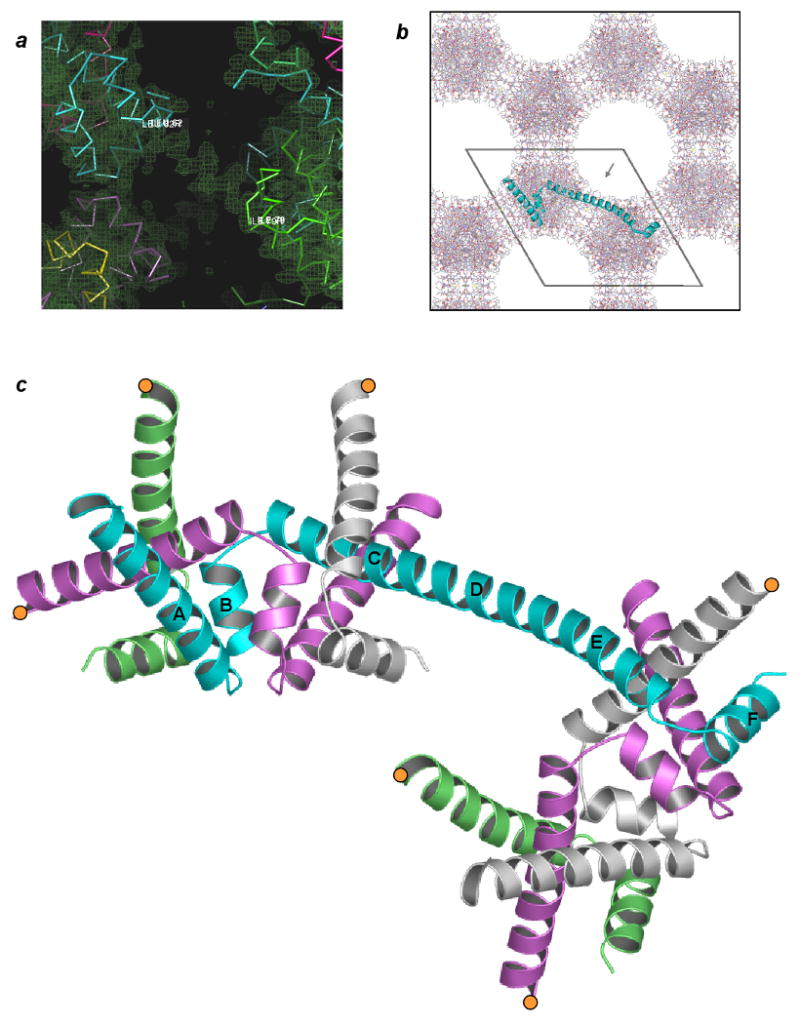
Domain-swapped trpR. (a) Electron density derived from initial molecular replacement phases. “Solvent-flipped” map (Abrahams and Leslie, 1996) is shown overlaid on C∀ traces of several crystal-symmetry equivalent subunits of the dimer molecular replacement search model (varied colors). Residues Leu62 (cyan subunit) and Ile79 (green subunit) define the boundaries of a helical span of density between crystallographically related copies of the search model. Search model D-helices sit outside of electron density. (b) The ds-trpR array hexagonal crystal lattice, ab plane view. All protein atoms within one unit cell c repeat are shown. The polypeptide backbone path of a single trpR subunit, representing one asymmetric unit of the P6122 symmetry structure, is represented with a cyan ribbon. Pores within the lattice are ∼50 Å in diameter (grey arrow in pore indicates view orientation of panel (a)). (c) Domain-swapped trpR, schematic ribbon view. The central cyan subunit, shown in same orientation as cyan subunit in (b), bridges two “nodes” of the array. Truncated segments of equivalent subunits that complete the two nodes are shown in alternating colors, with positions of truncation indicated by orange circles. The orientation of the upper, left node is equivalent to the orientation of the trpR dimer in Fig. 1a. Helices of the cyan subunit are labelled according to dimer convention. Helices C, D and E of the dimer coalesce to form a long, central helix in ds-trpR.
The full domain-swapped array is generated by application of crystallographic symmetry operators (space group P6122) to the ds-trpR single-chain model (Fig. 2b). We define the dimer-like structure formed by segments of four crystallographically equivalent subunits as a “node,” following the definition of this term as a knot or connecting point. Two subunits contribute intertwined N-terminal segments, and two contribute C-terminal segments (Fig. 2c; segment residue ranges are defined in Fig. 3a). The C∀ atom coordinates of the ds-trpR node (two N + two C segments) differ from corresponding coordinates of trpR dimer structures (Lawson et al., 1988; Schevitz et al., 1985; Zhang et al., 1987) (dashed-line brackets in Fig. 3a) by root-mean-square positional deviations of only 0.8-1.2 Å. Each ds-trpR polypeptide chain connects two nodes through a long helical linker between N and C-terminal segments, and each ds-trpR node is linked to four neighboring nodes in a roughly tetrahedral arrangement (Fig. 3b). The alignment of the nodes on 3-fold screw axes of the hexagonal lattice yields an interlocking honeycomb with 50 Å diameter pores (Fig. 2b). The array structure is reminiscent of hexagonal ice, also built from tetrahedral units and characterized by large channels.
Figure 3.
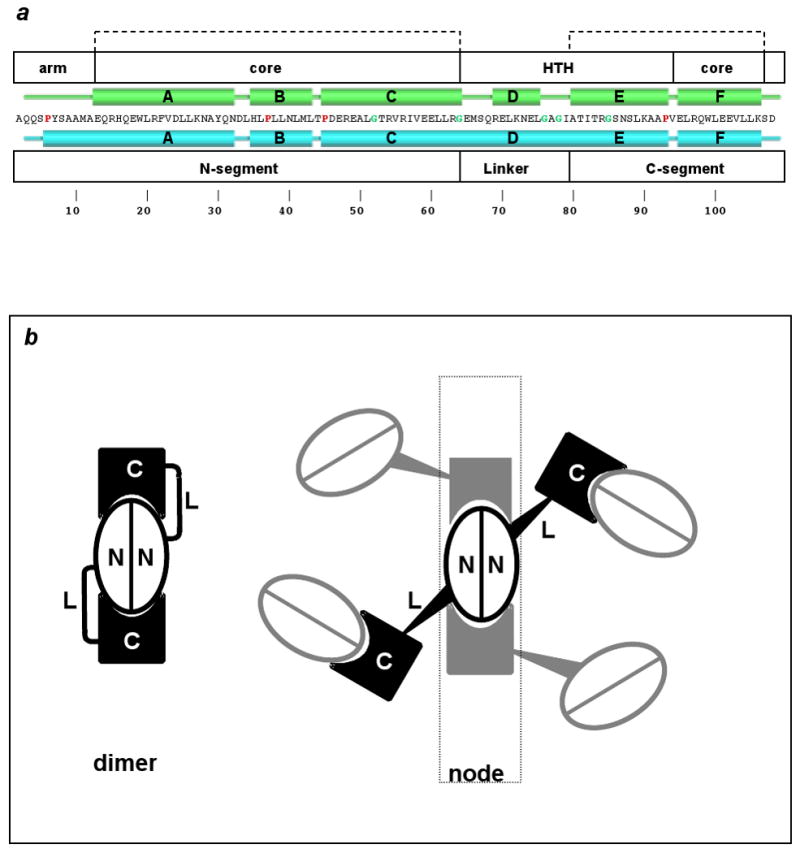
Dimeric vs. domain-swapped trpR. (a) TrpR primary structure (one-letter code) is shown at center with secondary structural elements above in green for the dimer, and below in cyan for ds-trpR. Proline and glycine residues are highlighted with red and green characters, respectively. White boxes above indicate structural elements of the dimer: N-terminal arm (arm), core, and helix-turn-helix (HTH); white boxes below define domain-swap elements: N- and C-segments, linker. Dashed brackets at top indicate residues with conserved fold in dimeric and domain-swapped trpR. (b) Schematic representation of domain-swapping by trpR. At left, the dimer fold is shown. Each subunit is represented as an N and C segment connected by flexible linker L. At right, an array node is generated when four neighbors swap C-terminal segments with a central dimer (we use the term “node” following its definition as a knot or connecting point). Tapered lines represent the approximate tetrahedral geometry of node coordination.
Ds-trpR crystals differ from dimeric trpR crystals in one further respect: L-tryptophan has no apparent influence on ds-trpR crystal formation, growth, or stability, even though a structurally equivalent corepressor-binding pocket is present in the ds-trpR structure. No linker residues are involved in binding L-tryptophan. In the ds-trpR structure, the pocket is filled with solvent, including at least one isopropanol molecule. In all dimeric holo-trpR crystals, the corepressor is represented by strong, unambiguous electron density. In contrast, L-tryptophan or 5-bromotryptophan soaked ds-trpR crystals yield weak corepressor density. Poor binding of L-tryptophan by ds-trpR may result from the high concentration of competing ligand (isopropanol).
Alcohol and trpR structure
Biophysical studies of myoglobin, lysozyme, -lactoglobulin, and other proteins in solution indicate that alcohols can significantly increase helical content, and/or destabilize tertiary structures (Babu et al., 2001; Buck, 1998). The solution behavior of trpR in TFE likely involves similar conformational changes. Titration of trpR with 0 to 12% trifluoroethanol (TFE) results in an increase in helical content from 77 to 87%, as measured by circular dichroism (Jin et al., 1999). A similar increase (from 81% to 89%) is observed between dimer and ds-trpR crystal structures (Fig. 3a), and is due to conversion of residues near the N-terminus and the linker turns. The domain-swapped crystal structure appears to have achieved the maximum helical content permitted by primary structure, with proline residues punctuating the first turn of each ds-trpR helix.
Increased helical content in ds-trpR compared to dimeric trpR is correlated with reduced exposure of polar backbone atoms to solvent. The total solvent-accessible surface area of a trpR subunit is quite similar in the context of the dimer and the ds-trpR lattice (5420 vs. 5260 Å2); however, accessibility of the main chain is nearly halved in ds-trpR (960 vs. 590 Å2), with the bulk of reduction due to burial of main-chain carbonyl oxygens. At the only solvent-exposed interhelical turn of the ds-trpR array, a conformational change that introduces slight backbone strain also increases main-chain polar atom burial (Fig. 4a). The overall reduction in main-chain polar-atom accessibility is balanced by an increase in accessibility of non-polar side-chain atoms, mainly from exposure of aliphatic groups located between the linker and the dimer core. Polar atoms contribute 46% of the accessible surface area in the dimer, but only 38% in ds-trpR.
Figure 4.
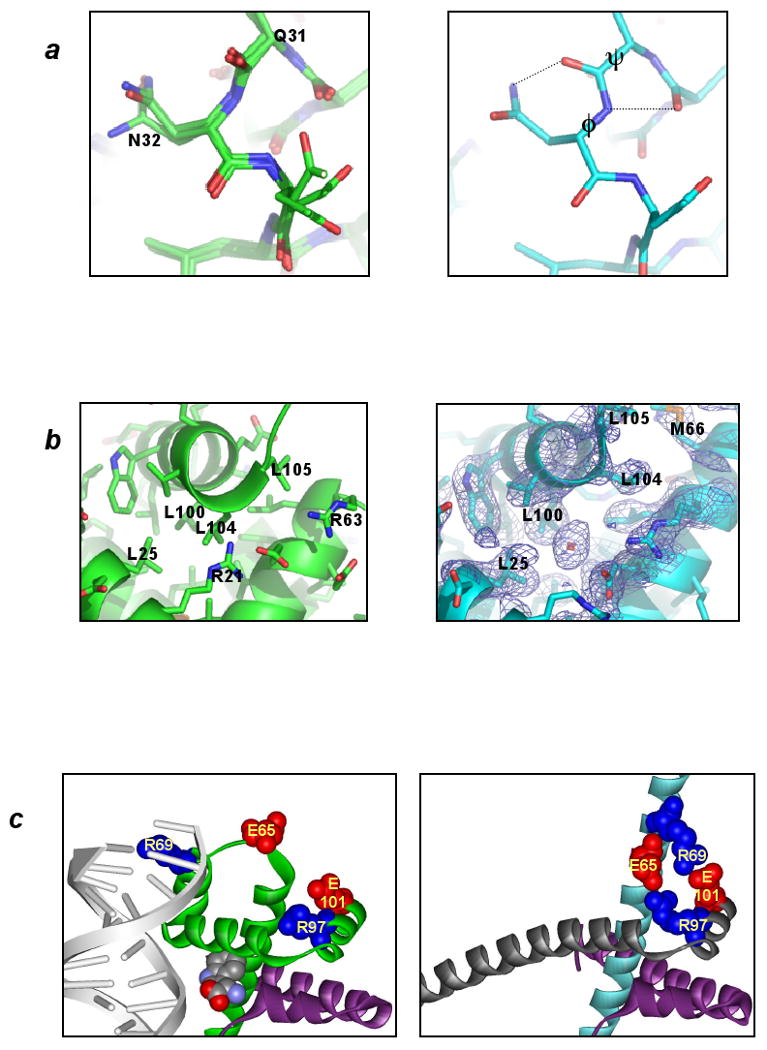
Dimeric vs. domain-swapped trpR: structural differences. In each panel the dimeric structure is represented at left and ds-trpR array is represented at right. (a) Conformational change at a solvent-exposed interhelical turn. In trpR dimer structures overlaid at left (1WRP (Schevitz et al., 1985), 2WRP (Lawson et al., 1988) and 3WRP (Zhang et al., 1987)), main-chain torsion angles of residues in the turn between helices A and B are consistently in most-favored regions of the Ramachandran plot; in ds-trpR, Gln31 P and Asn32 N angles (labeled at right) are rotated by +60E and -60E respectively, moving these residues to less-favored (though allowed) regions. Dotted lines indicate new hydrogen bonds that may form as a consequence of the conformational change. (b) Solvent intrusion at the C-terminus. In all known dimeric trpR structures, a bulge in the final turn of helix F permits the side-chain of Leu 104 to occupy a hydrophobic pocket within the subunit interface (left). In ds-trpR, the final turn adopts a regular helix structure, and the isopropyl side-chain of Leu 104 is displaced by solvent (right, red crosshatch, ds-trpR model shown with (2Fo-Fc), model phased map, contour level = 1 Φ). The solvent density is roughly spherical, but its position with respect to atom neighbors is more consistent with isopropanol than water. (c) Linker-core charge interactions in ds-trpR. Conversion of the first linker turn to helix in ds-trpR (compare green subunit at left with cyan subunit at right) brings Glu65 and Arg69 in close proximity both to each other and to complementary charged residues (Arg97, Glu101) in the swapped F helix. The left panel also highlights the role of the linker in DNA recognition by dimeric trpR (trp repressor/DNA complex 1TRO (Otwinowski et al., 1988)). Sidechains (labeled) and L-tryptophan (unlabeled) are shown in space-fill.
Crystallization of trpR in alcohol appears to promote opening of the dimer to permit tertiary rearrangement coupled to linker conformational change. The interface between one C-segment and the remainder of the dimer fold (1600 Å2 surface burial) is entirely hydrophobic and is openly accessible to solvent at the position of the ligand-binding pocket. Isopropanol also binds at the helix interface near the ds-trpR C-terminus (Fig. 4b). New ionic interactions between the linker and core (Fig 4c) produced in the swapped state are likely to be favored in the lowered dielectric environment.
Alcohol and trpR dimer-dimer association
Small angle X-ray scattering (SAXS) measurements were undertaken to investigate the effects of alcohol on the self-association properties of trpR in solution (Figs. 5, 6). To our knowledge, this is the first SAXS study of trpR. Scattering was measured under conditions representing the protein state both before and after the initial step of ds-trpR crystallization (mixing of protein and reservoir solutions in a 1:1 volume ratio to yield trpR in ∼16% (v/v) isopropanol), for both apo- and holo-trprR. Sample scattering curves are shown in Fig. 5a.
Figure 5.


Small angle X-ray scattering of trpR: intensity vs. resolution profiles. (a) Representative scattering of trpR in solution, ± 2 mM L-tryptophan and ± 16% (v/v) isopropanol. Scattering intensities are plotted as log I(q) vs. q (q=4π sin θ/λ; θ is 1/2 of the scattering angle; λ is the wavelength of the incident radiation). Profiles shown correspond to measurements at 28 mg/ml protein; similar profiles were obtained at 7, 14, and 19 mg/ml. Inset: Guinier plot (ln I(q) vs. q2) of the lowest resolution region, where scattering is dominated by the largest particles in solution. The linear fit was used to estimate the radius of gyration, Rg, and forward scatter, I(0), from the slope and intercept, respectively (see Methods). Y-axis values are offset in both plots for ease of comparison. (b) Distribution of Guinier-derived Rg. (c) Distribution of Guinier-derived forward scatter normalized to protein concentration. In both (b) and (c), vertical bars and brackets indicate the average and standard deviation of four measurements (one each at 7, 14, 19, and 28 mg/ml trpR) for each solution condition. Key: “apo” = trp aporepressor; “holo” = trp aporepressor + 2 mM L-tryptophan; the “+I” extension indicates inclusion of 16% (v/v) isopropanol (see Methods for further experimental details).
Figure 6.

Small angle X-ray scattering of trpR: pair-distance distribution functions (p(r)). P(r) is obtained by Fourier transformation directly from scattering curves and it describes the distribution of distances r between pairs of scattering centers within particles present in solution. The function provides a measure of particle dimensions and shape. (a) and (b): P(r) of experimental scattering profiles presented in Fig. 5. (c): P(r) calculated for models derived from crystal structures. DNA was excluded from all calculations. The “1-dimer” and “2-dimer” models (Rg = 21 Å and 32 Å, respectively) are as illustrated in Fig. 1 panels a and b. The “16-dimer” model corresponds to 4-turns of the left-handed (43) superhelix that forms around DNA in tandem trpR/DNA crystals (Rg = 125 Å, length ≈ 440 Å, model not shown) (Lawson and Carey, 1993). A similar superhelix is formed by trpR dimers without DNA (A. Chin, B.B., and C.L.L., manuscript in preparation). The p(r) curve for the 16-dimer model (dotted line) shows the characteristic sharp rise followed by linearly decreasing slope that is typical for rod-shaped particles (Glatter and Kratky, 1982). For ease of comparison, p(r) curves are shown with their peak values set to a common, arbitrary y value.
All measured samples were polydisperse, i.e., they contained mixtures of several different oligomeric species. At low resolution (q between .08 and .12 nm-1), scattering is dominated by the largest particles in solution. Low-resolution measurements were evaluated in a Guinier plot (Fig 5a, inset) to yield estimates of the radius of gyration (Rg) of the largest trpR aggregates. (Rg is the mean-square distance between scattering atoms and the center of gravity of a particle.) Rg ranged from 82 Å to 194 Å for all measured samples; Rg of the trpR dimer, calculated from atomic coordinates of holo-trpR (2WRP) (Lawson et al., 1988), is only 21 Å.
For both apo- and holo-trpR, isopropanol strongly affects low-resolution scattering. Guinier-derived Rg is increased, on average, ∼50% (Fig 5b). This change can be interpreted as increased size and/or a more extruded shape for the largest aggregate particles. On the other hand, Guinier-derived forward scatter I(0), proportional to the sum of the squared volumes of all scattering particles, is increased, on average, by only ∼25% (Fig 5c). This change can be interpreted as a modest increase in the number of dimers within large aggregate particles and/or a modest increase in the volume occupied per dimer within large aggregate particles. The combined parameter changes are most consistent with models in which the volume occupied per dimer within the aggregates is increased by isopropanol.
Pair-distance distribution functions (p(r)) obtained by Fourier transformation of the scattering curves define the distribution of distances r between pairs of atoms within scattering particles, yielding information about particle dimensions and shape (Glatter and Kratky, 1982). Experimental p(r) profiles (Fig. 6a,b) were compared with calculated p(r) profiles generated from trpR models (Fig. 6c) to evaluate the likely distribution of oligomeric species present in each sample.
Without isopropanol, p(r) reaches a maximum for both apo- and holo-trpR at r ≈ 34 Å, indicating predominance of tetramer species (Fig. 6a,b, black curves)--compare to calculated p(r) curve for 2 dimers arranged as in the tandem trpR-DNA complex (Fig. 6c, black line). P(r) decreases for r above ∼40 Å, with approximately linear decrease above ∼80 Å, indicating rod-shape for higher-order aggregates--compare to the p(r) curve for a 16 dimer superhelical model, also based on the tandem trpR-DNA complex (Fig. 6c, dotted line; The model curve illustrates the characteristic sharp rise followed by linearly decreasing slope for p(r) that is typical for rod-shaped particles). Steeper decrease in slope and introduction of ∼40 Å periodicity in p(r) for holo-trpR compared to apo-trpR indicates that L-tryptophan alters the arrangement of dimers within aggregates, producing periodic variation in electron density along the long aggregate dimension. The periodicity suggests a model in which dimer-dimer associations alternate within the aggregate between compact and extended forms, e.g., “tetramer-beads on a string.”
After addition of isopropanol, the p(r) maximum for both apo- and holo-trpR is clearly shifted downward to r ≈ 22 Å, and distance pairs that correspond uniquely to tetrameric association (40 Å < r < 80 Å) are strongly diminished, indicating the appearance of unassociated dimers at the expense of tetramers (Fig. 6a,b, grey curves). The p(r) profiles also suggest that aggregates with r > 180 Å have altered shape in the presence of isopropanol.
The combined observations of Rg, I(0), and p(r) for trpR in the absence of isopropanol are consistent with a model in which trpR exists as tetrameric and larger rod-shaped aggregates with compact dimer-dimer associations. After isopropanol addition, the major species are unassociated dimers together with larger aggregates having complex shapes and increased volumes per dimer. Formation of ds-trpR crystals thus appears to proceed through an initial step where trpR dimers dissociate from each other, and the mode of association in aggregate particles is altered. The limited, predominantly hydrophobic interaction between the N-terminal arm and HTH observed in several different crystal structures of dimeric trpR (e.g., as in Fig. 1b) is a logical candidate for disruption by isopropanol, particularly in light of the structural changes observed for these regions in the ds-trpR structure. A domain-swapped association (e.g., as in Fig. 2c) is a logical alternative association motif for aggregate particles in 16% isopropanol.
Discussion
Despite wide diversity in primary, secondary, and tertiary structures, proteins that domain-swap share two key linked structural elements: a fold composed of at least two subdomains, and a flexible linker joining the subdomains that can adopt different conformations in swapped and unswapped states (Bennett et al., 1995; Liu and Eisenberg, 2002; Newcomer, 2002; Schlunegger et al., 1997). Dimeric trpR possesses these elements in duplicate. The ds-trpR structure presented here, together with complementary results on the trpR dimer from NMR (Zhao et al., 1993), proteolytic analysis (Carey, 1989), fragment reassembly (Tasayco and Carey, 1992), computational studies (Wallqvist et al., 1999), and crystal structure comparisons of the trpR dimer (Lawson et al., 1988) all support a four-subdomain structural organization for the protein (shown schematically in Fig. 3b), and indicate that the linker is dispensable for folding. In the dimer, flexible linker design affords adaptability to bind L-tryptophan and operator DNA (Fig. 4c, left panel) with physiologically appropriate affinity and specificity (Gryk et al., 1996; Jin et al., 1993; Szwajkajzer and Carey, 1997). The intertwined four-subdomain fold can alternately act as a nexus connecting up to four polypeptide chains, permitting native-like structures to assemble even with the linker completely displaced. The ds-trpR structure demonstrates that propagation of domain swapping from a multiple-subdomain fold can lead to formation of extended aggregates.
Many proteins crystallize in aqueous alcohols, particularly isopropanol and MPD, without apparent structural effect, and some enzyme crystals can be bathed in neat alcohols or other organic solvents without significant conformational change (English et al., 1999; Mattos and Ringe, 2001). However, two proteins, cyanovirin (Yang et al., 1999) and cyclophilin 40 (Taylor et al., 2001), share features with trpR that may be general to alcohol-mediated domain-swapping. Both are monomeric in solution but crystallize in aqueous alcohols as domain-swapped dimers. These proteins differ from each other with regard to swapped elements (β-sheet vs. α-helix) and linker structural conversions (turn⇒coil vs. turn⇒helix), but like trpR both have reduced polar atom exposure in the domain-swapped form (for cyanovirin, burial is through linker-linker contacts) and rearrangement of highly hydrophobic interfaces.
How are trpR domain-swapped arrays formed? Prior to alcohol addition, concentrated trpR exists as tetramers and higher-order oligomers likely mediated by N-terminal arm contacts, as when dimers associate on DNA (Fig. 1b) (Lawson and Carey, 1993). Formally, domain-swapped associations might also form limited oligomeric species. (For example, a hypothetical domain-swapped tetramer can be constructed in which the swapping subunits have CD turns in ds-trpR conformation and DE turns in dimer conformation, C.L.L., unpublished observations.) Alcohol addition frees individual dimers from tetrameric species and substantially alters the character of the largest aggregated species. Aggregates are the likely locus of cooperative nucleation and subsequent propagation of domain-swapped arrays.
Methods
Crystallization
trpR apoprotein was purified as described (Carey et al., 1993; Joachimiak et al., 1983a) and crystallized by standard hanging-drop vapor diffusion at 295 K. Protein stock (5-20 mg/ml) was mixed in a 1:1 volume ratio with reservoir composed of 30-35% (v/v) isopropanol, 100 mM sodium HEPES, pH 7.5, and 100 mM NaCl. Diamond-shaped hexagonal bipyramids typically appear within 24 hours.
Data collection and processing
X-ray diffraction data were collected at the National Synchrotron Light Source (NSLS) with CCD detectors. For native data, a single crystal (maximum dimension ∼0.2 mm) was soaked for two minutes in reservoir solution plus 25 % (v/v) ethylene glycol, then flash-cooled to 100 K in a nitrogen gas stream. A derivative was prepared by soaking a second crystal for 36 hours in reservoir solution containing 10 mM 5-Bromo-(DL)-tryptophan. Integration and merging were performed with Denzo/HKL (Otwinowski and Minor, 1997). Statistics and additional experimental details are provided in Table I.
Structure solution, modeling and refinement
An unambiguous molecular replacement solution was obtained with AmoRe (Navaza, 1994) using PDB entry 2WRP (Lawson et al., 1988) as a search model, with one subunit (½ dimer) per asymmetric unit. Iterative cycles of manual model-building were followed by maximum-likelihood refinement using CNS (Adams et al., 1997). Final refinement was performed in REFMAC (see below). All but 4 of 107 residues in the trpR subunit are included in the final model; N- and C-terminal residues Ala 2, Lys 106, Ser 107, and Asp 108 are disordered. Of 48 solvent sites identified during refinement, only one within the corepressor binding pocket had a non-spherical electron density shape and was modeled as isopropanol. The remaining solvent sites were modeled as single water oxygen atoms; however, the geometry and chemistry of as many as half of the sites are consistent with at least partial occupancy by isopropanol (see e.g., Fig 4b). Conventional statistics that express the agreement of the final model to diffraction data after standard positional and individual B-factor refinement (with overall anisotropic B and bulk solvent correction) are within suggested limits for a correct model, though values are higher than is typical for this resolution (Table I, lower left). Improved statistics are obtained in REFMAC by inclusion of twenty additional parameters defining translation-libration-screw (TLS) positional displacements of the entire model as a single rigid body (Winn et al., 2001) (Table I, lower right). The major disorder component identified by TLS refinement is a libration with 8.0 degrees2 mean-square displacement perpendicular to the longest rigid body axis (roughly, the subunit helix C-D-E axis). This disorder may be a symptom of subtle deviations of the crystal lattice from true hexagonal symmetry. A few weak 00l reflections that should be extinct (l=3n, n odd) as well as refinement tests with CNS point to possible twinning of a lower symmetry lattice (P3121 or P3112). Relative occupancy refinement with CNS indicates that no more than 1% of protein chains exist in dimer (helix-turn-helix) conformation (test performed with overall B, no bulk solvent correction). An omit electron density map of the linker region is shown in Fig. 7.
Figure 7.
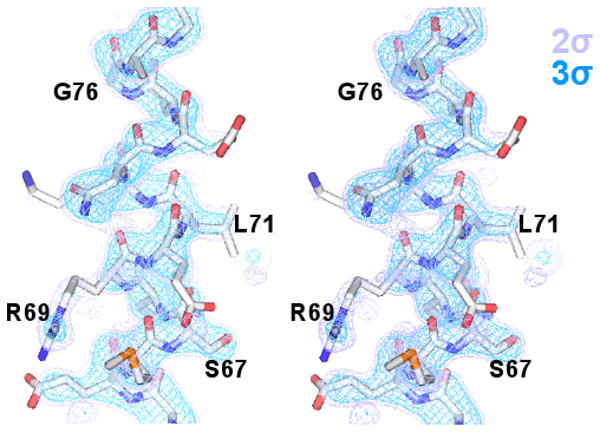
Final “omit” map of the ds-trpR linker (stereo view). A sigmaa-weighted (Fo-Fc) difference map (purple, 2Φ; blue, 3Φ) is shown overlaid on the region of the ds-trpR model excluded from calculation of map phases (residues 68-74).
Validation
The quality of the final model was assessed with Procheck (Laskowski et al., 1993) and Sfcheck (Vaguine et al., 1999). All phi/psi angle combinations are in allowed regions of the Ramachandran plot (including residues illustrated in Fig. 3a), with 97.8% in most favored regions. Electron density correlation is >0.72 for all residues, >0.90 for most. There are no breaks in electron density along the backbone of the entire protein chain. To validate model phases and at the same time examine the integrity of the corepressor binding pocket in the ds-trpR array, a (F5-bromotryptophan - Fnative), model-phased difference Fourier map was examined. A single strong 13.7 Φ difference peak was found at the expected position for the bromine atom. The final model has been deposited in the Protein Data Bank (1MI7).
Surface-area calculations
Accessible surface area was calculated in CNS (Adams et al., 1997) using a 1.4 Å probe. Polar (N+O) and non-polar (C+S) atom accessibilities were summed separately. Accessible surface area of one trpR subunit (residues 6-104) was calculated in the context of (a) the orthorhombic trp holorepressor dimer (2WRP) with corepressor omitted, or (b) the ds-trpR array.
SAXS measurements and processing
Small angle X-ray scattering measurements of trpR in solution were carried out at NSLS bending magnet beamline X27C. All measurements were based on a primary stock of trpR at 57 mg/ml (equivalent to 2.3 mM dimer) in 100 mM sodium HEPES, pH 7.5, 400 mM NaCl (SAXS buffer). The NaCl concentration chosen is near the high end of the range under which ds-trpR crystals will form (50-500 mM). Secondary stocks of 38, 28, and 14 mg/ml were prepared by direct dilution of the primary stock in the same buffer. Each SAXS sample was prepared just prior to measurement by mixing primary or secondary protein stock with buffer of varied composition (± 4 mM L-tryptophan, ±32% (v/v) isopropanol) in a 1:1 volume ratio to a final volume of 20 μl. Thus, for each protein concentration, four different experimental conditions were tested consisting of SAXS buffer ± 2 mM L-tryptophan, ±16% (v/v) isopropanol. Measurements were also taken of SAXS buffer alone ±16% (v/v) isopropanol for background subtraction. Mixed samples were loaded into a 1.5 mm diameter glass capillary after brief centrifugation. Scattering data were collected on a Fuji image plate placed approximately 1700 mm from the sample, with monochromatic incident radiation (λ= 1.366 Å, Δλ/λ=0.006 Å), and with 5-minute exposures. Integrated ion chamber measurements of the direct beam were recorded both in front of and behind the sample. Scanned images were processed with POLAR (Stonybrook Technology and Applied Research) to obtain raw intensities for q in the range 0.08 to 2.6 nm-1 (q=4π sin θ/λ). A silver standard was used to calibrate q. All raw curves were normalized relative to incident radiation dose. Normalization and background subtraction were performed with SIGMAPLOT.
SAXS analyses
The radius of gyration, Rg, and relative forward scatter, I(0), were obtained for each curve from the slope, -(Rg2/3), and intercept, ln I(0), of a line fit to the seven lowest resolution points in a Guinier plot, q2 vs. ln I(q). Pair-distance distribution functions (p(r)) were evaluated with GNOM (Svergun, 1992). Based on the low angle limit of the data, p(r) could be examined for r to 450 Å. Model Rg and scattering curves were calculated using CRYSOL (Svergun et al., 1998).
Graphics
Images were generated using PyMol (www.pymol.org), WebLabViewer, and Turbo-Frodo.
Acknowledgments
We are grateful to Andy Napoli, Mike Becker, Dieter Schneider and Bob Sweet for assistance at BNL-NSLS beamlines X25 and X12B, to Valerie Copié and Rob Tyler for wild-type trpR used in the SAXS study, to Lizhu Liu for assistance at beamline X27C, and to Ben Hsaio, Lizhi Liu and Dieter Schneider for guidance in interpretation of the SAXS data. This work was supported by NIH GM21589 to H.M.B. and C.L.L. and NSF MCB01-36094 to J.C.
References
- Abrahams JP, Leslie AGW. Methods used in the structure determination of bovine mitochondrial F-1 ATPase. 5. 1996;2:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- Adams PD, Pannu NS, Read RJ, Brunger AT. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc Natl Acad Sci U S A. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu KR, Moradian A, Douglas DJ. The methanol-induced conformational transitions of beta-lactoglobulin, cytochrome c, and ubiquitin at low pH: a study by electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2001;12:317–328. doi: 10.1016/s1044-0305(00)00226-9. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M. Trifluoroethanol and colleagues: cosolvents come of age. Recent studies with peptides and proteins. Q Rev Biophys. 1998;31:297–355. doi: 10.1017/s003358359800345x. [DOI] [PubMed] [Google Scholar]
- Carey J. trp repressor arms contribute binding energy without occupying unique locations on DNA. J Biol Chem. 1989;264:1941–1945. [PubMed] [Google Scholar]
- Carey J, Combatti N, Lewis DE, Lawson CL. Cocrystals of Escherichia coli trp repressor bound to an alternative operator DNA sequence. J Mol Biol. 1993;234:496–498. doi: 10.1006/jmbi.1993.1602. [DOI] [PubMed] [Google Scholar]
- Chae YK, Abildgaard F, Royer CA, Markley JL. Oligomerization of the EK18 mutant of the trp repressor of Escherichia coli as observed by NMR spectroscopy. Arch Biochem Biophys. 1999;371:35–40. doi: 10.1006/abbi.1999.1394. [DOI] [PubMed] [Google Scholar]
- English AC, Done SH, Caves LS, Groom CR, Hubbard RE. Locating interaction sites on proteins: the crystal structure of thermolysin soaked in 2% to 100% isopropanol. Proteins. 1999;37:628–640. [PubMed] [Google Scholar]
- Fernando T, Royer C. Role of protein--protein interactions in the regulation of transcription by trp repressor investigated by fluorescence spectroscopy. Biochemistry. 1992;31:3429–3441. doi: 10.1021/bi00128a018. [DOI] [PubMed] [Google Scholar]
- Glatter O, Kratky O. Small Angle X-ray Scattering. New York: Academic Press; 1982. [Google Scholar]
- Graddis TJ, Klig LS, Yanofsky C, Oxender DL. Formation of heterodimers between wild type and mutant trp aporepressor polypeptides of Escherichia coli. Proteins. 1988;4:173–181. doi: 10.1002/prot.340040304. [DOI] [PubMed] [Google Scholar]
- Gryk MR, Jardetzky O, Klig LS, Yanofsky C. Flexibility of DNA binding domain of trp repressor required for recognition of different operator sequences. Protein Sci. 1996;5:1195–1197. doi: 10.1002/pro.5560050624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson M, Linse S. Protein reconstitution and 3D domain swapping. Curr Protein Pept Sci. 2002;3:629–642. doi: 10.2174/1389203023380459. [DOI] [PubMed] [Google Scholar]
- Hurlburt BK, Yanofsky C. Analysis of heterodimer formation by the Escherichia coli trp repressor. J Biol Chem. 1993;268:14794–14798. [PubMed] [Google Scholar]
- Jancarik K, Kim SH. Sparse matrix sampling: a screening method for crystallization of proteins. J Appl Crystallogr. 1991;24:409–411. [Google Scholar]
- Jaskolski M. 3D domain swapping, protein oligomerization, and amyloid formation. Acta Biochim Pol. 2001;48:807–827. [PubMed] [Google Scholar]
- Jin L, Fukayama JW, Pelczer I, Carey J. Long-range effects on dynamics in a temperature-sensitive mutant of trp repressor. J Mol Biol. 1999;285:361–378. doi: 10.1006/jmbi.1998.2311. [DOI] [PubMed] [Google Scholar]
- Jin L, Yang J, Carey J. Thermodynamics of ligand binding to trp repressor. Biochemistry. 1993;32:7302–7309. doi: 10.1021/bi00079a029. [DOI] [PubMed] [Google Scholar]
- Joachimiak A, Kelley RL, Gunsalus RP, Yanofsky C, Sigler PB. Purification and characterization of trp aporepressor. Proc Natl Acad Sci USA. 1983a;80:668–672. doi: 10.1073/pnas.80.3.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachimiak A, Schevitz RW, Kelley RL, Yanofsky C, Sigler PB. Functional inferences from crystals of Escherichia coli trp repressor. J Biol Chem. 1983b;258:12641–12643. [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JS. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- Lawson CL, Carey J. Tandem binding in crystals of a trp repressor/operator half-site complex. Nature. 1993;366:178–182. doi: 10.1038/366178a0. [DOI] [PubMed] [Google Scholar]
- Lawson CL, Zhang RG, Schevitz RW, Otwinowski Z, Joachimiak A, Sigler PB. Flexibility of the DNA-binding domains of trp repressor. Proteins. 1988;3:18–31. doi: 10.1002/prot.340030103. [DOI] [PubMed] [Google Scholar]
- Liu Y, Eisenberg D. 3D domain swapping: As domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gotte G, Libonati M, Eisenberg D. Structures of the two 3D domain-swapped RNase A trimers. Protein Sci. 2002;11:371–380. doi: 10.1110/ps.36602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002;3:759–768. doi: 10.1038/nrg907. [DOI] [PubMed] [Google Scholar]
- Mackintosh SG, McDermott PF, Hurlburt BK. Mutational analysis of the NH2-terminal arms of the trp repressor indicates a multifunctional domain. Mol Microbiol. 1998;27:1119–1127. doi: 10.1046/j.1365-2958.1998.00721.x. [DOI] [PubMed] [Google Scholar]
- Martin KS, Royer CA, Howard KP, Carey J, Liu YC, Matthews K, Heyduk E, Lee JC. Electrostatic forces contribute to interactions between trp repressor dimers. Biophys J. 1994;66:1167–1172. doi: 10.1016/S0006-3495(94)80898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattos C, Ringe D. Proteins in organic solvents. Curr Opin Struct Biol. 2001;11:761–764. doi: 10.1016/s0959-440x(01)00278-0. [DOI] [PubMed] [Google Scholar]
- Navaza J. AMoRe: an automated package for molecular replacement. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- Newcomer ME. Protein folding and three-dimensional domain swapping: a strained relationship? Curr Opin Struct Biol. 2002;12:48–53. doi: 10.1016/s0959-440x(02)00288-9. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. In: Sweet RM, editor. Methods in enzymology. Vol. 276. San Diego: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Luisi BF, Sigler PB. Crystal structure of trp repressor/operator complex at atomic resolution. Nature. 1988;335:321–329. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Guo S, Tabor S, Richardson CC, Ellenberger T. Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell. 1999;99:167–177. doi: 10.1016/s0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- Schevitz RW, Otwinowski Z, Joachimiak A, Lawson CL, Sigler PB. The three-dimensional structure of trp repressor. Nature. 1985;317:782–786. doi: 10.1038/317782a0. [DOI] [PubMed] [Google Scholar]
- Schlunegger MP, Bennett MJ, Eisenberg D. Oligomer formation by 3D domain swapping: a model for protein assembly and misassembly. Adv Protein Chem. 1997;50:61–122. doi: 10.1016/s0065-3233(08)60319-8. [DOI] [PubMed] [Google Scholar]
- Sharp AM, Stein PE, Pannu NS, Carrell RW, Berkenpas MB, Ginsburg D, Lawrence DA, Read RJ. The active conformation of plasminogen activator inhibitor 1, a target for drugs to control fibrinolysis and cell adhesion. Structure Fold Des. 1999;7:111–118. doi: 10.1016/S0969-2126(99)80018-5. [DOI] [PubMed] [Google Scholar]
- Story RM, Weber IT, Steitz TA. The structure of the E. coli recA protein monomer and polymer. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- Strop P, Smith KS, Iverson TM, Ferry JG, Rees DC. Crystal structure of the “cab”-type beta class carbonic anhydrase from the archaeon Methanobacterium thermoautotrophicum. J Biol Chem. 2001;276:10299–10305. doi: 10.1074/jbc.M009182200. [DOI] [PubMed] [Google Scholar]
- Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
- Svergun DI, Richard S, Koch MH, Sayers Z, Kuprin S, Zaccai G. Protein hydration in solution: experimental observation by x-ray and neutron scattering. Proc Natl Acad Sci U S A. 1998;95:2267–2272. doi: 10.1073/pnas.95.5.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwajkajzer D, Carey J. Molecular and biological constraints on ligand-binding affinity and specificity. Biopolymers. 1997;44:181–198. doi: 10.1002/(SICI)1097-0282(1997)44:2<181::AID-BIP5>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Tasayco ML, Carey J. Ordered self-assembly of polypeptide fragments to form nativelike dimeric trp repressor. Science. 1992;255:594–597. doi: 10.1126/science.1736361. [DOI] [PubMed] [Google Scholar]
- Taylor P, Dornan J, Carrello A, Minchin RF, Ratajczak T, Walkinshaw MD. Two structures of cyclophilin 40: folding and fidelity in the TPR domains. Structure (Camb) 2001;9:431–438. doi: 10.1016/s0969-2126(01)00603-7. [DOI] [PubMed] [Google Scholar]
- Tyler R, Pelczer I, Carey J, Copie V. Three-dimensional solution NMR structure of Apo-L75F-TrpR, a temperature-sensitive mutant of the tryptophan repressor protein. Biochemistry. 2002;41:11954–11962. doi: 10.1021/bi020304t. [DOI] [PubMed] [Google Scholar]
- Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 1):191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- Vangala S. Protein folding and protein-protein interactions: exploring issues of function and bioavailability. University of Wisconsin; Madison: 1998. [Google Scholar]
- Wallqvist A, Lavoie TA, Chanatry JA, Covell DG, Carey J. Cooperative folding units of escherichia coli tryptophan repressor. Biophys J. 1999;77:1619–1626. doi: 10.1016/S0006-3495(99)77010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Yang F, Bewley CA, Louis JM, Gustafson KR, Boyd MR, Gronenborn AM, Clore GM, Wlodawer A. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. J Mol Biol. 1999;288:403–412. doi: 10.1006/jmbi.1999.2693. [DOI] [PubMed] [Google Scholar]
- Zerovnik E. Amyloid-fibril formation. Proposed mechanisms and relevance to conformational disease. Eur J Biochem. 2002;269:3362–3371. doi: 10.1046/j.1432-1033.2002.03024.x. [DOI] [PubMed] [Google Scholar]
- Zhang RG, Joachimiak A, Lawson CL, Schevitz RW, Otwinowski Z, Sigler PB. The crystal structure of trp aporepressor at 1.8 A shows how binding tryptophan enhances DNA affinity. Nature. 1987;327:591–597. doi: 10.1038/327591a0. [DOI] [PubMed] [Google Scholar]
- Zhao D, Arrowsmith CH, Jia X, Jardetzky O. Refined solution structures of the Escherichia coli trp holo- and aporepressor. J Mol Biol. 1993;229:735–746. doi: 10.1006/jmbi.1993.1076. [DOI] [PubMed] [Google Scholar]