

Abstract
Objective
Chromosomal loss within the region of 18q and loss of SMAD4 expression have been reported to be frequent somatic events during colorectal cancer tumour progression; however, their associations with age at onset have not been widely studied.
Method
We analysed 109 tumours from a population-based case-family study based on colorectal cancers diagnosed before the age of 45 years. These patients with early-onset colorectal cancer had been previously screened for germ-line mismatch repair gene mutations, microsatellite instability (that included the mononucleotide repeat in TGFβRII) and somatic k-ras mutations. We measured SMAD4 protein expression using immunohistochemistry and SMAD4 copy number using quantitative real-time PCR.
Results
Loss of SMAD4 protein expression was observed in 27/109 (25%) of cancers tested and was more commonly observed in rectal tumours (15/41, 36%) when compared with tumours arising in the colon (11/66, 17%) (P = 0.04). There was no association between SMAD4 protein expression and TGFβR11 mutation status, SMAD4 copy number, family history, MSI status, tumour stage or grade.
Conclusion
Loss of SMAD4 expression is a common feature of early-onset colorectal tumours as it is in colorectal cancers diagnosed in other age-groups. Taken together, the molecular pathways (genetic and epigenetic) now known to be involved in early-onset colorectal cancer only explain a small proportion of the disease and require further exploration.
Keywords: Early-onset colorectal cancer, SMAD4, 18q, copy number
Introduction
A large number of studies have investigated the frequency, histopathological and clinical features and outcomes of colorectal cancers that have acquired chromosomal loss in the 18q region. Several studies have focussed on SMAD4 (on 18q21), and examined loss of expression, or change in copy number of this gene during tumourigenesis [1–3]. The SMAD family of genes is involved in mediating cellular responses and plays an essential role in the downstream conduction of the transforming growth factor-beta (TGF-β) signalling pathway [4]. Reports of loss of SMAD4 protein expression in colorectal cancer range from 9% to 66% (9.3% [5], 14% [6], 66% [7]). Recent studies have reported loss of, or reduced level of, SMAD4 expression to be more commonly observed in poorly differentiated carcinoma and signet-ring cell carcinoma (31% and 80% respectively) [1], to be associated with reduced disease-free years and overall survival after surgery with [3] and without 5-FU-based adjuvant treatment [2] and to be associated with the process of colorectal cancer metastasis to the liver [8]. These reports are typically from small studies, and from studies of colorectal cancer unselected for age at diagnosis (therefore mostly late age of onset). There have been no specific reports of the role of SMAD4 protein expression or SMAD4 copy number in early-onset disease.
In a population-based study of early-onset colorectal cancer (diagnosed before the age of 45 years) conducted in Victoria, Australia, we have previously reported the prevalence of mismatch repair (MMR) gene germ-line mutations to be 17%. Only a small proportion (3%) had hMLH1 silencing via a mechanism other than germ-line mutation [9,10] and the frequency of somatic K-ras mutations was small [11]. The molecular events that predispose to and progress the majority of early-onset colorectal cancers are yet to be well-described. SMAD4 can act as a tumour suppressor gene, via interruption of the TGF-β signalling pathway and its loss can be a marker for loss of heterozygosity at chromosome 18q, which is a common feature of the multistep pathway of accumulated genetic mutations and gene silencing described for colorectal cancer. We sought to examine the possibility that disruption to the SMAD4/TGF-β signalling pathway was a common event in early-onset colorectal cancer by assessing SMAD4 protein expression (via immunohistochemistry) in 109 early-onset colorectal cancers and SMAD4 copy number (via real-time PCR methodology) in a subset of these cancers selected for SMAD4 protein expression status.
Method
Subjects
The Victorian Colorectal Cancer Family Study (VCCFS) is a population-based, case-family study of early-onset colorectal cancer, conducted from 1993 to 1997 on 131 patients of histologically verified primary adenocarcinomas of the colon or rectum registered on the Victorian Cancer Registry [9,12]. The case-probands (all diagnosed between 18 and 44 years of age) consisted of 66 female- and 65 male patients, with a mean age at diagnosis of 39.5 years (SD = 4.7 years). Case-probands and some of their living relatives were asked to provide a blood sample and if diagnosed with cancer sign an informed consent to allow access to their tumour tissues from pathology laboratories for use in molecular analyses.
Clinicopathological data was also collected. The site of primary tumour was found to be the right side of the colon in 33% of the patients with early-onset colorectal cancer, at the splenic flexure (2%), in the left side of the colon (27%) and in the rectum (38%). Eleven per cent of the tumours were well-differentiated, 60% were moderately differentiated and 29% were poorly differentiated. The American Joint Committee on Cancer stage was stage I, II, III and IV in 18%, 43% 31% and 9% of these cancers respectively. Tumours proximal to the splenic flexure were classified as right colon, those distal to the splenic flexure as left colon, and tumours arising distal to the peritoneal reflection were classified as rectal cancers.
The study was approved by the ethics committees of The University of Melbourne and The Cancer Council Victoria.
Tumour specimens
Samples of invasive tumours from primary colorectal adenocarcinoma were obtained from hospitals and private pathology laboratories for 118 case probands (90%). Six case-probands did not consent to release of tissue to the study and two laboratories had not agreed to release the remaining seven samples at the time of this study [9,10]. Nine tumour specimens were found to be unsuitable or insufficient for IHC studies resulting in 109 specimens being included (83%).
Previous molecular characterization
Testing for tumour microsatellite instability (MSI) using a 10-microsatellite marker panel that included the mononucleotide repeat in TGFβRII, testing for germ-line MMR gene mutations and testing for tumour expression of MMR genes in these specimens from the VCCFS has been reported previously [9,10].
Immunohistochemistry
Immunohistochemistry for SMAD4 was performed on 3-μm sections (coated slides) of archived invasive carcinoma tissue from 109 participants. Sections were deparaffinized in three 5-min changes of xylene, followed by a series of alcohol changes and re-hydrated in tap-water. Antigen retrieval included submitting slides to 30 min at pressure in 0.1 M citrate buffer (pH 6.0), before the same being incubated with monoclonal antibody B-8 to full length SMAD4 protein (Santa Cruz Biotechnology, Santa Cruz, California, USA) for 120 min. Streptavidin horseradish peroxidase detection reagents (LSAB+ system; Dako, Carpinteria, California, USA) and the chromagen 3-3′diaminobenzidine (DAKO) were used to detect sites of bound antibody. Sections were counterstained with haematoxylin. Immunohistochemical procedures were performed using the DAKO Autostainer (DAKO Corporation; Serial No. DC 3400-6912-03). Presence and absence of SMAD4 protein expression was scored after being observed under a light microscope. SMAD4 nuclear staining was classified as positive, while absence of nuclear staining, or cytoplasmic staining only was scored as negative.
SMAD4 copy number
All participants that presented negative SMAD4 staining via immunohistochemistry (n = 27) were analysed for SMAD4 gene copy number, as well as 29 randomly selected participants that were found to express SMAD4 via immunohistochemistry. The copy number of the SMAD4 gene was determined using quantitative (real-time) PCR, as per Boulay et al. (1999) [14]. Primer sequences for the SMAD4 gene (5′: gcagacagaaactggattaaaacaatt; 3′: gaatgtgtttctcctaatcttcaagct), and 36B4 (autosomal reference gene) (5′: agcaagtgggaaggtgtaatcc; 3′: ccattctatcatcaacgggtacaa) were derived after correspondence with the authors, ordered from GeneWorks and re-hydrated in 1 × TE buffer to 100 uM. Probes were TaqMan, with FAM fluorescent dyes and TAMRA quencher dyes (SMAD4: 6FAMtcctcttcagaacttgtcaggcatggctcTAMRA; 36B4: 6FAMtctccacagacaaggccaggactcgTAMRA) Micro-dissected tumour-enriched and normal DNA (prepared as previously described [13]) and genomic DNA (from peripheral blood, where available) were analysed. Each reaction was performed in triplicate on a RotorGene 2000 Real-time Cycler (Corbett Research, Sydney, Australia) (60°C hold for 2 min; 94°C hold for 10 min; followed by 45 cycles of 95°C for 15 s; 60°C for 60 s. Signals were acquired at 60°C).
To determine the gene copy number, the cycle threshold (Ct) for each of the reactions was calculated by the software. The Ct is defined as the threshold cycle; the fractional cycle number at which the fluorescence emitted is greater than the fixed threshold [14]. To determine the fixed threshold for each of the genes, a standard curve was determined using a control genomic DNA (15 μl/ml) as a template. For an internal control (to monitor and compare the efficiency of reactions), a 1:1 control DNA sample was included in each run and compared with the values given on the standard curve.
The Ct of the SMAD4 gene from the histologically normal micro-dissected tissue of a participant was subtracted from the Ct of the SMAD4 gene from the tumour-enriched tissue taken from the same individual (ΔCtSMAD4). A similar calculation was made for the Ct values of the 36B4 gene (ΔCt36B4). To make adjustments caused by the differing DNA concentrations, ΔCtSMAD4 was subtracted from ΔCt36B4, giving ΔCt. A ΔCt between −0.45 and 0.45 was interpreted to indicate no change in the gene copy number (2n). Following Boulay et al., 2002, a ΔCt less than −0.55 was interpreted to indicate gene loss (n, or zero n), and a ΔCt greater than 0.55 was interpreted to indicate a possible gain of a gene copy (>2n) [15].
Results
SMAD4 protein expression in early-onset colorectal cancer
Table 1 shows that, of the 109 early-onset colorectal cancers tested, 82 (75%) showed staining for SMAD4 protein throughout the normal colonic epithelium and tumour tissue via immunohistochemical analysis. Six of these 82 patients with early-onset colorectal cancer (7%) showed focal positive and negative staining suggesting clonal differences in SMAD4 expression. Twenty-seven patients with early-onset colorectal cancer (25%) were negative for SMAD4 protein expression (Fig. 1). Lack of SMAD4 protein expression in tumour specimens was found to be related to tumour site and was more common in rectal than colonic carcinomas (P = 0.04), but was not associated with tumour stage or degree of tumour differentiation.
Table 1.
Loss of SMAD4 protein expression in early-onset colorectal cancers.
| Total | SMAD4 staining (%)
|
||
|---|---|---|---|
| Positive | Negative | ||
| Tumour site | |||
| colonic tumours | 66 | 55 (83) | 11 (17) |
| Right colon | 35 | 30 (86) | 5 (14) |
| Splenic flexure | 2 | 1 (50) | 1 (50) |
| Left colon | 29 | 24 (83) | 5 (17) |
| Rectal tumours | 41 | 26 (63) | 15 (36)* |
| Site unknown | 2 | 1 | 1 |
| Total tumours | 109 | 82 (75) | 27 (25) |
| TGFβR11 | |||
| Frameshift mutation | 21 | 18 (86) | 3 (14) |
| No mutations | 82 | 61 (74) | 21 (26) |
| No data | 6 | 3 | 3 |
| Total | 109 | 82 | 27 |
| SMAD4 gene copy number | |||
| Loss of copy number | 27 | 14 (51) | 13 (48) |
| No change | 21 | 9 (42) | 12 (544) |
| Gain of copy number | 4 | 4 (100) | 0 |
| No data | 4 | 2 (50) | 2 (50) |
| Total | 56 | 29 | 27 |
| MSI | |||
| MSI-High | 17 | 14 (82)* | 3 (18)‡ |
| MSI-Low | 18 | 13 (72)† | 5 (28) |
| MSS | 70 | 52 (74) | 18 (26) |
| No data | 4 | ||
| Total | 109 | 82 | 27 |
Loss of SMAD4 protein expression was more common in tumours diagnosed in the rectum than in the colon (P = 0.04).
Ten patients with early-onset colorectal cancer had germ-line mutations in MMR genes (8 in MLH1 and 2 in MSH2).
Four patients with early-onset colorectal cancer had germ-line mutations in MSH6.
All three patients with early-onset colorectal cancer had germ-line mutations in MMR genes (1 in MLH1 and 2 in MSH2).
Figure 1.
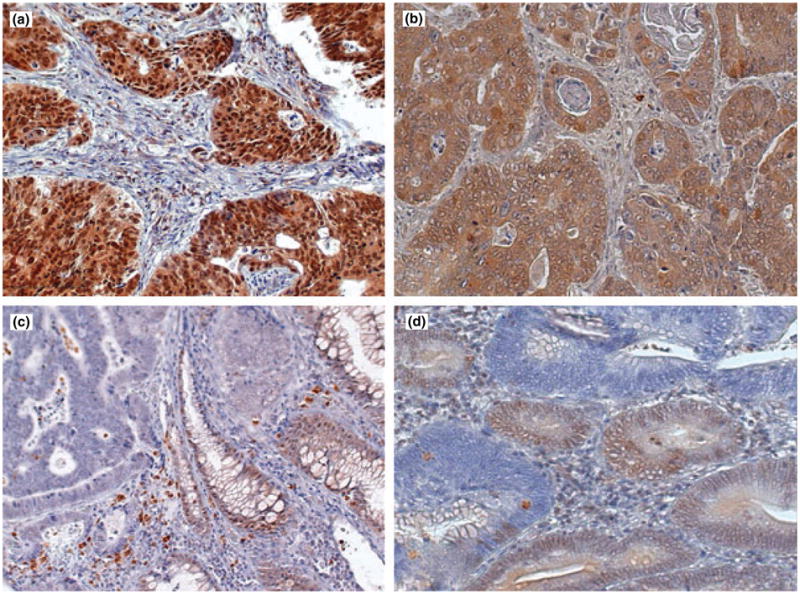
Immunohistochemical staining for SMAD4 performed using mAb B-8 in formalin-fixed paraffin-embedded sections of colorectal carcinomas. (a) Tumour showing strong expression of SMAD4 protein in the nucleus of tumour cells. (b) Tumour showing strong diffuse staining localized to cytoplasm with weaker nuclear staining. (c) Adenocarcinoma with loss of SMAD4 protein in tumour cells. Staining is present in adjacent normal colonic mucosa (arrow). (d) Representative section of tumour with focal positive and negative staining suggesting clonal differences in SMAD4 expression. Original magnification ×45.
SMAD4 immunohistochemistry and TGF-βRII mutations
Of the 21 tumours with a frameshift mutation at the TGF-βRII mononucleotide repeat, 18 (86%) expressed SMAD4 protein, which was no different to the proportion of tumours without TGF-βRII mutations that expressed SMAD4 protein (61 of 82, 74%) (Table 1).
SMAD4 copy number and protein expression
Thirty-one tumours shown to express the SMAD4 protein and all 27 tumours that lacked SMAD4 expression were analysed to determine SMAD4 copy number using quantitative (real-time) PCR. Ten showed loss of one copy of the gene, and three showed loss of both copies [total of 13 (48%) with gene loss]. Twelve had no change in SMAD4 copy number and the gene copy status of two patients with early-onset colorectal cancer was unable to be determined for technical reasons related to the quality of the DNA extracted from the fixed pathology material.
We found a similar frequency of SMAD4 copy loss in 31 tumours that expressed SMAD4 protein (14/31, 45%) to that observed in SMAD4 negative participants (Table 2).
Table 2.
Loss of SMAD4 copy number, protein expression and MSI status in early-onset colorectal cancers.
| SMAD4 copy number | Loss
|
No change | Gain | Undeterminable | ||
|---|---|---|---|---|---|---|
| Total | Hemizygosity | Loss of both copies | ||||
| SMAD4 IHC | ||||||
| Protein present | 31 | 13 | 1 | 12 | 4 | 1 |
| Protein absent | 27 | 10 | 3 | 12 | 0 | 2 |
| Total | 58 | 23 | 4 | 24 | 4 | 3 |
| MSI‡ | ||||||
| MSI-high | 10 | 1† | 0 | 8* | 1 | 0 |
| MSI-low | 11 | 4 | 0 | 5 | 1 | 1 |
| MSS | 35 | 16 | 4 | 11 | 2 | 2 |
| Unknown | 2 | |||||
| Total | 58 | 21 | 4 | 16 | 4 | 3 |
Four patients with early-onset colorectal cancer have germ-line mutations in MSH2 and 2 patients with early-onset colorectal cancer have germ-line mutations in MLH1 as reported in Southey et al., 2005 [9].
Case has a germ-line mutation in MLH1 as reported in Southey et al., 2005 Ref [9].
Southey et al., 2005 [9].
SMAD4 copy number and MSI status
Table 2 shows that only one of the 27 (4%) tumours that displayed loss of SMAD4 was MSI-high. This MSI-high tumour with SMAD4 loss did not arise in an individual whose family cancer history met the Amsterdam criteria II for Hereditary Nonpolyposis Colorectal Cancer (HNPCC) [16] (data not shown). Four tumours of varying MSI status, with positive SMAD4 protein staining, showed gain of SMAD4 copy number. A larger proportion of the MSI-positive tumours (including the majority of the MSI-high tumours) showed no change in SMAD4 copy number (13/21, 62%), compared with MSS tumours (11/35, 31%).
Table 2 also shows that previous germ-line screening for MMR gene mutations in these individuals had identified that seven of the 10 MSI-high patients with early-onset colorectal cancer studied for SMAD4 copy number were known to have arisen in individuals with germ-line mutations (four in MSH2 and three in MLH1, as indicated).
Discussion
To our knowledge, this is the first report of SMAD4 protein expression in early-onset colorectal cancer. We observed that 25% of colorectal cancers diagnosed before the age of 45 years lacked protein expression of SMAD4 and another 7% showed focal positive and negative staining suggesting clonal differences in SMAD4 expression. Therefore, the frequency of loss of SMAD4 protein expression in early-onset colorectal cancers falls within the broad range that studies of colorectal cancer of later onset have found for SMAD4 protein loss (~9–66%; 9.3% [5], 14% [6], 66% [7]). This suggests that loss of SMAD4 expression in tumours is a common feature of all colorectal cancers irrespective of age at diagnosis.
A total of 45 of the 109 tumours (41%) analysed had at least one identifiable molecular disruption to the TGF-β signalling pathway. Twenty-seven tumours lacked the expression of SMAD4 and 18 of the SMAD4 protein-expression-positive tumours had frameshift mutations in TGFβR11. We also found that disruption of the TGF-β pathway is therefore involved in a large proportion of early-onset colorectal cancer.
We observed SMAD4 staining in the nucleus in both normal tissue and in positive-staining tumour tissue, indicative of a tumour suppressor gene active within the nucleus and that this nuclear staining was usually maintained even when TGFβR11 was lost because of a frameshift mutation. This observation is consistent with the findings of Montgomery et al., who demonstrated positive nuclear staining for SMAD4 in xenografted tumours from TGFβR11 negative SMAD4 +/+ cell lines [17]. We did not observe an association between SMAD4 expression and TGFβR11 mutation status supporting other reports that these are independent molecular events that target the TGF-β signalling pathway. We observed only three patients with early-onset colorectal cancer whose colorectal cancers lacked the expression of SMAD4 and also carried a frameshift mutation in TGFβR11 (and all three of these patients with early-onset colorectal cancer carried germ-line mutations in a MMR gene, see below). This suggests that any transforming or tumourigenic advantage conveyed by this dual hit is probably at best, small. TGFβR11 frameshift mutations have been measured as part of a 10-microsatellite marker panel to measure MSI in these early onset colorectal cancers [9,10]. Similar to TGFβR11 status alone, we found no association between MSI status and SMAD4 expression (Table 1). This is consistent with other studies that have reported high rates of SMAD4 expression in MSI-positive tumours [7].
These early-onset colorectal cancer patients have been screened previously for germ-line mutations in MMR genes [9]. Of the tumours that lacked SMAD4 expression, three were MSI-high and all three patients with early-onset colorectal cancer carried germ-line MMR gene mutations (two in MSH2 and one in MLH1). Of the tumours that expressed SMAD4, 14 were MSI-high and 10 of these carried germ-line mutations in a MMR gene (two in MSH2 and eight in MLH1). Thirteen tumours were SMAD4 positive and MSI-low and four of these patients with early-onset colorectal cancer had germ-line mutations in MSH6.
We also observed no association between SMAD4 expression status and SMAD4 copy number. Thirteen of the 27 (48%) of the tumours lacking SMAD4 expression showed some degree of genetic loss of SMAD4 (three had loss of both copies). This suggests there are other genetic or epigenetic events responsible for the loss of SMAD4 in tumours that have lost only one copy of the gene (Table 2). In addition, 14 tumours that were determined to have no change in copy number at the gene loci, despite an absence of staining for SMAD4, have possibly undergone two of these alternate molecular events. The work of others supports a role for somatic mutation of SMAD4, which, while reported to be at high frequency in pancreatic cancers (20%) [18], is relatively rare in colorectal cancer (3%) [9]. A similar frequency of gene copy loss (14/29, 48%) was observed in tumours that expressed SMAD4. This could be because of a discrepancy in the IHC, but is most likely indicative of loss at 18q being frequent in colorectal cancer and that the retention of one copy is sufficient for normal protein expression, as measured by IHC (the biological implications of possible haploinsufficiency are unknown).
Loss of SMAD4 expression is a common feature of early-onset colorectal tumours as it is in colorectal cancer diagnosed in other age groups. The molecular mechanisms giving rise to loss of SMAD4 expression require further exploration as our data shows that it is not attributable exclusively to either SMAD4 copy number loss or MSI. Emerging and future genome-wide studies of somatic mutation, methylation and germ-line variation (GWASs) may give further insight into the possible mechanisms of loss of SMAD4 expression as well as identifying the molecular mechanisms underlying the majority of early-onset colorectal cancer.
Acknowledgments
We thank Judi Maskiell and the research interviewers; Gillian Dite and the data management staff. We also thank the men and women who participated in this study.
Contract grant sponsor: National Health and Medical Research Council, Australia.
Footnotes
Competing interest
The authors have no competing interest to declare.
References
- 1.Seshimo I, Yamamoto H, Mishima H, et al. Expression and mutation of SMAD4 in poorly differentiated carcinoma and signet-ring cell carcinoma of the colorectum. J Exp Clin Cancer Res. 2006;25:433–42. [PubMed] [Google Scholar]
- 2.Alazzouzi H, Alhopuro P, Salovaara R, et al. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res. 2005;11:2606–11. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- 3.Alhopuro P, Alazzouzi H, Sammalkorpi H, et al. SMAD4 levels and response to 5-fluorouracil in colorectal cancer. Clin Cancer Res. 2005;11:6311–6. doi: 10.1158/1078-0432.CCR-05-0244. [DOI] [PubMed] [Google Scholar]
- 4.Zhou S, Kinzler KW, Vogelstein B. Going mad with Smads. N Engl J Med. 1999;341:1144–6. doi: 10.1056/NEJM199910073411510. [DOI] [PubMed] [Google Scholar]
- 5.Isaksson-Mettävainio M, Palmqvist R, Forssell J, Stenling R, Oberg A. SMAD4/DPC4 expression and prognosis in human colorectal cancer. Anticancer Res. 2006;26:507–10. [PubMed] [Google Scholar]
- 6.Korchynskyi O, Landstrom M, Stoika R, Funa K, Heldin CH, ten Dijke P, Souchelnytskyi S. Expression of Smad proteins in human colorectal cancer. Int J Cancer. 1999;82:197. doi: 10.1002/(sici)1097-0215(19990719)82:2<197::aid-ijc8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Salovaara R, Roth S, Loukola A, et al. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 2002;51:56–9. doi: 10.1136/gut.51.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka T, Watanabe T, Kazama Y, et al. Chromosome 18q deletion and Smad4 protein inactivation correlate with liver metastasis: a study matched for T- and N- classification. Br J Cancer. 2006;95:1562–7. doi: 10.1038/sj.bjc.6603460. Epub 2006 Nov 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Southey MC, Jenkins MA, Mead L, et al. Mismatch repair gene mutation testing, immunohistochemistry and micro-satellite instability testing in a population-based series of unselected early-onset colorectal cancers. J Clin Oncol. 2005;23:6524–32. doi: 10.1200/JCO.2005.04.671. [DOI] [PubMed] [Google Scholar]
- 10.Mead LJ, Jenkins MA, Young J, et al. Microsatellite instability markers for identifying early-onset colorectal cancers caused by germ-line mutations in DNA mismatch repair genes. Clin Cancer Res. 2007;13:2865–9. doi: 10.1158/1078-0432.CCR-06-2174. [DOI] [PubMed] [Google Scholar]
- 11.Alsop K, Mead L, Smith LD, et al. Low somatic K-ras mutation frequency in colorectal cancer diagnosed under the age of 45 years. Eur J Cancer. 2006;42:1357–61. doi: 10.1016/j.ejca.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins MA, Baglietto L, Dite GS, et al. After hMSH2 and hMLH1–what next? Analysis of three-generational, population-based, early-onset colorectal cancer families. Int J Cancer. 2002;102:166–71. doi: 10.1002/ijc.10670. [DOI] [PubMed] [Google Scholar]
- 13.Armes JE, Trute L, White D, et al. Distinct molecular pathogeneses of early-onset breast cancers in BRCA1 and BRCA2 mutation carriers: a population-based study. Cancer Res. 1999;59:2011–7. [PubMed] [Google Scholar]
- 14.Boulay JL, Reuter J, Ritschard R, Terracciano L, Herrmann R, Rochlitz C. Gene dosage by quantitative real-time PCR. BioTechniques. 1999;27:228–30. doi: 10.2144/99272bm03. [DOI] [PubMed] [Google Scholar]
- 15.Boulay JL, Mild G, Lowy A, et al. SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy in patients with colorectal cancer. Br J Cancer. 2002;87:630–4. doi: 10.1038/sj.bjc.6600511. Erratum in: Br J Cancer. 2003 Jul 21;89(2):422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 17.Montgomery E, Goggins M, Zhou S, et al. Nuclear localization of Dpc4 (Madh4, Smad4) in colorectal carcinomas and relation to mismatch repair/transforming growth factor-beta receptor defects. Am J Pathol. 2001;158:537–42. doi: 10.1016/s0002-9440(10)63995-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–6. [PubMed] [Google Scholar]