

Abstract
To understand the structure-function relationship of muscle-regulatory-protein isoforms, mutations, and posttranslational modifications, it is necessary to probe functional effects at the level of the protein-protein interaction. Traditional methodologies assessing such protein-protein interactions are laborious and require significant amounts of purified protein, while many current methodologies require costly and specialized equipment or modification of the proteins, which may affect their interaction. To address these issues, we developed a novel method of microplate-based solid-phase protein-binding assay over the recent years. This method assesses specific protein-protein interactions at physiological conditions, utilizes relatively small amounts of protein, is free of protein modification, and does not require specialized instrumentation. Here we present detailed methodology for the solid-phase protein-binding assay with examples that we have successfully applied to quantify interactions of myofilament-regulatory proteins. We further provide considerations for optimization of the assay conditions and its broader application in studies of other protein-protein interactions.
1. Introduction
To ultimately understand the structure-function relationship resulting from protein isoform variation, mutation, and posttranslational modification, one must be able to quantify the functional effect of the structural alteration on the interaction of the protein with its binding proteins. Traditional methodologies used to investigate these interactions, such as equilibrium dialysis and affinity chromatography, rely on large amounts of proteins, are time consuming, and are labor intensive. While newer methodologies such as Förster resonance energy transfer or surface plasmon resonance utilize less protein and can be of high throughput, they rely on specialized, costly equipment and/or modification of the target protein with labeling that by itself may alter the protein-protein interaction to be investigated.
Over the past number of years, we have developed a novel microplate-based solid-phase protein-protein binding assay. This assay requires no specialized equipment, uses a minimal amount of protein, is rapid throughput, does not rely on modification of the target protein, and results in quantitative measurements. In this assay one of the proteins of interest is noncovalently immobilized to a solid phase followed by incubation with a soluble binding partner protein dissolved in a physiological solution. Binding is then detected via an antibody against the soluble partner protein using enzyme-linked immunosorbent assay (ELISA). Here we present the detailed methodology for this novel high-throughout protein-binding assay that we have successfully employed for investigating myofilament protein binding, including troponin T to tropomyosin [1–3] and troponin T to troponin I [1, 2, 4]. The assay is also highly effective in revealing the functional effects of muscle myofilament protein alternative splicing variants [1, 2], phosphorylation [5], restrictive proteolysis [4, 6], point mutations [7], as well as the effects of solution salt, metal ions, or pH on myofilament protein binding [8–11]. In addition, this methodology has also been used to study the binding of calponin [12, 13] and titin motifs [14] to F-actin. Beyond these applications this assay can readily be extended to study the interactions of nonmuscle proteins. Any protein binding pair can be analyzed provided that a specific antibody against one of the proteins is available.
In this paper we first discuss traditional protein-binding assays and use this background to present the general concepts of the microplate ELISA-based solid-phase protein-binding assay. We then provide detailed methodology to conduct a simple binary solid-phase binding assay. Finally, we will discuss modifications expanding on the simple binary binding experiment and optimization of the assay conditions.
2. Traditional Protein-Protein Binding Assays
Classical assays to measure the interaction and binding of one protein to another largely consist of two main methodologies: (1) equilibrium dialysis and (2) affinity chromatography. These two methodologies rely on different principals to separate bound from nonbound interacting proteins.
To determine the affinity of two proteins for each other by equilibrium dialysis, the experimental proteins of known concentration are placed in two chambers separated from each other by a membrane permeable to only one of the proteins. The permeable protein is then allowed to diffuse across the membrane and bind the nonpermeable protein. Once the permeable protein achieves equilibrium between the two chambers, its free concentration is determined in the chamber lacking the impermeable protein. Following dialysis of the protein pair at appropriate concentrations, the binding affinity of the pair can be determined. The dialysis can be conducted with one or more variants of one of the two proteins for comparison. Equilibrium dialysis, thus, provides the affinity of one protein for another at equilibrium between association and disassociation in solution. Although the data generated by equilibrium dialysis is quantitatively informative, a major limitation of this method is that it requires a size difference between the two binding proteins to be distinguishable by the dialysis membrane. The downfall of this method also includes its labor-intensive nature and its requirement for large amounts of the proteins.
The other commonly used traditional protein binding assay is affinity chromatography. Affinity chromatography requires immobilization of one protein to a support resin that is usually packed into a column for chromatographic analysis. The binding partner protein in solution is then incubated with the protein-resin conjugate at sufficient concentration and contact time to saturate its binding to the immobilized protein. The binding affinity between the two proteins is then assessed by step or continuous gradient elution with a buffer condition that weakens the protein-protein interaction and the strength of elution necessary to achieve peak dissociation of the bound protein determined. Once the bound protein is completely eluted, the column can be reequilibrated and a similar measurement conducted for another or variant partner protein. Unlike the solution steady-state binding measured in the equilibrium dialysis method, affinity chromatography measures a nonequilibrium disassociation rate from a maximally bound state. Therefore, by its nature this assay only investigates the disassociation characteristic of the two proteins. Further drawbacks of this methodology include the measurement of only relative affinity and the necessity of chemically coupling one of the proteins to a support resin, which by itself may affect the structure and function of the immobilized protein.
3. The Microplate Solid-Phase Protein-Binding Assay
The solid-phase protein-binding assay was developed as an alternate methodology to asses association and disassociation of protein-protein interactions using relatively small amounts of protein without specialized equipment [9]. The basis for the solid-phase protein-binding assay is derived from ELISA-based methodology. The rational for designing the protein-binding assay based on ELISA, a well-established immunological assay, is that the antibody-antigen interaction measured by various immunological detection methods is basically a protein-protein interaction. The assay consists of three main components. The first is immobilization of a protein to the wells of an assay plate by noncovalent coating. The second is the protein-protein interaction. The third is detection of the bound protein. As a basis for our discussion, we will first describe details of a standard solid-phase protein-binding assay followed by variants of the assay for extending its applications.
The simplest and most widely used format is the binary binding assay that measures the interaction between two proteins, one immobilized and the other free in solution. This assay format is commonly used to compare the binding interactions between an immobilized protein to two or more variants of a soluble partner protein. However, the assay can be designed such that the variant proteins are either in the solution or immobilized position.
Although simple in its design, the binary assay format (Figure 1(a)) demonstrates the basis of the solid phase protein binding assay. In this experimental system a single protein is noncovalently coated through absorption to the well surface of a 96-well polystyrene (not polypropylene) assay plate. Excess free protein in solution is subsequently removed and the wells are washed. The remaining available binding surfaces of the wells are blocked with a non-interacting protein such as bovine serum albumin (BSA) often together with a nonionic detergent. The plate is then washed again followed by adding serial dilutions of the binding partner protein to the wells and incubated allowing equilibrium binding with the immobilized protein to be reached. Remaining free protein in solution is then removed followed by washes, and the portion of the partner protein bound to the immobilized protein is quantified through detection via an antibody specific to the binding partner protein.
Figure 1.
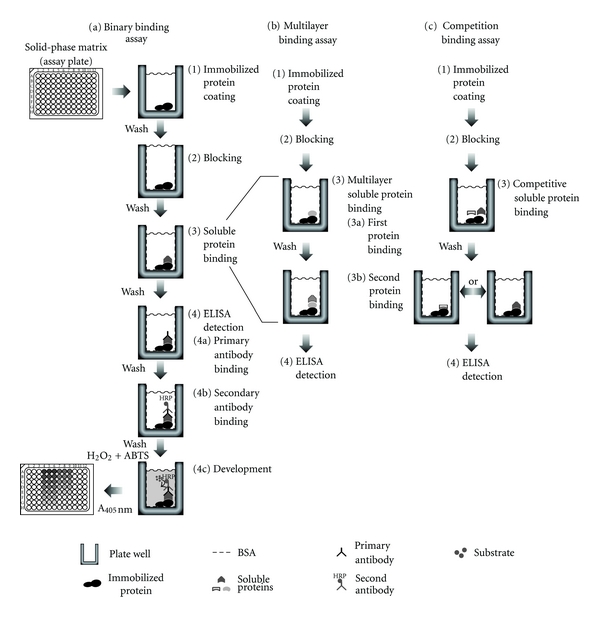
Solid-phase protein-binding assay method. (a) Binary binding assay to quantify the interaction between an immobilized protein and an interacting soluble protein. (1) The immobilized protein of interest is maximally coated into the wells of a 96 well assay plate. (2) Unbound protein is removed by washing, and remaining surfaces blocked with a nonspecific protein. (3) The plate is washed, and binding is evaluated by incubation with serial dilutions of a soluble interacting protein. (4) Following another wash to remove unbound interacting protein, bound protein is detected by ELISA with a primary antibody against the interacting protein, a HRP-conjugated second antibody and substrate development to quantify bound protein by absorbance. (b) Multilayer binding assay to quantify the interaction between an immobilized protein and two related interacting soluble proteins. The multilayer protein binding is conducted similar to the binary binding assay in (a) with the exception that, following binding of the first soluble protein and washes, the wells are incubated with another protein that binds the first protein bound to the immobilized protein followed by ELISA quantification. (c) Competitive binding assay to quantify the interaction between an immobilized protein and an interacting soluble binding protein in the presence of varied amounts of a second binding protein. Protein coating and blocking are conducted as in (a) and (b) with the exception that during the soluble protein binding step, addition of the first interacting protein of interest is conducted at a constant concentration in the presence of serial concentrations of a competitive binding protein. Resultant binding of the constant concentration of the first binding protein of interest to the immobilized protein is then quantified by ELISA with a specific antibody. ELISA quantification of the degree of competition will be detected by decreases in the final reading of absorbance.
The detection employs an enzyme-linked antibody approach. The enzyme, for example, horse radish peroxidase (HRP), can be directly conjugated to the detecting antibody; however, it is more convenient to use an enzyme-conjugated second antibody. In this setting a primary antibody specifically recognizing the bound protein is used as the detecting antibody, and excess antibody is removed by washes. Subsequently an enzyme-conjugated second antibody recognizing the detecting primary antibody is added for another step of incubation followed by washes to remove excess second antibody. Finally, a substrate development reaction is conducted and quantified using a microplate reader. The amount of the bound enzyme-linked second antibody is in direct proportion to the amount of bound primary detecting antibody that is determined by the amount of the soluble partner protein remaining bound to the immobilized protein. As the result of this direct relationship between the bound protein and enzyme-linked secondary antibody, the intensity of the color or fluorescence product produced from the substrate reaction is quantitatively dependent on the amount of the soluble protein remaining bound to the immobilized protein. By plotting the microplate readings against the concentration of the free protein used in the equilibrium binding step, the association AND disassociation features can be compared between two or more variants of the proteins studied.
4. Experimental Procedure
As outlined above, Figure 1(a) shows the basic design of the solid-phase protein-protein binding assay consisting of a binary protein-binding experiment. The design of the binary binding experiment investigates the binding characteristics between two proteins, one of which is immobilized in the wells of microplate and allowed to incubate with the binding partner protein in solution. A thorough understanding of this basic experimental setting will provide the foundation for understanding more sophisticated designs derived from it. For this reason we will first present the binary binding assay protocol and then the application of this methodology to other designs.
As routinely employed in our laboratories, the binary protein-binding experiment consists of coating the immobilized protein through noncovalent absorption, binding of the partner protein, and indirect ELISA detection of the protein binding. This section will describe the detailed protocol to conduct the binary binding assay similar to that described in our previous publications [2, 9, 15].
4.1. Specific Materials
96-well polystyrene microtitering plates.
Multichannel pipette in the 100–200 μL range.
Squishing wash bottle. A standard 500 mL laboratory wash bottle that can be used to provide a consistent stream of wash buffer from a spout to fill the wells of the assay plate.
Buffer A (Buf A; 100 mM KCl, 3 mM MgCl2, and 10 mM PIPES, pH 7.0) can be made as a 5x stock, stable at room temperature. This buffer is used for the study of myofilament proteins. It should be modified according to the proteins to be studied. Neutral or alkaline pH is required for proper coating of the immobilized protein to the plate. A concentration of 3–6 M urea can be included for the coating of less soluble proteins. Neutral pH should be used for the urea-coating buffer to avoid carbamylation of the protein. Most proteins will refold well following immobilization during the blocking step after urea is removed.
Buffer T (Buf T; buffer A + 0.1% Tween-20).
Buffer B (Buf B; buffer T + 0.1% BSA).
Substrate solution (2,2′-azino-bis(3-ethylbenzo-thiazoline-6-sulfonic acid (ABTS)/H2O2 is used for horseradish peroxidase-conjugated first or second antibody). 0.04% ABTS diammonium is dissolved in 65.7 mM citric acid monohydrate, 34 mM sodium citrate dihydrate, pH 4.0, adjusted with sodium citrate dihydrate. The substrate stock should be made in autoclaved, deionized water with autoclaved tools to avoid peroxidase contamination and stored at −20°C in aliquots of the volume for individual experiments. Before use the substrate solution is brought to room temperature and immediately prior to its application, 0.03% H2O2 added.
Microplate reader capable of reading absorbance at 415 nm or 405 nm (for the ABTS substrate).
Primary antibody against the soluble protein to be studied. Although it is necessary to have a specific antibody against one of the proteins to be studied, current methods of generating recombinant fusion proteins with a tag, such as FLAG or His6-8, and the availability of antibodies against such tags allow this approach to be applied to a wide range of protein interactions, overcoming the restriction from the availability of a specific antibody.
Horseradish peroxidase-conjugated second antibody that recognizes the primary antibody species (e.g., goat anti-mouse IgG horseradish peroxidase conjugated secondary antibody) was used in our studies employing mouse monoclonal primary antibodies.
4.2. Assay Design
A practical binary protein-binding assay design consists of eight dilutions of the soluble binding protein in triplicate. The 8 × 12 well format of the 96-well microplate should be considered in the assay design. For example, the immobilized protein can be arranged in columns (A to H) and incubated with eight serial dilutions of the soluble variant proteins. The assay design should also include control columns for each variant protein consisting of the serial diluted soluble protein incubated in triplicate wells without immobilized protein. Figure 2 illustrates the 96-well microplate layout of a typical binary binding experiment conducted in triplicate for two variant soluble proteins.
Figure 2.
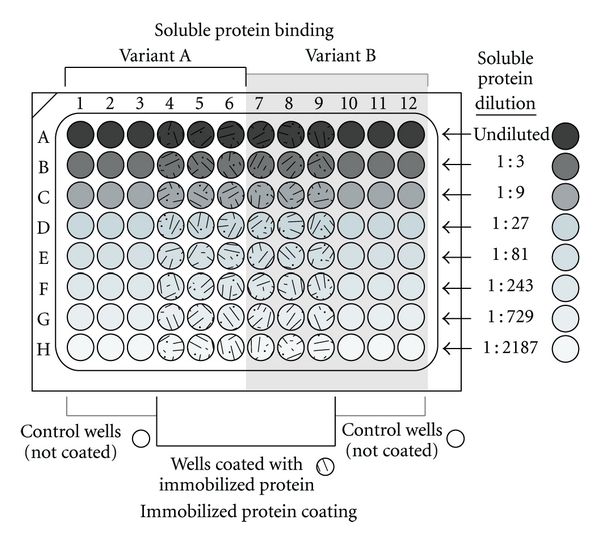
Representative design of a 96-well microtitering plate assay for binary binding consisting of an immobilized protein incubated with two variant soluble interacting proteins in 8 serial concentrations of triplicate wells. Immobilized protein is equally and maximally coated in the experimental wells, rows A to H and columns 4 to 9. Columns 1 to 3 and 10 to 12 are left uncoated (incubated with coating buffer only) as controls. Following coating all wells are blocked, and the wells in columns 1 to 6 are incubated with serial dilutions, one dilution per row, of one of the soluble proteins, and wells in columns 7 to 12 are incubated with serial dilutions of the other soluble protein variant. All wells are then processed uniformly for ELISA detection.
4.3. Immobilization of Protein to the Microplate
It is preferred that the single partner protein is selected to be the immobilized protein with the two or more variant binding proteins incubated in the soluble phase. This strategy will ensure a uniform level of the coated protein for the comparison among the variants of the binding protein. However, multiple variants of the proteins to be studied may be coated under similar conditions for incubation with a single soluble partner protein as well. Standard coating of the immobilized protein occurs through absorption to the wells of the assay plate based on hydrophobic interactions between the plastic surface of the assay plate and the nonpolar amino acid residues of the protein to be immobilized.
The coating of the immobilized protein onto the wells of a microtiter plate is performed at 100 μL/well with 2–5 μg/mL protein in Buf A by incubation at 4°C sealed overnight. Coating conducted at these protein concentrations is in excess of the amount able to bind the plate well for most proteins of 10–200 kDa in size. The use of excess amount of protein for coating exploits conditions to ensure saturated coating to minimize variation of the assay.
Most small proteins or peptide fragments are sufficiently immobilized to the plate well to allow a robust assay. In the case that simple hydrophobic absorption does not result in a sufficient amount and strength of coating; microplates with added reactive groups for covalent conjugation may be considered (see Section 4.10).
4.4. Blocking of the Coated Plate
To begin protein binding, the well contents of the microplate are emptied by shaking into a sink, and the remaining solution removed by tapping onto a stack of paper towels. The plate is then washed once rapidly to avoid protein drying by the addition of a volume of Buf T sufficient to fill each well using a squishing bottle (see Section 4.6). Following filling of the wells, the washing buffer is emptied as above, tapped dry, and the wells are blocked with 150 μL/well 1% BSA in Buf T by incubation at room temperature for 1 hr sealed. When dealing with limited protein material, the coating protein solution may be recovered for reuse, although the resulting coating capacity may decrease.
4.5. Protein Binding
Following empting of the blocking solution from the wells and tapping, the plate is washed three times with Buf T to remove free protein (see Section 4.6). Serial dilutions of the binding partner protein solution in Buf B are then added to the wells at 100 μL/well and incubated with the immobilized protein at room temperature for 2 hrs sealed. A typical binding assay designed for myofilament proteins consists of free protein concentrations starting from 0.5–1 μM and eight 3-fold serial dilutions (Figure 2).
4.6. Washing
Following protein-binding incubation, the plate is washed as follows. The well contents are emptied by shaking into the sink, the remaining solution is removed by tapping onto paper towels, and the plate is washed a total of three times with Buf T in a total period of 10 min. The lag time between emptying the well to filling with Buf T should be as short as possible to avoid drying of the binding protein on the plate resulting in high background.
Washing is conducted using a squishing bottle by applying a consistent stream of Buf T to sequentially fill the wells without overfilling. Accurate filling is critical for the first wash to avoid spilling over trace amount of the binding protein between wells, especially from high to low concentration and from positive to negative control wells. It is useful to start the wash stream outside of the plate and then move sequentially through the wells delivering a continuing stream of wash solution while moving from well to well. It is helpful to hold the plate slightly tilted, beginning filling from the lower-side rows. This will help avoid spillover from entering into empty wells without further dilution. After the first filling, the washing buffer is immediately removed followed by the second wash. This will limit the time for any spillover protein to interact at a significant concentration with the immobilized protein in unwanted wells. The plate is then allowed to incubate at room temperature for approximately 3 min. The wash procedure is repeated for the 3rd time and allowed to incubate approximately 4 min before final removal of the washing buffer as above.
The washing condition is an important factor in determining the effectiveness and stringency of the assay by eliminating nonspecific binding while quantitatively evaluating the dissociation of specific binding between the protein pair. Using appropriate washing conditions will allow evaluating the coupling strength of the two proteins studied. More stringent separation by increasing the number of buffer changes, duration of the washes, and/or the detergent concentration will differentially affect weak and strong bindings as a sensitive method to reveal differences among protein variants of interest. It is worth noting that no single washing condition fits all protein-protein interactions, and the wash strength should be evaluated empirically.
4.7. ELISA Detection
The partner protein specifically bound to the immobilized protein is detected using an ELISA procedure in which an antibody recognizing the partner protein is the primary reagent (Figure 1(a)). Following the final wash to remove unbound partner protein a predetermined, constant dilution of the primary antibody in Buf B is added to all the wells at 100 μL/well using a multi-channel pipette and incubated sealed at room temperature for 1 hr. The working concentration of the primary antibody should be predetermined to give a final absorbance of around 1.0 by ELISA titration against the partner protein directly coated to a microplate in the same buffer system, incubation, and washing conditions. The antibody solution should be added quickly such that the well contents do not dry out. The plate is then washed 3 times in 10 min as described in the Washing Section. A constant dilution of HRP-conjugated second antibody against the primary antibody in Buf B is added to all the wells at 100 μL/well and incubated at room temperature for 45 min. Similar to that of the primary antibody, the second antibody's working concentration should be determined empirically. The plate is then washed 3 times in 10 min as described in the Washing Section (see Section 4.10) prior to adding 100 μL/well of the ABTS-H2O2 substrate solution.
The substrate reaction is incubated at room temperature with periodic mixing (e.g., 2-3 seconds shaking in the plate reader) and the color development monitored by absorbance reading at 415 nm or 405 nm using a microplate reader at several time intervals (e.g., 5, 10, 15, 20, 25, and 30 min). The well of highest absorbance is then plotted against time and a time point chosen for analysis before deviation from linear color development. Under typical assay conditions, this will be between 10 to 20 minutes of development. The time of development should also be such that the absorbance reading remains within the reliable reading range of the microplate reader, less than 2.0 for most instruments. If the color development is too quick and the absorbance values too high, the concentration of the primary and/or second antibody should be decreased.
4.8. Data Analysis
Background absorbance for each serial dilution of the binding protein is determined from 3 control wells not coated with immobilized protein but processed the same as the assays wells. Absorbance of the 3 control wells is averaged, and this value is subtracted from data readings for that dilution. Resultant absorbance values at the highest concentration of each binding protein are averaged as the maximum (100%) binding for that protein. It is important to only compare the absolute values of the maximal binding from experiments conducted at the same time to avoid the effect of day-to-day variations. Background-subtracted absorbance values for each dilution of the binding protein are then normalized to the average maximal binding value and the titration curve plotted against a log scale of the dilution concentrations. The curve is then fit to determine the soluble protein concentration required to reach 50% maximal binding. Although it is best to compare the 50% maximal binding values of protein variants from the same assay, it is possible to compare day-to-day results as long as the reagents, incubation times, washing stringency, and room temperature remain consistent (Figure 3).
Figure 3.
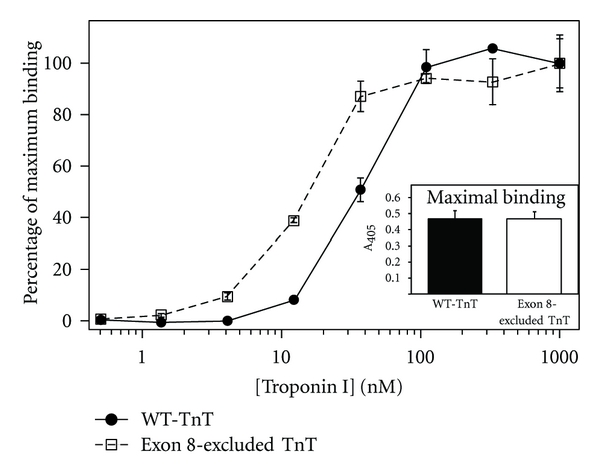
A set of typical, original protein-protein binding curves demonstrating the altered binding of an exon 8-excluded cardiac troponin T (exon 8-excluded TnT, white square) to the interacting protein troponin I in comparison with wild-type cardiac troponin T (WT-TnT, black circle). The troponin T variants are configured as the immobilized protein and troponin I the soluble protein. The normalized binding curves demonstrate exon 8-excluded TnT bound with higher affinity to troponin I compared to WT-TnT as evident by the lower concentration required for 50% maximal binding. Absolute absorbance of the maximal binding is shown in the inset demonstrating that both exon 8-excluded TnT and WT-TnT exhibited similar coupling strength to troponin I.
The above procedure provides relative comparisons between two or more binding protein variants for their interaction with the immobilized protein. With a standard curve produced with known concentrations of the binding partner protein, this approach can be employed to gain quantification of the partner in an unknown sample of purified protein, body fluid, or tissue homogenate.
4.9. Data Interpretation
The molar concentration required for 50% maximal binding of the soluble partner protein to the immobilized protein reflects the on-rate of their association at equilibrium and is a representative of the binding affinity (Ka). Although the strength and time of washing will affect the nonequilibrium disassociation of the binding protein, these conditions affect all samples similarly. Thus, the initial binding between the immobilized and free proteins during equilibrium incubation is determined by the intrinsic affinity between the protein pair.
On the other hand, the level of maximal binding of the soluble partner protein to a given amount of the immobilized protein represents their coupling strength or resistance to the washes. In contrast to the maximally saturated binding achieved during the equilibrium incubation step, during washes the absence of free soluble binding protein produces nonequilibrium dissociation. Thus, this coupling strength determined in the solid-phase protein-binding assay employing stringent washing-separation steps reflects the off-rate of the protein pair's binding. In the assay system, immobilized protein coating is conditioned to a maximum, and binding of the partner protein at high concentration reflects saturated binding. Therefore, similar to that in affinity chromatography, the disassociation resulting from wash separation conditions is a highly sensitive measure to provide information of the disassociation rate (Kd) of the protein pair studied. Washing strength can, therefore, be altered to exploit coupling strength differences between protein variants.
4.10. Optimizing Assay Conditions
A number of conditions can be modified to optimize the solid-phase protein-binding assay allowing investigation of a number of specific protein-protein interactions, including nonmuscle proteins. Unlike most traditional protein binding assays, a unique advantage of the microplat-based protein binding assay is that it allows different buffer conditions during the incubation and washing steps. For example, one may coat a low-solubility protein to the plate by dissolving it in a modified coating buffer containing high salt and/or 3–6 M urea. The salt and urea will then be washed away, and the following binding assay can be performed in physiological buffers. Another example is that reducing agents may be included for coating and protein-binding steps but removed before the antibody incubation steps (reducing agents will dissociate the quaternary structure of immunoglobulins). Further examples include the use of different buffer conditions for the equilibration protein-binding step and the washing steps that determine the stringency for separately investigating association and dissociation rates.
Other optimizations include adaptations for small peptides. Small peptides that are weak in hydrophobic absorption may be immobilized as a conjugate with a carrier protein. Small peptides may also be immobilized through a specific interaction such as the use of biotin-streptavidin system. Microtiter assay plates of varied surface chemistry can be used to enhance the coating of the immobilized protein. For example, covalent bonding of peptides can be achieved via precoating the plate with a hydrophilic polymer such as soluble dextran treated with 2,2,2-triflouroethanesulphonyl chloride (tresyl chloride) to activate hydroxyl groups [16].
As discussed above, separation strength can be varied by altering the number, time, or stringency of the washes to exploit the properties of the interaction between the protein pair studied. Issues of low amounts of protein binding can be overcome by employing detection methods of increased sensitivity. For example, higher concentrations of the primary and/or secondary antibodies may be used. Alternatively, instead of ABTS, o-phenylenediamine (OPD) or 3,3′,5′,-tetramethylbenzidine (TMB) can be employed as the substrate to increase detection sensitivity of weaker binding protein pairs. If labeling a protein does not interfere with its binding, the binding partner protein can directly be labeled by conjugation to a fluorescent tag and its binding to the immobilized protein detected by measuring fluorescence of the bound protein.
Taken together the adaptability of the solid-phase protein-binding assay offers significant flexibility to develop specifically optimized experiments allowing for the investigation of a wide range of proteins under varied conditions.
5. Extended Assay Configurations
As presented above, the simple binary protein-binding assay can provide significant information regarding differences in the interaction of one protein with another. Additionally the solid-phase protein-binding assay is amendable to provide information on more than binary protein-protein interactions. With appropriate optimization, other protein-protein interactions of increased complexity can be studied using this approach. One or both of the binding partners may be a protein complex as long as a suitable antibody is available for the ELISA detection [17]. This methodology can also be readily applied to multilayer binding assays and competition assays. In the multilayer binding assay an additional protein binding step can be added to evaluate the binding among the subunits of a multiprotein complex (Figure 1(b)). We have successfully employed a similar multilayer assay in our laboratory investigating the binding of tropomyosin and troponin T in the presence of filamentous actin (unpublished data). Likewise, other multi-protein complexes or binding cofactors could similarly be investigated.
In the competition assay, binding of a partner protein at a constant concentration to the immobilized protein is carried out in the presence of serial concentrations of a competitive protein (Figure 1(c)). Using a specific primary antibody to detect the partner protein, its affinity to the immobilized protein relative to that of the competitive protein can be assessed. The competition assay design can be modified to study the blocking effect of a protein, a peptide, or a small-molecule ligand on the binding of the partner protein to the immobilized protein. The competition assay is also applicable not only to two proteins but also to two isoforms of the same protein [18] or different posttranslational modifications [19] to asses the effect of the modification on binding.
The competition assay is also amendable to study the spatial relationship between a protein-binding site and an antibody-binding site [19]. Using a monoclonal or anti-peptide antibody against a known epitope of structural and/or functional interests, the assay can be extended to identify binding partners of the protein in a complex mixture through competitive blocking of the antibody epitope by protein binding. Likewise, substrate-enzyme interactions could be investigated using a similar competitive approach by employing an antibody against the catalytic site. When an antibody probe is available, the competitive assay configuration is likewise applicable to study the interactions between proteins and cofactors.
6. Special Cautions
Although the solid-phase protein-binding assay is readily amendable to a number of different situations, a few key points must be observed to ensure success. Importantly, reducing agents must be excluded from all buffers for the primary antibody incubation and subsequent steps. The inclusion of reducing agents in these steps will disrupt immunoglobulin disulfide bonds destroying the ability of the antibody to detect the bound protein and, thus, rendering no signal. Furthermore, although employing methods of increased sensitivity can help with detection of low-affinity binding, this methodology, applying high stringent washes, is most effective in studying high-affinity binding events.
7. Conclusions
We have presented detailed methodology of a novel solid-phase binding assay to assess protein-protein interactions emphasizing examples using muscle regulatory proteins. This assay expands on previous methodologies to provide a simple and high-throughput assay to assess protein-protein interactions in solution. In contrast to many other current methods that process a few samples a time, this approach readily handles hundreds of assay wells by a single operator in a days time using common laboratory equipment. By employing the adaptations and optimizations discussed, this assay should be readily applicable to the quantitative assessment of other nonmuscle proteins interactions.
Acknowledgment
This work was supported by the National Institutes of Health Grants HL-091056 (to B. J. Biesiadecki), AR048816, HL-078773, HL-086720, and HL-098945 (to J.-P. Jin).
Abbreviations
- ELISA:
Enzyme-linked immunosorbent assay
- HRP:
Horse radish peroxidase
- ABTS:
2,2′-Azino-bis(3-ethylbenzo-thiazoline-6-sulfonic acid
- BSA:
Bovine serum albumin.
References
- 1.Biesiadecki BJ, Elder BD, Yu ZB, Jin JP. Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. Journal of Biological Chemistry. 2002;277(52):50275–50285. doi: 10.1074/jbc.M206369200. [DOI] [PubMed] [Google Scholar]
- 2.Biesiadecki BJ, Jin JP. Exon skipping in cardiac troponin T of turkeys with inherited dilated cardiomyopathy. Journal of Biological Chemistry. 2002;277(21):18459–18468. doi: 10.1074/jbc.M200788200. [DOI] [PubMed] [Google Scholar]
- 3.Jin JP, Chong SM. Localization of the two tropomyosin-binding sites of troponin T. Archives of Biochemistry and Biophysics. 2010;500(2):144–150. doi: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biesiadecki BJ, Schneider KL, Yu ZB, Chong SM, Jin JP. An R111C polymorphism in wild turkey cardiac troponin I accompanying the dilated cardiomyopathy-related abnormal splicing variant of cardiac troponin T with potentially compensatory effects. Journal of Biological Chemistry. 2004;279(14):13825–13832. doi: 10.1074/jbc.M314225200. [DOI] [PubMed] [Google Scholar]
- 5.You B, Yan G, Zhang Z, et al. Phosphorylation of cardiac troponin I by mammalian sterile 20-like kinase 1. Biochemical Journal. 2009;418(1):93–101. doi: 10.1042/BJ20081340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Biesiadecki BJ, Jin JP. Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated μ-calpain cleavage. Biochemistry. 2006;45(38):11681–11694. doi: 10.1021/bi060273s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engel PL, Kobayashi T, Biesiadecki B, et al. Identification of a region of troponin I important in signaling cross-bridge-dependent activation of cardiac myofilaments. Journal of Biological Chemistry. 2007;282(1):183–193. doi: 10.1074/jbc.M512337200. [DOI] [PubMed] [Google Scholar]
- 8.Jin JP, Chong SM, Hossain MM. Microtiter plate monoclonal antibody epitope analysis of Ca2+- and Mg2+-induced conformational changes in troponin C. Archives of Biochemistry and Biophysics. 2007;466(1):1–7. doi: 10.1016/j.abb.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin JP, Wang K. Nebulin as a giant actin-binding template protein in skeletal muscle sarcomere. Interaction of actin and cloned human nebulin fragments. FEBS Letters. 1991;281(1-2):93–96. doi: 10.1016/0014-5793(91)80366-b. [DOI] [PubMed] [Google Scholar]
- 10.Ogut O, Jin JP. Developmentally regulated, alternative RNA splicing-generated pectoral muscle-specific troponin T isoforms and role of the NH2-terminal hypervariable region in the tolerance to acidosis. Journal of Biological Chemistry. 1998;273(43):27858–27866. doi: 10.1074/jbc.273.43.27858. [DOI] [PubMed] [Google Scholar]
- 11.Ogut O, Jin JP. Expression, zinc-affinity purification, and characterization of a novel metal-binding cluster in troponin T metal-stabilized alpha-helical structure and effects of the NH2-terminal variable region on the conformation of intact troponin T and its association with tropomyosin. Biochemistry. 1996;35(51):16581–16590. doi: 10.1021/bi961712y. [DOI] [PubMed] [Google Scholar]
- 12.Jin JP, Walsh MP, Sutherl C, Chen W. A role for serine-175 in modulating the molecular conformation of calponin. Biochemical Journal. 2000;350(2):579–588. [PMC free article] [PubMed] [Google Scholar]
- 13.Jin JP, Wu D, Gao J, Nigam R, Kwong S. Expression and purification of the h1 and h2 isoforms of calponin. Protein Expression and Purification. 2003;31(2):231–239. doi: 10.1016/s1046-5928(03)00185-2. [DOI] [PubMed] [Google Scholar]
- 14.Jin JP. Cloned rat cardiac titin class I and class II motifs. Expression, purification, characterization, and interaction with F-actin. Journal of Biological Chemistry. 1995;270(12):6908–6916. [PubMed] [Google Scholar]
- 15.Wang J, Jin JP. Conformational modulation of troponin T by configuration of the NH2- terminal variable region and functional effects. Biochemistry. 1998;37(41):14519–14528. doi: 10.1021/bi9812322. [DOI] [PubMed] [Google Scholar]
- 16.Gregorius K, Mouritsen S, Elsner HI. Hydrocoating: a new method for coupling biomolecules to solid phases. Journal of Immunological Methods. 1995;181(1):65–73. doi: 10.1016/0022-1759(94)00330-y. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Akhter S, Mottl S, Jin JP. Calcium-regulated conformational change in the C-terminal end segment of troponin I and its binding to tropomyosin. FEBS Journal. 2011;278(18):3348–3359. doi: 10.1111/j.1742-4658.2011.08250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogut O, Jin JP. Cooperative interaction between developmentally regulated troponin T and tropomyosin isoforms in the absence of F-actin. Journal of Biological Chemistry. 2000;275(34):26089–26095. doi: 10.1074/jbc.M910360199. [DOI] [PubMed] [Google Scholar]
- 19.Akhter S, Zhang Z, Jin JP. The NH2-terminal extension regulates molecular conformation and function of cardiac troponin I. Biophysical Journal. 2011;100(3, supplement):370a–371a. doi: 10.1152/ajpheart.00637.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]