

Abstract
We characterized the effects of a newly developed STAT3 inhibitor, LLL12 in multiple myeloma (MM) cells. LLL12 specifically inhibited STAT3 phosphorylation, nuclear localization, DNA binding activity, down-regulated STAT3 downstream genes, and induced apoptosis in MM cells. Importantly, LLL12 significantly inhibited STAT3 phosphorylation, induced apoptosis in primary MM cells which came from patients that were clinically resistant to lenalidomide and bortezomib. LLL12 is a potent inhibitor of cell proliferation with IC50 values ranging between 0.26μM and 1.96μM in MM and primary MM cells. LLL12 also inhibited STAT3 phosphorylation induced by interleukin-6 (IL-6) and interferon-α but not STAT1, STAT2, STAT4, and STAT6 phosphorylation induced by interferon-α, interferon-γ, and interleukin-4 indicating the selectivity of LLL12 for STAT3. The selectively of LLL12 on STAT3 was further demonstrated on 21 protein kinases, which LLL12 had IC50 values 73.92μM. In addition, the pre-treatment of LLL12 blocked the promotion of the cell proliferation and resistance to lenalidomide by IL-6. Furthermore, LLL12 significantly blocked tumor growth of MM cells in mouse model. Our results indicate that LLL12 blocks constitutive STAT3 and IL-6 induced STAT3 signaling and may be a potential therapeutic agent for MM.
Keywords: STAT3, multiple myeloma, small molecule inhibitors
Introduction
Multiple Myeloma (MM) is the second most common hematologic malignancy and accounts for about 20,000 new diagnoses annually in the United States 1. The incidence of the disease is rising and, despite the advent of novel agents including lenalidomide and bortezomib, the disease remains mostly incurable and new therapies are desperately needed.
Signal Transducers and Activators of Transcription (STAT) proteins are transcription factors which normally mediate orderly and tightly regulated signaling processes initiated through extracellular cytokines and growth factors 2–4. On the other hand, constitutive activation of STATs has been demonstrated to contribute to oncogenesis 2, 5, and STAT3 in particular, is considered to be an oncogene due to its ability to promote malignancy 3, 5, 6. STAT3 activation occurs through phosphorylation of the tyrosine 705 (Tyr705) residue, leading to dimerization and translocation from the cytoplasm to the nucleus 5, 7, 8. In the nucleus, STAT3 binding to target genes induces the transcription and up regulation of proliferation and anti-apoptotic associated proteins 3, 5, 6, 9. STAT3 can also dimerize via reversible lysine acetylation which is independent of tyrosine phosphorylation and thus could also be essential for cell transformation, particularly for IL-6 independent tumors 10.
Prior work has demonstrated that constitutively active STAT3 is sufficient for inducing cellular transformation 6 and resistance to transformation was observed in STAT3 deficient cells 11, 12. STAT3 is frequently activated in many types of human solid and blood cancer and contribute to cancer progression 2, 4. The STAT3 signaling pathway is especially important in the proliferation, chemoresistance, and survival of MM cells through constitutive phsophorylation of STAT3 or in response to interleukin (IL)-6 produced by cells in the bone marrow microenvironment or by MM cells, per se 13, 14. Inhibition of constitutive STAT3 signaling by a dominant-negative mutant, a JAK2 inhibitor (AG490), and other strategies leads to apoptosis in MM cells 13, 14.
While STAT3 may be important for normal embryologic development, it appears to be less important for the function of differentiated tissues 11, 12, 15–17. For example, no obvious deleterious effects were observed when STAT3 antisense therapy was used to deplete protein from normal cells in mice 15. Furthermore, fibroblasts deficient in STAT3 exhibited similar proliferative capacities compared to their wild-type counterparts, similar survival in vitro, and responded appropriately to several growth factors 12. This supports the notion that in relatively differentiated cell types, STAT3 function is not required to maintain basal cell survival. Moreover, inhibition of STAT3 signaling in immune cells may enhance surveillance and cytotoxicity against MM tumor targets 18. Taken together, these findings suggest that development of agents directly inhibiting the STAT3 pathway could represent a novel form of “targeted therapy” for MM.
Using structure-based drug design, we developed a novel STAT3 inhibitor, named LLL12 (Chemical structure depicted in Supplemental Figure 1). LLL12 is an optimal structural analog of our previously reported STAT3 inhibitors LLL319 and STA-2120. Computer models with docking simulation showed that LLL12 binds directly to the phosphoryl tyrosine 705 (pTyr705) binding site of the STAT3 monomer 21. Herein, we characterize the effects of LLL12 on MM tumor cells. We demonstrate that LLL12 exhibits high specificity for inhibiting STAT3 phosphorylation and leads to down regulation of STAT3-modulated proliferation and survival genes. LLL12 inhibits proliferation and induces apoptosis of primary, human MM cells in vitro, even in samples procured from patients with relapsed/refractory MM, including prior therapies with lenalidomide and bortezomib 22. LLL12 suppresses in vivo MM tumor growth in a mouse xenograft model. These findings strongly support further development of LLL12 as a novel therapeutic agent for MM.
Materials and Methods
Cell lines and primary MM tumor cells
Human MM cell lines (U266, ARH-77, IM-9, MM.1S and RPMI8226) were purchased from the American Type Culture Collection (Manassas, VA). MM cell lines were maintained in RPMI1640 medium supplemented with 10% Fetal Bovine Serum (FBS), 4.5 g/L L-glutamine, sodium pyruvate, and 1% penicillin/streptomycin and maintained in a humidified 37°C incubator with 5% CO2. CD138(+) cells from patients with MM were obtained with written informed consent under Ohio State University IRB-approved procurement protocol and isolated by positive selection utilizing EasySep CD138(+) magnetic nanoparticles per manufacturer’s instructions (StemCell Technologies, Vancouver, BC). The majority of CD138+ cells in the marrow of MM patients are myeloma cells.
Small molecular JAK2, STAT3 inhibitors and Lenalidomide
LLL12, a new STAT3 inhibitor 21, and WP1066 23, a JAK2 inhibitor, were synthesized at The Ohio State University (P-K Li, College of Pharmacy). AG490, a JAK2 inhibitor 24, Stattic 25 and S3I-201 26, two STAT3 SH2 inhibitors, were purchased from Calbiochem (Darmstadt, Germany). Lenalidomide was purchased from LC Laboratories (Woburn, MA). Drugs were dissolved in sterile dimethyl sulfoxide (DMSO) to make 20mM stock solution, stored at −20° C until use.
Protein kinase activity assay
The effects of LLL12 on twenty one purified human protein kinases were performed at Millipore UK Limited (Dundee, UK) using a validated kinase profiler assay as described in detail by the manufacturer. In short, assays contained a peptide substrate, purified recombinant human protein kinases to be tested, and gamma-labeled ATP, magnesiumion. Radioactive phosphorylated product was measured and quantitated via a scintillation counter. Appropriate kinase inhibitor, which gave half-maximal inhibitory concentrations (IC50) values at nM ranges was used as a positive control. The IC50 inhibitory values of LLL12 on the kinase activity of each protein kinase were determined using 10 different concentrations of LLL12 (up to 100 μM).
Cell viability assay
U266, ARH-77, and primary MM cells (3,000–5,000/well in 96-well plates) incubated with indicated concentrations of compounds in triplicate at 37 C for 72 hours. 3-(4,5-Dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) viability assay was performed according to manufacturer’s protocol (Roche Diagnostics, Mannheim, Germany). The absorbance was recorded at 595 nm. IC50 values were determined using Sigma Plot 9.0 Software (Systat Software Inc., San Jose, CA). CD138+ and CD138- marrow fraction were from a patient with MM and were cultured in DMSO or LLL12 (10μM) for 24h. Cell viability was measured by MTS assay (5 replicates per condition). The synergy of LLL12 and lenalidomide in the inhibition of U266 cell growth was determined as previously21. Calcusyn software (Biosoft, Ferguson, MO) was used to determine the combinational index (CI) for combination. A CI value of less than 1 represents synergism. A CI value equal to 1 represents additive effects. A CI value greater than 1 represents antagonistic effects.
Nuclear staining
To examine whether LLL12 inhibits STAT3 nuclear localization, the immunofluroence staining was performed according to the protocol from the Cell Signaling Technology. Briefly, after wash once with Phosphate Buffered Saline (PBS) buffer, U266 MM cells were blocked with 5% normal goat serum for 1 hour and incubated with rabbit polyclonal anti-humanSTAT3 antibody (Cell Signaling Technology, Beverly, MA, 1/100 dilution) overnight. Then cells were washed and incubated with goat anti-rabbit IgG Alexa Fluor 594 (1/100) for 1 hour and counterstained for nuclei with Hoechst (50 ng/ml) for 5 min. Stained slides were mounted with mounting medium (Vector Laboratories, Burlingame, CA) and analyzed under a fluorescence microscope (Zeiss Axioskop microscope, Carl Zeiss, Gottingen, Germany). Pictures were captured using an AxioCam HRc camera and Axiovision 3.1 software (Carl Zeiss, Gottingen, Germany).
Western blot analysis
MM cells were treated with LLL12 (2.5, 5 or 10μM) or DMSO for 24 hours before cells were collected for Western blot analysis. For interferon-α (IFN-α), interferon-γ (IFN-γ), IL-4, and IL-6 stimulation experiments, IM-9 and MM.1S MM cells were serum-starved for 24 hours and left untreated or pre-treated with LLL12 (2.5–10 μM), Stattic (2.5–20 μM) or DMSO for 2 hours. Then, 25ng/ml IFN-α, IFN-γ, or IL-4 was added, the cells were harvested for Western Blot analysis 30 minutes later. MM cells were lysed in cold RIPA lysis buffer containing protease inhibitors and subjected to SDS-PAGE. Proteins were transferred on to PVDF membrane and probed with antibodies. Antibodies (Cell Signaling Technology, Beverly, MA) against phospho-specific STAT1 (Tyrosine 701), phospho-specific STAT2 (Tyrosine 690), phospho-specific STAT3 (Tyrosine 705), phospho-specific STAT4 (Tyrosine 693), phospho-specific STAT6 (Tyrosine 641), phospho-specific ERK1/2 (Threonine 202/Tyrosine 204), phospho-specific Src (Tyrosine 416), phospho-specific mTOR (Serine 2448), cleaved Poly (ADP-ribose) polymerase (PARP), cleaved caspase-3, cleaved caspase-8, phospho-independent STAT3 cyclin D, Bcl-2, DNMT1, survivin, and GAPDH were used for western blots. Membranes were analyzed using enhanced chemiluminescence Plus reagents and scanned with a Storm Scanner (Amersham Pharmacia Biotech Inc, Piscataway, NJ).
STAT3 DNA binding activity
U266 and ARH-77 MM cells were treated with LLL12 (2.5μM and 5μM) or DMSO for 24 hours. The nuclear extracts were analyzed for STAT3 DNA binding activity using a STAT3 Transcription Factor Kit (Clontech Inc, Mountain View, CA). Statistical significance (p < 0.05) relative to DMSO (vehicle control) is designated by an asterisk in figures.
Reverse transcriptase-Polymerase chain reaction (RT-PCR)
U266 and ARH-77 MM cells were treated with LLL12 (2.5 and/or 5 μM) or DMSO for 24 hours. RNAs of ARH77, U266 and MM.1S cells were collected using RNeasy Kits (Qiagen, Valencia, CA). Primer sequences and source information of STAT3 downstream target genes can be found in Supplemental Table 1. PCR amplification was done under the following conditions: 5 min at 94°C followed by 25 cycles of 30 seconds at 94°C, 30 sec at 55°C, and 30 seconds at 72°C with a final extension of 5 min at 72°C. Primer sequences are shown in Supplemental Table 1.
Flow cytometry
Apoptotic cell death induced by LLL12 was quantified by flow cytometry with Annexin-V/propidium iodide (PI) double staining (BD Pharmingen, San Jose, CA). After treatment with LLL12 or DMSO for 24 hours, U266 and ARH-77 MM cells were harvested and washed with cold PBS. The cell pellet was re-suspended in 1× binding buffer at a concentration of 1 × 106 cells/ml. 100 μl of cell suspension was transferred into another tube. 5 μl of Annexin V-FITC and 5 μl of PI were added for 15 minutes at room temperature (RT) in darkness, and then analysed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ) within 1 hour.
Mouse xenograft model
Animal studies were conducted in accordance with the principles and standard procedures approved by IACUC at the Research Institute at Nationwide Children’s Hospital. 2× 107 ARH-77 MM tumor cells were injected subcutaneously into the right flank of 4- to 5- week-old female NOD/SCID mice (Harlan Laboratories, Indianapolis, IN, USA). After 12 days, mice were divided into two treatment groups (n=6 each): (a) control vehicle (100% DMSO) and (b) 5mg/kg of LLL12. LLL12 or DMSO was administered via intraperitoneal injection. Tumor growth was determined by caliper-measured the length (L) and width (W) every other day, and tumor volume was calculated on the basis of the following formula: volume = (π/6) LW2. Bodyweights of mice were measured daily during 14 days period treatments.
Statistical analysis
The Mann-Whitney non-parametric test was used for the evaluation of differences of STAT3 DNA binding activity, percentage of apoptosis, and tumor volume between DMSO- and LLL12-treated experiments. IC50 values were determined using Sigma Plot 9.0 Software (Systat Software Inc., San Jose, CA).
Results
LLL12 specifically inhibited STAT3 phosphorylation and induces apoptosis in MM cells
In the human MM cell lines U266 and ARH-77, which express elevated levels of STAT3 phosphorylation, LLL12 inhibited STAT3 phosphorylation at tyrosine residue 705 (Tyr 705) (Figures 1A and 1B). Inhibition of STAT3 phosphorylation was associated with induction of apoptosis, as evidenced by PARP, caspase-3, and caspase-8 cleavage (Figures 1A and Figure 1B). LLL12 also inhibited STAT3 phosphorylation and induced apoptosis in primary, human MM cells from three different patients (Figure 1C). Importantly, patients M.M.P. 2 and M.M.P. 3 were clinically resistant to prior therapy with lenalidomide and bortezomib. These primary MM cells were all found to have constitutively active STAT3. Thus, therapy targeting STAT3 inhibition may be an effective treatment for patients with lenalidomide and bortezomib resistant disease.
Figure 1. LLL12 inhibited constitutive and cytokine-induced STAT3 phosphorylation, reduced expression of downstream STAT3 targets, and exhibitd high specificity for STAT3.

(A, B) The human MM cell lines U266 and ARH-77 exhibit constitutively phosphorylated STAT3. LLL12 (2.5μM or 5μM) inhibited STAT3 phosphorylation at Tyr705, resulting in induction of apoptosis as indicated by cleaved PARP, caspase-3 and caspase 8. Other signaling pathways were not affected by LLL12 (mTOR, Src, and ERK1/2). GAPDH is shown as a loading control. (C) LLL12 blocked STAT3 phosphorylation in purified, freshly isolated primary, human MM tumor cells from multiple myeloma patient (M.M.P., representative result in n=3 independent patient samples) leading to PARP and caspase-3 cleavage. (D) LLL12 was more potent than Stattic (2 hours pre-treatment) in inhibiting IL-6-induction of STAT3 phosphorylation in the MM.1S cell line. (E) LLL12 inhibited STAT3 but not STAT1 and STAT2 phosphorylation induced by interferon-α(IFN-α, 25ng/ml) in the MM.1S cell line. The potency of LLL12 was better than Stattic. Cells were pre-treated with LLL12 and/or Stattic for 2 hours, then were stimulated with IL-6 or interferon-αfor 30-minutes in the presence or absence of LLL12.
We also detected the effects of LLL12 on the phosphorylation of mTOR, Src and ERK(1/2). The phosphorylation of mTOR and Src were not reduced significantly. In U266 MM cells, the phosphorylation of ERK(1/2) was even increased (Figure 1A). In ARH-77 MM cells, the phosphorylation of ERK(1/2) and mTOR were slightly reduced by LLL12 at higher concentration (5μM) (Figure 1B). In order to characterize the specificity of these effects of LLL12 in MM cells further, we examined whether LLL12 exhibited inhibitory effects on 21 other human protein kinase activities using a standardized, validated kinase profile. LLL12 did not inhibit BMX, BRK, CSK, Fgr, Fyn, JAK3, LcK, Lyn, or ZAP-70 which contain an SH2 domain (Table 1) at achievable, relevant in vivo concentrations (IC50 are at least 57.19 μM to > 100 μM). Furthermore, LLL12 exhibited no relevant inhibition against other protein kinases, including AKT2, CDK1/cyclin B, FAK, FGFR2, IKKβ, JNK1, JNK2, PAK1, PAK2, PDGFR-α, PKC-α, and PKC-δ (IC50 are 91.76 μM to > 100 μM, Table 1). Staurosporine was used as positive control on these kinase assays (IC50 between <0.001 and 0.456 μM). These results support the selectivity of LLL12 to inhibit STAT3 and thus induce pro-apoptotic effects in MM specifically through STAT3 inhibition.
Table 1. The effect of LLL12 on the activity of human protein kinases.
The IC50 inhibitory values of LLL12 on the kinase activity were determined using 10 different concentrations of LLL12 with 100 μM as the highest concentration in kinase assays. Results suggested LLL12 had little or much less effect on other human kinase proteins than STAT3. Staurosporine was used as positive control on these kinase assay (IC50 between <0.001 and 0.456 μM).
| Protein Kinases | IC50 (μM) | Protein Kinases | IC50 (μM) | Protein Kinases | IC50 (μM) | |
|---|---|---|---|---|---|---|
| Tyrosine kinases contain SH2 Domain | BMX | >100 | Fgr | 57.19 | LcK | >100 |
| BRK | >100 | Fyn | >100 | Lyn | >100 | |
| CSK | 73.92 | JAK3 | 89.70 | ZAP-70 | >100 | |
| Other human protein kinases | AKT2 | 91.76 | IKKb | >100 | PAK2 | >100 |
| CDK1/CyclinB | >100 | JNK1 | >100 | PDGFR-a | >100 | |
| FAK | >100 | JNK2 | >100 | PKC-a | >100 | |
| FGFR2 | >100 | PAK1 | >100 | PKC-d | >100 |
LLL12 inhibited STAT3 but not other STATs phosphorylation induced by interleukin-6, interferon-α, interferon-γ, and IL-4
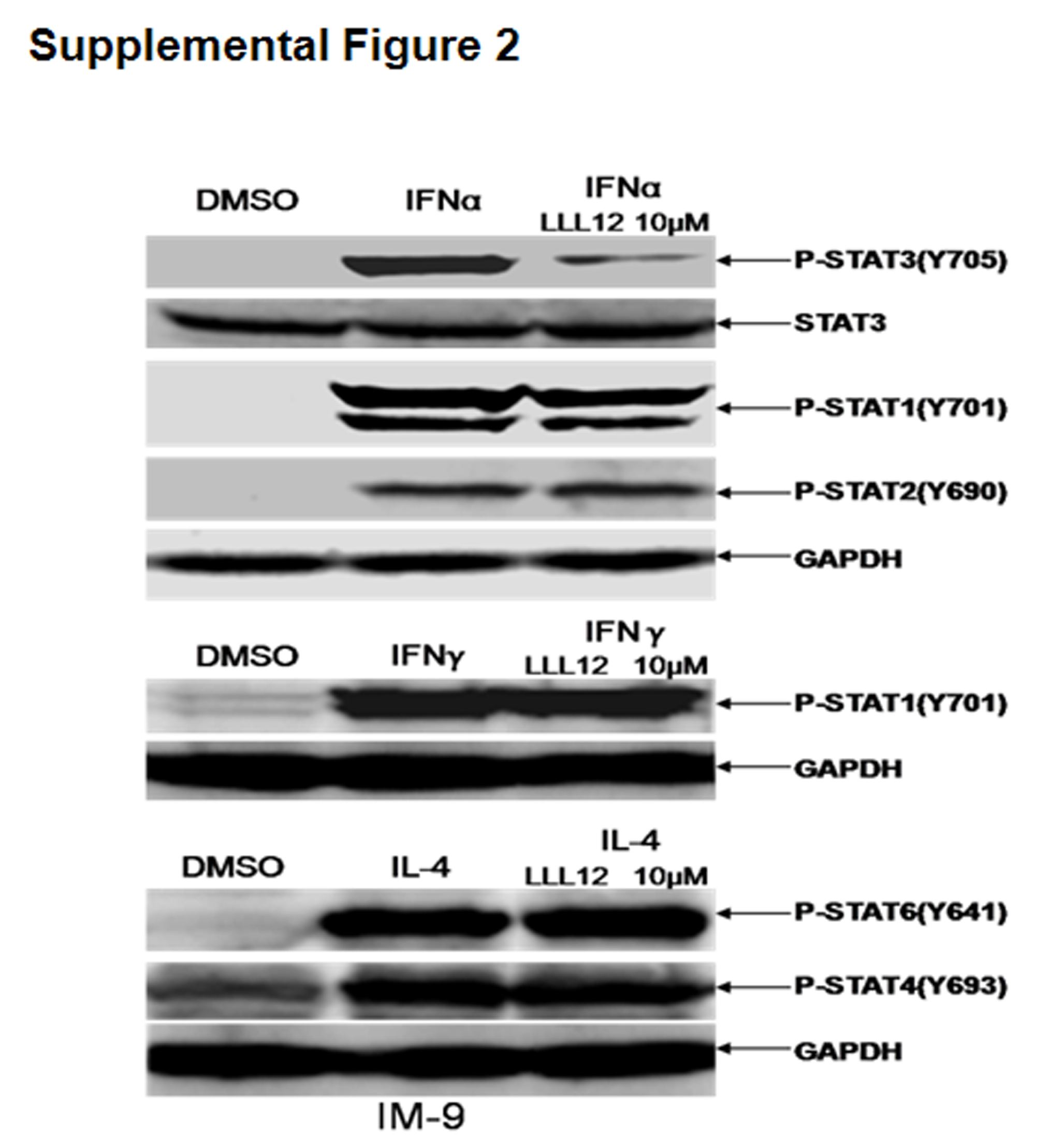
The relationship between the ambient microenvironment and MM tumor cells is crucial to maintaining and perpetuating the tumor cell clone via cytokine-mediated signaling in particular, IL-6 13, 27. The MM1.S human MM cell line, which expresses very lower constitutively phosphorylated STAT3, was utilized to determine whether or not LLL12 is capable of inhibiting cytokine-induced STAT3 phosphorylation. Pretreatment of MM.1S multiple myeloma cells stimulated by IL-6 and IFN-α respectively with LLL12 prevented phosphorylation of STAT3, but not STAT1 or STAT2 (Figure 1D and Figure 1E). Another reported STAT3 inhibitor, Stattic 25 was found to be much less potent than LLL12 in inhibiting IL-6 or IFN-α mediated STAT3 phosphorylation (Figure 1D and Figure 1E). LLL12 also inhibited STAT3 phosphorylation induced by IFN-α but did not or much less to inhibit the stimulation STAT1 and/or STAT2 phosphorylation by IFN-α or IFN-γ in IM-9 multiple myeloma cells (Supplemental Figure 2). Similarly, LLL12 did not prevent phosphorylation of STAT4 or STAT6 by IL-4 in IM-9 human MM cells (Supplemental Figure 2). These results further highlight the specificity of LLL12 for STAT3 inhibition without associated effects on other members of the STAT signaling protein family.
LLL12 inhibited STAT3 nuclear localization, STAT3 DNA binding activity and the expression of STAT3 downstream target genes
To confirm the inhibition of STAT3 signaling by LLL12, we examined whether or not LLL12 inhibits STAT3 nuclear localization, because the main function of STAT3 is to function as a transcription factor in nucleus. In U266 MM cells treated with DMSO, STAT3 was mainly observed localized in nuclei (Left panels, Supplemental Figure 3). However, in U266 cells treated with LLL12, most STAT3 was retained in cytoplasm and absent in nuclei (Right panels, Supplemental Figure 3). We also examined the effect of LLL12 on STAT3 DNA binding activity in U266 and ARH-77 MM cells. LLL12 caused a statistically significant inhibition (approximately 30–33% reduction with 2.5μM and 58–69% reduction with 5μM) of STAT3 DNA binding activity in U266 and ARH-77 MM cells (Figure 2A). These findings demonstrated that inhibition of STAT3 phosphorylation by LLL12 consequently impaired STAT3 function in MM cells.
Figure 2. LLL12 inhibited STAT3 DNA binding activity and expression of downstream targets associated with proliferation and survival of MM tumor cells.

(A) U266 and ARH-77 MM cells were treated in LLL12 (2.5μM) or DMSO for 24 hours and nuclear extracts were examined for DNA binding activity. LLL12 induced statistically significant (*) reductions in STAT3 DNA binding activity, results are representative of two independent experiments in each cell line. (B) U266 and ARH-77 human MM cell lines were cultured in LLL12 (5 μM) or DMSO for 24 hours. Reverse transcriptase PCR revealed decreased expression of STAT3 target genes in LLL12-treated cells as compared to DMSO control following treatment with LLL12. (C) Downstream STAT3 target proteins, Cyclin D1, Survivin, Bcl-2, and DNMT1 were downregulated by LLL12 as shown by western blot analysis. (D) MM.1S cells were stimulated with IL-6 (2.5–10 ng/ml) for 48 hours in the presence or absence of LLL12 (2.5μM) for the last 24 hours. Reverse transcriptase PCR showed IL-6 enhanced expression of STAT3 target genes, which was blocked by LLL12.
To characterize further the functional effects of LLL12-induced STAT3 inhibition, we examined the transcription of STAT3 downstream target genes involved in proliferation and survival of MM cells by reverse transcriptase PCR, including cyclin D1, survivin, DNMT1, and Bcl-2 6, 28–30. U266 and ARH-77 MM cell lines were treated with LLL12 or DMSO for 24-h. Reverse Transcriptase PCR was run for cyclin D1, survivin, Bcl-2, Bcl-XL, and DNMT1. LLL12 treatment resulted in an inhibition of the transcription of all five of these STAT3-regulated genes in both MM cell lines (Figure 2B). These downstream target proteins of STAT3 were also down regulated by LLL12 (Figure 2C), which confirm the results of the inhibition at mRNA levels (Figure 2B). The expression of STAT3 downstreatm target genes, such as survivin, Bcl-2, Bcl-XL, was increased after stimulated by IL-6 (2.5–10 ng/ml) for 48 hours (Figure 2D). 2.5μM of LLL12 also reduced the expression of these genes to lower than the basal level (Figure 2D).
LLL12 impaired MM cell proliferation and viability
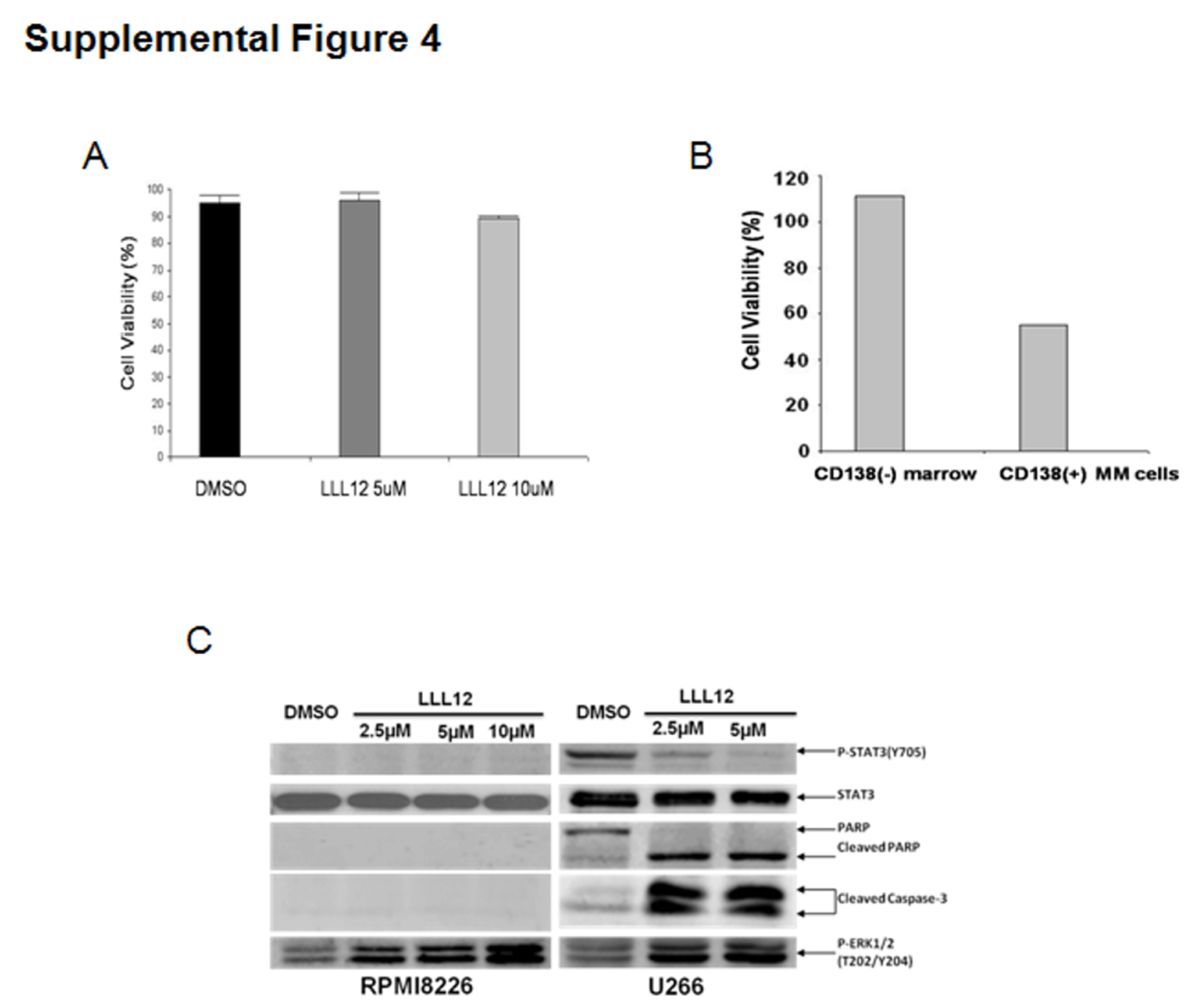
Cell viability assays were conducted to examine the inhibitory affect of LLL12 on U266 ARH-77 human MM cell lines and primary MM cells from two patients. IC50 values after 72 hours of treatment were calculated for LLL12 and compared to other previously characterized compounds: JAK2 inhibitors: WP1066 23 and AG490 24, and STAT3 inhibitors: Stattic 25, LLL3 31, and S3I-201 26. Results summarized in Supplemental Table 2 show that IC50 values for LLL12 are 0.49μM and 1.96 μM in U266 and ARH-77 MM cells respectively, suggesting greater or comparable inhibitory potency of LLL12 than other inhibitors tested and comparable to the activity of Stattic (Supplemental Table 2). LLL12 is also potent in primary multiple myeloma cells (M.M.P.4 and M.M.P5) isolated from two patients that are clinically resistant to lenalidomide and bortezomib (Supplemental Table 2). In contrast, LLL12 has little effects on normal human peripheral blood lymphocytes (Supplemental Figure 4A) and normal human marrow [CD138(−)] cells (Supplemental Figure 4B). We also examined the effect of LLL12 on survival of IL-6 independent MM cell lines RPMI-8226. LLL12 did not induce cleaved caspase-3 and cleaved PARP in this cell line (Supplemental Figure 4C). The phosphorylation of STAT3 at tyrosine residue 705 (Tyr 705) of this cell line was too low to be detected (Supplemental Figure 4C).
To quantify the apoptotic effects of LLL12, U266 and ARH-77 MM cell lines were cultured in LLL12 or DMSO for 24 hours and stained with Annexin V and PI. Representative findings are shown in Figure 3A. LLL12 led to a dose-dependent increase in apoptosis as shown in Figure 3B and Figure 3C. Apoptosis was observed in 38.2% (+/− 7.5%, p < 0.05 compared to DMSO) in U266 MM cells at LLL12 (2.5μM) and 53.3% (+/− 7.1%, p < 0.05 compared to DMSO) at 5μM (Figure 3B). Apoptosis of 70.8% (+/− 1.7%, p < 0.05 compared to DMSO) of ARH-77 cells was seen at LLL12 (2.5μM) and 73.5% (+/− 4.1, p < 0.05 compared to DMSO) at 5μM (Figure 3C).
Figure 3. LLL12 induced apoptosis in U266 and ARH-77 multiple myeloma cells.

(A). Representative histograms of U266 and ARH-77 MM cells cultured for 24 hours in DMSO or LLL12 (2.5μM or 5μM) following annexin V/PI staining and flow cytometric analysis. (B, C) LLL12 led to dose-dependent, statistically significant (*p < 0.05) increase in apoptosis of MM cells, in all instances, results are representative of at least 3 independent experiments.
IL-6 promoted cell viability, confers resistance to growth inhibition induced by lenalidomide and is suppressed by LLL12 in MM1.S human MM cells
To explore the proliferation/cell viability promoting role of IL-6 in MM cells, MM1.S MM cells were stimulated with IL-6 for 48 hours and cells viability was detected by MTT assay. IL-6 can increase cell proliferation/viability by two-fold which could be blocked by small molecular STAT3 inhibitor, LLL12 (Figure 4A). This is consistent with the stimulation of STAT3 phosphorylation by IL-6 and inhibited by LLL12 in MM1.S cells (Figure 1D). Treatments of MM1.S cells with lenalidomide caused a dose-dependent reduction of cell viability (Figure 4B). This inhibition of cell viability was reversed with IL-6 treatment. However, pre-treatment of MM1.S cells with LLL12, further reversed the rescue of lenalidomide-mediated inhibition of cell viability by IL-6 (Figure 4B). Therefore, these results suggest that IL-6 conferred resistance to growth inhibition by lenalidomide and this activity was blocked by small molecular STAT3 inhibitor, LLL12.
Figure 4. LLL12 inhibited cell proliferation induced by IL-6 and blocked IL-6 mediated drug resistance in human MM cells.

(A) MM.1S cells were treated with IL-6 (25ng/ml) with or without LLL12. IL-6 can increase cell proliferation, which could be blocked by LLL12. (B) Treatments of MM1.S cells with lenalidomide caused a dose-dependent reduction of cell viability. IL-6 reversed lenalidomide induced inhibition of cell viability. Pre-treatment of MM1.S cells with LLL12, further reversed the rescue of lenalidomide-mediated inhibition of cell viability by IL-6. (C) U266 cells secreted higher levels of IL-6 than MM1.S cells. (D) U266 cells expressed higher levels of IL-6 than MM1.S cells. Both MM cell lines expressed similar levels of IL-6R and Gp130. (E) U266 cells expressed higher levels of STAT3 phosphoryltion than MM1.S cells. (F) U266 cells were more resistance to lenalidomide than MM1.S cells. (G) LLL12 enhanced lenalidomide-mediated inhibition of cell proliferation in U266 MM cells.
LLL12 enhanced the activity of lenalidomide in U266 human MM cells with higher levels of IL-6/P-STAT3
We also examined the effects of lenalidomide on U266 human MM cells. It was reported that IL-6 plays an important role in myeloma cell proliferation, survival, and drug resistance 13, 32. So we compared the level of IL-6/P-STAT3 pathway in U266 and MM.1S cell lines. ELISA assay showed that the level of IL-6 in the medium of U266 cells is higher than MM.1S cells (Figure 4C). U266 cell line also has higher level of IL-6 gene expression as detected by RT-PCR (Figure 4D). Western blot results also showed the phosphorylation of STAT3 (Tyr 705) in U266 is higher than MM.1S cell line (Figure 4E). Because U266 cells express higher levels of IL-6 and STAT3 phosphorylation than MM.1S cells, we predicted that U266 cells may be more resistant to certain drugs. Our results demonstrated that U266 cells were indeed more resistant to lenalidomide compared to MM.1S cells (Figure 4F). Furthermore, LLL12 could increase the sensitivity of U266 cells to lenalidomide treatment (Figure 4G). The Combinational Index (CI) for LLL12 and lenalidomide was calculated. The CI value of all the combinations of treatments were less than 1, indicating synergism between LLL12 and lenalidomide. Therefore, LLL12 is likely to enhance the effects of lenalidomide through the inhibition of IL-6/STAT3 pathway in U266 multiple myeloma cells.
LLL12 suppressed MM tumor growth in vivo
A murine xenograft model was utilized to determine the in vivo anti-MM effects of LLL12. 10 days after subcutaneous implantation of 2×107 ARH-77 MM tumor cells in NOD/SCID mice, subjects were randomized to receive daily via intraperitoneal administration of 5 mg/kg LLL12 (6 mice each group) or DMSO vehicle control (6 mice each group). As shown in Figure 5, LLL12 significantly suppressed tumor volume (Figure 5A and 5C, p < 0.05 for all comparisons indicated), tumor weight (Figure 5B, p < 0.05), and inhibited STAT3 phosphorylation (Figure 5D) compared with DMSO-treated controls. The bodyweights of LLL12-treated mice were not reduced and similar to that of the vehicle-treated mice suggesting that LLL12 toxicity was minimal (Figure 5E). These results demonstrate that LLL12 is potent in suppressing tumor growth of human MM cells in vivo.
Figure 5. LLL12 abrogated in vivo MM tumor cell growth.

ARH-77 MM flank tumors were established in 12 NOD/SCID mice, then randomized to receive intraperitoneal injection with 5mg/kg of LLL12 or DMSO. Tumor volume was measured every other day and tumor mass was determined after 26 days. (A–C) LLL12 statistically significantly (*, all comparisons shown p < 0.05) impaired MM tumor cell growth as assessed by serial volume measurements and tumor mass. (D) Western blotting demonstrated inhibition of STAT3 phosphorylation in response to LLL12 therapy. (E) The body weights of LLL12-treated mice were not decreased and similar to that of the vehicle-treated mice over 15-day of treatments.
Discussion
MM remains mostly incurable despite the advent of novel therapies and, with the incidence of the disease rising, new treatments are urgently needed. STAT3 plays a crucial role in MM, proliferation, resistance to apoptosis, and survival 33–35 and represents an important molecular target for the development of novel therapies 2, 4, 33–35. However, to date, the translation of anti-STAT3 therapies into clinical trials has been difficult 33, 36. Previous methods aimed at blocking STAT3 have included the use of RNA interference, STAT3 antisense oligonucleotides, and dominant negative STAT3 7, 37, 38. Although the stated approaches have been successful limitations apply to the RNA or antisense oligonucleotides delivery and stability 39, 40. To our knowledge, no direct STAT3 inhibitors have begun assessment in clinical studies to date. The purpose of the present work was to characterize a novel STAT3 inhibitor, LLL12, as a potential therapy for MM, and as research tools to study the STAT3 pathway in cancer.
LLL12 is a novel small molecule inhibitor of STAT3 with high specificity to the Tyr705 residue mediating activation of STAT3 signaling via phosphorylation. Herein, we present the first data characterizing LLL12 as a promising, novel therapy for MM. We show that LLL12 prevents phosphorylation of STAT3 and this effect is associated with caspase-mediated apoptosis in MM cells. The phosphorylation of ERK(1/2) was increased in U266 cells but slightly reduced in ARH-77 cells by LLL12. In addition to suppressing constitutive activity of STAT3 in MM cell lines and primary tumor samples, our data show that LLL12 prevents STAT3 activation induced through cytokine stimulation, including IFN-α and IL-6. These effects appear STAT3-specific, in that no inhibitory effects of LLL12 on other STAT family members such as STAT1 induced by IFN-α and IFN-γ, STAT2 by IFN-α, and STAT4, STAT6 by IL-4 stimulation.
In blocking STAT3 phosphorylation, we demonstrate that LLL12 prevents STAT3 nuclear localization as well as DNA binding activity. Additionally, LLL12 leads to down regulation of expression of downstream targets of STAT3 known to be involved in proliferation and survival of MM cells providing further mechanistic detail regarding the pro-apoptotic effects of LLL12 in MM. These effects culminate in statistically significant, deleterious effects on MM cell in vitro viability as well as impaired in vivo MM tumor cell growth in a murine model.
Two limitations of the present dataset include the relatively high in vitro concentrations of LLL12 utilized as well as the lack of pharmacodynamic and pharmacokinetic data in the in vivo model. Further, ongoing work with LLL12 will address these issues which could represent limitations to clinical translation. None-the-less, these data provide additional proof-of-principle that STAT3 may be an attractive target for drug development in MM and LLL12 does warrant further research as a potentially promising novel therapy for patients with MM.
IL-6 is a key cytokine mainly produced by myeloma cells and a verity of other cells in the marrow microenvironment. A high IL-6 serum level is often associated to worse progression-free survival and overall survival in myeloma. IL-6 exerts its biological effects through binding to two signal transducing receptor subunits gp130 and IL-6R. IL-6 binding results in gp130 and IL-6R dimerization and in the subsequent activation of Janus kinases/STAT3 pathway. We observed that IL-6 can increase cell proliferation/viability in MM1.S cells and this is inhibited by small molecule, LLL12. This is consistent with the stimulation of STAT3 phosphorylation by IL-6 and inhibited by LLL12 in MM1.S cells. Treatments of MM1.S cells with lenalidomide also caused a dose-dependent reduction of cell viability and were reversed with IL-6 treatment. However, pre-treatment with LLL12 reversed the rescue of lenalidomide-mediated inhibition of cell viability by IL-6. We also observed U266 cells express higher levels of IL-6 and STAT3 phosphorylation compared to the MM.1S cells. Interestingly, U266 cells are also more resistant to lenalidomide than MM.1S cells. Treatment of LLL12 blocked lenalidomide-resistant in U266 cells suggesting that LLL12 is likely to enhance the effects of lenalidomide through the inhibition of IL-6/P-STAT3 pathway in U266 cells. Therefore, these results support that IL-6 conferred resistance to lenalidomide and this promoting activity was blocked by STAT3 inhibitor, LLL12.
Constitutive STAT3 activity has also been implicated in chemoresistance against a number of effective anti-MM therapies 13, 34, 35, 41. More recently, it has been suggested that STAT3 is also implicated in emergent resistance to lenalidomide and bortezomib 42, 43. A number of studies, by members of our group and others, have demonstrated that curcumin, a natural product derived from Curcuma longa, may enhance the effects of anti-cancer agents against MM and other cancers through STAT3 inhibition 35, 43, 44 However, curcumin also has a number of off-target effects in addition to STAT3 inhibition and doses associated with biologic activity are associated with clinical toxicities 44. In contrast, LLL12 is not a derivative from curcumin and the inhibitory effects of LLL12 appear STAT3-selective, in that no effects of LLL12 on twelve other human protein kinases were observed. Interestingly, in samples procured from patients with disease clinically-resistant to lenalidomide and bortezomib, we still observed potent anti-STAT3 effects with LLL12 and related induction of apoptosis, suggesting by inhibiting STAT3 survival pathway, LLL12 is active in lenalidomide and bortezomib resistant myeloma cells, at least in the in vitro experiments.
In conclusion, our findings suggest LLL12 holds promise for further development as a novel therapy for MM. As constitutive and/or IL-6-mediated activation of STAT3 is a common occurrence in MM, the pathway represents an attractive target for development of targeted therapies. Inhibition of STAT3 in normal cells yields no apparent deleterious effects 13, 20, 45–47, suggesting that blocking STAT3 signaling may not be grosslytoxic in normal cells. Moreover, STAT3 inhibition in immune effector subsets may enhance anti-tumor cytotoxicty 48. These data support ongoing translational work with LLL12 as a novel therapy for MM.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This research was partly funded by The National Foundation for Cancer Research, and NIHR21 grant (R21CA133652-01) to Jiayuh Lin and funding from Multiple Myeloma Opportunities for Research and Education (MMORE) to Don M Benson, Jr. We thank Dr. Rong Bai for doing the Mann-Whitney non-parametric test.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54. [PubMed] [Google Scholar]
- 3.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–73. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 4.Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–26. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- 5.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 6.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 7.Kaptein A, Paillard V, Saunders M. Dominant negative stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J Biol Chem. 1996;271:5961–4. doi: 10.1074/jbc.271.11.5961. [DOI] [PubMed] [Google Scholar]
- 8.Faruqi T, Gomez D, Bustelo X, Bar-Sagi D, Reich N. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci U S A. 2001;98:9014–9. doi: 10.1073/pnas.161281298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM, Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002;21:7611–8. doi: 10.1038/sj.onc.1206004. [DOI] [PubMed] [Google Scholar]
- 10.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–73. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 11.Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell cycle. 2005;4:1131–3. doi: 10.4161/cc.4.9.1985. [DOI] [PubMed] [Google Scholar]
- 12.Schlessinger K, Levy DE. Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res. 2005;65:5828–34. doi: 10.1158/0008-5472.CAN-05-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–15. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 14.Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin’s lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin Cancer Res. 2003;9:316–26. [PubMed] [Google Scholar]
- 15.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, Karras JG, Levy DE, Inghirami G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11:623–9. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 16.Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, Yoshikawa K, Akira S, Takeda J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. Embo J. 1999;18:4657–68. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda K, Akira S. Multi-functional roles of Stat3 revealed by conditional gene targeting. Arch Immunol Ther Exp (Warsz) 2001;49:279–83. [PubMed] [Google Scholar]
- 18.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr W, Jove R, Pardoll D, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 19.Bhasin D, Cisek K, Pandharkar T, Regan N, Li C, Pandit B, Lin J, Li PK. Design, synthesis, and studies of small molecule STAT3 inhibitors. Bioorg Med Chem Lett. 2008;18:391–5. doi: 10.1016/j.bmcl.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102:4700–5. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S, Foust E, Sobo M, Friedman L, Bhasin D, Cen L, Li C, et al. A Novel Small Molecule, LLL12, Inhibits STAT3 Phosphorylation and Activities and Exhibits Potent Growth-Suppresive Activity in Human Cancer Cells. Neoplasia. 2010;12:39–50. doi: 10.1593/neo.91196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durie BG, Harousseau JL, Miguel JSBJ, Barlogie B, Anderson K, Gertz M, Dimopoulos M, Westin J, Sonneveld P, Ludwig H, Gahrton G, Beksac M, et al. International uniform response criteria for multiple myeloma. leukemia. 2006;20:1467–73. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 23.Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, Kondo S, Priebe W, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007;26:2435–44. doi: 10.1038/sj.onc.1210031. [DOI] [PubMed] [Google Scholar]
- 24.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder J, Freedman M, Cohen A, Gazit A, Levitzki A, Roifman C. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–8. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 25.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–42. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 26.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–6. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shain K, Yarde D, Meads M, Huang M, Jove R, Hazlehurst L, Dalton W. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69:1009–15. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–64. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen M, Kaestel C, Eriksen K, Woetmann A, Stokkedal T, Kaltoft K, Geisler C, Ropke C, Odum N. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia. 1999;13:735–8. doi: 10.1038/sj.leu.2401415. [DOI] [PubMed] [Google Scholar]
- 30.Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–76. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuh B, Sobo M, Cen L, Josiah D, Hutzen B, Cisek K, Bhasin D, Regan N, Lin L, Chan C, Caldas H, DeAngelis S, et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma multiforme cell growth and prolongs survival in a mouse glioblastoma multiforme model. Br J Cancer. 2009;100:106–12. doi: 10.1038/sj.bjc.6604793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shain K, Yarde D, Meads M, Huang M, Jove R, Hazlehurst L, Dalton W. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69:1009–15. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhardwaj A, Sethi G, Vadhan-Raj S, Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S, Aggarwal BB. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood. 2007;109:2293–302. doi: 10.1182/blood-2006-02-003988. [DOI] [PubMed] [Google Scholar]
- 34.Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, Estrov Z, Talpaz M, Aggarwal BB. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–84. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 35.Bharti A, Donato N, Aggarwal B. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol. 2003;171:3863–71. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 36.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005;65:2532–6. doi: 10.1158/0008-5472.CAN-04-2425. [DOI] [PubMed] [Google Scholar]
- 38.Barton BE, Karras JG, Murphy TF, Barton A, Huang HF. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol Cancer Ther. 2004;3:11–20. [PubMed] [Google Scholar]
- 39.Sledz CA, Williams BR. RNA interference in biology and disease. Blood. 2005;106:787–94. doi: 10.1182/blood-2004-12-4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stein CA. The experimental use of antisense oligonucleotides: a guide for the perplexed. J Clin Invest. 2001;108:641–4. doi: 10.1172/JCI13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alas S, Bonavida B. Inhibition of Constitutive STAT3 Activity Sensitizes Resistant Non-Hodgkin’s Lymphoma and Multiple Myeloma to Chemotherapeutic Drug-mediated Apoptosis. Clin Cancer Res. 2003;9:316–26. [PubMed] [Google Scholar]
- 42.van der Spek E, Bloem AC, Lokhorst HM, van Kessel B, Bogers-Boer L, van de Donk NW. Inhibition of the mevalonate pathway potentiates the effects of lenalidomide in myeloma. Leuk Res. 2009;33:100–8. doi: 10.1016/j.leukres.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Park J, Ayyappan V, Bae EK, Lee C, Kim BS, Kim BK, Lee YY, Ahn KS, Yoon SS. Curcumin in combination with bortezomib synergistically induced apoptosis in human multiple myeloma U266 cells. Mol Oncol. 2008;2:317–26. doi: 10.1016/j.molonc.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bill MA, Bakan C, Benson DM, Jr, Fuchs J, Young G, Lesinski GB. Curcumin induces proapoptotic effects against human melanoma cells and modulates the cellular response to immunotherapeutic cytokines. Mol Cancer Ther. 2009;8:2726–35. doi: 10.1158/1535-7163.MCT-09-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niu G, Heller R, Catlett-Falcone R, Coppola Jaroszeski M, Dalton W, Jove RHY. Gene Therapy with Dominant-Negative STAT 3 Suppresses Growth of the Murine Melanoma B16 Tumor in Vivo. Cancer Res. 1999;59:5059–63. [PubMed] [Google Scholar]
- 46.Burke W, Jin X, Liu R, Huang M, Reynolds RK, Lin J. Inhibition of constitutively active Stat3 pathway in ovarian and breast cancer cells. Oncogene. 2001;20:7925–34. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 47.Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, Jove R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001;98:7319–24. doi: 10.1073/pnas.131568898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr W, Jove R, Pardoll D, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–21. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.