

Abstract
The recent crystal structure determinations of druggable class A G protein-coupled receptors (GPCRs) has opened up excellent opportunities in structure-based ligand discovery for this pharmaceutically important protein family. We have developed and validated a customized structure-based virtual fragment screening method against the recently determined human histamine H1 receptor (H1R) crystal structure. The method combines molecular docking simulations with a protein-ligand interaction fingerprint (IFP) scoring method. The optimized in silico screening approach was successfully applied to identify a chemically diverse set of novel fragment-like (≤ 22 heavy atoms) H1R ligands with an exceptionally high hit rate of 73%. Of the 26 tested fragments, 19 compounds had affinities ranging from 10 μM to 6 nM. The current study shows the potential of in silico screening against GPCR crystal structures to explore novel, fragment-like GPCR ligand space.
Keywords: Human histamine H1 receptor (hH1R), structure-based virtual fragment screening (SBVFS), fragment-based drug design (FBDD), docking, interaction fingerprints (IFPs), G protein-coupled receptor (GPCR), Protein-Ligand ANT System (PLANTS)
INTRODUCTION
G protein-coupled receptors (GPCRs) comprise the largest family of transmembrane proteins, mediating the intracellular signals of a wide array of signaling molecules and playing an essential role in numerous cellular and physiological effects.1 Knowledge of the three-dimensional structure of GPCRs provides valuable insights into receptor function and receptor-ligand interactions,2, 3 and is key for the in silico discovery of new bioactive molecules that can target this family of pharmaceutically relevant drug targets.4, 5 The human histamine H1 receptor (hH1R) is a key player in allergic responses and so-called ‘antihistamines’ are widely used to relieve the symptoms of allergic rhinitis by inhibiting the constitutive activity of H1R, as well as antagonizing histamine binding to the H1R.6 Recently the first inverse agonist bound H1R crystal structure was solved,7 opening up new possibilities in the rational design and in silico discovery of novel H1R ligands. GPCR homology models have already been used successfully to identify new ligands,5, 8–19 but the increasing number of GPCR X-ray structures solved in the last few years offers unique opportunities to push the limits of structure-based virtual screening (SBVS).4, 20–23 With more and detailed structural information, one should be able to increase hit rates of SBVS campaigns and to specifically apply the in silico approach in the field of fragment-based drug discovery (FBDD).24, 25 FBDD is a new paradigm in drug discovery that utilizes small molecules (≤ 22 heavy atoms) as starting points for efficient hit optimization.24 While previous GPCR SBVS campaigns have mainly identified larger molecules,5 the aim of the current study was to overcome the challenges of structure-based virtual fragment screening; the in silico discovery of smaller, fragment-like molecules, based on the recently elucidated crystal structure of the H1R. Although more than 70% of H1R ligands have a heavy atom count higher than 22 (Fig. 1A), doxepin, the co-crystallized high affinity inverse agonist in the H1R X-ray structure,7 can be considered as a large fragment-like compound, containing 21 heavy atoms.24 We have developed and validated a target-customized, docking-based virtual screening method which combines molecular docking with a novel protein-ligand interaction scoring method.26 This optimized SBVS method was successfully applied to identify novel fragment-like H1R ligands with an exceptionally high hit rate. The current study shows the potential of in silico screening against GPCR crystal structures to explore novel fragment-like ligand space and to investigate the fine atomic details of molecular recognition by this pharmaceutically relevant family of protein targets.
Figure 1.

A four panel overview of the preparation, validation, selection and screening process. (A)29 Distribution of the heavy atom count for known H1R ligands (ChEMBLdb (red) and CNS active drugs (orange)), decoys used for retrospective validation (gray), fragment-like compounds from ZINC used for prospective virtual screening (black), and in silico hits selected by our structure-based virtual screening method (blue) is shown. (B) Scatter plot of PLANTS-scores versus IFP-scores for known actives from the ChEMBLdb (orange) and CNS drugs29 (cyan) and physicochemically similar decoys (gray). (C) Overview of the structure-based virtual screening post-processing steps of 108790 fragment-like, basic compounds, which resulted in final selection of 26 fragment-like compounds: D3.32 filter: docking poses making an ionic interaction with D1073.32; model cutoff: compound for which docking poses are generated with IFP Tc ≥ 0.75 and PLANTS ≤ 90 (not necessarily the same pose); consistency cutoff: only compounds with an IFP-score ≥ 0.7 according to the best PLANTS pose as well as a PLANTS-score ≤ −75 according to the best IFP pose were selected; novelty filter: ECFP-4 Tanimoto similarity < 0.40 to any known H1R ligand; visual inspection: close analogues with highest IFP score are kept, compounds for which buried polar groups are placed in hydrophobic parts of the binding site in all filtered docking poses are discarded). The number indicates the number of compounds present at each step. (C) SCA-plot of the PLANTS-scores versus the IFP-scores for the fragment screening dataset (gray) with the selected compounds (blue). The dotted lines in B and D indicate the selected model cutoffs (IFP Tc ≥ 0.75 and PLANTS ≤ 90).
RESULTS AND DISCUSSION
Development and validation of a customized structure-based virtual screening approach
We performed docking experiments with PLANTS27, using the H1R crystal structure on 543 known H1R ligands from the ChEMBL database28 (Ki ≤ 10 μM) and 59 CNS active drugs acting as inverse agonists on H1R29, as well as 7088 decoys with physicochemical properties similar to the ChEMBLdb actives (Fig. 1A, Supplementary Table 1). PLANTS combines an ant colony optimization algorithm with an empirical scoring function30 for the prediction and scoring of binding poses in a protein structure. The resulting docking poses were post-processed using molecular interaction fingerprints (IFPs).31 The IFP scoring method determines ligand binding mode similarity to experimentally supported ligand binding poses (Fig. 2). IFPs have been used as an efficient alternative post-processing method of docking poses26, 31 to overcome target dependent scoring problems.32 Seven different interaction types (negatively charged, positively charged, H-bond acceptor, H-bond donor, aromatic face-to-edge, aromatic face-to-face, and hydrophobic interactions) were used to define the IFP. A Tanimoto coefficient (Tc-IFP) measuring IFP similarity with the reference doxepin pose in the H1R crystal structure (Fig. 2), was used to score the docking poses of known actives and decoys.31 The scatterplot in Fig. 1B shows that active compounds can be discriminated from decoys by considering both the best IFP score and best PLANTS score for each compound, and the Kernel density plot for the CHEMBLdb actives in Supplementary Figure 2 clearly demonstrates that most actives can obtain binding modes in the H1R binding site which: i) are similar to the binding mode of doxepin (indicated by high IFP Tanimoto similarity scores), and ii) are energetically favorable (high (negative) PLANTS docking scores). Based on this analysis we determined IFP (Tc ≥ 0.75) and PLANTS (≤ −90) cutoffs to discriminate H1R ligands from decoys (Table 1). This combination of binding mode similarity and docking scores resulted in high retrospective virtual screening enrichments33 of known H1R ligands from the ChEMBLdb (39-fold enrichment of over random picking) and CNS drugs (58-fold enrichment) test sets over decoys with similar physical-chemical properties (Table 1).
Figure 2.

The binding pose of doxepin (magenta carbon atoms, panel A) in the H1R structure (PDB ID 3RZE) and the predicted binding poses of the novel fragment-like H1R ligands identified by prospective structure-based virtual screening: 3 (orange, B), 4 (gold, C) and 5 (green, D). The IFPs corresponding to the compounds in the displayed pose are partially presented (E). Parts of the backbone of transmembrane (TM) helices 3 4, 5, 6 and 7 are represented by transparent light yellow ribbons. Important binding residues are depicted as ball-and-sticks with grey carbon atoms. Oxygen, nitrogen, and hydrogen atoms are coloured red, blue and cyan, respectively. H-bonds described in the text are depicted by black dots. The IFP bit strings of the docking poses of the hits 3–5 (B–D) are compared to the reference IFP of doxepin 1 (A) in panel E, encoding different interaction types with each residue in the binding site. For reasons of clarity, the bit strings of only 6 residues (out of 33) are shown as an example.
Table 1.
Retrospective virtual screening accuracy of different SBVFS scoring methods. Percentage of known H1R ligands (from ChEMBLdb and CNS drugs29 sets) and decoys in the regions defined by IFP-Tc (≥ 0.75), PLANTS docking score (≤ −90), and combined IFP-Tc and PLANTS docking score cutoffs (Fig. 1B, D)
| Combination IFP (≥ 0.75) and PLANTS (≤ −90) filters | IFP (≥ 0.75) filter only | PLANTS (≤ −90) filter only | ||||
|---|---|---|---|---|---|---|
| ChEMBLdb | CNS drugs | ChEMBLdb | CNS drugs | ChEMBLdb | CNS drugs | |
| Actives (%)a | 39.3 | 57.6 | 48.0 | 69.5 | 50.7 | 76.3 |
| Decoys (%)b | 1.0 | 1.0 | 3.4 | 3.4 | 6.4 | 6.4 |
| Enrichmentc | 39.3 | 57.6 | 14.1 | 20.4 | 7.9 | 11.9 |
Actives in the ChEMBLdb set are defined by an pKi≥5 and in the CNS actives set by a pIC50 ≥5.
The decoy set consists of ~7000 compounds, selected based on the physicochemical properties of the ChEMBLdb dataset.
The enrichment reported here is the percentage of retrieved true positives (TP) divides by the percentage of retrieved false positives, TP(%)/FP(%), at the specified cutoffs.
Structure-based identification of novel fragment-like H1R ligands with exceptionally high virtual screening hit rates
The validated SBVFS method was subsequently used to identify new fragment-like H1R ligands from a subset of 108790 molecules extracted from 13 milion commercially available compounds (Fig. 1C) stored in the ZINC database34 which: i) obey fragment rules based on rule-of-three35, 36 (number of heavy atoms ≤ 22, logP < 3, number of H-bond donors ≤ 3, number of H-bond acceptors ≤ 3, number of rotatable bonds ≤ 5, number of rings ≥ 1), and ii) contain a basic moiety (to enable ionic interactions with the essential D1073.32 residue37 in the H1R binding pocket (Fig. 2)). The docking poses of 354 fragments were capable of making an ionic interaction with D1073.32 (ref 37) and complied with the optimized IFP and PLANTS score cutoffs as well as the consistency cutoffs (see Methods, Fig. 1C and Supplementary Fig. S3). Interestingly, this set contained 9 compounds with known activity on the H1 receptor (Ki ≤ 10 μM in ChEMBLdb28), of which 7 compounds are FDA approved drugs (Epinastine, Mianserin, Triprolidine, Promazine, Amoxapine, Imipramine and Desipramine, see Supplementary Table 2). In fact, 282 of the 354 compounds were chemically dissimilar to any known H1R ligand (ECFP-4 Tanimoto similarity < 0.4038). This set of novel fragments was visually clustered and for each cluster the fragment with highest IFP and/or PLANTS score was selected. Fragments for which buried polar groups were placed in hydrophobic parts of the H1R binding site in all filtered docking poses were discarded based on visual inspection. This resulted in a final selection of 30 compounds, of which 26 were actually available and experimentally tested for their binding affinity to H1R (Fig. 1C). Of the 26 experimentally tested compounds 19 had affinities ranging from 10 μM to 6 nM for H1R (Fig. 2, Table 2). Seven fragments have submicromolar affinity (3–9, Fig. 3A–B) and fragment 3 (Ki = 6 nM) has one of the highest affinities reported for a GPCR ligand identified by structure-based virtual screening.12, 39 Nine of the 19 H1R binders (3–8, 10, 13, and 15) were characterized as inverse agonists in histamine stimulated inositol phosphate (InsP) accumulation assays, while fragment 9 was determined to be a very weak partial agonist (intrinsic activity of 0.07). When tested against histamine (0.1 μM), doxepin (1), mepyramine (2) and fragment 3 were able to completely inhibit the histamine-induced effects. Also the other fragment hits with submicromolar affinities were able to interfere with the agonist response (Supplementary Fig. S6). Similar to the doxepin binding mode in the H1R crystal structure7, (Fig. 2A), the predicted docking poses of the novel inverse agonists in H1R (Fig. 2B–D) make extensive hydrophobic interactions with W6.48, a highly conserved key residue in GPCR activation.40 This observation is in line with the hypothesis that inverse agonists of H1R should be able to lock W6.48 in an inactive conformation to reduce H1R basal activity.7, 41
Table 2.
Histamine H1 (H1R) receptor binding affinities of validated fragment-like hits (Ki ≤ 10μM) selected by an optimized structure-based virtual screening protocol (Fig. 2) against the H1R crystal structure. PLANTS docking scores, interaction fingerprint similarities (IFP) and 2D (ECFP-4) and 3D (ROCS) chemical similarities to doxepin (binding mode), as well as closest chemical similarity to any known H1R ligand are given for each validated hit.
| cpd | pKi H1Ra,b | IFP (rank)c | PLANTS (rank)d | ROCSDOX (rank)e | ECFP-4DOX (rank)f | ECFP4g | closest known H1R ligandf | |
|---|---|---|---|---|---|---|---|---|
| 1 |

|
9.75 ± 0.14 | - | - | - | - | - | - |
| 2 |

|
8.68 ± 0.03 | - | - | - | - | - | - |
| 3 |
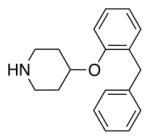
|
8.20 ± 0.10 | 0.75 (1836) | −102.14 (429) | 1.416 (146) | 0.16 (8677) | 0.34 |
|
| 4 |

|
7.21 ± 0.03 | 0.77 (1326) | −90.19 (6205) | 1.263 (3601) | 0.06 (102773) | 0.34 |

|
| 5 |

|
6.37 ± 0.06 | 0.75 (1926) | −92.3 (4270) | 1.287 (2819) | 0.09 (68145) | 0.25 |

|
| 6 |

|
6.27 ± 0.07 | 0.79 (500) | −109.00 (31) | 1.305 (2240) | 0.11 (45055) | 0.23 |

|
| 7 |

|
6.15 ± 0.08 | 0.78 (754) | −92.21 (4346) | 1.379 (699) | 0.21 (1206) | 0.28 |
|
| 8 |

|
6.15 ± 0.10 | 0.77 (1319) | −90.44 (5944) | 1.406 (399) | 0.07 (17047) | 0.32 |

|
| 9 |

|
6.10 ± 0.28 | 0.77 (1229) | −97.43 (1416) | 1.374 (707) | 0.14 (17047) | 0.38 |

|
| 10 |

|
5.75 ± 0.04 | 0.81 (312) | −98.61 (1095) | 1.311 (2061) | 0.13 (27979) | 0.25 |

|
| 11 |

|
5.72 ± 0.09 | 0.79 (518) | −100.13 (757) | 1.125 (17913) | 0.04 (108215) | 0.27 |

|
| 12 |

|
5.64 ± 0.03 | 0.77 (1294) | −91.51 (4934) | 1.402 (425) | 0.14 (16574) | 0.24 |

|
| 13 |

|
5.58 ± 0.14 | 0.78 (689) | −98.13 (1240) | 1.178 (10007) | 0.13 (29867) | 0.32 |

|
| 14 |

|
5.49 ± 0.04 | 0.82 (183) | −97.56 (1419) | 1.356 (972) | 0.11 (47280) | 0.30 |

|
| 15 |

|
5.38 ± 0.03 | 0.85 (83) | −93.98 (3060) | 1.325 (1605) | 0.11 (40224) | 0.34 |

|
| 16 |

|
5.34 ± 0.14 | 0.77 (1278) | −92.53 (4082) | 1.356 (972) | 0.16 (7054) | 0.26 |

|
| 17 |

|
5.27 ± 0.04 | 0.78 (744) | −92.93 (3775) | 1.106 (22077) | 0.12 (39372) | 0.31 |

|
| 18 |

|
5.20 ± 0.13 | 0.81 (308) | −100.63 (675) | 1.296 (2535) | 0.16 (9372) | 0.28 |

|
| 19 |

|
5.09 ± 0.07 | 0.83 (115) | −102.04 (446) | 1.375 (690) | 0.14 (17769) | 0.29 |

|
| 20 |

|
4.97 ± 0.10 | 0.85 (43) | −102.63 (372) | 1.301 (2368) | 0.15 (11737) | 0.24 |
|
| 21 |

|
4.96 ± 0.02 | 0.78 (719) | −95.17 (2397) | 1.016 (47198) | 0.13 (11736) | 0.34 |

|
pKi values are calculated from at least three independent measurements as the mean ± SEM.
Measured by displacement of [3H]-mepyramine binding using membranes of HEK293T cells transiently expressing the human H1R.
IFP Tanimoto similarity with doxepin pose in the H1R crystal structure. Optimized IFP score cutoff ≥ 0.75. IFP ranking is given between brackets.
Score and rank accoriding to PLANTS scoring function.30 Optimized PLANTS score cutoff ≤ −90. PLANTS ranking is given between brackets.
ROCS shape-based 3D similarity to doxepin based on Comboscore. 48 ROCS ranking is given between brackets.
ECFP-4 2D Tanimoto similarity to doxepin. A similarity higher than 0.40 is considered as significative.38 ECFP-4 ranking is given between brackets.
ECFP-4 circular fingerprint Tanimoto similarity to closest known H1R active in ChEMBLdb. A similarity higher than 0.40 is considered as significative.38
Figure 3.

Radioligand displacement by (A, B) and functional effects of (C) cpds 1–9. A, B: Displacement curves of [3H]-mepyramine in HEK293T cells transiently transfected with hH1R (n=3, each performed in triplicate). C: Inhibition of histamine-stimulated inositol phosphate accumulation assay in HEK293T cells transiently transfected with hH1R by cpds 1–9 (n=2, each performed in triplicate).
The hit rate of 73% (19 out 26 tested compounds) of our SBVS study is exceptionally high compared to other prospective studies.42 In fact, to the best of our knowledge, this is the highest hit rate reported for any prospective SBVS campaign reported for GPCRs5 as shown in Fig. 4. It should be noted that our in silico screening approach combines a docking scoring function with a molecular interaction fingerprint scoring method using knowledge of the co-crystallized ligand binding mode, while many of the other recent SBVS runs against GPCR crystal structures used only energy-based scoring functions to rank the docking poses.20–22 However, like in our study, all these virtual screening exercises also included additional selection criteria (e.g., complementarity to the binding site, clustering/novelty, polarity) to select ca. 10% of the top hit list. Furthermore, our analysis of 15 previously published GPCR SBVS campaigns8–22 indicates that our experimentally validated hit set also has the lowest average heavy count of all GPCR SBVS studies (Fig. 4). This makes the high virtual screening hit rate even more remarkable because of the typically low-affinity fragment-protein interactions.
Figure 4.

Hit rate and size of hits identified in prospective structure-based virtual screening studies against GPCR crystal structures20–22 and homology models8–19. The bars shown for the heavy atom count indicate the minimum and maximum heavy atom count for all hits of each SBVS. The labels indicate the screening on the following receptors adenosine alpha-2a receptor (A2A_121 and A2A_222), adrenergic alpha-1a receptor (ADA1A10), adrenergic beta-2 receptor (ADRB220), complement component 3a receptor 1 (C3A19), C-C chemokine receptor type 5 (CCR514), cannabinoid receptor 2 (CNR213), dopamine receptor D3 (DRD38), free fatty acid receptor 1 (FFAR118), formyl peptide receptor 1 (FPR111), histamine receptor H1, histamine receptor H4 (H417), melanin-concentrating hormone receptor 1 (MCH1_115 and MCH1_29), neurokinin 1 receptor (NK1R12) and transferrin receptor protein 1 (TRFR116). The maximum heavy atom count of FPR1 (41) is not shown for clarity purposes. Only hits for which: i) binding affinity (Ki ≤ 15 μM) or potency (EC50 ≤ 15 μM) was experimentally determined; and ii) for which a molecular structure was reported are included in the analysis.
While IFP rankings are in most cases higher than PLANTS score ranking (Table 2), it should be noted that our optimized screening approach enabled the selection of novel H1R ligands from a small subset of fragments (Fig. 1D). Moreover, most of the validated hits would not have been selected using only one of the two SBVS techniques applied. The top 300 compounds of the IFP and PLANTS ranking lists (of the 2274 and 6416 fragments passing the IFP and PLANTS filters) contain only four and one of the validated fragments, respectively (see Figure 1C, Table 2 and Supplementary Figure 3). Compound 4 for example, one of the 7 validated hits with submicromolar affinity for H1R (Ki = 62 nM), is ranked only 1326th and 6205th in the IFP and PLANTS hits list, respectively, but is one of the few (354) fragments (corresponding to 0.325% of the initial fragment dataset) which passed the combined IFP and PLANTS filter (see Figure 1C and Supplementary Figure 3). Apparently the combination of binding mode similarity with the experimentally supported H1R-doxepin pose (determined by IFP) and an energetically favorable H1R-ligand configuration (assessed by the PLANTS docking score) was required to obtain this high hit rate of novel in silico predicted fragment-like H1R ligands. Further research is needed to determine whether this customized virtual screening approach is generally applicable to other targets as well. While general energy-based scoring functions have been successfully applied in recent virtual screening studies against GPCR crystal structures,20–22 the recent community wide GPCR DOCK 2010 challenge showed that a customized and experimentally supported modeling methodology can improve the prediction of GPCR-ligand interactions.43 Moreover, concensus scoring strategies44, 45 as well as target-customized scoring strategies,5, 46 have been successfully applied in structure-based virtual screening exercises (and in GPCR-based in silico screening studies8–19 in particular).
Interaction fingerprint (IFP) based virtual screening explores novel fragment-like ligand space
Interestingly, 18 out of the 19 experimentally validated hits do not rank within the top 300 (a selection comparable to the 282 fragments we clustered and visually inspected) of a 2D topological (ECFP-4)47 or 3D shape-based (ROCS)48 similarity searches of the fragment library against doxepin, the co-crystallized ligand in the H1R X-ray structure (Table 2). A combination of previously defined, minimal ECFP-4 (Tanimoto ≥ 0.2649 and ROCS Comboscore score ≥ 1.2050) cutoffs yields a large hit list of 1536 compounds (1.5% of the initial fragment dataset), containing none of the validated hits (Supplementary Fig. 4). Only the high affinity compound 3 (Ki = 6.2 nM) is ranked relatively high (146th) in the ROCS list with a Comboscore of 1.416 (but is ranked 8677th in the ECFP-4 list). This indicates that 3 has a similar three-dimensional pharmacophore and can adopt a similar shape as doxepin in H1R (Fig. 2A). This is confirmed by the docking pose presented in Fig. 2B. Compound 3, containing a piperazine ring and a linear chain connecting two benzene rings, is chemically dissimilar from doxepin (and any other known H1R ligand) which combines a linear amine head with a tricyclic ring system (Table 2). Both ligands, however, make the same ionic and aromatic interactions with D1073.32 (ref 37, 51) and W1584.56/F1995.47/W4286.48/F4326.52/F4356.55 (ref 37, 51, 52), respectively, by placing their amine group and benzene rings in almost exactly the same locations in the H1R binding pocket (Fig. 2A–B). Another high affinity hit, compound 4 (Ki = 62 nM), combines a basic piperazine group with a pyrazolo[3,4-d]pyrimidine ring system and has a low 2D and 3D similarity with doxepin or any other known H1R ligand (Table 2, dFig. 2C). Data mining of the ChEMBLdb indicates that the chemically complex pyrazolo[3,4-]pyrimidine scaffold is included in adenosine receptor53 and corticotropin receptor (CRFR1)54 ligands, but has so far not yet been incorporated in ligands of bioaminergic GPCRs. In the docking pose of 4, the pyrazolopyrimidine group and its pyrrolidine substituent mimic the benzene rings of doxepin by binding in the same aromatic cavity between TM helices 3, 4, 5, and 6 (Fig. 2C). Chemically complex bicyclic or tricyclic aromatic ring systems which have been not yet included in histamine H1R ligands are also present in validated virtual screening hits 5 (dihydrobenzo-imidazo-triazine), 7 (tetrazoloquinazoline), 8 (tienopyrimidine), 9 (pyrolopyridine), 13 (benzoamidazotriazole), 15 (triazoloindazole) and 17 (benzofuropyrimidine) and play the same role in aromatic pi stacking with W1584.56/F1995.47/W4286.48/F4326.52/F4356.55. While non-cyclic amine groups (6–7, 9, 12, 16, 18–21), (homo)piperidine (3, 10, 13, 15), and piperazine (4, 8, 11, 14, 17) are typical basic groups in bioaminergic GPCR ligands, compound 6 forms an ionic link with the negatively charged carboxylate group of the conserved D3.32 residue55, 56 via a dihydrobenzo-imidazo-triazine-amine group (Fig. 2D). This demonstrates the potential of the IFP scoring method to identify novel ligands with alternative chemical groups which allow formation of the same protein-ligand interactions.57 This scaffold hopping potential of our structure-based virtual fragment screening approach is further emphasized by the fact that none of the hits are chemically similar to any known H1R ligand (ECFP-4 Tanimoto similarity < 0.4038, Table 2). Scaffold diversity analysis58 indicated that the validated fragment hits cover the complexity vs. cyclicity space of known fragment-like H1R ligands (Fig. 5). Many of the hits have a high complexity and cyclicity score, including high affinity hits 3–5, while submicromolar hit 9 has relatively low complexity and cyclity scores compared to other fragment-like H1R ligands. Together with the ECFP-4 score reported in Table 2 this clearly indicates that our SBVFS approach is able to successfully retrieve a diverse set of ligands for the H1R receptor.
Figure 5.

SCA plot showing the distribution of experimentally validated structure-based virtual screening hits of H1R (blue dots) in the chemical space covered by previously fragment-like H1R ligands in the ChEMBLdb. The positions of all sub-micromolar affinity hits are indicated by the numbers (see Table 2).
Two of the H1R hits, compounds 13 and 14, have affinity for H3R, with Ki values of 0.6 and 0.1 μM, respectively (Supplementary Table 3). Two other H1R hits, 4 and 18 have medium affinity for H4R, (Ki values of 2.9 and 2.4 μM, respectively). On one hand this shows that our structure-based virtual screening protocol yields mainly histamine H1R subtype selective fragment-like ligands. Many known H3R ligands contain homopiperidines and substituted piperazines59, a scaffold present in H3R binders 13 and 14, respectively. Compounds 10 and 15 however demonstrate that homopiperidine ligands (without H3R affinity) can also bind H1R selectively. The discovery of fragment 4 and 18 offer new opportunities to develop a dual H1R-H4R ligand with synergistic anti-inflammatory properties.60 For example, the pyrazolo methyl group of fragment 4 could be used as a handle to grow the ligand towards the extracellular region of the binding site between TM5 and TM6 (Fig. 2C) to fine tune H1R and H4R affinity by introducing groups matching previously identified interaction hot spots for these receptors (e.g., K1915..39 in H1R, and L1755.39 in H4R).51, 61, 62 Moreover, experimentally determined (or modelled) binding modes of more selective H1R ligands than doxepin, like the zwitterion ceterizine which is expected to bind to K1915.39 in the “anion” binding pocket7, 51 might serve as IFP26 references for future structure-based virtual screening studies.
CONCLUSION
In conclusion, we have developed a structure-based virtual fragment screening method with which we efficiently identified new fragment-like H1R ligands with an exceptionally high hit rate of 73%, the highest reported for GPCRs. Many of the identified fragments are both promising and challenging new starting points for structure-based ligand optimization. The current study shows the potential of in silico screening against GPCR crystal structures to explore novel, fragment-like GPCR ligand space.
METHODS
Residue numbering and nomenclature
The Ballesteros–Weinstein residue numbering scheme29 was used throughout this manuscript. For explicitely numbered residues in specifc receptors, the UniProt63 residue number is given before the Ballesteros–Weinstein residue number in superscript (e.g. D1073.32 in H1R).
Preparation of retrospective validation databases
For the retrospective validation of our structure-based virtual screening protocol, two independent H1R ligand test sets were prepared: 543 known active H1R compounds from the ChEMBL database with Ki ≤ 10 μM (test set 1) and 59 CNS drugs with inverse agonist activity on H1R29 (test set 2). In order to avoid biasing virtual screening results, caution was given to select 7088 decoys from the BioInfo database64 covering similar property ranges (molecular weight, number of rotatable bonds, number of rings, hydrogen bond donor/acceptor counts, at least one positively charged atom) as H1R ligand test set 1 (Supplementary Table S1). SMILES (available as supporting information) were retrieved from the ChEMBLdb and plausible tautomers and protonation states were computed for these compounds with ChemAxon’s Calculator65 and converted into Mol2 format with Molecular Networks’ Corina.66
Preparation of prospective virtual screening database
From 15 vendors we downloaded their commercial compound datasets in SMILES format from the ZINC website (~ 13 million compounds). With use of Openeye’s filter (version 2.1.1)67, only fragment-like compounds were selected (757.728 compounds). Plausible tautomers and protonation states were computed for these compounds with Tauthor (version 1.4.90) and Blabber (version 1.4.90) respectively (both part of MolDiscovery’s MoKa package68). A second filter was applied to select only compounds with a formal charge of at least +1, this selection ensures that all selected compounds have the possibility for an ionic bond with key residue D1073.32 in the pocket (108790 compounds).
Automated docking
All virtual screenings were performed by docking program PLANTS (version 1.1)27. PLANTS combines an ant colony optimization algorithm with an empirical scoring function30 for the prediction and scoring of binding poses in a protein structure. For each compound, 25 poses were calculated and scored by the chemplp scoring function at speed setting 2. The binding pocket of H1R was defined by the coordinates of the center of co-crystallized doxepin in the 3RZE structure and a radius of 10.8 Å (which is the maximum distance from the center defined by a 5 Å radius around doxepin). All other options of PLANTS were left at their default setting.
IFP post-processing
The doxepin binding mode in the original H1R X-ray structure7 was used to generate reference interaction fingerprints (IFPs) as previously described.31 Seven different interaction types (negatively charged, positively charged, H-bond acceptor, H-bond donor, aromatic face-to-edge, aromatic-face-to- face, and hydrophobic interactions) were used to define the IFP. The cavity used for the IFP analysis consisted of the same set of 33 residues used in a previous retrospective structure-based virtual screening study26 (30 residues earlier proposed to define a consensus TM binding pocket56 plus three additional residues at positions 3.37, 5.47, and 7.40): L1.35, L1.39, I1.42, T1.46, V2.57, M2.58, N2.61, L2.65, W3.28, L3.29, D3.32, Y3.33, S3.36, T3.37, I3.40, W4.56, I4.60, F5.38, K5.39, T5.42, A5.43, N5.46, F5.47, F6.44, W6.48, Y6.51, F6.52, F6.55, H7.35, I7.39, W7.40, Y7.43, N7.45. Note that for each PLANTS docking pose, a unique subset of protein coordinates with rotated hydroxyl hydrogen atoms were used to define the IFP. Standard IFP scoring parameters, and a Tanimoto coefficient (Tc-IFP) measuring IFP similarity with the reference molecule pose (doxepin in the H1R crystal structure (Fig. 2A), was used to filter and rank the docking poses of 543 known active H1R compounds from the ChEMBLdb, 59 CNS drugs with inverse agonist activity on H1R, 7088 decoys and the focused database of 108790 fragment like molecules (only poses forming an H-bond and ionic interaction with D1073.32 are considered). The reference IFP bit string is available as Supporting Information.
Retrospective virtual screening analysis
H1R ligand test set 1 (543 known H1R ligands from ChEMBLdb), test set 2 (59 CNS active drugs with inverse agonist activity on H1R29) and the focused database of 7088 similar decoys were docked into H1R and scored with PLANTS and IFP. Based on optimal virtual screening enrichment of test set 1 (true positives) against the decoy set (false positives), IFP (Tanimoto similarity to doxepin ≥ 0.75) and PLANTS (≤ −90) score cutoffs were defined. In an independent retrospective virtual screening study, the enrichment of test set 2 against the decoy set was determined at these IFP and PLANTS cutoff values.
Prospective virtual screening
The screening database was docked with the same PLANTS protocol used for the retrospective validation. After post-processing the results using IFPs and filtering for the ionic and H-bond interaction with D1073.32, the previously mentioned cutoffs (Tc IFP ≥ 0.75 and PLANTS ≤ −90) were applied (see Fig. 1C–D). To further focus the dataset, we selected only compounds that had a consistently high PLANTS and IFP score for the best poses according to PLANTS and IFP; only compounds with an IFP-score ≥ 0.7 according to the best PLANTS pose as well as a PLANTS-score ≤ −75 according to the best IFP pose were selected (see Fig. 1C). To assess the novelty of the selected compounds, all remaining 354 compounds were compared using Pipeline Pilot’s ECFP-469 to the known active compounds and only compounds with an ECFP-4 score below 0.40 were selected (282 compounds).
ROCS 3D similarity search
The conformer database was generated using standard settings OMEGA70 and searched with ROCS71 using standard settings as well. The conformations of doxepin found in the H1R X-ray structure were used as query molecules for independent ROCS runs. Compounds were ranked by decreasing Comboscore71 (combination of shape Tanimoto and the normalized color score in this optimized overlay).
ECFP-4 2D similarity search
Two-dimensional similarity searches were carried out using ECFP-4 (extended connectivity fingerprints47) descriptors available in Pipeline Pilot69 and compared using the Tanimoto coefficient.
Scaffold diversity analysis
Scaffold diversity58 of the novel fragment hits in known H1R fragment-like ligand space was determined using the publicly available sca.svl script in MOE72. In this analysis, fragments are indexed by two parameters, that is, cyclicity and complexity. Cyclicity is the ratio between ring atoms and side chain atoms (thus, if all the atoms of the molecule belong to the ring structure cyclicity equals one). In addition, the complexity was calculated as a descriptor of the size and shape of the scaffold, taking into account the smallest set of smallest rings, the number of heavy atoms, the number of bonds between the heavy atoms, and the sum of heavy atoms atomic number.58
Compounds selected by virtual screening
The compounds selected by virtual screening were purchased from available screening collections of six vendors (Supplementary Tabe 4), Asinex (www.asinex.com), Chembridge (www.Hit2Lead.com), Enamine (www.enamine.com), IBScreen (www.ibscreen.com), Matrix Scientific (www.matrixscientific.com), Vitas-M (www.vitasmlab.com). Purity of compounds was verified by liquid chromatography-mass spectrometry (LC-MS) or nuclear magnetic resonance (NMR) experiments performed by the vendors.
Plasmids
Human H1R cDNA was kindly provided by Dr. H Fukui (Japan)73. Human H3R, H4R, and ADRB2 cDNA were obtained from the Missouri S&T cDNA Resource Center (www.cdna.org).
Cell culture and transfection
HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 50 IUml penicillin and 50 μg/ml streptomycin at 37°C and 5% CO2. Approximately 4 × 106 cells in 10-cm dishes were transiently transfected with 0.5 or 5 μg receptor DNA using 25-kDa linear polyethylenimine (PEI; Polysciences, Warrington, USA) as transfection reagent (1:4 DNA/PEI ratio), for inositol phosphate (InsP) accumulation or radioligand displacement assays, respectively.
Radioligand displacement assay
Cells were harvested 2 days after transfection and homogenized in 50 mM Tris-HCl binding buffer (pH7.4). Cell homogenates were co-incubated with indicated concentrations of fragment-like ligands and ~3 nM [3H]-mepyramine (H1R), ~1 nM [3H]-N-α-methylhistamine (H3R), ~10 nM [3H]-histamine (H4R), or ~2 nM [3H]-dihydroalprenolol (ADRB2) in a total volume of 100 μl/well. The reaction mixtures were for 1–1.5 hrs at 25°C on a microtiter shaker (750 rpm). Incubations were terminated by rapid filtration through Unifilter glass fiber C plates (PerkinElmer Life Sciences) that were presoaked in 0.3% polyethylenimine and subsequently washed three times with ice-cold binding buffer (pH7.4 at 4°C). Retained radioactivity was measured by liquid scintillation using a MicroBeta Trilux (PerkinElmer Life Sciences). Nonlinear curve fitting was performed using GraphPad Prism 4.03 software. The Ki values were calculated using the Cheng-Prusoff equation Ki = IC50/(1+[radioligand]/Kd)74.
Inositol phosphate accumulation assay
Twenty-four hours after transfection, cells were collected and seeded in Earle’s inositol-free minimal essential medium (Invitrogen) supplemented with 10% fetal bovine serum, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 1 μCi/ml myo-[2-3H]-inositol in poly-L-lysine-coated 48-well plates. The next day, cells were washed with DMEM supplemented with 25 mM HEPES, pH7.4, and 20 mM LiCl, and subsequently incubated with the indicated concentrations of fragment-like ligands in the absence or presence of 0.1 μM histamine for 1 hr at 37°C. Incubations were terminated by replacing the assay buffer with 10 mM formic acid. Next, accumulated inositol phosphates were isolated using anion exchange chromatography (Dowex AG1-X8 columns; Bio-Rad) and counted by liquid scintillation.
Supplementary Material
Acknowledgments
The authors thank Herman D. Lim for technical assistance with the H4R binding assays. This research was financially supported by the Netherlands Organization for Scientific Research (NWO) through a VENI grant (700.59.408 to C. de G.), by TI-Pharma through grant D1-105 (GPCR Forum to A.J.K.), and a NIGMS PSI:Biology grant (U54 GM094618 to R.C.S. and V.K.).
Footnotes
Additional analyses of the retrospective and prospective virtual screening studies, H1R, H3R, and H4R radioligand displacement curves, and InsP accumulation barplots, molecular structures of ligand test set (SMILES) and receptor mol2 coordinates (H1R crystal structure), PLANTS docking conf(iguration) file, reference IFP bit-strings and cavity coordinates used for IFP calculations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nature reviews Drug discovery. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein- coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Topiol S, Sabio M. X-ray structure breakthroughs in the GPCR transmembrane region. Biochemical pharmacology. 2009;78:11–20. doi: 10.1016/j.bcp.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Congreve M, Langmead CJ, Mason JS, Marshall FH. Progress in structure based drug design for g protein-coupled receptors. Journal of medicinal chemistry. 2011;54:4283–4311. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Graaf C, Rognan D. Customizing G Protein-coupled receptor models for structure-based virtual screening. Current pharmaceutical design. 2009;15:4026–4048. doi: 10.2174/138161209789824786. [DOI] [PubMed] [Google Scholar]
- 6.Leurs R, Church MK, Taglialatela M. H1-antihistamines: inverse agonism, anti- inflammatory actions and cardiac effects. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2002;32:489–498. doi: 10.1046/j.0954-7894.2002.01314.x. [DOI] [PubMed] [Google Scholar]
- 7.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, Wang S. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. Journal of medicinal chemistry. 2003;46:4377–4392. doi: 10.1021/jm030085p. [DOI] [PubMed] [Google Scholar]
- 9.Clark DE, Higgs C, Wren SP, Dyke HJ, Wong M, Norman D, Lockey PM, Roach AG. A virtual screening approach to finding novel and potent antagonists at the melanin-concentrating hormone 1 receptor. Journal of medicinal chemistry. 2004;47:3962–3971. doi: 10.1021/jm040762v. [DOI] [PubMed] [Google Scholar]
- 10.Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. Journal of medicinal chemistry. 2004;47:5381–5392. doi: 10.1021/jm0311487. [DOI] [PubMed] [Google Scholar]
- 11.Edwards BS, Bologa C, Young SM, Balakin KV, Prossnitz ER, Savchuck NP, Sklar LA, Oprea TI. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Molecular pharmacology. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 12.Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the alpha1A adrenergic receptor. Journal of medicinal chemistry. 2005;48:1088–1097. doi: 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- 13.Salo OM, Raitio KH, Savinainen JR, Nevalainen T, Lahtela-Kakkonen M, Laitinen JT, Jarvinen T, Poso A. Virtual screening of novel CB2 ligands using a comparative model of the human cannabinoid CB2 receptor. Journal of medicinal chemistry. 2005;48:7166–7171. doi: 10.1021/jm050565b. [DOI] [PubMed] [Google Scholar]
- 14.Kellenberger E, Springael JY, Parmentier M, Hachet-Haas M, Galzi JL, Rognan D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. Journal of medicinal chemistry. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 15.Cavasotto CN, Orry AJ, Murgolo NJ, Czarniecki MF, Kocsi SA, Hawes BE, O’Neill KA, Hine H, Burton MS, Voigt JH, Abagyan RA, Bayne ML, Monsma FJ., Jr Progress in structure based drug design for g protein-coupled receptorsProgress in structure based drug design for g protein-coupled receptors. Journal of medicinal chemistry. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 16.Engel S, Skoumbourdis AP, Childress J, Neumann S, Deschamps JR, Thomas CJ, Colson AO, Costanzi S, Gershengorn MC. A virtual screen for diverse ligands: discovery of selective G protein-coupled receptor antagonists. Journal of the American Chemical Society. 2008;130:5115–5123. doi: 10.1021/ja077620l. [DOI] [PubMed] [Google Scholar]
- 17.Kiss R, Kiss B, Konczol A, Szalai F, Jelinek I, Laszlo V, Noszal B, Falus A, Keseru GM. Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. Journal of medicinal chemistry. 2008;51:3145–3153. doi: 10.1021/jm7014777. [DOI] [PubMed] [Google Scholar]
- 18.Tikhonova IG, Sum CS, Neumann S, Engel S, Raaka BM, Costanzi S, Gershengorn MC. Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. Journal of medicinal chemistry. 2008;51:625–633. doi: 10.1021/jm7012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klabunde T, Giegerich C, Evers A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. Journal of medicinal chemistry. 2009;52:2923–2932. doi: 10.1021/jm9001346. [DOI] [PubMed] [Google Scholar]
- 20.Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlsson J, Yoo L, Gao ZG, Irwin JJ, Shoichet BK, Jacobson KA. Structure-based discovery of A2A adenosine receptor ligands. Journal of medicinal chemistry. 2010;53:3748–3755. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katritch V, Jaakola VP, Lane JR, Lin J, Ijzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R. Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. Journal of medicinal chemistry. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabio M, Jones K, Topiol S. Use of the X-ray structure of the beta2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Bioorganic & medicinal chemistry letters. 2008;18:5391–5405. doi: 10.1016/j.bmcl.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 24.de Kloe GE, Bailey D, Leurs R, de Esch IJ. Transforming fragments into candidates: small becomes big in medicinal chemistry. Drug discovery today. 2009;14:630–646. doi: 10.1016/j.drudis.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Rognan D. Fragment-Based Approaches and Computer-Aided Drug Discovery. Topics in current chemistry. 2011 doi: 10.1007/128_2011_182. [DOI] [PubMed] [Google Scholar]
- 26.de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. Journal of medicinal chemistry. 2008;51:4978–4985. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- 27.Korb O, Stützle T, Exner TE. An ant colony optimization approach to flexible protein-ligand docking. Swarm Intelligence. 2007;1:115–134. [Google Scholar]
- 28.Overington J. ChEMBL. An interview with John Overington, team leader, chemogenomics at the European Bioinformatics Institute Outstation of the European Molecular Biology Laboratory (EMBL-EBI). Interview by Wendy A. Warr. Journal of computer-aided molecular design. 2009;23:195–198. doi: 10.1007/s10822-009-9260-9. [DOI] [PubMed] [Google Scholar]
- 29.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosciences. 1995;25:366 – 428. [Google Scholar]
- 30.Korb O, Stützle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. Journal of chemical information and modeling. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 31.Marcou G, Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. Journal of chemical information and modeling. 2007;47:195–207. doi: 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., 3rd Assessing scoring functions for protein-ligand interactions. Journal of medicinal chemistry. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 33.Jain AN, Nicholls A. Recommendations for evaluation of computational methods. Journal of computer-aided molecular design. 2008;22:133–139. doi: 10.1007/s10822-008-9196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. Journal of chemical information and modeling. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verheij MH, de Graaf C, de Kloe GE, Nijmeijer S, Vischer HF, Smits RA, Zuiderveld OP, Hulscher S, Silvestri L, Thompson AJ, van Muijlwijk-Koezen JE, Lummis SC, Leurs R, de Esch IJ. Fragment library screening reveals remarkable similarities between the G protein-coupled receptor histamine H(4) and the ion channel serotonin 5-HT(3A) Bioorganic & medicinal chemistry letters. 2011;21:5460–5464. doi: 10.1016/j.bmcl.2011.06.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegal G, Ab E, Schultz J. Integration of fragment screening and library design. Drug discovery today. 2007;12:1032–1039. doi: 10.1016/j.drudis.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Bruysters M, Pertz HH, Teunissen A, Bakker RA, Gillard M, Chatelain P, Schunack W, Timmerman H, Leurs R. Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. European journal of pharmacology. 2004;487:55–63. doi: 10.1016/j.ejphar.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 38.Wawer M, Bajorath J. Similarity-potency trees: a method to search for SAR information in compound data sets and derive SAR rules. Journal of chemical information and modeling. 2010;50:1395–1409. doi: 10.1021/ci100197b. [DOI] [PubMed] [Google Scholar]
- 39.Becker OM, Dhanoa DS, Marantz Y, Chen D, Shacham S, Cheruku S, Heifetz A, Mohanty P, Fichman M, Sharadendu A, Nudelman R, Kauffman M, Noiman S. An integrated in silico 3D model-driven discovery of a novel, potent, and selective amidosulfonamide 5-HT1A agonist (PRX-00023) for the treatment of anxiety and depression. Journal of medicinal chemistry. 2006;49:3116–3135. doi: 10.1021/jm0508641. [DOI] [PubMed] [Google Scholar]
- 40.Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, Frimurer TM, Schwartz TW. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. The Journal of biological chemistry. 2010;285:3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jongejan A, Bruysters M, Ballesteros JA, Haaksma E, Bakker RA, Pardo L, Leurs R. Linking agonist binding to histamine H1 receptor activation. Nature chemical biology. 2005;1:98–103. doi: 10.1038/nchembio714. [DOI] [PubMed] [Google Scholar]
- 42.Rognan D. Virtual Screening. Wiley-VCH Verlag GmbH & Co. KGaA; 2011. Docking Methods for Virtual Screening: Principles and Recent Advances; pp. 153–176. [Google Scholar]
- 43.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR Modeling and Docking as Reflected by Community-wide GPCR Dock 2010 Assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Boyle NM, Liebeschuetz JW, Cole JC. Testing assumptions and hypotheses for rescoring success in protein-ligand docking. Journal of chemical information and modeling. 2009;49:1871–1888. doi: 10.1021/ci900164f. [DOI] [PubMed] [Google Scholar]
- 45.Kellenberger E, Foata N, Rognan D. Ranking targets in structure-based virtual screening of three-dimensional protein libraries: methods and problems. Journal of chemical information and modeling. 2008;48:1014–1025. doi: 10.1021/ci800023x. [DOI] [PubMed] [Google Scholar]
- 46.Knox AJ, Meegan MJ, Sobolev V, Frost D, Zisterer DM, Williams DC, Lloyd DG. Target specific virtual screening: optimization of an estrogen receptor screening platform. Journal of medicinal chemistry. 2007;50:5301–5310. doi: 10.1021/jm0700262. [DOI] [PubMed] [Google Scholar]
- 47.Rogers D, Hahn M. Extended-connectivity fingerprints. Journal of chemical information and modeling. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 48.Grant JA, Gallardo MA, Pickup BT. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. Journal of Computational Chemistry. 1996;17:1653–1666. [Google Scholar]
- 49.Steffen A, Kogej T, Tyrchan C, Engkvist O. Comparison of molecular fingerprint methods on the basis of biological profile data. Journal of chemical information and modeling. 2009;49:338–347. doi: 10.1021/ci800326z. [DOI] [PubMed] [Google Scholar]
- 50.Fan Y, Lai MH, Sullivan K, Popiolek M, Andree TH, Dollings P, Pausch MH. The identification of neurotensin NTS1 receptor partial agonists through a ligand-based virtual screening approach. Bioorganic & medicinal chemistry letters. 2008;18:5789–5791. doi: 10.1016/j.bmcl.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 51.Wieland K, Laak AM, Smit MJ, Kuhne R, Timmerman H, Leurs R. Mutational analysis of the antagonist-binding site of the histamine H(1) receptor. The Journal of biological chemistry. 1999;274:29994–30000. doi: 10.1074/jbc.274.42.29994. [DOI] [PubMed] [Google Scholar]
- 52.Ohta K, Hayashi H, Mizuguchi H, Kagamiyama H, Fujimoto K, Fukui H. Site-directed mutagenesis of the histamine H1 receptor: roles of aspartic acid107, asparagine198 and threonine194. Biochemical and biophysical research communications. 1994;203:1096–1101. doi: 10.1006/bbrc.1994.2295. [DOI] [PubMed] [Google Scholar]
- 53.Gillespie RJ, Cliffe IA, Dawson CE, Dourish CT, Gaur S, Jordan AM, Knight AR, Lerpiniere J, Misra A, Pratt RM, Roffey J, Stratton GC, Upton R, Weiss SM, Williamson DS. Antagonists of the human adenosine A2A receptor. Part 3: Design and synthesis of pyrazolo[3,4-d]pyrimidines, pyrrolo[2,3-d]pyrimidines and 6-arylpurines. Bioorganic & medicinal chemistry letters. 2008;18:2924–2929. doi: 10.1016/j.bmcl.2008.03.072. [DOI] [PubMed] [Google Scholar]
- 54.Miller DC, Klute W, Calabrese A, Brown AD. Optimising metabolic stability in lipophilic chemical space: the identification of a metabolically stable pyrazolopyrimidine CRF-1 receptor antagonist. Bioorganic & medicinal chemistry letters. 2009;19:6144–6147. doi: 10.1016/j.bmcl.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 55.Shi L, Javitch JA. The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annual review of pharmacology and toxicology. 2002;42:437–467. doi: 10.1146/annurev.pharmtox.42.091101.144224. [DOI] [PubMed] [Google Scholar]
- 56.Surgand JS, Rodrigo J, Kellenberger E, Rognan D. A chemogenomic analysis of the transmembrane binding cavity of human G-protein-coupled receptors. Proteins. 2006;62:509–538. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- 57.Venhorst J, Nunez S, Terpstra JW, Kruse CG. Assessment of scaffold hopping efficiency by use of molecular interaction fingerprints. Journal of medicinal chemistry. 2008;51:3222–3229. doi: 10.1021/jm8001058. [DOI] [PubMed] [Google Scholar]
- 58.Xu J. A new approach to finding natural chemical structure classes. Journal of medicinal chemistry. 2002;45:5311–5320. doi: 10.1021/jm010520k. [DOI] [PubMed] [Google Scholar]
- 59.Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJ. Keynote review: histamine H3 receptor antagonists reach out for the clinic. Drug discovery today. 2005;10:1613–1627. doi: 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- 60.Westly E. Nothing to sneeze at. Nature medicine. 2010;16:1063–1065. doi: 10.1038/nm1010-1063. [DOI] [PubMed] [Google Scholar]
- 61.Lim HD, de Graaf C, Jiang W, Sadek P, McGovern PM, Istyastono EP, Bakker RA, de Esch IJ, Thurmond RL, Leurs R. Molecular determinants of ligand binding to H4R species variants. Molecular pharmacology. 2010;77:734–743. doi: 10.1124/mol.109.063040. [DOI] [PubMed] [Google Scholar]
- 62.Gillard M, Van Der Perren C, Moguilevsky N, Massingham R, Chatelain P. Binding characteristics of cetirizine and levocetirizine to human H(1) histamine receptors: contribution of Lys(191) and Thr(194) Molecular pharmacology. 2002;61:391–399. doi: 10.1124/mol.61.2.391. [DOI] [PubMed] [Google Scholar]
- 63.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Mazumder R, O’Donovan C, Redaschi N, Suzek B. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic acids research. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. [accessed at 26–06–2011]; http://bioinfo-pharma.u-strasbg.fr/bioinfo.
- 65.Calculator, version 5.1.4. ChemAxon; Budapest, Hungary: [Google Scholar]
- 66.Corina, version 3.46. Molecular Networks GmbH; Erlangen, Germany: [Google Scholar]
- 67.FILTER, version 2.1.1. OpenEye Scientific Software; Santa Fe, NM: [Google Scholar]
- 68.MoKa, version 1.1.0. Molecular Discovery Ltd; Pinner, Middlesex, UK: [Google Scholar]
- 69.Pipeline Pilot, version 6.1.5. Accelrys; San Diego, CA: [Google Scholar]
- 70.Omega, version 2.3.2. OpenEye Scientific Software; Santa Fe, NM: [Google Scholar]
- 71.ROCS, version 2.3.1. OpenEye Scientific Software; Santa Fe, NM: [Google Scholar]
- 72.MOE, version 2010.10. Chemical Computing Group, Inc; Montreal, Canada: [Google Scholar]
- 73.Fukui H, Fujimoto K, Mizuguchi H, Sakamoto K, Horio Y, Takai S, Yamada K, Ito S. Molecular cloning of the human histamine H1 receptor gene. Biochemical and biophysical research communications. 1994;201:894–901. doi: 10.1006/bbrc.1994.1786. [DOI] [PubMed] [Google Scholar]
- 74.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical pharmacology. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.