

Abstract
Coxsackie and adenovirus receptor (CAR) was first known as a virus receptor. Recently, it is also known to have tumor suppressive activity such as inhibition of cell proliferation, migration, and invasion. It is important to understand how CAR expression can be regulated in cancers. Based on an existence of putative Sp1 binding site within CAR promoter, we investigated whether indeed Sp1 is involved in the regulation of CAR expression. We observed that deletion or mutation of Sp1 binding motif (−503/−498) prominently impaired the Sp1 binding affinity and activity of CAR promoter. Histone deacetylase inhibitor (TSA) treatment enhanced recruitment of Sp1 to the CAR promoter in ChIP assay. Meanwhile, Sp1 binding inhibitor suppressed the recruitment. Exogenous expression of wild-type Sp1 increased CAR expression in CAR-negative cells; meanwhile, dominant negative Sp1 decreased the CAR expression in CAR-positive cells. These results indicate that Sp1 is involved in regulation of CAR expression.
1. Introduction
Coxsackie and adenovirus receptor (CAR) has been known only as a viral receptor [1], and a lot of efforts to increase its expressional level have been endeavored to improve gene delivery efficiency for virus-mediated cancer gene therapy. Moreover, recently, CAR is also known to have tumor suppression activity. To support this, deficiency of CAR expression has been observed in many cancer cell lines and gastric cancer tissue, and restoration of CAR expression leads to growth inhibition of those cancer cell lines [2–5]. To further substantiate the functional role of CAR, deficiency of CAR is closely associated with downregulation of α-catenin, which is involved in inhibition of cancer cell growth and motility [6]. Additionally, in gastric cancer patients loss of CAR expression significantly correlates with poor prognosis of gastric cancer, suggesting its functional contribution to cancer pathogenesis [6, 7]. Despite of such importance in cancer biology as well as in cancer gene therapy, little is known about how CAR is regulated at expression level.
A few regulation mechanisms including epigenetic control and oncogenic signaling pathway have been suggested to be involved in CAR expression. Histone deacetylase (HDAC) inhibitors, FR901228 [8] and trichostatin A (TSA) [9], induce CAR mRNA expression through epigenetic chromatin remodeling such as histone acetylation. Raf-MEK-ERK signaling pathway is also known to be involved in CAR expression regulation [10]. In urogenital cancer cells, Pong et al. group [8] characterized a distinct region (−585 and −400 bp) of CAR promoter, so-called core promoter that exerts strong promoter activity, containing several binding motifs including Sp1 transcription factor. Based on this report, we investigated whether indeed Sp1 is involved in the regulation of CAR expression in wide range of cell types. Here, using various cancer cell lines we demonstrated that Sp1 plays an important role in the regulation of CAR expression.
2. Materials and Methods
2.1. Cell Culture and Reagents
Cell culture reagents were purchased from GIBCO BRL (Rockville, MD, USA). HCT116 (CAR-positive colon cancer), PC-3 (CAR-negative prostate cancer), and SW480 (CAR-positive colon cancer) were cultured in RPMI 1640 supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL) at 37°C in 5% CO2. HepG2 (CAR-positive liver cancer) and HeLa cells (CAR-positive cervical cancer) were cultured in DMEM, and MCF-7 cells (CAR-negative breast cancer cell line) were cultured in MEM. Mithramycin (MTM) and TSA were purchased from Sigma (St. Louis, MO).
2.2. Plasmid and Transfection
A set of CAR promoter reporter constructs was kindly provided by Pong et al. [8]. 1213/−552 and −585/−400 constructs were generated by cloning it into pGL3-Basic (at Xho I and HindIII restriction sites) using a PCR-based technique. Mutant of the −585/−400 core reporter construct was made by replacing the Sp1 binding sequence (5′-CGGGGCGGAG-3′→ CGTTTTTGAG). Sp1 expression vector (pCMV-WT-Sp1) was kindly supplied by Dr. Robert Tjian (University of California, Berkeley, CA) and dominant negative Sp1 (DN-Sp1) was from Dr. Gerald Thiel (University of the Saarland Medical School Hamburg, Germany). To match the same vector backbone, DN-Sp1 was subcloned into pCMV vector. Transfection and promoter activity assay were performed according to our previous report [11].
2.3. Semiquantitative RT-PCR
Extraction of total cellular RNA and synthesis of first strand were performed as previously reported [11]. The primer sequences of CAR used for Semiquantitative PCR were as follows: forward, 5′-TGCTGTCGTAAAAAGCGCAG-3′; reverse, 5′-CTATACTATAGACCCATCCT-3′. The PCR conditions were as follows: hot starts at 95°C for 3 min followed by 25 cycles of amplification at 94°C for 15 sec, 55°C for 20 sec, and 72°C for 30 sec, and an extension step at 72°C for 7 min. GAPDH was used as a control: forward, 5′-TCGTGGAAGGACTCATGACC-3′; reverse, 5′-TCCACCACCCTGTTGCTGTA-3′. PCR products were analyzed on 1% agarose gels.
2.4. Western Blotting
Western blotting was performed as previously reported [11] using primary antiacetylated H3 antibody (Ac-H3; Upstate Biotechnology, Lake Placid, NY) (1 : 3000) and HRP-conjugated secondary antibody.
2.5. Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed according to our report [11], and for the preparation of the probe, 26 bp oligonucleotides comprising Sp1 binding site (−513 to −488: 5′-GGTGCAGGCGGGGCGGAGGGTAGGAG-3′) were annealed and labeled with [γ-32P] ATP. For the mutant probe, the underlined Sp1 binding sequence was replaced by TTTTTG. For competition experiments, unlabeled probes were added to the reaction mixture at 200-fold excess molar concentration. In the supershift assays, 1.5 μg of Sp1 antibody (Santa cruz) was preincubated for 20 min at room temperature. MTMs were preincubated for 1 h at 4°C with the labeled probe prior to mixing with nuclear proteins [12].
2.6. Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as described previously [11] with minor modification. Chromatin was precipitated with anti-Sp1 (Santa Cruze Biotechnology, Santa Cruz, CA) or anti-acetyl histone H3 (Upstate Biotechnology, Lake Placid, NY). Purified DNA fragments were subjected to PCR reaction [98°C (3 min), 29 cycles of 98°C (30 s), 62°C (30 s), and 72°C (1 min), then 72°C (7 min)] with a primer set, Forward: 5′-ACAGGTCGCATCCCGTGAG-3′, Reverse: 5′-CAGCCCGTCTCCCACATACTG-3′ [8].
2.7. Ad-β-Gal Transduction
For the analysis of adenoviral transduction efficiency, MCF-7 cells were treated by TSA (25 ng/mL) for 24 h with or without pretreatment of mithramycin for 30 min (50 and 100 nM) and transferred to Sonic Seal Slide 4-well plates (Nalge Nunc, North Aurora Road, Naperville, IL). Cells were infected at an MOI of 50 with Ad-β-gal (Neurogenex, Gwanak-Gu, Seoul, Republic of Korea) for 1 h in the serum free medium. The infected cells were maintained in complete medium for an additional 24 h and washed twice with PBS and fixed for 15 minutes with PBS containing 3.7% formaldehyde. After washing twice with PBS for 5 minutes, the fixed cells were stained with X-gal staining solution (2 mM MgCl2, 5 mM potassium ferri/ferro cyanide, 1 mg/mL X-gal, 0.02% NP-40) in the dark room at 37°C for 6 hours. Photographs of the whole or magnified cells were taken. β-Galactosidase-positive cells were counted from three nonoverlapping fields.
2.8. Statistical Analysis
All values are expressed as mean ± S.D. Statistical comparison between experimental groups was carried out using Student's t-test. P values <0.05 were considered to be statistically significant.
3. Results and Discussion
3.1. A Putative Sp1 Binding Site within the Core Promoter Is Required for CAR Promoter Activity
We first examined the promoter activity of CAR gene with a series of reporter gene constructs (Figure 1(a)) containing serially deleted CAR promoter region, similar to previous report [8], and we further tested it in several different cancer cell types beyond urogenital cancer cell lines. Consistent with previous report [8], deletion of a common region that comprises the core promoter containing Sp1 binding motif (−503/−498) severely impaired the CAR promoter activity in HCT116 and HeLa cell lines (Figures 1(b) and 1(c)). Additionally, mutation of Sp1 binding motif within the core promoter was enough to impair the promoter activity in CAR-positive cell lines, such as HCT116, HeLa, SW480, and HepG2 cell line (Figure 1(d)).
Figure 1.
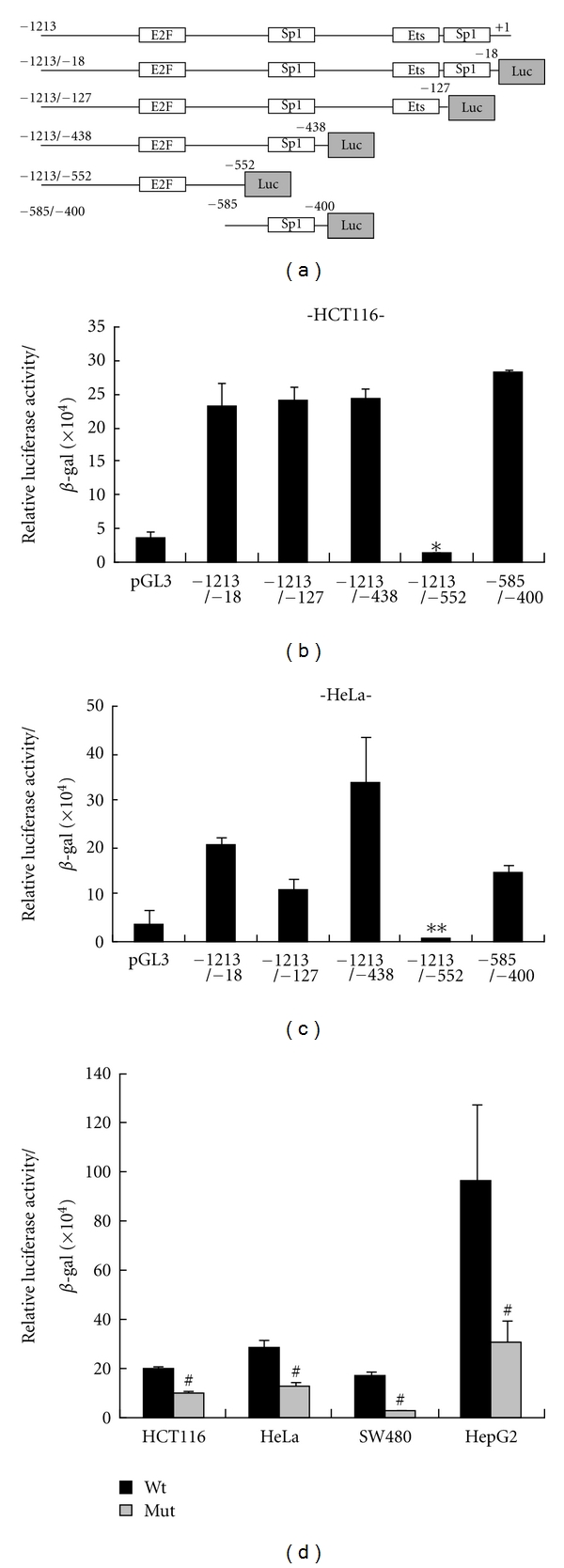
Comparative analysis of CAR promoter activity. (a) Schematic description of CAR gene promoter containing specific transcription factor binding sites and its deletion constructs. The luciferase reporter gene constructs were cloned into pGL3 vector. (b, c) Each CAR promoter-luciferase reporter construct was cotransfected into two different CAR-positive cells with β-galactosidase gene. *P = 0.0003, **P < 0.001 relative to other constructs, not pGL3. Bars represent SD. (d) Either Wt or mut of the core promoter (−585/−400) was cotransfected into four different CAR-positive cells with β-galactosidase gene. From total cell extracts, luciferase activities were examined and normalized by β-galactosidase activity. The relative luciferase activities were determined by statistical analysis of three independent experiments. #P < 0.05 relative to each of wild-type. Bars represent the mean ± SD.
3.2. Exogenous Expression of Sp1 Transactivates the Core Promoter Activity
Next, to examine how important the Sp1 transcription factors are involved in the transactivation, we compared the effects of ectopic expressions of Sp1 and dominant negative Sp1 (Sp1 DN) on the core promoter activity. The exogenous expression of Sp1 increased the activities more than 2-fold, compared with vector-transfected control cells in MCF-7 and PC-3 cell lines (Figures 2(a) and 2(b)). On the contrary, the ectopic expression of Sp1 DN couldnot regulate the activity. However, mithramycin treatment, which is known as blocking transcriptional activity of Sp1 family transcription factors through binding to G/C-rich region [12, 13], significantly suppressed the promoter activity in the cells expressed by ectopic Sp1, but not by Sp1 DN (Figures 2(a) and 2(b)). Likeness, the ectopic expression of Sp1 enhanced the core promoter activities more than 10-fold, compared with pGL3 control vector in the same cell lines (Figures 2(c) and 2(d)). However, under the mutation of Sp1 binding site in the core promoter, it exerted very low activity despite of ectopically Sp1 expression. Mithramycin treatment also significantly suppressed the core promoter activity (Figures 2(c) and 2(d)), suggesting that Sp1 binding inhibition to CAR promoter has effect on transactivation activity of CAR gene.
Figure 2.
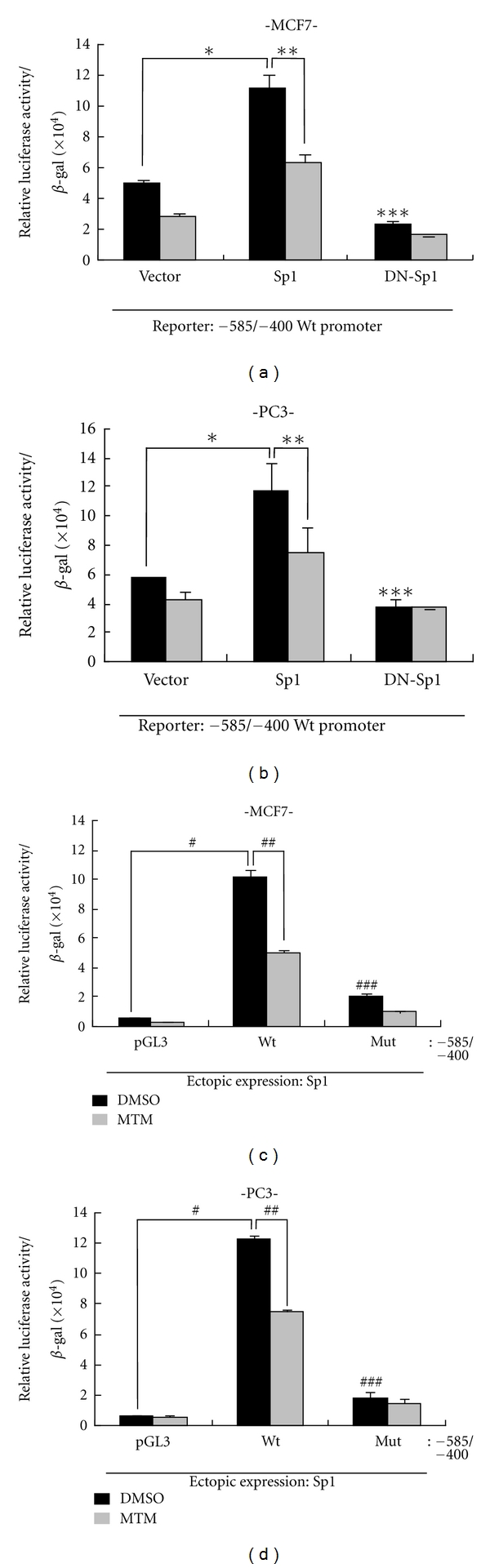
Effect of the mutation in Sp1 binding motif or Sp1 on CAR promoter activity. (a, b) For the examination of the effect of Sp1 on the promoter activity, either Sp1 or its dominant negative form (Sp1 DN) was cotransfected into MCF-7 (a) and PC-3 cells (b) with reporter construct of core promoter and β-galactosidase gene in the presence or absence of mithramycin. *P < 0.05, ***P > 0.05 relative to pCMV vector control, **P < 0.05 relative to Sp1 control. (c, d) For the examination of the effect of Sp1 binding site on the core promoter activity in response to ectopic expression of Sp1, either wild type or mutant type of core promoter was cotransfected with Sp1 and β-galactosidase into MCF-7 (c) and PC-3 (d), and the following procedures were identical to above case. From total cell extracts, luciferase activities were examined and normalized by β-galactosidase activity. The relative luciferase activities were determined by statistical analysis of three independent experiments. #P = 0.0003, # # #P < 0.05 relative to pGL3 vector control, # #P < 0.05 relative to wild-type core promoter (−585/−400) DMSO. Bars represent the mean ± SD.
3.3. Sp1 Directly Binds to CAR Promoter
To directly demonstrate the involvement of Sp1, we performed EMSA and observed complex formation between DNA probe and Sp1 protein (lane 2 in Figure 3), which was abolished by wild-type cold competitor, but not by mutated one (lanes 3 and 4 in Figure 3). Additionally, the complex was supershifted by Sp1 antibody (lane 5 in Figure 3). Meanwhile, mutant probe did not show any complex formation (lane 6 in Figure 3). Treatment with mithramycin (MTM), which is known to inhibit Sp1 binding by targeting GC-rich region of DNA [12, 14], prevented the complex formation between Sp1 and DNA probe in a dose-dependent manner (Figure 3(b)). To demonstrate Sp1 directly binding to CAR gene promoter, we also performed ChIP assay using Sp1 antibody. TSA treatment capable of inducing CAR expression increased Sp1 binding to CAR promoter, as reflected on enhanced PCR product in MCF-7 cells (Figure 3(c)). Furthermore, under open chromatin structure by TSA, we scrutinized relationship between acetyl-histone H3 and Sp1 binding inhibition with CAR promoter into MCF-7 cells or CAR-negative cell line. As shown in Figure 3(d), even if enhancing Sp1 binding to CAR promoter, along with acetyl-histone H3 (lane 4), in the presence of TSA, the association between Sp1 and acetyl-histone H3 was disrupted by Sp1 binding inhibition, mithramycin A, indicating that Sp1 can regulate CAR gene under open-chromatin structure by TSA, and Sp1 cannot bind CAR promoter in the presence of both TSA and Sp1 binding inhibitor into CAR-negative cell, MCF-7 cells. Besides, in CAR-positive cell, HepG2 cell line, in which CAR expression is basically high, Sp1 binding inhibitor itself led to dissociate binding between Sp1 and CAR promoter (Figure 3(e)). Taken together, Sp1 directly regulates CAR gene through binding at CAR promoter.
Figure 3.
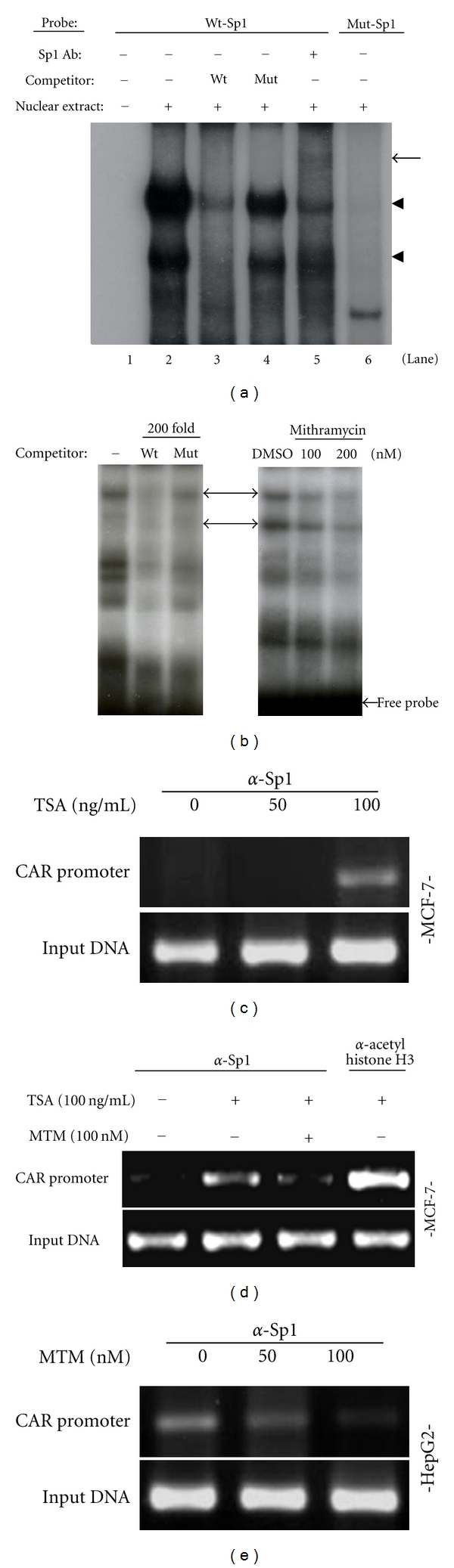
Sp1 binds to its putative binding motif within the core promoter. (a) Labeled probe and cold wild or mutant type probes used for competition assay were used as described in Section 2. The closed triangle indicates the Sp1-DNA probe complex. (b) Labeled probe was preincubated with either MTM or DMSO (vehicle) for 1 h at 4°C before incubation with the nuclear proteins. (c, d) Chromatin immunoprecipitation assay. Each of cell lysates was extracted from MCF-7 cells or HepG2 (e) cells after treatment either by TSA or MTM. Chromatin immunoprecipitation was performed with anti-Sp1, anti-acetyl histone H3, and then PCR was carried out with primers flanking Sp1 binding site.
3.4. Mithramycin Prevents TSA-Induced CAR mRNA Expression
Based on the observation that MTM could effectively prevent Sp1 binding to CAR promoter, as shown in the previous EMSA and ChIP assay, we examined cellular effect of MTM on CAR mRNA expression. As expected, MTM dose dependently suppressed CAR mRNA expression in a similar pattern to its effect on promoter activity in all cell lines tested (Figure 4). To further substantiate the involvement of Sp1 in transcriptional regulation of CAR gene, we identified the effect of MTM under combinational TSA treatment, which is known to induce CAR expression through epigenetic regulation, such as histone acetylation, in CAR-negative cells [9]. MTM suppressed both the enhanced CAR promoter activity and CAR mRNA expression, which were induced by TSA treatment, in a dose-dependent manner (Figures 5(a), 5(c), and 5(d)). Furthermore, as shown in Figure 5(c), CHIP assay (Figure 3(d)) reflected on CAR mRNA expression through the association between Sp1 and acetyl-histone H3, indicating that Sp1 can express CAR mRNA under open-chromatin structure by TSA, and Sp1 cannot regulate CAR mRNA in the presence of both TSA and Sp1 binding inhibitor into CAR-negative cell, MCF-7 cells. However, the induction effect by TSA was not prominent in HepG2 cell, which might be due to high expressional level at basal state. Furthermore, the inhibition effect of MTM was constant to a certain fixed level determined by the concentration of MTM treated, regardless of how much histone acetylation was induced by TSA as shown in hyperacetylated histone 3 (Figure 5(b)), suggesting that Sp1 plays critical role in the CAR regulation, in addition to chromatin remodeling, although which allows free access of many transcription factors.
Figure 4.
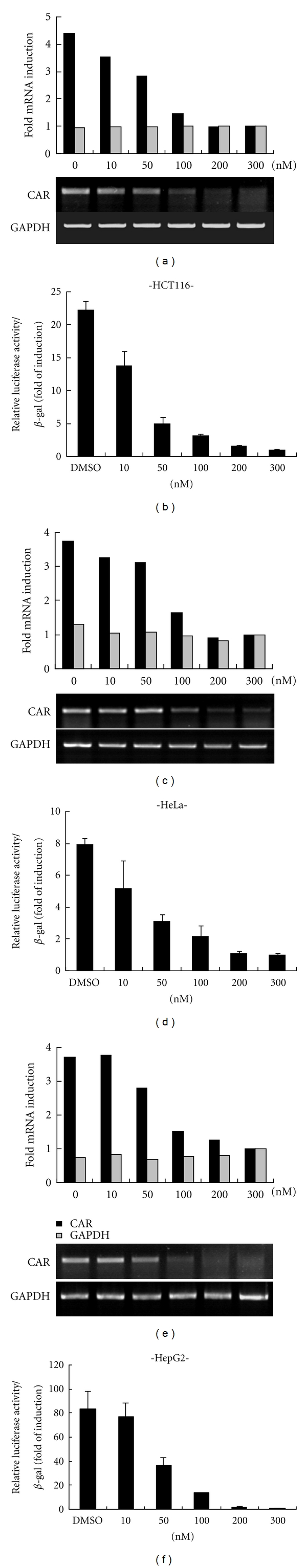
Effects of MTM on CAR promoter activity and mRNA expressions. HCT116 (a), HeLa (c), and HepG2 (e) were treated with either MTM or DMSO (vehicle) for 24 h at the concentration indicated. The mRNA expressions of CAR and GAPDH used as an internal control were examined by Semiquantitative RT-PCR. Core promoter reporter construct was cotransfected with β-galactosidase into the three different cells and followed by treatment with either MTM or DMSO for 24 h at the concentration indicated (b, d, and f). The relative luciferase activity was determined by statistical analysis of three independent experiments. Bars represent the mean ± SD.
Figure 5.
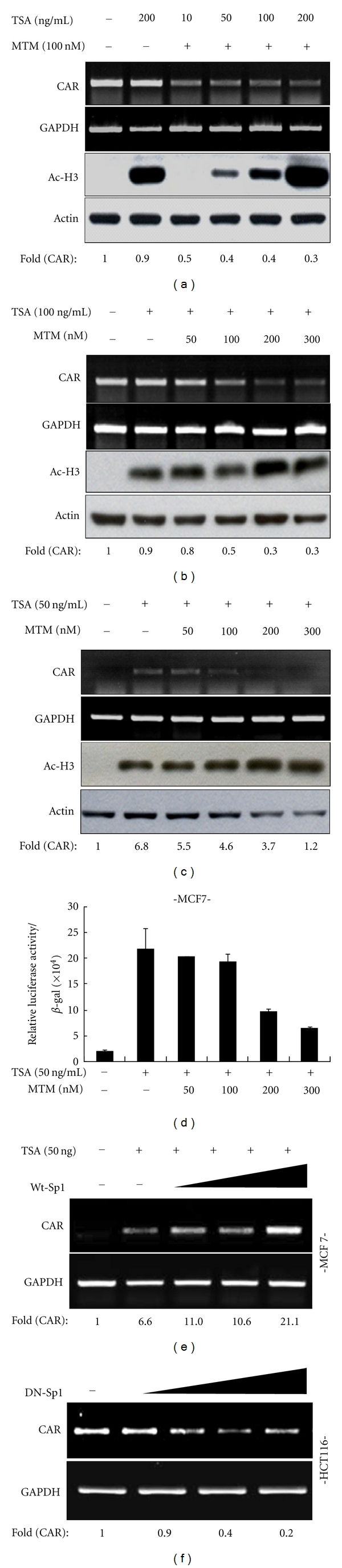
Effects of Sp1 or MTM on TSA-induced CAR promoter activity and mRNA expression. HepG2 (a, b) and MCF-7 (c) cells were pretreated with either MTM or DMSO for 30 min and followed by TSA treatment for 24 h. The mRNA expressions of CAR and GAPDH were examined by Semiquantitative RT-PCR. The extent of histone3 acetylation with actin used as a loading control was analyzed by western blotting. In contrary to (a), CAR mRNA expression was examined depending on TSA concentration with fixed concentration of MTM (c). The mRNA levels of lane1 were set to 1.0. (d) MCF-7 cells were transfected with the core promoter reporter construct and β-galactosidase and treated as in (c). The relative luciferase activity was determined. Bars represent S. D. Effects of WT-Sp1 and DN-Sp1 overexpression on CAR expression. (e) MCF-7 cells were transfected with WT-Sp1 (0.1, 0.25, 0.5, and 1 μg), treated with TSA (50 ng/mL) for 24 h, and examined for CAR mRNA expression by Semiquantitative RT-PCR. Relative expression of CAR mRNA to GAPDH was calculated based on the band intensity. (f) Similarly, HCT116 cells were transfected with DN-Sp1 (0.1, 0.25, 0.5, and 1 μg) in the absence of TSA, and relative expression of CAR mRNA was determined. The mRNA levels of lane1 were set to 1.0.
3.5. Sp1 Regulates the Expression of CAR mRNA to Carry out Function as Adenovirus Receptor in Cancer Cell
We checked the effect of Sp1 overexpression on CAR expression. Ectopic expression of wild-type Sp1 (WT-Sp1) induced CAR mRNA expression in CAR-negative MCF-7 cells in the presence of TSA (Figure 5(e)) but not absence of TSA (data not shown). Meanwhile, ectopic expression of dominant negative Sp1 (DN-Sp1) suppressed the mRNA expression in CAR-positive HCT116 cells (Figure 5(f)), in a dose-dependent manner. Eventually, the expression level of CAR regulated by Sp1 may have effect on its function as receptor for adenoviral entry in the cancer cells. Therefore, based on data shown previously, we could practically substantiate the importance of Sp1 binding through in vivo study, by demonstrating that MTM treatment prevented the enhanced uptake of adenovirus vector containing β-gal transgene in MCF-7 cells, which was mediated by TSA-induced CAR expression (Figure 6). Consistent with the previous report [15–17], TSA treatment by itself made adenoviral infection increase up to 70% into MCF-7 cell line (Figure 6). However, by mithramycin treatment, the inhibition of Sp1 binding at CAR gene promoter eventually reduced the effect of adenoviral infection into MCF-7 cell line below 20%, indicating that the decline of CAR expression by Sp1 binding inhibition was not enough to infect adenoviral containing transgene. Therefore, we could further confirm that Sp1 was essential for enhancing the mRNA level of CAR gene to carry out function as adenovirus receptor in cancer cell.
Figure 6.
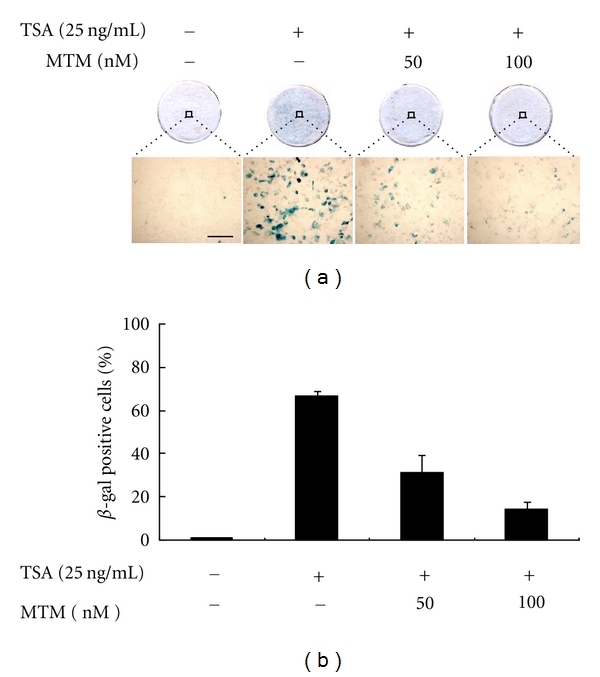
Effect of Sp1 binding on infectivity of adenoviral containing β-gal. (a) MCF-7 cells were treated by TSA with or without pretreatment of mithramycin and infected by Ad-β-gal (50 MOI). Photographs of the cells stained for β-galactosidase activity. (a) represents whole cells and some part of the whole cells under a higher magnification, respectively, scale bar: 50 μm. (b) Quantitation of β-galactosidase-positive cells after the virus infection. β-galactosidase-positive cells were counted from three nonoverlapping fields of the cells treated as indicated. Bars represent the mean ± SD.
CAR has recently attracted much attention as being regarded as tumor suppressor in addition to as a primary receptor of adenoviral vector used for cancer gene therapy. Nonetheless, little is known about the regulation mechanism of CAR expression. Through this study, we could be able to further substantiate HDAC inhibitor-induced CAR expression, which comprises Sp1 activation for its binding to open chromatin structure, and also we could directly demonstrate the involvement of Sp1 transcription factor by EMSA, mithramycin treatment, and ChIP assay. Eventually, we could further clarify the underlying mechanism of HDAC inhibitors by demonstrating that the Sp1 plays a critical role in the regulation of CAR expression.
4. Conclusions
Transcription factor Sp1 plays common role in regulating CAR expression in diverse cancer cells, such as HCT116 (CAR-positive colon cancer), PC-3 (CAR-negative prostate cancer), SW480 (CAR-positive colon cancer), HepG2 (CAR-positive liver cancer), HeLa cells (CAR-positive cervical cancer), and MCF-7 cells (CAR-negative breast cancer cell line). In case of CAR-positive cancer cell lines, CAR expression was downregulated through Sp1 binding inhibitor. Meanwhile, under open chromatin structure by HDAC inhibitor TSA, Sp1 also regulated CAR expression in CAR-negative cancer cell lines. In view of regulating CAR expression, it would be possible to be crosstalk between HDAC inhibitor and Sp1. Therefore, it would be of interest to study whether HDAC inhibitor could make Sp1 induce CAR expression through Sp1 acetylation.
Acknowledgments
The authors thank Dr. JT Hsieh for the CAR promoters and Dr. Robert Tjian, and Dr. Gerald Thiel for pCMV-Sp1, Sp1 DN. This study was supported by Korea National Institute of Health Intramural Research Grant (4800-4845-300-210-13).
References
- 1.Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275(5304):1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 2.Okegawa T, Pong RC, Li Y, Bergelson JM, Sagalowsky AI, Hsieh JT. The mechanism of the growth-inhibitory effect of coxsackie and adenovirus receptor (CAR) on human bladder cancer: a functional analysis of car protein structure. Cancer Research. 2001;61(17):6592–6600. [PubMed] [Google Scholar]
- 3.Okegawa T, Li Y, Pong RC, Bergelson JM, Zhou J, Hsieh JT. The dual impact of coxsackie and adenovirus receptor expression on human prostate cancer gene therapy. Cancer Research. 2000;60(18):5031–5036. [PubMed] [Google Scholar]
- 4.Zhang LL, He DL, Li X, et al. Overexpression of coxsackie and adenovirus receptor inhibit growth of human bladder cancer cell in vitro and in vivo. Acta Pharmacologica Sinica. 2007;28(6):895–900. doi: 10.1111/j.1745-7254.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 5.Kim M, Sumerel LA, Belousova N, et al. The coxsackievirus and adenovirus receptor acts as a tumour suppressor in malignant glioma cells. British Journal of Cancer. 2003;88(9):1411–1416. doi: 10.1038/sj.bjc.6600932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stecker K, Koschel A, Wiedenmann B, Anders M. Loss of Coxsackie and adenovirus receptor downregulates α-catenin expression. British Journal of Cancer. 2009;101(9):1574–1579. doi: 10.1038/sj.bjc.6605331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anders M, Vieth M, Röcken C, et al. Loss of the coxsackie and adenovirus receptor contributes to gastric cancer progression. British Journal of Cancer. 2009;100(2):352–359. doi: 10.1038/sj.bjc.6604876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pong RC, Lai YJ, Chen H, et al. Epigenetic Regulation of Coxsackie and Adenovirus Receptor (CAR) gene promoter in urogenital cancer cells. Cancer Research. 2003;63(24):8680–8686. [PubMed] [Google Scholar]
- 9.Sachs MD, Ramamurthy M, Van Der Poel H, et al. Histone deacetylase inhibitors upregulate expression of the coxsackie adenovirus receptor (CAR) preferentially in bladder cancer cells. Cancer Gene Therapy. 2004;11(7):477–486. doi: 10.1038/sj.cgt.7700726. [DOI] [PubMed] [Google Scholar]
- 10.Anders M, Christian C, McMahon M, McCormick F, Korn WM. Inhibition of the Raf/MEK/ERK pathway up-regulates expression of the coxsackievirus and adenovirus receptor in cancer cells. Cancer Research. 2003;63(9):2088–2095. [PubMed] [Google Scholar]
- 11.Lim JY, Won HK, Kim J, Sang IP. Induction of Id2 expression by cardiac transcription factors GATA4 and Nkx2.5. Journal of Cellular Biochemistry. 2008;103(1):182–194. doi: 10.1002/jcb.21396. [DOI] [PubMed] [Google Scholar]
- 12.Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. Journal of Clinical Investigation. 1991;88(5):1613–1621. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenwel P, Inagaki Y, Hu W, Walsh M, Ramirez F. Sp1 is required for the early response of α2(I) collagen to transforming growth factor-β1. Journal of Biological Chemistry. 1997;272(32):19738–19745. doi: 10.1074/jbc.272.32.19738. [DOI] [PubMed] [Google Scholar]
- 14.Ray R, Snyder RC, Thomas S, Koller CA, Miller DM. Mithramycin blocks protein binding and function of the SV40 early promoter. Journal of Clinical Investigation. 1989;83(6):2003–2007. doi: 10.1172/JCI114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitazono M, Goldsmith ME, Aikou T, Bates S, Fojo T. Enhanced adenovirus transgene expression in malignant cells treated with the histone deacetylase inhibitor FR901228. Cancer Research. 2001;61(17):6328–6330. [PubMed] [Google Scholar]
- 16.Kitazono M, Rao VK, Robey R, et al. Histone deacetylase inhibitor FR901228 enhances adenovirus infection of hematopoietic cells. Blood. 2002;99(6):2248–2251. doi: 10.1182/blood.v99.6.2248. [DOI] [PubMed] [Google Scholar]
- 17.Goldsmith ME, Kitazono M, Fok P, Aikou T, Bates S, Fojo T. The histone deacetylase inhibitor FK228 preferentially enhances adenovirus transgene expression in malignant cells. Clinical Cancer Research. 2003;9(14):5394–5401. [PubMed] [Google Scholar]