

Abstract
An intact immune system is essential to prevent the development and progression of neoplastic cells in a process termed immune surveillance. During this process the innate and the adaptive immune systems closely cooperate and especially T cells play an important role to detect and eliminate tumor cells. Due to the mechanism of central tolerance the frequency of T cells displaying appropriate arranged tumor-peptide-specific-T-cell receptors is very low and their activation by professional antigen-presenting cells, such as dendritic cells, is frequently hampered by insufficient costimulation resulting in peripheral tolerance. In addition, inhibitory immune circuits can impair an efficient antitumoral response of reactive T cells. It also has been demonstrated that large tumor burden can promote a state of immunosuppression that in turn can facilitate neoplastic progression. Moreover, tumor cells, which mostly are genetically instable, can gain rescue mechanisms which further impair immune surveillance by T cells. Herein, we summarize the data on how tumor cells evade T-cell immune surveillance with the focus on solid tumors and describe approaches to improve anticancer capacity of T cells.
1. Introduction
The theory of immune surveillance was introduced in the early 1900s by Ehrlich, who hypothesized that one critical function of the immune system was to detect and eliminate tumors from the host [1]. As a logical consequence, tumor development should more likely occur when the innate and/or adaptive immunity is impaired or repressed. This hypothesis could be tested in a variety of knock-out mice that were deficient in one or more components of the innate or adaptive immune system. And indeed, the elimination of perforin, interferon (IFN)-γ, or STAT1 genes (thus lacking interferon-mediated pathways) in mice resulted in increased incidence and growth of spontaneous and chemically-induced tumors [2–5]. Further evidence that the adaptive immune system is involved in immune surveillance of tumors was provided by experiments using RAG-2-deficient mutant mice [6]. The RAG genes encode DNA repair enzymes which are essential for B-cell receptor (i.e., antibody) and T-cell receptor (TCR) rearrangement. Mice with homozygous deletion of the RAG-2 alleles completely lack NKT, T, and B cells and have an increased incidence and growth of spontaneous tumors and chemically induced cancer lesions [5]. In humans these findings are reflected by the fact that immunocompromised patients, in particular transplant recipients and patients suffering from acquired immunodeficiency syndrome (AIDS), are more susceptible to certain types of neoplasms [7, 8].
Although the theory of immune surveillance will remain a matter of debate, it is meanwhile accepted that T cells play a crucial role in controlling the development of neoplastic lesions in vivo. T cells are activated via their T-cell receptor, which binds to antigen peptides presented on major histocompatibility complex class molecules (MHCs). Following the recognition of peptides presented by MHC class I molecules, activated CD8+ cytotoxic T cells (CTLs) can efficiently destroy target cells using death cell ligands such as TRAIL (TNF-related apoptosis-inducing ligand) or by execution of the perforin/granzyme pathway [9, 10]. Another fraction of T cells, the CD4+ T cells, recognizing peptides presented by MHC class II molecules, play also an important role in adaptive anticancer immunity [11]. CD4+ T cells can improve the capacity of dendritic cells (DCs) to induce CTLs by crosslinking the costimulatory molecule CD40 on DCs with the CD40 ligand on activated CD4+ T cells [12]. Furthermore, by secreting cytokines such as interleukin-2 (IL-2), activated CD4+ T cells support the clonal expansion of activated CTLs [13]. Besides this, activated CD4+ T cells can significantly boost cellular components of the innate immunity, such as macrophages and NK cells by enhanced IFN-γ secretion [14]. Concomitantly, increased IFN-γ levels improve the recognition capacity of T cells through induction of higher expression levels of MHC class I molecules on the target cells [14, 15].
Despite ongoing surveillance by T cells and other components of the immune system, tumors develop even in presence of an intact immune system and become eventually clinically detectable. Schreiber and colleagues have put forward the hypothesis of cancer immunoediting to explain this discrepancy [16]. According to their theory, cancer development can be divided in three phases. In the first phase, immune surveillance is intact and cells of the innate and adaptive immune system destroy neoplastic cells. During the second phase a long-winded ongoing campaign between the immune system and cancer cells establishes a dynamic equilibrium. The third phase is characterized by genetic and epigenetic instability of tumor cells which eventually give rise to variants escaping from immune surveillance (for reviewing see [16, 17]) and develop to clinical apparent tumors. Due to the constant selective pressure by the immune system, these variants display a multitude of evasion mechanisms from immune recognition and destruction. In the following paragraphs, we will focus in particular on evasion strategies which outmanoeuvre the immune recognition by T cells. Only a better understanding of the manifold interactions between tumors and T cells will help to improve current T-cell-based immunotherapy strategies.
2. Central Tolerance and Peripheral Tolerance Mechanisms Restrict Tumor-Specific T-Cell Responses
T cell surveillance of neoplastic development and growth primarily depends on recognition of processed so-called tumor-associated-antigen-(TAA-) derived peptides presented by MHC class I molecules on the surface of tumor cells. Conceptually, there are three different types of TAAs: the first group are “neoantigens” which originate from transforming viruses or are due to mutations or chromosomal aberrations in the tumor cells and to which the host is not tolerant, and secondly, “self-antigens” which are mainly proliferation and differentiation markers overexpressed in tumors or normal embryonic antigens aberrantly expressed in the course of epigenetic changes and cellular dedifferentiation of the tumor cells. Finally, the third group are “modified self-antigens” representing self-antigens having different tumor-specific posttranslational modifications due to metabolic disturbances (for reviewing see [18]). Most TAAs of solid tumors correspond to self-antigens and modified self-antigens which are re-expressed or overexpressed in tumors and are barely detected in normal tissues. However, T cells with high affinity receptors for MHC/self-peptides are eliminated during development in the thymus to establish central tolerance. Therefore, the repertoire of T cells specific for self-peptides in the periphery is restricted to those T cells displaying a low affinity T-cell receptor. Furthermore, under normal circumstances T cells reactive against self-peptides are in an ignorant state called peripheral tolerance and need to be activated by professional antigen-presenting cells (APCs), usually DCs, in a process termed cross-priming, before they can exert their effector functions [19]. Briefly, immunogenic responses of T cells require that DCs must encounter antigens that are associated with evolutionary conserved “pathogenic-associated molecular patterns” (PAMPs) or in other words “danger signals” derived from pathogenic microorganisms or viruses [20, 21]. After sensing such danger signals (i.e., LPS, CpG-rich DNA, and viral RNA) via toll-like receptors (TLRs) or other sensor molecules such as members of the retinoid inducible gene I (RIG-I) family the DCs upregulate costimulatory molecules such as CD80 and CD86 and now can fully activate CTLs in concert with activated CD4+ T cells [12, 22–25] (Figure 1). Despite the control mechanism of central tolerance, self-reactive CD4+ T cells and CTLs exist in the periphery and under certain conditions DCs can provide sufficient co-stimulation to activate these cells as exemplified in human autoimmune diseases such as psoriasis and systemic lupus erythematodes [26]. In particular, it has been shown that the antimicrobial peptide LL37 from psoriasis patients activates TLRs 7 and 9 when associated with self-nucleic acids [27]. Meanwhile, lines of evidence indicate that under certain circumstances tumor cell death can also deliver danger signals required for DC priming via TLRs. In fact, it has been shown that release of several factors such as calreticulin and high-mobility group box1 protein (HMGB1) from dying cancer cells can codeliver so-called “damage-associated molecular pattern” (DAMP) signals to DCs which in turn can break peripheral tolerance of T cells [28, 29] (Figure 1). Especially HMGB1 which associates with DNA from necrotic cells has been shown to play a pivotal role during sepsis and induction of TLRs [30]. Furthermore, the effects of some anticancer drugs, in particular anthracyclines and platinum-based drugs and also radiotherapy, suggest an immunogenic cancer cell death via calreticulin exposure and HMGB1 release [31]. Anthracycline-based chemotherapy limits tumor growth in wild-type mice, whereas mice with homozygous deletion of TLR4 or its downstream effector molecule MYD88 (myeloid differentiation primary response protein 88) failed to efficiently respond to chemotherapy. In addition, it has been reported that colon cancer patients having a loss of function allele of TLR4 exhibited a reduced progression-free survival compared to patients carrying the normal TLR4 allele after treatment with oxaliplatin (OXP) [28]. DCs which have not been primed by PAMP or DAMP danger signals can also display self-antigens in association with MHC molecules, but unlike in the primed state, they cannot provide sufficient costimulation for T-cell activation. Consequently, T cells recognizing MHC/peptide complexes on the surface of nonactivated DCs become anergic and eventually undergo apoptosis in a process termed cross-tolerance [32] (Figure 1). Cross-tolerance is a major hurdle for the generation of potent CTL immune responses against self-antigens as seen in various tumors where immature DCs mostly lacking costimulatory receptors are not able to elicit T-cell responses [33–36]. In summary, evidence is accumulating that immunogenic cell death of tumor cells provides tumor antigens and concomitant danger signals to DCs which are now able to activate tumor-specific T cells which can appear as so-called tumor-infiltrating lymphocytes (TILs) at the tumor site. So far, TILs, which were proven to specifically react against tumor cells or which were associated with a better clinical outcome, were reported for advanced ovarian cancer [37], breast cancer [38, 39], melanoma [40], and colorectal cancer [41]. Yet, the appearance of TILs in patients per se did not predict an efficient immune response or improved clinical outcome against the tumor since various further mechanisms, commonly defined as “tumor escape mechanisms,” can impair the effector function of tumor-reactive T cells.
Figure 1.
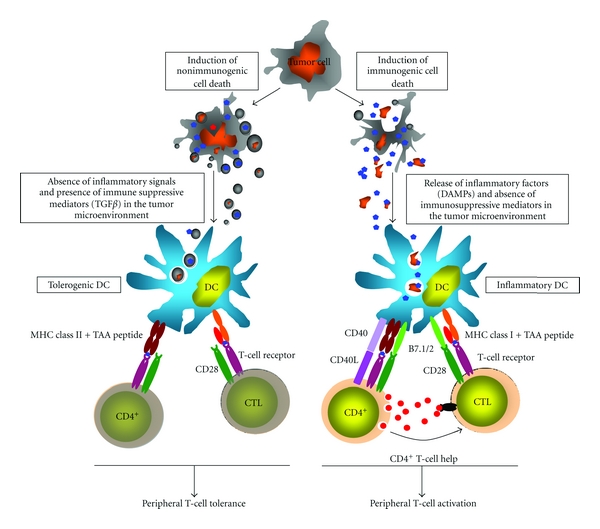
T-cell conversion from anergic to an activated status upon immunogenic cell death of tumor cells. The activation of tumor-specific T cells is dependent on DCs, which endocytose tumor cell debris and apoptotic vesicles. After intracellular processing the DCs present peptides derived from tumor-associated-antigens in complex with MHC class I molecules to T cells. Without the stimulation by danger-associated molecular patterns (DAMPs), DCs remain immature in a hostile immunosuppressive milieu and anergize CD4+ T cells and cytotoxic T cells (CTLs) resulting in peripheral tolerance. The release of inflammatory factors and the appearance of DAMPs lead to activation of DCs (inflammatory DCs) which subsequently upregulate costimulatory molecules of the B7 family. Inflammatory DCs are able to activate naïve CTLs through MHC I tumor peptide/TCR and B7/CD28 crosslinking. Furthermore, inflammatory DCs can activate naïve CD4+ T cells after MHC class II tumor peptide/TCR and B7/CD28 crosslinking. Activated CD4+ T cells in turn support clonal expansion and activity of CTLs by CD40/CD40L interaction and release of inflammatory cytokines such as IL-2.
3. Defective Recognition of Tumors by Activated T Cells
One of the best studied evasion mechanisms of tumor cells to escape recognition by CTLs are abnormalities in the antigen presentation machinery (APM). This includes downregulation or complete loss of MHC class I molecules as observed in head and neck squamous carcinoma [42], human esophageal squamous carcinoma [43], lung cancer [44], and prostate carcinoma [45]. Downregulation of MHC class I molecules can be caused by point mutations or by large deletions correlating with loss of heterozygosity (LOH) on chromosome 6p21 [46, 47]. Another reason for defective MHC class I surface expression as detected in colorectal tumors and melanoma is due to mutated β2-microglobulin (β2m) which severely impairs the transport of MHC class I molecules to the cell surface [48, 49]. Moreover, disturbed transcriptional regulation, such as decreased levels or loss of locus-specific transcription factors as well as epigenetic alterations like DNA hypermethylation can contribute to the decrease of MHC class I expression [43, 50, 51].
Assembly and transport of HLA class I molecules to the cell membrane crucially depends on the APM. The APM includes the proteasome subunits of low-molecular mass polypeptides 2 and 7 (LMP2 and LMP7), transporter associated with antigen processing 1 and 2 (TAP1 and TAP2), and several chaperons such as tapsin (for reviewing see [52]). In melanoma and renal cancer, cell lines promotor methylation has been demonstrated as a mechanism to inhibit or impair expression of tapsin, TAP2, and TAP1 that finally leads to a decrease or loss of MHC class I surface expression [53, 54].
IFN-γ plays a major role in the regulation of the APM. Particularly, TAP1 and LMP2 genes are controlled by a shared bidirectional IFN-γ-dependent promoter. In the presence of IFN-γ, the transcription factor interferon regulatory factor-1 (IRF-1) binds the interferon regulatory element (IRF-E) within the TAP1/LMP2 promoter and enhances the expression of TAP1 and LMP2 [55]. Epigenetic silencing of IRF-1 transactivation results in IFN-γ unresponsiveness and subsequently in lower expression levels of MHC class I molecules [56]. Further defects within the IFN-γ signaling pathway such as deletion or mutations in Janus kinases 1 and 2 (Jak1 and JAK2) genes, downstream of IFN-γ receptor and upstream of STAT1 have been described in several IFN-γ-resistant melanoma cell-lines and uterine leiomyosarcoma [57, 58]. Therefore, in certain tumors lack of IFN-γ sensitivity supports immune evasion from CTL recognition.
Besides the impaired MHC-class-I-dependent recognition, tumor cells can avoid confrontation with T cells by inhibiting extravasation of lymphocytes to the tumor site. Defects in adhesion of molecule expression on blood vessels have been described in human breast cancer [59], melanoma [60], and human squamous cell carcinoma [61]. Noteworthy, it was shown that decreased expression levels of addressins E-Selectin, P-Selectin, and intercellular adhesion molecule-1 (ICAM-1) in blood vessels of melanoma resulted in impaired T-cell extravasation at the tumor site when compared to accumulation of T cells in adjacent healthy tissue [62].
4. Resistance to T-Cell-Mediated Killing Mechanism
In the last years evidence accumulated that altered or defective apoptotic pathways might contribute to immune evasion of tumors. There are two distinct effector mechanisms, how T cells can eliminate target cells such as virus-infected cells or malignant cells. Generally, they induce apoptosis by the calcium-dependent “perforin/granzyme pathway” or by the calcium-independent “death receptor pathway”. During the last years, in vivo and in vitro studies have demonstrated that tumor cells can resist killing by CTLs through interference with the perforin/granzyme pathway, caused by the expression of the granzyme B inhibiting serine protease inhibitor PI-9/SPI-6. In particular melanoma, cervical carcinoma, and breast carcinoma were demonstrated to express PI9/SPI-6 [63]. A recent study revealed a poorer clinical outcome of vaccinated melanoma patients when the tumors expressed PI9/SPI-6 [64].
Death receptors are members of the tumor necrosis factor (TNF) receptor superfamily and are characterized by an intracellular death domain (DD). The crosslinking of death receptors such as CD95 (Fas; Apo-1) and TRAIL receptor 1 and 2 on tumor cells with their natural ligands CD95L and TRAIL induce formation of the death-inducing signaling complex (DISC). Within the DISC, procaspase-8 is recruited by the DD-associated adaptor protein Fas-associated death domain protein (FADD/MORT-1) and activated by autocatalytic cleavage. This event initiates downstream apoptotic processes causing mitochondrial cytochrome C efflux and subsequent activation of effector caspases-3, -6, and -7 [65]. The induction of apoptosis through death receptor signaling can be blocked in tumor cells at several steps. For example, in melanoma cells high levels of antiapoptotic regulator FLICE inhibitory protein (c-FLIP) have been shown to correlate with increased resistance to TRAIL-mediated apoptosis [66]. Due to its structural homology to procaspase-8, but lacking a catalytic site, c-FLIP binds to DISC and inhibits cleavage of procaspase-8 [67]. C-FLIP has been detected in different types of cancer and cancer cell lines [68–71]. In particular its increased expression in colorectal cancer predicts a poorer clinical outcome [72]. Moreover, tumor cells can obtain further apoptotic resistance by downregulation or inactivation of death receptors. Loss of CD95/FAS can occur at the transcriptional level which seems to be affected by oncogenic ras [73] or loss of functional p53 [74]. A decreased expression of CD95/FAS also has been found in colon cancer [75]. Noteworthy, expression levels of CD95/FAS in colon cancer cell lines can be improved by TNF-α and IFN-γ [75]. Besides transcriptional downregulation, a variety of tumor-associated mutations and deletions can lead to a loss of function of CD95/FAS and TRAIL receptors. For example, lack of cytoplasmic signaling domains of CD95/FAS, TRAIL-R1, and -R2 was detected in multiple myeloma [76], adult T-cell leukemia [77], gastric cancer [78], and breast cancer [79].
Another mechanism by which tumors gain resistance to apoptosis is the expression of transmembrane and soluble decoy receptors with a truncated nonfunctional or a missing death domain, respectively. Interactions of ligands with their respective decoy receptors have been shown to competitively inhibit death receptor signaling. So far, different decoy receptors for CTL's death ligands CD95L and TRAIL have been characterized: soluble CD95/FAS (sCD95) in various malignancies [80–82], decoy receptor 3 (DcR3) in lung cancer [83], colon cancer [83], as well as glioblastoma [84], and TRAIL-R3 (DcR-1) in primary gastrointestinal tract (GIT) cancers [85], TRAIL-R4 (DcR-2) in colon cancer [86], and TRAIL-R5 (Osteoprotegerin; OPG) in breast [87] and prostate cancer [88].
5. Induction of Anergy in Activated T Cells
Inhibitory coreceptors, including “cytotoxic T lymphocyte antigen-4” (CTLA-4) and “programmed death-1” (PD1), play a major role in maintaining peripheral T-cell tolerance. These inhibitory coreceptors are upregulated during T-cell activation and interact with molecules of the B7 family that are found on DCs but are also expressed in many tumor tissues [89]. CTLA-4 competes with the costimulatory receptor CD28 in binding to CD80 (B7.1) and CD86 (B7.2) molecules and activates protein phosphatase 2A (PP2A) [90]. PPA2 dephosphorylates Akt kinase and therefore antagonizes the TCR/CD28 signaling pathway resulting in decreased IL-2 production, impaired TCR signalling, and cell cycle arrest [91, 92]. The inhibitory T-cell receptor PD1 which is upregulated by IFN-γ [93] interacts with the B7 family member B7-homolog 1 (B7-H1) which is found in many tumor types [94–98]. Increased B7-H1 levels correlate with a poor outcome in ovarian cancer, esophageal cancer, and urothelial cancer [94–96]. Mechanistically PD1/B7-H1 interaction leads to recruitment of SH2-containing protein tyrosine phosphatases-1 and -2 (SHP-1, -2) at the immunoreceptor tyrosine-based switch motif of PD1 which ultimately results in downstream signals inhibiting phosphoinositide 3 kinase (PI3K) activity and disruption of TCR/CD28 signaling [99]. Furthermore, an induction of IL-10 was observed which might influence suppressive activity of Tregs (discussed in Section 7) [89]. A soluble form of B7-H1 was detected in aggressive renal cell carcinoma which could further promote immunosuppression [100].
Furthermore, anergy in T cells can be induced by high levels of intratumoral TGF-β and of the arachidonic acid derivative prostaglandin E2 (PGE2). TGF-β controls T-cell homeostasis by inhibiting T-cell activation, proliferation, and differentiation [101, 102]. Numerous in vivo and in vitro studies demonstrated that expression of TGF-β at the tumor site correlates with poor prognosis in many human cancer types such as colorectal cancer, esophageal adenocarcinoma, glioblastoma, breast cancer, and lung cancer [103–107]. However, the molecular mechanisms which eventually lead to TGF-β-mediated T-cell inhibition are still not well characterized. It is hypothesized that the TGF-β-induced mRNA downregulation of TCR components and signaling molecules like IL2-inducible T-cell kinase (ITK), ZAP70, and CD3-zeta chain results in an insufficient T-cell signal transduction [108]. Furthermore, TGF-β is also known for blocking the cytotoxic reaction of CTLs by transcriptional repression of genes encoding the cytotoxic mediators perforin, granzyme A and B, FasL, and INF-γ [109]. More recently, accumulating data suggests that under certain circumstances TGF-β also impairs the function of tumor-reactive CD4+ T cells by inducing them to become FoxP3-positive regulatory T cells (“Tregs”, discussed in Section 7) [110].
The inducible isoform of the cyclooxygenase enzyme COX2 is overexpressed in several tumor types and produces PGE2 which affects various processes relevant to tumorigenesis such as apoptosis, angiogenesis, and migration [111]. It is proposed that PGE2 shifts the immune system to a Th2-type response. Supporting evidence came from the observation that breast cancer patients showing increased intratumoral COX2/PGE2 expression had impaired DC and T-cell function, reduced Th1-type, and increased Th2-type cytokine levels in the serum [112]. In addition, it has also been proposed that PGE2 augments the induction of FoxP3 in CD4+ T cells [113]. In particular, mice with homozygous deletion of the PGE2 receptor P4 gene showed significantly reduced intratumoral levels of FoxP3+ Tregs, increased serum level of Th1-type cytokines, and an increase in IFN-γ-dependent antitumor reactivity of T cells [114].
Another immune evasion mechanism of tumors is based on protein-glycan interactions involving galectins. Galectins, defined as glycan-recognizing proteins with an affinity for β-galactosides, affect many cellular functions including adhesion, migration, chemotaxis, proliferation, apoptosis, and differentiation [115–117]. During the last years, galectins have been recognized as natural immunosuppressive proteins which also inhibit antitumor immune responses. Their overexpression in tumors such as melanoma [118], glioblastoma [119], prostate cancer [120], bladder cancer [121], ovarian cancer [122], and breast cancer [123] often correlates with tumor aggressiveness. Especially galectin-1 might play an important role in immune evasion by inducing T-cell apoptosis [124, 125]. So far little is known about how galectins impair T-cell functions. Yet, some evidence suggests that galectin-1 interferes with the correct assembly of the T-cell receptor complex in lipid rafts [126].
6. Tumor Cell “Counter Attack”
In an immunocompetent organism, T-cell activation by pathogens leads to a clonal expansion of antigen-specific T cells. Once they have accomplished their mission, T-cell expansion bearing the inherent danger of autodestruction is terminated by T-cell activation-induced cell death (AICD) resulting in clonal contraction of antigen-specific T cells [127]. The CD95/FAS death receptor, which is upregulated in activated T cells, is playing a key role in AICD. It is well known that upon TCR engagement in the absence of costimulation, T cells express high amounts of Fas ligand (FasL), which is assumed to induce apoptosis of these T cells (“suicide”) and between T cells (“fratricide”). Yet, some tumors such as melanoma [128], lung cancer [129], pancreatic cancer [130], and breast cancer [131] express FAS ligand (FasL), which might accelerate AICD and therefore could contribute to tumor immune evasion. First evidence that FasL can confer resistance to T-cell-mediated tumor rejection was demonstrated by delayed growth of FasL-positive melanoma cells in lpr-positive mice (lpr, lymphoproliferation gene, encoding mutated CD95/FAS with loss of function) when compared to congenic controls, supporting the concept that tumor cells can eliminate effector T cells by FasL counter attack [128]. However, despite accumulating data in support of FasL counter attack, many conflicting studies have reported that FasL can also induce proinflammatory and antitumoral effects in vivo. In particular, tumor xenografts genetically modified to express membrane-bound FasL showed an accelerated rejection which was accompanied by neutrophil infiltration [132–134]. In contrast, a more recent study revealed that antisense-mediated downregulation of FasL in colon cancer cells reduced tumor growth in syngeneic and immune competent mice [135]. It remained to be clarified whether other factors such as cytokines, tumor microenvironment, or technical peculiarities account for the different outcomes. The situation becomes far more complex by a recent report describing the appearance of so called microvesicles containing FasL which were found to be released by melanoma cells [136]. These microvesicles are endosome-derived small particles of 50 to 100 nm in size and are secreted through exocytosis pathways [137]. These microvesicles were able to induce cell death in lymphoid cells and furthermore might cause systemic immunosuppressive effects in patients [136]. In particular, it is assumed that besides the effects of death receptor ligands the microvesicles can carry various immunomodulatory factors that exert immunosuppressive effects on lymphocytes in draining lymph nodes but also modulate myeloid-derived cells to become myeloid-derived suppressor cells (MDSC) [137] (which are further discussed in Section 7).
In addition to FasL-mediated apoptosis induction in antitumor effector cells, a recent report also described expression of TRAIL in hepatocellular carcinoma which induced cell death of Jurkat leukemia T cells [138].
Another mechanism that may contribute to tumor evasion from tumor-specific T cells is based on the expression of the enzyme indoleamine 2,3-dioxygenase (IDO). This heme-containing enzyme degrades the essential amino acid tryptophan and catalyzes the initial and rate-limiting step of the kynurenine pathway leading to nicotinamide dinucleotide (NAD) biosynthesis. Initially, IDO was thought to represent a defence mechanism against bacteria, but it soon became evident that IDO plays a physiological role in the establishment of an immune privilege at the fetoplacental border. IDO expression at this site is proposed to block T-cell attack and therefore protects the embryo [139]. This effect was confirmed by in vitro studies showing that tryptophan depletion led to cell cycle arrest in T cells [140]. So far IDO expression was found in several primary tumors such as gastric cancer, colon cancer, and renal cell carcinoma as well as in derived cell lines [141]. Interestingly, some of the tumor cell lines expressed IDO only after IFN-γ treatment and the activity of IDO could be blocked by administration of the specific inhibitor levo-1-methyl-trypthopan [141]. Thus, in certain tumors pharmacological blockade of IDO may impair the development of an immune privileged tumor site and may improve immunotherapy.
7. Immunosuppressive Roles of FoxP3+ Regulatory T Cells and of Myeloid-Derived Suppressor Cells (MDSCs)
The existence of peripheral T cells exerting a suppressor function has been a matter of debate for many years. Nowadays it is widely accepted that a distinct population of CD4+ cells constitutively expressing the surface markers CD25 (the alpha-chain of the IL-2 receptor) and accounting for approximately 5–10% of all peripheral T cells can suppress responses of tumor-specific CD4+ T cells and CTLs [142]. Moreover, these cells, designated Tregs, are characterized by the expression of the transcription factor forkhead box P3 (FoxP3), CTLA-4, glucocorticoid-induced TNF receptor (GITR), and by their ability to suppress the activation of other T-cell subpopulations [110, 143, 144].
In humans, high numbers of CD4+ CD25+ Tregs were found in head and neck cancer [145], lung cancer [146, 147], pancreatic cancer [148], breast cancer [149], liver cancer [150], ovary cancer [151], and gastrointestinal cancer [152] either in the circulation or in the tumor itself. The appearance of Tregs at the tumor sites often correlates with poor prognosis of cancer patients [148, 151, 153, 154]. So far, it is not clarified why and how Tregs accumulate at the tumor site. As a mechanism of Treg recruitment, it has been proposed that tumor cells or tumor-infiltrating macrophages secrete the chemokine CCL22 which chemoattracts CD4+CD25+ Tregs [151, 155]. On the other hand, it is also likely that the immunosuppressive milieu of the tumor, that is, high concentrations of TGF-β, induces tumor infiltrating CD4+ T cells to become FoxP3+ Tregs [110]. Tregs, either regular Tregs or induced Tregs prevent antitumoral immune responses by inhibiting tumor-specific CD4+ cells and CTLs.
Another recently described immunosuppressive cell population in tumors are the MDSCs [156, 157]. These cells are generated in response to a variety of tumor-derived cytokines and appear as heterogenous populations representing myeloid cells at different stages of differentiation. In advanced cancer stages peripheral blood MDSCs were found to inhibit IFN-γ production by autologous CD8+ T cells stimulated with peptide-pulsed DCs [156, 158]. It is hypothesized that the appearance of MDSCs in tumors and in peripheral blood might be due to increased granulocyte macrophage colony-stimulating factor (GM-CSF) levels produced by human tumor cells [159–162] and that the immunosuppressive phenotype is induced by tumor-derived microvesicles [137]. Meanwhile, some evidence is accumulating that MDSCs might inhibit T cells by the production of amino acid metabolizing enzymes such as arginase, nitric oxide synthetase and the production of reactive oxygen species (ROS) [157, 158, 163]. Especially, the chemical modification of T-cell receptors by reactive nitrogenous species was demonstrated to impair binding to the cognate MHC/peptide complex [163].
8. Immunotherapeutical Approaches to Induce Antitumor T-Cell Responses
A number of different immunotherapeutic approaches focus on the generation of an effective T-cell-mediated antitumor response in patients (Table 1). Exploiting T cells as a target tool for cancer treatment seems to be advantageous as T cells have an exact specificity for their target, can home into antigen-expressing tumor sites and can generate a long-lasting immune response against reoccurrence of the disease. However, so far none of the various T-cell-dependent approaches has been established as a routine clinical treatment strategy for cancer as, for example, monoclonal antibody-based pharmaceuticals have been in the last years. The failure of various T-cell-based immunotherapeutic approaches in clinical studies is related in many aspects to escape mechanisms of tumors from T cell surveillance and the immunosuppressive effects mediated by tumors described in the preceding chapters (summarized in Figure 2). While in this section we will describe immunotherapeutic approaches using DCs and TILs, in the following sections we will give an overview on new strategies to enhance T-cell-mediated anti-tumor response.
Table 1.
Selected approaches to tumor immunotherapy.
| Approach | Target | Agent | Immune modulation of host | Phase of experimentation | Main findings |
|---|---|---|---|---|---|
| Vaccination | Glioma | Tumor-cell lysate pulsed DCs | — | Phase I clinical trial completed (NCT00576537) | T-cell responses, detection of infiltrating T cells in recurrent glioma associated with prolonged survival [210] |
| Vaccination | Adenocarcinoma of the prostate expressing “prostatic acid phosphatase” (PAP) | DCs ex vivo primed with PAP-GM-CSF fusion protein (Sipuleucel T, FDA-approved for treatment of hormone-refractory prostate cancer) | — | Phase III clinical trial completed (NCT00065442) | Increased overall survival, but no increase in progression-free survival of patients [169] |
| Vaccination and chemotherapy (Doxetacel) | Breast cancer with expression of MUC-1, CEA | Recombinant Vaccinia and Fowlpox virus (PANVAC) encoding MUC-1 and CEA | GM-CSF treatment during vaccinations | Phase II clinical trial ongoing (NCT00179309) | Induction of T-cell specific immune responses [211] |
| Vaccination | Adenocarcinoma of the prostate expressing “TCRγ Alternative Reading frame Protein” | Vaccination with TARP peptides | Use of Montanide ISA-51 VG as adjuvant and concomitant GM-CSF treatment | Preclinical study and phase-I clinical trial ongoing (NCT00908258) | Preclinical study demonstrated induction of T-cell-specific immune responses [212] |
| Vaccination | Melanoma | MART-1-, gp100-, tyrosinase- peptides | Subcutaneous injection of IFN-α2b and/or GM-CSF | Phase II clinical trial completed (ECOG E1696) | Neither IFN-α2b nor GM-CSF improved immune responses, 35% of patients developed T-cell responses associated with prolonged median overall survival [213] |
| Vaccination | Melanoma | MAGE-3.A1 peptide and/or NA17.A2 peptide | peritumoral injection of IL-2, IFN-α and GM-CSF, and topical application of imiquimod | Phase I/II clinical trial ongoing (NCT01191034) | — |
| Vaccination | CEA expressing cancers | Ex vivo activation of DCs using recombinant Fowlpox virus encoding CEA | Denileukin diftitox-mediated depletion of Tregs | Phase I clinical trial ongoing (NCT00128622) | Increased T-cell responses to CEA-positive target cells [171] |
| Vaccination | Melanoma | Tyrosinase/gp100/MART-1 Peptides | Use of Montanide ISA-51 VG as adjuvant, treatment with anti-CTLA4 antibody (MDX-010) | Phase I/II clinical trial ongoing (NCT00028431) | T-cell reactivity against gp100 and MART-1. CTLA-4 antibody dose-related adverse autoimmune effects in skin and gastrointestinal tract [214] |
| Melanoma | gp100 peptides | Vaccination using incomplete Freund's adjuvant, treatment with anti-CTLA4 antibody (Ipilimumab) | Phase II clinical trial completed (NCT00094653) | Improved overall survival when applying Ipilimumab irrespective of gp100 vaccination, adverse immunological site effects [185] | |
| Vaccination | Glioma | Tumor cell lysate pulsed dendritic cells | TLR agonist Imiquimod | Phase II clinical trial ongoing (NCT01204684) | Improved survival of a subset of glioma patients [168] |
| Vaccination | Leukemia | Autologous leukemia cells | Autologous skin fibroblasts transduced with recombinant adenoviral vectors encoding CD40L and IL-2 | Phase I/II clinical trial ongoing (NCT00058799) | Observed immune responses including antibodies against leukemic blasts, prolonged survival time of patients [215] |
| TLR agonist monotherapy | TLR 9 in Melanoma | CpG 7909 | — | Phase II clinical trial completed (NCT00070642) | Increased frequencies of T cells reactive against target cells expressing melanoma-associated-antigens in sentinel lymph nodes [166] |
| TLR agonist monotherapy | TLR7 in basal cell carcinoma | Imiquimod | — | Phase II clinical trial completed (NCT00189241) | FDA approved for treatment of superficial basal cell carcinoma [167] |
| Immunotoxin monotherapy | Renal cell carcinoma | Denileukin diftitox (IL-2-diphteria toxin fusion protein) | Complementary IL-2 treatment | Phase I clinical trial completed (NCT00278369) | Partial depletion of Tregs, no increase in antitumor response rates when compared to controls [216] |
| TGFβ antisense (AS) oligonucleotide monotherapy | TGFβ expression in patients with recurrent high grade glioma | TGFβ AS (AP-12009, Trabedersen) | Local depletion of TGFβ | Phase IIb clinical trial completed (NCT00431561) | Well tolerated, increased median survival time of patients [195] |
| Anti-TGFβ antibody monotherapy | TGFβ expression in patients with renal cell carcinoma, melanoma | Blocking anti-TGFβ antibody (GC-1008) | Systemic depletion of TGFβ | Phase I clinical trial safety and efficacy study (NCT00356460) | — |
| Anti-PD-1 antibody monotherapy | Refractory or relapsed malignancies (solid tumors) | Anti-PD-1 antibody (MDX-1106) | Blocking extrinsic self-regulatory pathways of T cells | Phase I clinical trial (NCT00441337) | Complete and partial responses, induction of inflammatory colitis [179] |
| Anti-CD137/4-1BB monotherapy | Melanoma | Anti-CD137/4-1BB antibody (BMS-663513) | Systemic co-stimulation of T cells | Phase II clinical trial completed (NCT00612664) [188] | — |
| Adoptive cell therapy | Melanoma | Ex vivo expanded TILs | Chemotherapy-mediated lymphodepletion prior infusion of TILs, high dose IL-2 treatment | Phase II clinical trial ongoing (NCT00513604) | Objective clinical responses, in some cases durable complete responses, toxic side effects after chemotherapy and high IL-2 doses [174, 176] |
| Adoptive cell therapy | Melanoma | Ex vivo expanded TILs enriched for CD8* T cells genetically engineered to express IL-12 | Chemotherapy-mediated lymphodepletion prior infusion of TILs | Phase I/II clinical trial ongoing (NCT01236573) | — |
| Genetic manipulation of T cells for immune therapy | Neuroblastoma cells expressing L1-CAM (CD171) | Ex vivo generated anti-CD171-CAR engineered T cells | — | Phase I/II clinical trial completed (BB-IND#9149, FDA) | Weak tumor responses and limited persistence of CD171-CAR [201] |
| Genetic manipulation of T cells for immunotherapy | Non-Hodgkin lymphoma and mantle cell lymphoma | Ex vivo generated anti-CD20-CAR engineered T cells | Low dose IL-2 treatment | Phase I/II clinical trial completed (NCT00012207) | No side effects, regression of tumors, persistence of modified T cells for 9 weeks [202] |
| Genetic manipulation of T cells for immunotherapy | Metastatic cancers that express NY-ESO-1 | Ex vivo generated anti-NY ESO-1 TCR-gene engineered T cells | Chemotherapy-mediated lymphodepletion prior infusion of modified T cells. Modified T cells are further stimulated using ALVAC–ESO-1 vaccine, G-CSF and high dose IL-2 treatment | Phase II clinical trial ongoing (NCT00670748) | — |
| Genetic manipulation of T cells for immunotherapy | B-cell malignancies with expression of CD19 | Ex vivo generated anti-CD19-CAR engineered T cells | Chemotherapy-mediated lymphodepletion prior infusion of TILs, high dose IL-2 treatment | Phase I/II clinical trial ongoing (NCT00924326) | Regression of B-cell lymphoma and elimination of B-cell precursors in patients [203] |
| Genetic manipulation of T cells for immunotherapy | Metastatic cancers expressing Her2 | Ex vivo generated anti-Her2-CAR engineered T cells | Chemotherapy-mediated lymphodepletion prior infusion of modified T cells, IL-2 treatment | Phase I/II clinical trial completed (NCT00924287) | — |
ClinicalTrials.gov identifier, Eastern Cooperative Oncology Group (ECOG) identifier, and FDA-authorized pilot clinical study identifier are given in brackets.
Figure 2.
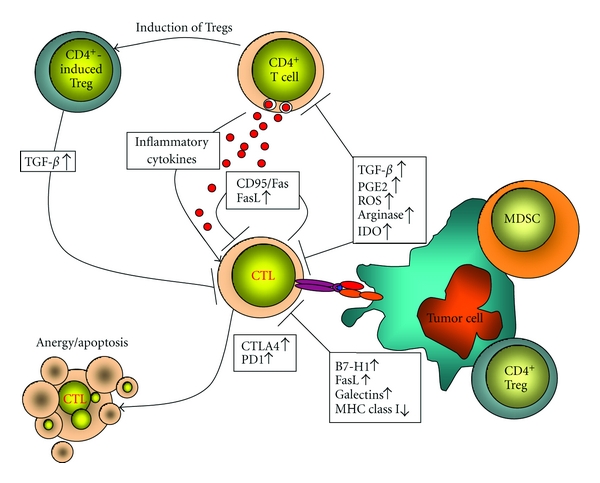
Overview of immunosuppressive evasion mechanisms. Tumor cells and tumor-infiltrating Tregs and MDSC can employ a plethora of immunosupressive factors and molecules that impair T-cell function or lead to T-cell anergy and/or apoptosis. Depicted are immunosuppressive self-inhibitory circuits of activated CTLs and also immunosuppressive mechanisms of tumors.
Strategies based on vaccination with tumor cell lysates, TAAs, or tumor peptides can elicit antitumoral responses through priming of DCs and subsequent activation of tumor-specific T cells [164]. However, many clinical trials were disappointing in terms of tumor regression or increased survival rates [164]. Potential reason for failure of this vaccination strategy can be alterations in number as well as functional defects in DCs which has been observed in a number of patients [33, 35]. In addition, lack of immunological response seemed to be more severe in patients bearing a large tumor burden and is likely caused by immunosuppressive factors released by the tumor, Tregs [101] and MDSCs [165]. It is assumed that induction of T-cell tolerance by immature or dysfunctional DCs is mostly responsible for the observed unresponsiveness of T cells [164]. Therefore, new vaccine strategies concentrate on the targeting of certain DC subsets and codelivery of PAMP or DAMP signals to DCs to improve the T-cell priming capacities. In particular, activators of TLRs, for instance the cytosine-phosphate-guanosine (CpG) dinucleotide CpG 7909, which induces TLR9 activation, came into the focus of tumor immunotherapy. Intradermally injected CpG 7909 near the tumor site obviously provided sufficient danger signals to plasmacytoid DCs which in turn were able to elicit melanoma responsive CTLs in sentinel lymph nodes of patients, whereas in patients treated with saline no melanoma-reactive CTLs were detected [166]. The chemical compound imiquimod, a potent TLR7 agonist, has been approved for treatment of basal cell carcinoma [167] and was shown to improve survival of glioblastoma patients bearing tumors with mesenchymal gene expression profile and vaccinated with ex vivo tumor cell lysate pulsed DCs [168].
Another strategy to enhance vaccination-induced T-cell activation is the ex vivo modulation of autologous or allogeneic DCs. This is often accomplished by incubation with factors like type-1 and type-2 interferons, TLR ligands or GM-CSF, which all enhance the antigen-presenting and T-cell activating capacities of DCs [164]. As an encouraging result of these efforts, Sipuleucel-T, a dendritic-cell-based vaccine recently approved by the FDA and based on peripheral blood cells ex vivo stimulated with a fusion protein of prostatic acid phosphatase and GM-CSF was shown to improve survival of prostate carcinoma patients in a phase III trial [169]. Furthermore, depletion of Tregs prior to vaccination of ex vivo-modulated DCs can be used to improve immune responses. In particular, Denileukin diftitox, an IL-2 peptide fused to diphtheria toxin which is approved for treatment of cutaneous T-cell lymphoma [170] enhanced T-cell activation of patients when applied before vaccination with DCs ex vivo transduced with a fowlpox vector encoding the TAA carcinoembryonic antigen [171].
Furthermore, the direct transfer of tumor-specific either allogeneic or ex vivo expanded patient-derived autologous T cells emerged as a promising approach for cancer immunotherapy (adoptive cell therapy, ACT). The rationale behind this strategy is to circumvent or break tolerance against the tumor by the transferred T cells. Direct clinical evidence has accumulated that adoptively transferred T cells can delay or even prevent tumor relapse after initial standard treatment [172]. Yet, in this first clinical trial the response rate was only transient, and transferred tumor-specific T cells vanished rapidly [173]. Recently, improvements of ex vivo cultivation conditions and more important lymphodepletion prior to infusion of T cells led to remarkable cancer regression [174–176]. One probable explanation for the efficiency of ACT after lymphodepletion might be the removal of immunosuppressive Tregs.
9. Improving Antitumoral Responses by Blocking Self-Restricting Inhibitory Circuits of T Cells
To further improve the outcome of immunotherapy immune-restrictive signals to T cells can be either blocked or eliminated. In particular, blocking antibodies can be utilized for switching off self-restricting inhibitory circuits of T cells directly at the site of the tumor, resulting in enhanced antitumor immune responses. Promising candidates are antibodies inhibiting CTLA-4 [177] which blocks the intrinsic T-cell regulatory pathway or anti-B7-H1 antibodies which block extrinsic T-cell inhibitory pathways driven by ligands on DCs and tumor cells, respectively [98, 178]. In preclinical studies, blockade of B7-H1 has resulted in enhanced antitumoral immune responses [98, 178]. Recently, a blocking anti-PD1 antibody (MDX-1106), the receptor for B7-H1 on T cells, has been evaluated in a clinical phase I study. So far, MDX-1106 was well tolerated in most patients bearing different solid tumor types. A small proportion of patients showed complete and partial responses to treatment whereas one case was associated with development of inflammatory colitis [179]. So far no clinical studies using PD-1 blockade during immunotherapeutic approaches have been reported. In preclinical mouse models CTLA-4 blockade-based immunotherapy enhances the T-cell-mediated killing of tumor cells such as prostate [180] and colon cancer [181]. Also anti-CTLA-4 antibodies have entered the clinic and have been used as monotherapy or in combination with vaccination strategies targeting melanoma [182]. Although clinical responses were observed in some patients with metastatic melanoma or renal cell cancer in a phase I clinical trial with humanized anti-CTLA-4 antibody (ipilimumab), caution is advised since CTLA-4 blockade in some patients caused immune-mediated site effects such as hypophysitis, enterocolitis, and nephritis [183–185]. Alternatively, agonistic antibodies specific for costimulatory receptors can be applied to improve T-cell responses to tumors. An example is CD137 (4-1BB) antibodies which trigger via their receptor the upregulation of antiapoptotic BclXL, Bfl-1, and c-FLIP through the PI3K/Akt and NFκB pathways and therefore rendered reactive T cells more resistant to AICD [186, 187]. Notably, a humanized monoclonal anti-CD137/4-1BB antibody (BMS-663513) has entered a clinical phase II trial as a second line monotherapy for previously treated unresectable melanoma, but so far no combinations with immunotherapeutic approaches have been reported [188]. Other attempts focus on OX40 (CD134), a co-receptor playing a central role in generating CD4+ and CD8+ memory cells [189]. A preclinical study using OX40L or an agonistic antibody during vaccination was shown to augment protection against melanoma, breast and colon carcinoma, and sarcoma in mice [190]. Currently, the role of OX40-OX40L interaction in the induction, maintenance, and function of Tregs came into focus of interest. Some lines of evidence support the view that OX40-OX40L interaction inhibit the induction of Tregs in the tumor, which might further enhance immunotherapy of cancer [189].
10. Targeting Tregs and the Tumor Micromilieu in order to Improve Antitumoral T-Cell Responses
To date, it became very clear that the tumor microenvironment, consisting of immunosuppressive cells, especially Tregs, tumor metabolites, and tumor-derived cytokines critically affects T-cell-mediated immunotherapy. In particular, immunosuppressive effects of Tregs can nowadays be minimized by antibody-mediated (i.e., anti-CD25 [191], anti-GITR [192]) Treg depletion or chemotherapeutic lymphodepletion protocols prior to adoptive cell transfer of T lymphocytes and vaccination approaches. In addition, blocking antibodies or soluble receptors can be exploited for the blockade of cytokines that either suppress effector T cells directly or act via macrophages and MDSCs. Candidates include for instance TGF-β [102, 109] and IL-10 [193]. A further promising avenue to increase the effectiveness of antitumoral T cells might be the antisense oligodeoxynucleotide or siRNA-mediated silencing of TGF-βRI and RII receptor expression in the tumor-reactive T cells themselves or the knock down of TGF-β expression at the tumor site, in particular in Tregs [194]. Indeed a first clinical study targeting TGFβ using the antisense oligonucleotide trabedersen as single agent showed promising results when locally given in recurrent glioma patients [195]. A clinical phase I study for systemic antibody-mediated blockade of TGFβ is ongoing (see Table 1). It is foreseen that immunosuppressive enzymes such as COX2, IDO, arginase, and nitric oxide synthetase will be targeted by small chemical compounds and used in combination with immunotherapy. In particular, small molecule inhibitors targeting COX2 and decreasing intratumoral PGE2 levels showed promising results in preclinical studies [196].
11. Genetic Manipulation of T Cells for Immunotherapy of Tumors
Finally, T cells can be genetically manipulated to engraft polyclonal T cells with an additional antitumor specificity and/or improved antitumor response. Additional specificity can be provided by the engraftment of T cells with a second TCR recognizing a specific tumor-associated antigen. In first clinical trials, TCR gene transfer into peripheral blood T cells has been shown to be feasible and to reprogram polyclonal T cells towards tumor antigens [197, 198]. Another approach uses chimeric antigen receptors (CARs), which are fusion proteins of an antigen-binding moiety, usually an antibody-derived single chain fragment variable (scFv) specific to a surface tumor antigen, linked to an ITAM-bearing signaling receptor like the CD3 zeta chain [199, 200]. In general, the use of CARs is advantageous when treating tumors with IFN-γ unresponsiveness or defects in APM as the recognition of the tumor cell by the engrafted T cell is independent of MHC presentation. However, for a CAR approach, the targeted surface antigen should only be expressed on tumor cells in order to avoid off-target effects. And indeed, clinical trials have proven that CARs were safe and off-target effects were not apparent when a strong target antigen expression was restricted to tumor cells [201–203] (Table 1). Further signals can be provided upon antigen-binding by the incorporation of additional signaling subunits from costimulatory receptors like CD28 [204], OX40 (CD134) [205], or 4-1BB (CD137) [206]. The combined signaling can induce the expression of anti-apoptotic proteins, sustained proliferation, inflammatory cytokine secretion, and resistance to immunosuppression by soluble or cellular components. The genetic manipulation of T cells can be further extended by including expression cassettes for homeostatic T-cell cytokines [207], anti-apoptotic molecules [208], shRNAs against T-cell inhibitory molecules, and chemokine receptors [209] to guide T-cell migration to tumor sides.
12. Conclusions
The considerable progress made in understanding the interaction between tumor cells and T cells opens avenues for the development of improved clinical protocols for T-cell-based immunotherapy. In particular, the development of therapeutic tools for efficient cross-priming such as TLR agonists and sustaining antitumoral functions of T cells as for example modifying self-restricting circuits should enable rejection of the tumor and should guarantee engraftment of long-lived memory T cells. To further improve outcomes, immunotherapy approaches need to be combined with other therapies that might target various components of tumor development such as tumor cell metabolism, apoptosis, angiogenesis, tumor stroma, and inflammation to reduce the immunosuppressive environment shaped by the tumor. Last but not least recent studies suggest that the immune system might contribute mechanistically to the clinical efficiency of chemotherapeutic protocols [28, 31] which should be further pursued in future clinical trials. We envisage that in future advanced protocols for adjuvant immunotherapy will complement conventional therapies for the benefit of cancer patients.
Acknowledgments
The authors apologize to all colleagues who could not be cited due to space restrictions. A. Temme is supported by the Deutsche Krebshilfe e.V. (DKH 109377) and the Dr. Robert Pfleger-Stiftung.
References
- 1.Ehrlich P. Über den jetzigen Stand der Karzinomforschung. Nederlands Tijdschrift voor Geneeskunde. 1909;5:273–290. [Google Scholar]
- 2.Kaplan DH, Shankaran V, Dighe AS, et al. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(13):7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Street SEA, Trapani JA, MacGregor D, Smyth MJ. Suppression of lymphoma and epithelial malignancies effected by interferon γ. Journal of Experimental Medicine. 2002;196(1):129–134. doi: 10.1084/jem.20020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Broek MF, Kägi D, Ossendorp F, et al. Decreased tumor surveillance in perforin-deficient mice. Journal of Experimental Medicine. 1996;184(5):1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankaran V, Ikeda H, Bruce AT, et al. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 6.Shinkai Y, Rathbun G, Lam KP, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68(5):855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 7.Penn I. Post-transplant malignancy. The role of immunosuppression. Drug Safety. 2000;23(2):101–113. doi: 10.2165/00002018-200023020-00002. [DOI] [PubMed] [Google Scholar]
- 8.Bower M, Palmieri C, Dhillon T. AIDS-related malignancies: changing epidemiology and the impact of highly active antiretroviral therapy. Current Opinion in Infectious Diseases. 2006;19(1):14–19. doi: 10.1097/01.qco.0000200295.30285.13. [DOI] [PubMed] [Google Scholar]
- 9.Kayagaki N, Yamaguchi N, Nakayama M, Hiroshi E, Okumura K, Yagita H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: a novel mechanism for the antitumor effects of type I IFNs. Journal of Experimental Medicine. 1999;189(9):1451–1460. doi: 10.1084/jem.189.9.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rousalova I, Krepela E. Granzyme B-induced apoptosis in cancer cells and its regulation (review) International Journal of Oncology. 2010;37(6):1361–1378. doi: 10.3892/ijo_00000788. [DOI] [PubMed] [Google Scholar]
- 11.Wang RF. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends in Immunology. 2001;22(5):269–276. doi: 10.1016/s1471-4906(01)01896-8. [DOI] [PubMed] [Google Scholar]
- 12.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. Journal of Experimental Medicine. 1996;184(2):747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke SRM. The critical role of CD40/CD40L in the CD4−dependent generation of CD8+ T cell immunity. Journal of Leukocyte Biology. 2000;67(5):607–614. doi: 10.1002/jlb.67.5.607. [DOI] [PubMed] [Google Scholar]
- 14.Le Page C, Génin P, Baines MG, Hiscott J. Interferon activation and innate immunity. Reviews in Immunogenetics. 2000;2(3):374–386. [PubMed] [Google Scholar]
- 15.Seliger B, Ruiz-Cabello F, Garrido F. Chapter 7 IFN inducibility of major histocompatibility antigens in tumors. Advances in Cancer Research. 2008;101:249–276. doi: 10.1016/S0065-230X(08)00407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 17.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nature Immunology. 2002;3(11):999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palena C, Schlom J. Vaccines against human carcinomas: strategies to improve antitumor immune responses. Journal of Biomedicine and Biotechnology. 2010;2010 doi: 10.1155/2010/380697. Article ID 380697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nature Reviews Immunology. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 20.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 21.Burgdorf S, Schölz C, Kautz A, Tampé R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nature Immunology. 2008;9(5):558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 22.Maurer T, Heit A, Hochrein H, et al. CpG-DNA aided cross-presentation of soluble antigens by dendritic cells. European Journal of Immunology. 2002;32(8):2356–2364. doi: 10.1002/1521-4141(200208)32:8<2356::AID-IMMU2356>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 23.Schulz O, Diebold SS, Chen M, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433(7028):887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 24.Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. Journal of Immunology. 2008;181(3):1849–1858. doi: 10.4049/jimmunol.181.3.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annual Review of Immunology. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 26.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nature Immunology. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 27.Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 28.Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 29.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 30.Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462(7269):99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 31.Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunological Reviews. 2007;220(1):47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 32.Kurts C, Kosaka H, Carbone FR, Miller JFAP, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. Journal of Experimental Medicine. 1997;186(2):239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Troy AJ, Summers KL, Davidson PJT, Atkinson CH, Hart DNJ. Minimal recruitment and activation of dendritic cells within renal cell carcinoma. Clinical Cancer Research. 1998;4(3):585–593. [PubMed] [Google Scholar]
- 34.Enk AH, Jonuleit H, Saloga J, Knop J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. International Journal of Cancer. 1997;73(3):309–316. doi: 10.1002/(sici)1097-0215(19971104)73:3<309::aid-ijc1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Nestle FO, Burg G, Fäh J, Wrone-Smith T, Nickoloff BJ. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen- presenting cells. American Journal of Pathology. 1997;150(2):641–651. [PMC free article] [PubMed] [Google Scholar]
- 36.Gabrilovich DI, Corak J, Ciernik IF, Kavanaugh D, Carbone DP. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clinical Cancer Research. 1997;3(3):483–490. [PubMed] [Google Scholar]
- 37.Stumpf M, Hasenburg A, Riener MO, et al. Intraepithelial CD8−positive T lymphocytes predict survival for patients with serous stage III ovarian carcinomas: relevance of clonal selection of T lymphocytes. British Journal of Cancer. 2009;101(9):1513–1521. doi: 10.1038/sj.bjc.6605274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jerome KR, Barnd DL, Bendt KM, et al. Cytotoxic T-lymphocytes derived from patients with breast adenocarcinoma recognize an epitope present on the protein core of a mucin molecule preferentially expressed by malignant cells. Cancer Research. 1991;51(11):2908–2916. [PubMed] [Google Scholar]
- 39.Baxevanis CN, Dedoussis GVZ, Papadopoulos NG, Missitzis I, Stathopoulos GP, Papamichail M. Tumor specific cytolysis by tumor infiltrating lymphocytes in breast cancer. Cancer. 1994;74(4):1275–1282. doi: 10.1002/1097-0142(19940815)74:4<1275::aid-cncr2820740416>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 40.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. Journal of Immunotherapy. 2004;27(3):184–190. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deschoolmeester V, Baay M, Van Marck E, et al. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunology. 2010;11, article 19 doi: 10.1186/1471-2172-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meissner M, Reichert TE, Kunkel M, et al. Defects in the human leukocyte antigen class I antigen-processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clinical Cancer Research. 2005;11(7):2552–2560. doi: 10.1158/1078-0432.CCR-04-2146. [DOI] [PubMed] [Google Scholar]
- 43.Nie Y, Yang GY, Song Y, et al. DNA hypermethylation is a mechanism for loss of expression of HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis. 2001;22(10):1615–1623. doi: 10.1093/carcin/22.10.1615. [DOI] [PubMed] [Google Scholar]
- 44.Korkolopoulou P, Kaklamanis L, Pezzella F, Harris AL, Gatter KC. Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. British Journal of Cancer. 1996;73(2):148–153. doi: 10.1038/bjc.1996.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanda MG, Restifo NP, Walsh JC, et al. Molecular characterization of defective antigen processing in human prostate cancer. Journal of the National Cancer Institute. 1995;87(4):280–285. doi: 10.1093/jnci/87.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maleno I, Cabrera CM, Cabrera T, et al. Distribution of HLA class I altered phenotypes in colorectal carcinomas: high frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Immunogenetics. 2004;56(4):244–253. doi: 10.1007/s00251-004-0692-z. [DOI] [PubMed] [Google Scholar]
- 47.Maleno I, Romero JM, Cabrera T, et al. LOH at 6p21.3 region and HLA class altered phenotypes in bladder carcinomas. Immunogenetics. 2006;58(7):503–510. doi: 10.1007/s00251-006-0111-8. [DOI] [PubMed] [Google Scholar]
- 48.Bicknell DC, Rowan A, Bodmer WF. β2-Microglobulin gene mutations: a study of established colorectal cell lines and fresh tumors. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(11):4751–4755. doi: 10.1073/pnas.91.11.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hicklin DJ, Wang Z, Arienti F, Rivoltini L, Parmiani G, Ferrone S. β2-Microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. Journal of Clinical Investigation. 1998;101(12):2720–2729. doi: 10.1172/JCI498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fonsatti E, Sigalotti L, Coral S, Colizzi F, Altomonte M, Maio M. Methylation-regulated expression of HLA class I antigens in melanoma. International Journal of Cancer. 2003;105(3):430–431. doi: 10.1002/ijc.11077. [DOI] [PubMed] [Google Scholar]
- 51.Soong TW, Hui KM. Locus-specific transcriptional control of HLA genes. Journal of Immunology. 1992;149(6):2008–2020. [PubMed] [Google Scholar]
- 52.Koch J, Tampé R. The macromolecular peptide-loading complex in MHC class I-dependent antigen presentation. Cellular and Molecular Life Sciences. 2006;63(6):653–662. doi: 10.1007/s00018-005-5462-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manning J, Indrova M, Lubyova B, et al. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology. 2008;123(2):218–227. doi: 10.1111/j.1365-2567.2007.02689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunology, Immunotherapy. 2008;57(11):1719–1726. doi: 10.1007/s00262-008-0515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.White LC, Wright KL, Felix NJ, et al. Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity of CD8+ T cells in IRF-1(-/-) mice. Immunity. 1996;5(4):365–376. doi: 10.1016/s1074-7613(00)80262-9. [DOI] [PubMed] [Google Scholar]
- 56.Rodríguez T, Méndez R, Del Campo A, et al. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer. 2007;7, article 34 doi: 10.1186/1471-2407-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Respa A, Bukur J, Ferrone S, et al. Association of IFN-γ signal transduction defects with impaired HLA class I antigen processing in melanoma cell lines. Clinical Cancer Research. 2011;17(9):2668–2678. doi: 10.1158/1078-0432.CCR-10-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayashi T, Kobayashi Y, Kohsaka S, Sano K. The mutation in the ATP-binding region of JAK1, identified in human uterine leiomyosarcomas, results in defective interferon-γ inducibility of TAP1 and LMP2. Oncogene. 2006;25(29):4016–4026. doi: 10.1038/sj.onc.1209434. [DOI] [PubMed] [Google Scholar]
- 59.Madhavan M, Srinivas P, Abraham E, Ahmed I, Vijayalekshmi NR, Balaram P. Down regulation of endothelial adhesion molecules in node positive breast cancer: possible failure of host defence mechanism. Pathology and Oncology Research. 2002;8(2):125–128. doi: 10.1007/BF03033721. [DOI] [PubMed] [Google Scholar]
- 60.Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. Journal of Experimental Medicine. 1995;181(2):811–816. doi: 10.1084/jem.181.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clark RA, Huang SJ, Murphy GF, et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. Journal of Experimental Medicine. 2008;205(10):2221–2234. doi: 10.1084/jem.20071190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clinical Cancer Research. 2007;13(9):2549–2556. doi: 10.1158/1078-0432.CCR-06-2450. [DOI] [PubMed] [Google Scholar]
- 63.Medema JP, de Jong J, Peltenburg LTC, et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(20):11515–11520. doi: 10.1073/pnas.201398198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Houdt IS, Oudejans JJ, van den Eertwegh AJM, et al. Expression of the apoptosis inhibitor protease inhibitor 9 predicts clinical outcome in vaccinated patients with stage III and IV melanoma. Clinical Cancer Research. 2005;11(17):6400–6407. doi: 10.1158/1078-0432.CCR-05-0306. [DOI] [PubMed] [Google Scholar]
- 65.Mahmood Z, Shukla Y. Death receptors: targets for cancer therapy. Experimental Cell Research. 2010;316(6):887–899. doi: 10.1016/j.yexcr.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 66.Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. Journal of Immunology. 1998;161(6):2833–2840. [PubMed] [Google Scholar]
- 67.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. Journal of Biological Chemistry. 1999;274(3):1541–1548. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- 68.Nam SY, Jung GA, Hur GC, et al. Upregulation of FLIPs by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Science. 2003;94(12):1066–1073. doi: 10.1111/j.1349-7006.2003.tb01402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elnemr A, Ohta T, Yachie A, et al. Human pancreatic cancer cells disable function of Fas receptors at several levels in Fas signal transduction pathway. International Journal of Oncology. 2001;18(2):311–316. doi: 10.3892/ijo.18.2.311. [DOI] [PubMed] [Google Scholar]
- 70.Xiao CW, Yan X, Li Y, Reddy SAG, Tsang BK. Resistance of human ovarian cancer cells to tumor necrosis factor α is a consequence of nuclear factor κB-mediated induction of Fas-associated death domain-like interleukin-1β-converting enzyme-like inhibitory protein. Endocrinology. 2003;144(2):623–630. doi: 10.1210/en.2001-211024. [DOI] [PubMed] [Google Scholar]
- 71.Zhang X, Jin TG, Yang H, Dewolf WC, Khosravi-Far R, Olumi AF. Persistent c-FLIP(L) expression is necessary and sufficient to maintain resistance to tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in prostate cancer. Cancer Research. 2004;64(19):7086–7091. doi: 10.1158/0008-5472.CAN-04-1498. [DOI] [PubMed] [Google Scholar]
- 72.Ullenhag GJ, Mukherjee A, Watson NFS, Al-Attar AH, Scholefield JH, Durrant LG. Overexpression of FLIPL is an independent marker of poor prognosis in colorectal cancer patients. Clinical Cancer Research. 2007;13(17):5070–5075. doi: 10.1158/1078-0432.CCR-06-2547. [DOI] [PubMed] [Google Scholar]
- 73.Peli J, Schröter M, Rudaz C, et al. Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO Journal. 1999;18(7):1824–1831. doi: 10.1093/emboj/18.7.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Volkmann M, Schiff JH, Hajjar Y, et al. Loss of CD95 expression is linked to most but not all p53 mutants in European hepatocellular carcinoma. Journal of Molecular Medicine. 2001;79(10):594–600. doi: 10.1007/s001090100244. [DOI] [PubMed] [Google Scholar]
- 75.Moller P, Koretz K, Leithauser F, et al. Expression of APO-1 (CD95), a member of the NGF/TNF receptor superfamily, in normal and neoplastic colon epithelium. International Journal of Cancer. 1994;57(3):371–377. doi: 10.1002/ijc.2910570314. [DOI] [PubMed] [Google Scholar]
- 76.Landowski TH, Qu N, Buyuksal I, Painter JS, Dalton WS. Mutations in the Fas antigen in patients with multiple myeloma. Blood. 1997;90(11):4266–4270. [PubMed] [Google Scholar]
- 77.Maeda T, Yamada Y, Moriuchi R, et al. Fas gene mutation in the progression of adult T cell leukemia. Journal of Experimental Medicine. 1999;189(7):1063–1071. doi: 10.1084/jem.189.7.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park WS, Oh RR, Kim YS, et al. Somatic mutations in the death domain of the Fas (Apo-I/CD95) gene in gastric cancer. Journal of Pathology. 2001;193(2):162–168. doi: 10.1002/1096-9896(2000)9999:9999<::aid-path759>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 79.Shin MS, Kim HS, Lee SH, et al. Mutations of tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and receptor 2 (TRAIL-R2) genes in metastatic breast cancers. Cancer Research. 2001;61(13):4942–4946. [PubMed] [Google Scholar]
- 80.Cheng J, Zhou T, Liu C, et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263(5154):1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 81.Midis GP, Shen Y, Owen-Schaub LB. Elevated soluble Fas (sFas) levels in nonhematopoietic human malignancy. Cancer Research. 1996;56(17):3870–3874. [PubMed] [Google Scholar]
- 82.Ugurel S, Rappl G, Tilgen W, Reinhold U. Increased soluble CD95 (sFas/CD95) serum level correlates with poor prognosis in melanoma patients. Clinical Cancer Research. 2001;7(5):1282–1286. [PubMed] [Google Scholar]
- 83.Pitti RM, Marsters SA, Lawrence DA, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396(6712):699–703. doi: 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 84.Roth W, Isenmann S, Nakamura M, et al. Soluble decoy receptor 3 is expressed by malignant gliomas and suppresses CD95 ligand-induced apoptosis and chemotaxis. Cancer Research. 2001;61(6):2759–2765. [PubMed] [Google Scholar]
- 85.Sheikh MS, Huang Y, Fernandez-Salas EA, et al. The antiapoptotic decoy receptor TRID/TRAIL-R3 is a p53-regulated DNA damage-inducible gene that is overexpressed in primary tumors of the gastrointestinal tract. Oncogene. 1999;18(28):4153–4159. doi: 10.1038/sj.onc.1202763. [DOI] [PubMed] [Google Scholar]
- 86.Meng RD, McDonald ER, III, Sheikh MS, Fornace AJ, Jr., El-Deiry WS. The TRAIL decoy receptor TRUNDD (DcR2, TRAIL-R4) is induced by adenovirus-p53 overexpression and can delay TRAIL-, p53-, and KILLER/DR5-dependent colon cancer apoptosis. Molecular Therapy. 2000;1(2):130–144. doi: 10.1006/mthe.2000.0025. [DOI] [PubMed] [Google Scholar]
- 87.Fisher JL, Thomas-Mudge RJ, Elliott J, et al. Osteoprotegerin overexpression by breast cancer cells enhances orthotopic and osseous tumor growth and contrasts with that delivered therapeutically. Cancer Research. 2006;66(7):3620–3628. doi: 10.1158/0008-5472.CAN-05-3119. [DOI] [PubMed] [Google Scholar]
- 88.Holen I, Croucher PI, Hamdy FC, Eaton CL. Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Research. 2002;62(6):1619–1623. [PubMed] [Google Scholar]
- 89.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nature Reviews Immunology. 2008;8(6):467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 90.Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17(2):201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 91.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and Cellular Biology. 2005;25(21):9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clinical Cancer Research. 2007;13(18):5238–5242. doi: 10.1158/1078-0432.CCR-07-0813. [DOI] [PubMed] [Google Scholar]
- 93.Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Research. 2004;64(3):1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 94.Hamanishi J, Mandai M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(9):3360–3365. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ohigashi Y, Sho M, Yamada Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clinical Cancer Research. 2005;11(8):2947–2953. doi: 10.1158/1078-0432.CCR-04-1469. [DOI] [PubMed] [Google Scholar]
- 96.Nakanishi J, Wada Y, Matsumoto K, Azuma M, Kikuchi K, Ueda S. Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunology, Immunotherapy. 2007;56(8):1173–1182. doi: 10.1007/s00262-006-0266-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gadiot J, Hooijkaas AI, Kaiser ADM, Van Tinteren H, Van Boven H, Blank C. Overall survival and PD-L1 expression in metastasized malignant melanoma. Cancer. 2011;117(10):2192–2201. doi: 10.1002/cncr.25747. [DOI] [PubMed] [Google Scholar]
- 98.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature Medicine. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 99.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. Journal of Immunology. 2004;173(2):945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 100.Frigola X, Inman BA, Lohse CM, et al. Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clinical Cancer Research. 2011;17(7):1915–1923. doi: 10.1158/1078-0432.CCR-10-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kehrl JH, Wakefield LM, Roberts AB, et al. Production of transforming growth factor β by human T lymphocytes and its potential role in the regulation of T cell growth. Journal of Experimental Medicine. 1986;163(5):1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ranges GE, Figari IS, Espevik T, Palladino MA., Jr. Inhibition of cytotoxic T cell development by transforming growth factor β and reversal by recombinant tumor necrosis factor α. Journal of Experimental Medicine. 1987;166(4):991–998. doi: 10.1084/jem.166.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Robson H, Anderson E, James RD, Schofield PF. Transforming growth factor β1 expression in human colorectal tumours: an independent prognostic marker in a subgroup of poor prognosis patients. British Journal of Cancer. 1996;74(5):753–758. doi: 10.1038/bjc.1996.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.von Rahden BHA, Stein HJ, Feith M, et al. Overexpression of TGF-β1 in esophageal (Barrett’s) adenocarcinoma is associated with advanced stage of disease and poor prognosis. Molecular Carcinogenesis. 2006;45(10):786–794. doi: 10.1002/mc.20259. [DOI] [PubMed] [Google Scholar]
- 105.Bodmer S, Strommer K, Frei K, et al. Immunosuppression and transforming growth factor-β in glioblastoma. Preferential production of transforming growth factor-β2. Journal of Immunology. 1989;143(10):3222–3229. [PubMed] [Google Scholar]
- 106.Walker RA, Dearing SJ. Transforming growth factor beta1 in ductal carcinoma in situ and invasive carcinomas of the breast. European Journal of Cancer. 1992;28(2-3):641–644. doi: 10.1016/s0959-8049(05)80116-9. [DOI] [PubMed] [Google Scholar]
- 107.Hasegawa Y, Takanashi S, Kanehira Y, Tsushima T, Imai T, Okumura K. Transforming growth factor-β1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer. 2001;91(5):964–971. [PubMed] [Google Scholar]
- 108.di Bari MG, Lutsiak ME, Takai S, et al. TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunology, Immunotherapy. 2009;58(11):1809–1818. doi: 10.1007/s00262-009-0692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thomas DA, Massagué J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 110.Walker MR, Kasprowicz DJ, Gersuk VH, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25− T cells. Journal of Clinical Investigation. 2003;112(9):1437–1443. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Greenhough A, Smartt HJM, Moore AE, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30(3):377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 112.Pockaj BA, Basu GD, Pathangey LB, et al. Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Annals of Surgical Oncology. 2004;11(3):328–339. doi: 10.1245/aso.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 113.Baratelli F, Lin Y, Zhu L, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. Journal of Immunology. 2005;175(3):1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 114.Sharma S, Zhu L, Yang SC, et al. Cyclooxygenase 2 inhibition promotes IFN-γ-dependent enhancement of antitumor responses. Journal of Immunology. 2005;175(2):813–819. doi: 10.4049/jimmunol.175.2.813. [DOI] [PubMed] [Google Scholar]
- 115.Adams L, Scott GK, Weinberg CS. Biphasic modulation of cell growth by recombinant human galectin-1. Biochimica et Biophysica Acta—Molecular Cell Research. 1996;1312(2):137–144. doi: 10.1016/0167-4889(96)00031-6. [DOI] [PubMed] [Google Scholar]
- 116.Matsumoto R, Matsumoto H, Seki M, et al. Human ecalectin, a variant of human galectin-9, is a novel eosinophil chemoattractant produced by T lymphocytes. Journal of Biological Chemistry. 1998;273(27):16976–16984. doi: 10.1074/jbc.273.27.16976. [DOI] [PubMed] [Google Scholar]
- 117.Yamaoka K, Ingendoh A, Tsubuki S, Nagai Y, Sanai Y. Structural and functional characterization of a novel tumor-derived rat galectin-1 having transforming growth factor (TGF) activity: the relationship between intramolecular disulfide bridges and TGF activity. Journal of Biochemistry. 1996;119(5):878–886. doi: 10.1093/oxfordjournals.jbchem.a021325. [DOI] [PubMed] [Google Scholar]
- 118.Mourad-Zeidan AA, Melnikova VO, Wang H, Raz A, Bar-Eli M. Expression profiling of galectin-3-depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. American Journal of Pathology. 2008;173(6):1839–1852. doi: 10.2353/ajpath.2008.080380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rorive S, Belot N, Decaestecker C, et al. Galectin-1 is highly expressed in human gliomas with relevance for modulation of invasion of tumor astrocytes into the brain parenchyma. GLIA. 2001;33(3):241–255. doi: 10.1002/1098-1136(200103)33:3<241::aid-glia1023>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 120.van den Brûle FA, Waltregny D, Castronovo V. Increased expression of galectin-I in carcinoma-associated stroma predicts poor outcome in prostate carcinoma patients. Journal of Pathology. 2001;193(1):80–87. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH730>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 121.Cindolo L, Benvenuto G, Salavatore P, et al. Galectin-1 and galectin-3 expression in human bladder transitional-cell carcinomas. International Journal of Cancer. 1999;84(1):39–43. doi: 10.1002/(sici)1097-0215(19990219)84:1<39::aid-ijc8>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 122.Van den Brûle F, Califice S, Garnier F, Fernandez PL, Berchuck A, Castronovo V. Galectin-1 accumulation in the ovary carcinoma peritumoral stroma is induced by ovary carcinoma cells and affects both cancer cell proliferation and adhesion to laminin-1 and fibronectin. Laboratory Investigation. 2003;83(3):377–386. doi: 10.1097/01.lab.0000059949.01480.40. [DOI] [PubMed] [Google Scholar]
- 123.Jung EJ, Moon HG, Bok IC, et al. Galectin-1 expression in cancer-associated stromal cells correlates tumor invasiveness and tumor progression in breast cancer. International Journal of Cancer. 2007;120(11):2331–2338. doi: 10.1002/ijc.22434. [DOI] [PubMed] [Google Scholar]
- 124.Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378(6558):736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 125.Rabinovich GA, Iglesias MM, Modesti NM, et al. Activated rat macrophages produce a galectin-1-like protein that induces apoptosis of T cells: biochemical and functional characterization. Journal of Immunology. 1998;160(10):4831–4840. [PubMed] [Google Scholar]
- 126.Chung CD, Patel VP, Moran M, Lewis LA, Miceli MC. Galectin-1 induces partial TCR ζ-chain phosphorylation and antagonizes processive TCR signal transduction. Journal of Immunology. 2000;165(7):3722–3729. doi: 10.4049/jimmunol.165.7.3722. [DOI] [PubMed] [Google Scholar]
- 127.Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunological Reviews. 2003;193:70–81. doi: 10.1034/j.1600-065x.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 128.Hahne M, Rimoldi D, Schröter M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274(5291):1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 129.Niehans GA, Brunner T, Frizelle SP, et al. Human lung carcinomas express Fas ligand. Cancer Research. 1997;57(6):1007–1012. [PubMed] [Google Scholar]
- 130.Bernstorff WV, Glickman JN, Odze RD, et al. Fas (CD95/APO-1) and Fas ligand expression in normal pancreas and pancreatic tumors: implications for immune privilege and immune escape. Cancer. 2002;94(10):2552–2560. doi: 10.1002/cncr.10549. [DOI] [PubMed] [Google Scholar]
- 131.Müllauer L, Mosberger I, Grusch M, Rudas M, Chott A. Fas ligand is expressed in normal breast epithelial cells and is frequently up-regulated in breast cancer. Journal of Pathology. 2000;190(1):20–30. doi: 10.1002/(SICI)1096-9896(200001)190:1<20::AID-PATH497>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 132.Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of Fas ligand induces tumor regression in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(25):13862–13867. doi: 10.1073/pnas.94.25.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kang SM, Lin Z, Ascher NL, Stock PG. Fas ligand expression on islets as well as multiple cell lines results in accelerated neutrophilic rejection. Transplantation Proceedings. 1998;30(2):p. 538. doi: 10.1016/s0041-1345(97)01396-1. [DOI] [PubMed] [Google Scholar]
- 134.Drozdzik M, Qian C, Lasarte JJ, Bilbao R, Prieto J. Antitumor effect of allogenic fibroblasts engineered to express Fas ligand (FasL) Gene Therapy. 1998;5(12):1622–1630. doi: 10.1038/sj.gt.3300763. [DOI] [PubMed] [Google Scholar]
- 135.Ryan AE, Shanahan F, O’Connell J, Houston AM. Addressing the "Fas counterattack" controversy: blocking fas ligand expression suppresses tumor immune evasion of colon cancer in vivo. Cancer Research. 2005;65(21):9817–9823. doi: 10.1158/0008-5472.CAN-05-1462. [DOI] [PubMed] [Google Scholar]
- 136.Andreola G, Rivoltini L, Castelli C, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. Journal of Experimental Medicine. 2002;195(10):1303–1316. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Iero M, Valenti R, Huber V, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death and Differentiation. 2008;15(1):80–88. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 138.Shiraki K, Yamanaka T, Inoue H, et al. Expression of TNF-related apoptosis-inducing ligand in human hepatocellular carcinoma. International Journal of Oncology. 2005;26(5):1273–1281. [PubMed] [Google Scholar]
- 139.Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 140.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. Journal of Experimental Medicine. 2002;196(4):459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Löb S, Königsrainer A, Zieker D, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunology, Immunotherapy. 2009;58(1):153–157. doi: 10.1007/s00262-008-0513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+CD25+CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunological Reviews. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 143.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunological Reviews. 2008;224(1):166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 144.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 145.Schaefer C, Kim GG, Albers A, Hoermann K, Myers EN, Whiteside TL. Characteristics of CD4+CD25+ regulatory T cells in the peripheral circulation of patients with head and neck cancer. British Journal of Cancer. 2005;92(5):913–920. doi: 10.1038/sj.bjc.6602407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clinical Cancer Research. 2003;9(2):606–612. [PubMed] [Google Scholar]
- 147.Yannelli JR, Tucker JA, Hidalgo G, Perkins S, Kryscio R, Hirschowitz EA. Characteristics of PBMC obtained from leukapheresis products and tumor biopsies of patients with non-small cell lung cancer. Oncology Reports. 2009;22(6):1459–1471. doi: 10.3892/or_00000588. [DOI] [PubMed] [Google Scholar]
- 148.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of Foxp3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clinical Cancer Research. 2006;12(18):5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 149.Perez SA, Karamouzis MV, Skarlos DV, et al. CD4+CD25+ regulatory T-cell frequency in HER-2/neu (HER)-positive and HER-negative advanced-stage breast cancer patients. Clinical Cancer Research. 2007;13(9):2714–2721. doi: 10.1158/1078-0432.CCR-06-2347. [DOI] [PubMed] [Google Scholar]
- 150.Ormandy L, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Research. 2005;65(6):2457–2464. doi: 10.1158/0008-5472.CAN-04-3232. [DOI] [PubMed] [Google Scholar]
- 151.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Medicine. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 152.Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clinical Cancer Research. 2003;9(12):4404–4408. [PubMed] [Google Scholar]
- 153.Viguier M, Lemaître F, Verola O, et al. Foxp3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. Journal of Immunology. 2004;173(2):1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 154.Kobayashi N, Hiraoka N, Yamagami W, et al. Foxp3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clinical Cancer Research. 2007;13(3):902–911. doi: 10.1158/1078-0432.CCR-06-2363. [DOI] [PubMed] [Google Scholar]
- 155.Ishida T, Ishii T, Inagaki A, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Research. 2006;66(11):5716–5722. doi: 10.1158/0008-5472.CAN-06-0261. [DOI] [PubMed] [Google Scholar]
- 156.Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clinical Cancer Research. 2000;6(5):1755–1766. [PubMed] [Google Scholar]
- 157.Rodriguez PC, Hernandez CP, Morrow K, et al. L-arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. Journal of Immunology. 2010;185(9):5198–5204. doi: 10.4049/jimmunol.1001224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Research. 2001;61(12):4756–4760. [PubMed] [Google Scholar]
- 159.Mueller MM, Fusenig NE. Constitutive expression of G-CSF and GM-CSF in human skin carcinoma cells with functional consequence for tumor progression. International Journal of Cancer. 1999;83(6):780–789. doi: 10.1002/(sici)1097-0215(19991210)83:6<780::aid-ijc14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 160.Rochet N, Dubousset J, Mazeau C, et al. Establishment, characterisation and partial cytokine expression profile of a new human osteosarcoma cell line (CAL 72) International Journal of Cancer. 1999;82(2):282–285. doi: 10.1002/(sici)1097-0215(19990719)82:2<282::aid-ijc20>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 161.Wislez M, Fleury-Feith J, Rabbe N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor prolong the survival of neutrophils infiltrating bronchoalveolar subtype pulmonary adenocarcinoma. American Journal of Pathology. 2001;159(4):1423–1433. doi: 10.1016/S0002-9440(10)62529-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bronte V, Chappell DB, Apolloni E, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. Journal of Immunology. 1999;162(10):5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 163.Nagaraj S, Gupta K, Pisarev V, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Medicine. 2007;13(7):828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tuyaerts S, Aerts JL, Corthals J, et al. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunology, Immunotherapy. 2007;56(10):1513–1537. doi: 10.1007/s00262-007-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang L, Carbone DP. Tumor-host immune interactions and dendritic cell dysfunction. Advances in Cancer Research. 2004;92:13–27. doi: 10.1016/S0065-230X(04)92002-7. [DOI] [PubMed] [Google Scholar]
- 166.Molenkamp BG, Sluijter BJR, Van Leeuwen PAM, et al. Local administration of PF-3512676 CpG-B instigates tumor-specific CD8+ T-cell reactivity in melanoma patients. Clinical Cancer Research. 2008;14(14):4532–4542. doi: 10.1158/1078-0432.CCR-07-4711. [DOI] [PubMed] [Google Scholar]
- 167.Papadavid E, Stratigos AJ, Falagas ME. Imiquimod: an immune response modifier in the treatment of precancerous skin lesions and skin cancer. Expert Opinion on Pharmacotherapy. 2007;8(11):1743–1755. doi: 10.1517/14656566.8.11.1743. [DOI] [PubMed] [Google Scholar]
- 168.Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clinical Cancer Research. 2011;17(6):1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Di LG, Buonerba C, Kantoff PW. Immunotherapy for the treatment of prostate cancer. Nature Reviews Clinical Oncology. 2011;8:551–561. doi: 10.1038/nrclinonc.2011.72. [DOI] [PubMed] [Google Scholar]
- 170.Foss F. Clinical experience with denileukin diftitox (ONTAK) Seminars in Oncology. 2006;33(3):S11–S16. doi: 10.1053/j.seminoncol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 171.Morse MA, Hobeika AC, Osada T, et al. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood. 2008;112(3):610–618. doi: 10.1182/blood-2008-01-135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.June CH. Principles of adoptive T cell cancer therapy. Journal of Clinical Investigation. 2007;117(5):1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dudley ME, Wunderlich J, Nishimura MI, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. Journal of Immunotherapy. 2001;24(4):363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 174.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Journal of Clinical Oncology. 2005;23(10):2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Current Opinion in Immunology. 2006;18(2):206–213. doi: 10.1016/j.coi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 178.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(19):12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. Journal of Clinical Oncology. 2010;28(19):3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kwon ED, Foster BA, Hurwitz AA, et al. Elimination of residual metastatic prostate cancer after surgery and adjunctive cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blockade immunotherapy. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(26):15074–15079. doi: 10.1073/pnas.96.26.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 182.Sarnaik AA, Weber JS. Recent advances using anti-CTLA-4 for the treatment of melanoma. Cancer Journal. 2009;15(3):169–173. doi: 10.1097/PPO.0b013e3181a7450f. [DOI] [PubMed] [Google Scholar]
- 183.Blansfield JA, Beck KE, Tran K, et al. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. Journal of Immunotherapy. 2005;28(6):593–598. doi: 10.1097/01.cji.0000178913.41256.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. Journal of Clinical Oncology. 2006;24(15):2283–2289. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. Journal of Immunology. 2002;169(9):4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 187.Stärck L, Scholz C, Dörken B, Daniel PT. Costimulation by CD137/4-1BB inhibits T cell apoptosis and induces Bcl-xL and c-FLIPshort via phosphatidylinositol 3-kinase and AKT/protein kinase B. European Journal of Immunology. 2005;35(4):1257–1266. doi: 10.1002/eji.200425686. [DOI] [PubMed] [Google Scholar]
- 188.Tomillero A, Moral MA. Gateways to clinical trials. Methods and Findings in Experimental and Clinical Pharmacology. 2008;30(7):543–588. [PubMed] [Google Scholar]
- 189.Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annual Review of Immunology. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Weinberg AD, Rivera MM, Prell R, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. Journal of Immunology. 2000;164(4):2160–2169. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 191.Jacobs JFM, Punt CJA, Lesterhuis WJ, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clinical Cancer Research. 2010;16(20):5067–5078. doi: 10.1158/1078-0432.CCR-10-1757. [DOI] [PubMed] [Google Scholar]
- 192.Coe D, Begom S, Addey C, White M, Dyson J, Chai JG. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunology, Immunotherapy. 2010;59(9):1367–1377. doi: 10.1007/s00262-010-0866-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Moore KW, de Waal MR, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annual Review of Immunology. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 194.Hau P, Jachimczak P, Schlingensiepen R, et al. Inhibition of TGF-β2 with ap 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17(2):201–212. doi: 10.1089/oli.2006.0053. [DOI] [PubMed] [Google Scholar]
- 195.Bogdahn U, Hau P, Stockhammer G, et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro-Oncology. 2011;13(1):132–142. doi: 10.1093/neuonc/noq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.DeLong P, Tanaka T, Kruklitis R, et al. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Research. 2003;63(22):7845–7852. [PubMed] [Google Scholar]
- 197.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bachmann M, Cartellieri M, Feldmann A, et al. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. Journal of Biomedicine and Biotechnology. 2010;2010 doi: 10.1155/2010/956304. Article ID 956304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Park JR, DiGiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Molecular Therapy. 2007;15(4):825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 202.Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112(6):2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hombach A, Sent D, Schneider C, et al. T-cell activation by recombinant receptors: CD28 costimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Research. 2001;61(5):1976–1982. [PubMed] [Google Scholar]
- 205.Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Molecular Therapy. 2005;12(5):933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 206.Zhao Y, Wang QJ, Yang S, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. Journal of Immunology. 2009;183(9):5563–5574. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. Journal of Immunology. 2001;167(11):6356–6365. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Charo J, Finkelstein SE, Grewal N, Restifo NP, Robbins PF, Rosenberg SA. Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer Research. 2005;65(5):2001–2008. doi: 10.1158/0008-5472.CAN-04-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Di Stasi A, De Angelis B, Rooney CM, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–6402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Research. 2004;64(14):4973–4979. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 211.Arlen PM, Pazdur M, Skarupa L, Rauckhorst M, Gulley JL. A ramdomized phase II study of docetaxel alone or in combination with PANVAC-V (Vaccinia) and PANVAC-F (Fowlpox) in patients with metastatic breast cancer (NCI 05-C-0229) Clinical Breast Cancer. 2006;7(2):176–179. doi: 10.3816/CBC.2006.n.032. [DOI] [PubMed] [Google Scholar]
- 212.Oh S, Terabe M, Pendleton CD, et al. Human CTLs to wild-type and enhanced epitopes of a novel prostate and breast tumor-associated protein, TARP, lyse human breast cancer cells. Cancer Research. 2004;64(7):2610–2618. doi: 10.1158/0008-5472.can-03-2183. [DOI] [PubMed] [Google Scholar]
- 213.Kirkwood JM, Lee S, Moschos S, et al. Immunogenicity and antitumor effects of vaccination with peptide vaccine +/- granulocyte-monocyte colony-stimulating factor and/or IFIN-α2b in advanced metastatic melanoma: eastern cooperative oncology group phase II trial E1696. Clinical Cancer Research. 2009;15(4):1443–1451. doi: 10.1158/1078-0432.CCR-08-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and montanide ISA 51 for patients with resected stages III and IV melanoma. Journal of Clinical Oncology. 2005;23(4):741–750. doi: 10.1200/JCO.2005.01.128. [DOI] [PubMed] [Google Scholar]
- 215.Rousseau RF, Biagi E, Dutour A, et al. Immunotherapy of high-risk acute leukemia with a recipient (autologous) vaccine expressing transgenic human CD40L and IL-2 after chemotherapy and allogeneic stem cell transplantation. Blood. 2006;107(4):1332–1341. doi: 10.1182/blood-2005-03-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Atchison E, Eklund J, Martone B, et al. A pilot study of denileukin diftitox (DD) in combination with high-dose interleukin-2 (IL-2) for patients with metastatic renal cell carcinoma (RCC) Journal of Immunotherapy. 2010;33(7):716–722. doi: 10.1097/CJI.0b013e3181e4752e. [DOI] [PubMed] [Google Scholar]