

Summary
Bronchopulmonary dysplasia (BPD) remains the major morbidity of extreme preterm birth. The incidence of BPD has remained stable despite recent efforts to reduce postnatal exposures to volutrauma and hyperoxia. This review will focus on recent clinical and experimental insights that provide support for the concept that the ‘new BPD’ is the result of inflammation-mediated injury and altered lung development during a window of vulnerability in genetically susceptible infants that is modified by maternal and postnatal exposures.
Keywords: Bronchopulmonary dysplasia, Chorioamnionitis, Lung development, Preterm birth, Ureaplasma
Introduction
It is estimated that up to 90% of preterm births prior to 28 weeks’ gestation are associated with intrauterine infection and/or inflammation.1 Despite recent advances in neonatal care to minimize postnatal lung injury from mechanical ventilation,2,3 the incidence of bronchopulmonary dysplasia (BPD) remains high in extremely low gestational age newborns (ELGANs, <28 weeks) with antenatal infection/inflammation exposure. Although analysis of a national database indicates that the absolute incidence of BPD diagnosis decreased 3.3% per year from 1993 to 2006,4 the National Institutes of Child Health Neonatal Network reported stable BPD rates from 2003 to 2007 for ~10 000 ELGANs regardless of BPD definition used.5 The severity-based definition of BPD6 (including mild cases) classified 68% of infants in the network cohort with BPD compared to 42% according to the traditional definition of BPD [supplemental oxygen at 36 weeks’ postmenstrual age (PMA)] and 40% for physiologic-defined BPD.5,7 The lung pathologic findings that characterize the ‘new BPD’ in ELGANs include more uniform inflation, fewer but larger alveoli, mild fibrosis, and less fulminant but persistent inflammation.8
Bronchopulmonary dysplasia is associated with longer hospital stays,4,5 higher medical costs,4 other morbidities of prematurity including neurodevelopmental delays,9 and adverse pulmonary outcomes in childhood10 and possibly beyond. This review will focus on recent clinical and experimental insights that provide support for the concept that the ‘new BPD’ is the result of inflammation-mediated injury and altered lung development during a window of vulnerability in genetically susceptible infants that is modified by maternal and postnatal exposures (Fig. 1).
Figure 1.

Proposed scheme of pre- and postnatal inflammation altering lung development during the vulnerable saccular stage of lung development in genetically susceptible extremely low gestational age newborns. ROS, reactive oxygen species.
Chorioamnionitis and altered lung development
Infection/inflammation in the intrauterine compartment has been defined by histological (presence of polymorphonuclear cells in choriodecidual space, fetal membranes, and/or cord), microbiologic (positive culture or molecular detection methods), and/or biochemical (elevated amniotic fluid cytokines, chemokines) criteria for chorioamnionitis.11 The fetal inflammatory response syndrome (FIRS) has been defined histologically by fetal vasculitis/funisitis characterized by polymorphonuclear infiltration of the chorionic vessels or umbilical cord11 and biochemically by elevated umbilical cord concentrations of cytokines [interleukin (IL)-1ß, IL-6 and TNF-α].12,13 The fetal inflammatory response also involves upregulation of chemokines (IL-8, MIP-1ß, and RANTES), adhesion molecules (ICAM-1, ICAM-3, and E-selectin), matrix metalloproteinases (MMP-1 and MMP-9), angiogenic factors such as vascular endothelial growth factor (VEGF), and acute phase protein (C-reactive protein, CRP) in venous blood in the first few days of life.14,15 These changes indicate that the fetus is capable of initiating a complex cascade of immune responses to microbial invasion.
Watterberg et al.16 observed that histologic chorioamnionitis was associated with a reduced risk for respiratory distress syndrome (RDS), but an increased risk for BPD in a small cohort of mechanically ventilated preterm infants <2000 g birth weight who were not exposed to antenatal steroids or exogenous surfactant. This observation suggested that in-utero infection/inflammation exposure accelerated functional lung maturation, but increased the vulnerability of the preterm lung to postnatal injury. However, subsequent studies over the past 15 years have found chorioamnionitis associated with reduced17–20 or no effect21–23 on RDS risk, and increased,13,21,23 decreased24 or no effect18,22,25,26 on BPD risk. This may be due to the imprecision of the clinical diagnoses of RDS and BPD, the multifactorial nature of the disease, and variability of the study populations and clinical practices.
Recently, Laughon and ELGAN study investigators27 described three different patterns of respiratory disease during the first 14 days of postnatal life in infants born at <28 weeks’ gestation. Approximately 20% received consistently low fractional inspired oxygen (FiO2) during this period, and 43% were classified as early and persistent pulmonary dysfunction. The remaining 38% of infants were classified with a pulmonary deterioration pattern if they received FiO2 <0.23 on any day between 3 and 7 days of life and received FiO2 >0.25 on day 14. The incidence of BPD defined as oxygen requirement at 36 weeks’ PMA was 17% in the consistently low group, 51% in the pulmonary deterioration group, and 67% in the early and persistent pulmonary dysfunction group. Although the rates of histologic chorioamnionitis and funisitis were similar among the ELGAN groups, there was a trend towards more frequent detection of mycoplasma species, including Ureaplasma species in placentas from infants who developed early and persistent pulmonary dysfunction. Infants in this group also were more likely to experience postnatal sepsis, suggesting that antenatal/postnatal infection/inflammation events contribute to the pathogenesis of early and persistent lung dysfunction and risk for BPD. This study underscores the importance of delineating early patterns of respiratory disease to identify specific groups of infants at high risk for pulmonary morbidity who may benefit from interventions to prevent or ameliorate further lung injury.
Experimental models of intrauterine infections
Animal models of intrauterine infection/inflammation provide experimental evidence for altered lung development that result in accelerated functional maturation and arrested alveolarization and vascular development. These models also demonstrate the central role of the developing immune system in the pathogenesis of neonatal lung injury.
Experimental models of inflammatory neonatal lung injury
Experimental sterile chorioamnionitis models caused by Escherichia coli lipopolysaccharide (LPS) have been developed in sheep28,29 and mice.30 In the sheep model, a single LPS injection stimulated pulmonary inflammation with increased expression of pro-inflammatory cytokines such as IL-1ß and IL-6 and chemokines IL-8 and monocyte chemoattractant protein (MCP-1), recruitment of polymorphonuclear cells and monocytes, maturation of monocytes to alveolar macrophages, increased secretion of surfactant proteins, and arrested alveolar and microvascular development. Surfactant phospholipid secretion increased contributing to increased pulmonary compliance. Hence, endotoxin exposure induces both maturational responses as well as aberrant lung structural changes. Repeated LPS doses induced immune tolerance to multiple stimuli.31 In BALB/cJ mice, intra-amniotic LPS injection at embryonic day 15 increased airspace volume of fetal lungs at time of harvest on E17 or E18.30 Lipopolysaccharide exposure of E15 lung explants inhibited distal airway branching by altering the expression pattern of mesenchymal fibronectin, thus identifying a potential mechanism linking immune responses to altered lung development pathways.
Saccular stage of lung development: window of vulnerability to inflammation
Interleukin (IL)-1ß is a central cytokine involved in the upregulation and maintenance of inflammation. It is elevated in infected amniotic fluid, fetal lung fluid and tracheal aspirates in preterm infants, and increased levels in these compartments are associated with the development of BPD.12,13,32 To analyze the role of IL-1ß in BPD pathogenesis, Bry et al.33 developed a bitransgenic mouse in which human IL-1ß is conditionally expressed in lung epithelial cells. When doxycycline was administered from the beginning of pregnancy through lactation, pulmonary IL-1ß expression increased from E14.5 until late gestation and decreased postnatally. Postnatal growth was impaired and mortality was higher in the IL-1ß-expressing pups. The newborn lungs demonstrated many features of the BPD phenotype, including disrupted alveolar septation and capillary development and disordered α-smooth muscle actin (myofibroblast marker) and elastin deposition in alveolar septa.33 To determine whether susceptibility to inflammation is developmental stage-specific, additional experiments were conducted in the bitransgenic IL-1ß model in which doxycycline was added to maternal drinking water beginning at different time points.34 All but the earliest IL-1ß exposures were associated with increased neutrophil and macrophage counts and chemokine expression at postnatal day 7. However, IL-1ß expression during the saccular stage, but not during the late canalicular–early saccular stage, caused a BPD phenotype, poor growth, and increased mortality. IL-1ß expression initiated postnatally during the late saccular–alveolar stage resulted in thinner alveolar walls, shorter alveolar chord length, less airway remodeling, and better survival compared to the saccular stage-exposed pups. This is consistent with the clinical observation that ELGANs (23–27 weeks, early saccular stage) are at highest risk for inflammation-mediated BPD whereas infants born at >32 weeks’ gestation (late saccular–alveolar stage) are much lower risk.8
Experimental models of Ureaplasma intrauterine infection
The genital mycoplasmas Ureaplasma parvum and U. urealyticum are the most common organisms isolated from the amniotic fluid in women with preterm labor, preterm and term premature rupture of the membranes, short cervix, and cervical insufficiency.35 The rate of vertical transmission increases with an increase in duration of membrane rupture,36 suggesting that an ascending infection at or near the time of delivery is the most common route of infection. However, ureaplasmas have been detected by culture or by polymerase chain reaction in up to 13% of amniotic fluid samples at the time of genetic amniocentesis at 16–20 weeks’ gestation in asymptomatic women, indicating possible prolonged subclinical infection.35 These organisms are the most common organisms isolated from cord blood,37,38 cerebrospinal fluid,38,39 and respiratory secretions40 of ELGANs. There is now considerable clinical and experimental evidence that these organisms contribute to chorioamnionitis,37,41 a fetal inflammatory response,37,38,41 preterm birth,35,41–43 and neonatal morbidities including BPD,29,40,44,45 intraventricular hemorrhage,38,46 and necrotizing enterocolitis.47 Pathologic changes in Ureaplasma-infected lungs of preterm infants are characterized by moderate–severe fibrosis, disordered elastin accumulation, myofibroblast accumulation, and chronic inflammation.48,49
Experimental models in non-human primates, sheep, and mice have provided insight into possible mechanisms of Ureaplasma-mediated lung injury and altered immune responses. Pneumonia models in preterm baboons50 and mice51–53 confirmed that direct inoculation of Ureaplasma into the airway leads to inflammation and lung injury. In 140-day preterm baboons, intratracheal Ureaplasma inoculation induced an acute bronchiolitis.50 Intranasal inoculation in newborn but not 14-day-old mice resulted in an acute interstitial pneumonitis.51 When combined with hyperoxia exposure, there was greater mortality, increased lung inflammation severity, and delayed pathogen clearance in the Ureaplasma-inoculated newborn mice.52
Intrauterine Ureaplasma infection models in non-human primates,41,45,54 sheep,29 and mice55 more closely mimic the human exposure during early stages of lung development. In the Rhesus macque model of antenatal infection, the severity of fetal pneumonitis worsened with longer duration of in-utero exposure following inoculation with U. parvum serovar 1 at 132–147 days’ gestation (early saccular stage).41 Intra-amniotic inoculation with U. parvum serovar 1 two days before delivery at 125 days’ gestation (early saccular stage, 67% of term) in the baboon initiated a robust inflammatory response in the amniotic and fetal lung compartments as well as vertical transmission to the fetal lung that persisted up to 2 weeks postnatally in half of the antenatal-exposed animals.45 The combination of brief antenatal infection and postnatal mechanical ventilation resulted in augmented inflammatory cell recruitment, extensive lung fibrosis, myofibroblast proliferation, immunoreactive TGF-ß1 staining, increased bronchoalveolar lavage fluid IL-1ß, and active TGF-ß1 but not IL-10 concentrations, and a trend towards greater activation of pro-fibrotic transcription factors Smad-2 and -3 relative to anti-fibrotic Smad-7 in lung homogenates.45,54 In the sheep model, prolonged exposure to intra-amniotic U. parvum was associated with pulmonary inflammation and improvement in indices of lung function.29 In contrast to the Rhesus model, inflammation and altered lung development in the sheep model were inversely related to duration of in-utero exposure. In CD-1 mice, antenatal exposure at E13.5 (pseudoglandular stage) to low dose U. parvum serovar 3 inoculum, but not postnatal hyperoxia exposure, increased lung homogenate IL-1ß concentrations.55 Antenatal Ureaplasma did not augment postnatal oxygen-induced lung injury in this model. In all intrauterine models, ureaplasma organisms established a persistent infection in the intrauterine compartment, indicating limited capacity to clear these organisms. These findings suggest that antenatal Ureaplasma infection causes an imbalance of pro-inflammatory, pro-fibrotic and anti-inflammatory, anti-fibrotic factors in the fetal lung that may be augmented by postnatal exposure to hyperoxia and mechanical ventilation in some experimental models.
Discrepancies in the effects of antenatal Ureaplasma exposure in animal models might be explained by differences in virulence of experimental isolates and host species differences. Humans are the specific host for U. parvum and U. urealyticum, so the organism may elicit a less robust response in less related species such as sheep and mice. Alternatively, as shown with the conditional IL-1ß-expressing mouse model, acute inflammation occurring during the saccular stage but not other stages of lung development results in arrested alveolarization, and airway remodeling typical of human BPD. This may explain the observed mild inflammation and limited changes in lung morphometry in the mouse model inoculated during the pseudoglandular period (E13.5) and sheep inoculated during the pseudoglandular period (day 55 gestation)56 or late canalicular period (days 110–121 of gestation).57
Maternal inflammation: modulator of fetal inflammation
Extrauterine infections such as periodontitis, pneumonia, and urinary tract infections are risk factors for preterm birth.58,59 Recently developed animal models shed light on the role of maternal inflammation on the developing lung. To model maternal systemic infection, pregnant rats were injected intraperitoneally with E. coli LPS on E20 and 21 (term, 22 days).59 This exposure resulted in prolonged pulmonary inflammation postnatally, a BPD phenotype, and altered gene expression of molecules implicated in alveologenesis.59 Specifically, IL-1ß mRNA was increased in newborn lungs up to 14 days after maternal exposure to LPS.
Timing of doxycycline introduction in the bitransgenic IL-1ß model differentiated maternal inflammation prior to fetal onset of IL-1ß vs combined maternal and fetal expression.60 Since the fetal CCSP promoter is inactive in the fetus until E14, IL-1ß expression in bitransgenic dams could be induced by doxycycline starting at E0, or simultaneously with fetal IL-1ß expression when doxycycline is introduced at E15. Compared to simultaneous maternal/fetal inflammation, prior maternal IL-1ß-induced inflammation improved survival, growth, and lung development. Human IL-1ß expression in the pulmonary epithelium of the dams was associated with local neutrophil and macrophage infiltration and a mild systemic inflammatory reaction. The benefit of prior maternal inflammation may have been due to maternal IL-1ß production, reducing expression of pro-inflammatory genes such as CXC and CC chemokines, murine IL-1ß, and Toll-like receptors 2 and 4 in fetal lungs. By contrast, simultaneous induction of human IL-1ß in the bitransgenic dams and their fetuses did not provide protection from fetal lung injury. Although this study suggests that maternal inflammation may reduce the risk for BPD by downregulating pulmonary inflammatory responses, suppression of the fetal lung’s inflammatory response may increase susceptibility to bacterial infections. These observations suggest that the fetal response to maternal inflammation and pulmonary outcomes are dependent on the timing and duration of exposure and the nature of the inflammatory stimuli.
Can modification of clinical practice reduce the contribution of postnatal inflammation?
The pulmonary outcomes for preterm infants exposed antenatally to infection/inflammation may depend on postnatal exposures that may augment lung injury. The detection of microbes in tracheal and gastric aspirates at any time postnatally was associated with an increased risk for BPD.61 Van Marter et al.62 demonstrated that prior exposure to chorioamnionitis alone decreased the incidence of BPD, but the combination of prenatal inflammation with postnatal sepsis or mechanical ventilation for >7 days increased the risk for BPD. We observed a similar interaction of Ureaplasma respiratory colonization and duration of mechanical ventilation in a cohort of 230 infants at <33 weeks’ gestation (unpublished data) (Fig. 2). Preterm infants mechanically ventilated for <12 days were less likely to be colonized with Ureaplasma. Regardless of colonization status, infants ventilated for ≤2 days had a very low risk for BPD while infants ventilated for >33 days had a high risk of BPD. In infants ventilated for >2 and <33 days, Ureaplasma-colonized infants were more likely than Ureaplasma-negative infants to develop BPD. Experimentally, postnatal hyperoxia exacerbated inflammation and fibrosis induced by prenatal LPS exposure in mice.63
Figure 2.
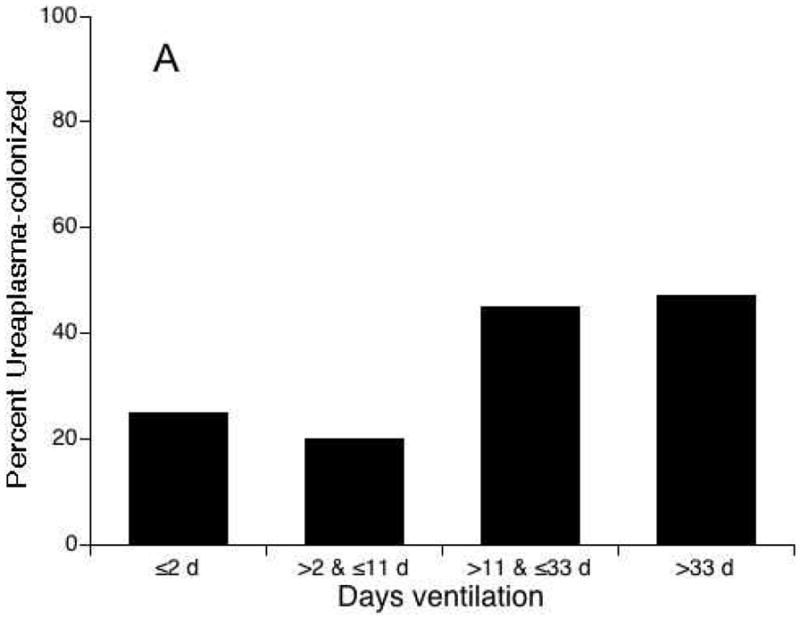
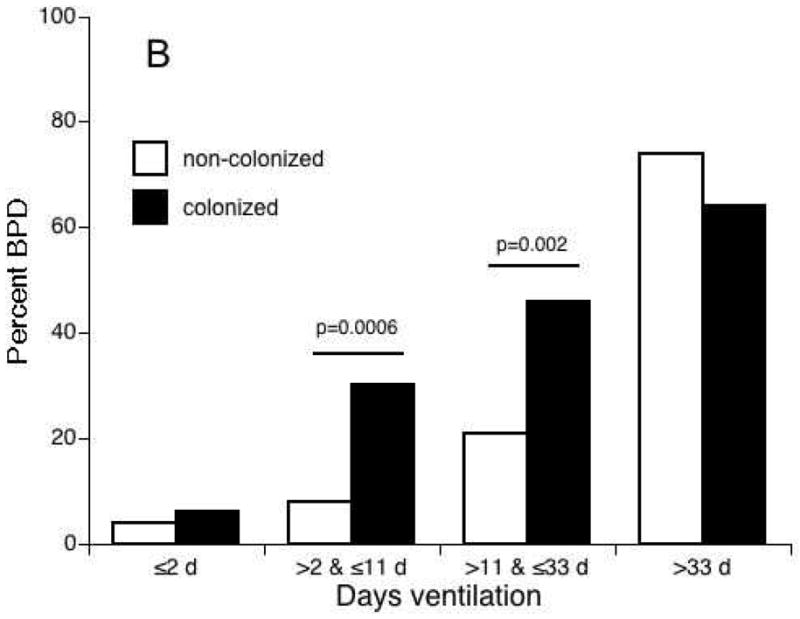
The relationship between Ureaplasma respiratory tract colonization and duration of mechanical ventilation and risk for bronchopulmonary dysplasia (BPD). In a cohort of 230 preterm infants <33 weeks’ gestation, infants with duration of ventilation <12 days compared to infants with longer duration of mechanical ventilation were less likely to be colonized with Ureaplasma (P < 0.01) (A). Ureaplasma-colonized infants ventilated for >2 and <33 days were more likely to develop BPD than non-colonized infants (B).
These data highlight the importance of analyzing the confounding effect of chorioamnionitis exposure in clinical trials of interventions to prevent BPD. For example, mortality and BPD incidence were decreased in the infants in the treatment group exposed to chorioamnionitis, but not in non-chorioamnionitis-exposed infants in a multicenter trial of hydrocortisone as prophylaxis of early adrenal insufficiency to prevent BPD.64 The data also predict that reducing mechanical ventilation in ELGAN infants should improve pulmonary outcomes. However, in the SUPPORT trial comparing early continuous positive airway pressure (CPAP) to intubation and surfactant in ELGANs, the early CPAP group experienced fewer days of mechanical ventilation and less frequent postnatal steroid use, but no difference in combined outcome of death or physiologic BPD.3 In a similar trial in New Zealand, there was similar BPD or death composite outcome in the CPAP compared to early intubation groups, but 46% of infants randomized to early CPAP were intubated in the first 5 days and the incidence of pneumothorax was higher in this group (9%) compared to the intubation group (3%).2 In these studies, groups were not stratified by chorioamnionitis exposure.
Since intrauterine and postnatal infections appear to be significant risk factors for BPD, antibiotic therapies have been proposed as interventions. However, antibiotics alone may be insufficient to prevent lung injury. In newborn rabbits exposed to intrauterine inoculation with live E. coli at the end of gestation followed by effective eradication with antibiotic therapy, postnatal growth and lung alveolarization were impaired despite absence of inflammation in the lungs at birth.65 In the Rhesus macque intrauterine Ureaplasma infection model, azithromycin alone or in combination with dexamthasone and indocin as anti-inflammatory agents prevented fetal lung damage.66 Although Ureaplasma spp. are susceptible to macrolide antibiotics in vitro, trials of erythromycin therapy in the first few weeks of life in Ureaplasma-colonized infants failed to demonstrate efficacy to prevent BPD or eradicate respiratory tract colonization.40 A phase I pharmacokinetics and safety trial of a single dose of 10 mg/kg azithromycin in ELGAN infants found that azithromycin was safe, but predicted that the dose as a single or multiple dose would be insufficient to eradicate Ureaplasma.67 Carefully designed clinical trials will be required to assess whether an appropriate dose of azithromycin can eradicate Ureaplasma and prevent BPD in colonized infants.68
Genetic susceptibility to inflammation-mediated lung injury
Although randomized trials have shown benefits of vitamin A69 and caffeine70 in reducing the risk for BPD, the effects were modest,72 suggesting that BPD risk in the smallest infants is relatively independent of postnatal events. Recently, research has focused on identifying the genetic contribution to risk of intrauterine infection, preterm birth, and BPD. Elevated mid-trimester vaginal IL-1ß is associated with increased risk for spontaneous preterm birth. Homozygous carriers of IL1RN*1, a single nucleotide polymorphism in the IL-1 receptor antagonist (IL-1ra) gene, a genotype associated with elevated IL-1ß, are at increased risk for preterm birth, suggesting that genetic polymorphisms that affect the innate immune system may contribute to genetic susceptibility to altered vaginal flora, and risk for preterm birth.73 In women who had a preterm birth, the combination of clinical chorioamnionitis and IL-10 (-1082)*G allele was associated with an increased risk for delivery before 29 weeks’ gestation, suggesting a gene–environment interaction.74 Twin concordance studies have suggested that the contribution of genetic risk to BPD is high, accounting for 35–65% risk for the outcome.75,76 To date, studies have focused on candidate genes that encode components of the innate immune system, antioxidants, and determinants of lung and vascular development, and surfactant proteins.76 Although a number of single nucleotide polymorphisms have been described as associated with an increased or decreased risk for BPD, the associations have not been replicated in subsequent studies. This may be due to the challenges of enrolling an adequate sample size in the preterm population, racial/ethnic heterogeneity in populations, variations in clinical practice, and the multifactorial pathogenesis of BPD.76
Implications for future therapies
Recent insights into the contribution of intrauterine infection/inflammation to preterm birth and BPD pathogenesis identify potential key factors in the design of future trials to prevent these conditions. Since the saccular stage of lung development appears particularly vulnerable to inflammation-mediated alterations in lung development, identifying women with subclinical intrauterine infection/inflammation mid-trimester using genomic/proteomic biomarkers for targeted antibiotic/anti-inflammatory therapy may prevent these outcomes. For future randomized trials to prevent BPD in ELGAN, stratification by exposure to chorioamnionitis will be important to assess possible response modification and effects on long-term outcomes.
Practice points.
The incidence of BPD in ELGANS remains high despite reduced use of mechanical ventilation.
Recognition of two distinct patterns of early lung disease in ELGANS characterized by early and persistent lung dysfunction (consistently high FIO2) and pulmonary deterioration (low FiO2 days 3–7 and increase FiO2 on day 14).
Benefits of evidence-based efficacious therapies to prevent BPD (e.g. vitamin A and caffeine) are modest (risk reduction ≤10%).
Multidisciplinary approach is recommended to optimize pulmonary, neurodevelopmental and growth outcomes of infants with BPD.
Research directions.
Identification of screening biomarkers for early detection of intrauterine infection.
Development of rapid, inexpensive molecular tests for mycoplasma/ureaplasma detection in amniotic fluid, blood, and respiratory secretions.
Well-powered clinical trials of azithromycin in Ureaplasma-colonized infants to determine whether pathogen clearance improves pulmonary outcomes.
Genetic studies will yield powerful tools to provide insight into disease processes, identify gene markers to identify at-risk infants for future clinical trials, and to assess response to new therapies.
Acknowledgments
Funding sources
This work was funded in part by NIH grants HL087166 and HD056424.
Footnotes
Conflict of interest statement
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morley CJ, Davis PG, Doyle LW, Brion LP, Hascoet JM, Carlin JB. Nasal CPAP or intubation at birth for very preterm infants. N Engl J Med. 2008;358:700–8. doi: 10.1056/NEJMoa072788. [DOI] [PubMed] [Google Scholar]
- 3.Finer NN, Carlo WA, Walsh MC, et al. Early CPAP versus surfactant in extremely preterm infants. N Engl J Med. 2010;362:1970–9. doi: 10.1056/NEJMoa0911783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stroustrup A, Trasande L. Epidemiological characteristics and resource use in neonates with bronchopulmonary dysplasia: 1993–2006. Pediatrics. 2010;126:291–7. doi: 10.1542/peds.2009-3456. [DOI] [PubMed] [Google Scholar]
- 5.Stoll BJ, Hansen NI, Bell EF, et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–56. doi: 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163:1723–9. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 7.Walsh MC, Yao Q, Gettner P, et al. Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics. 2004;114:1305–11. doi: 10.1542/peds.2004-0204. [DOI] [PubMed] [Google Scholar]
- 8.Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr. 2011;23:167–72. doi: 10.1097/MOP.0b013e3283423e6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Ehrenkranz RA, Walsh MC, Vohr BR, et al. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics. 2005;116:1353–60. doi: 10.1542/peds.2005-0249. [DOI] [PubMed] [Google Scholar]
- 10.Greenough A. Long-term pulmonary outcome in the preterm infant. Neonatology. 2008;93:324–7. doi: 10.1159/000121459. [DOI] [PubMed] [Google Scholar]
- 11*.Thomas W, Speer CP. Chorioamnionitis: important risk factor or innocent bystander for neonatal outcome? Neonatology. 2010;99:177–87. doi: 10.1159/000320170. [DOI] [PubMed] [Google Scholar]
- 12.Yoon BH, Romero R, Kim KS, et al. A systematic fetal inflammatory response and the development of bronchopulmonary dysplasia. Am J Obstet Gynecol. 1999;181:773–9. doi: 10.1016/s0002-9378(99)70299-1. [DOI] [PubMed] [Google Scholar]
- 13.Viscardi RM, Muhumuza CK, Rodriguez A, et al. Inflammatory markers in intrauterine and fetal blood and cerebrospinal fluid compartments are associated with adverse pulmonary and neurologic outcomes in preterm infants. Pediatr Res. 2004;55:1009–17. doi: 10.1203/01.pdr.0000127015.60185.8a. [DOI] [PubMed] [Google Scholar]
- 14.Gotsch F, Romero R, Kusanovic JP, et al. The fetal inflammatory response syndrome. Clin Obstet Gynecol. 2007;50:652–83. doi: 10.1097/GRF.0b013e31811ebef6. [DOI] [PubMed] [Google Scholar]
- 15.Hecht JL, Fichorova RN, Tang VF, Allred EN, McElrath TF, Leviton A. Relationship between neonatal blood protein concentrations and placenta histologic characteristics in extremely low GA newborns. Pediatr Res. 2011;69:68–73. doi: 10.1203/PDR.0b013e3181fed334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics. 1996;97:210–15. [PubMed] [Google Scholar]
- 17.Andrews WW, Goldenberg RL, Faye-Petersen O, Cliver S, Goepfert AR, Hauth JC. The Alabama preterm birth study: polymorphonuclear and mononuclear cell placental infiltrations, other markers of inflammation, and outcomes in 23- to 32-week preterm newborn infants. Am J Obstet Gynecol. 2006;195:803–8. doi: 10.1016/j.ajog.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 18.Kaukola T, Tuimala J, Herva R, Kingsmore S, Hallman M. Cord immunoproteins as predictors of respiratory outcome in preterm infants. Am J Obstet Gynecol. 2009;200:100.e1–8. doi: 10.1016/j.ajog.2008.07.070. [DOI] [PubMed] [Google Scholar]
- 19.Lahra MM, Beeby P, Jeffery HE. Maternal versus fetal inflammation and respiratory distress syndrome: a 10-year hospital cohort study. Arch Dis Child Fetal Neonatal Ed. 2009;94:F13–16. doi: 10.1136/adc.2007.135889. [DOI] [PubMed] [Google Scholar]
- 20.Lee J, Oh KJ, Park CW, Park JS, Jun JK, Yoon BH. The presence of funisitis is associated with a decreased risk for the development of neonatal respiratory distress syndrome. Placenta. 2011;32:235–40. doi: 10.1016/j.placenta.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Ogunyemi D, Murillo M, Jackson U, Hunter N, Alperson B. The relationship between placental histopathology findings and perinatal outcome in preterm infants. J Matern Fetal Neonatal Med. 2003;13:102–9. doi: 10.1080/jmf.13.2.102.109. [DOI] [PubMed] [Google Scholar]
- 22.Richardson BS, Wakim E, daSilva O, Walton J. Preterm histologic chorioamnionitis: impact on cord gas and pH values and neonatal outcome. Am J Obstet Gynecol. 2006;195:1357–65. doi: 10.1016/j.ajog.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 23.Zanardo V, Vedovato S, Suppiej A, et al. Histological inflammatory responses in the placenta and early neonatal brain injury. Pediatr Dev Pathol. 2008;11:350–4. doi: 10.2350/07-08-0324.1. [DOI] [PubMed] [Google Scholar]
- 24.Lahra MM, Beeby PJ, Jeffery HE. Intrauterine inflammation, neonatal sepsis, and chronic lung disease: a 13-year hospital cohort study. Pediatrics. 2009;123:1314–19. doi: 10.1542/peds.2008-0656. [DOI] [PubMed] [Google Scholar]
- 25.Redline RW, Wilson-Costello D, Hack M. Placental and other perinatal risk factors for chronic lung disease in very low birth weight infants. Pediatr Res. 2002;52:713–19. doi: 10.1203/00006450-200211000-00017. [DOI] [PubMed] [Google Scholar]
- 26.Kent A, Dahlstrom JE. Chorioamnionitis/funisitis and the development of bronchopulmonary dysplasia. J Paediatr Child Health. 2004;40:356–9. doi: 10.1111/j.1440-1754.2004.00366.x. [DOI] [PubMed] [Google Scholar]
- 27.Laughon M, Allred EN, Bose C, et al. Patterns of respiratory disease during the first 2 postnatal weeks in extremely premature infants. Pediatrics. 2009;123:1124–31. doi: 10.1542/peds.2008-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer BW, Kallapur S, Newnham J, Jobe AH. Prenatal inflammation and lung development. Semin Fetal Neonatal Med. 2009;14:2–7. doi: 10.1016/j.siny.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kramer BW. Chorioamnionitis – new ideas from experimental models. Neonatology. 2011;99:320–5. doi: 10.1159/000326620. [DOI] [PubMed] [Google Scholar]
- 30.Prince LS, Dieperink HI, Okoh VO, Fierro-Perez GA, Lallone RL. Toll-like receptor signaling inhibits structural development of the distal fetal mouse lung. Dev Dyn. 2005;233:553–61. doi: 10.1002/dvdy.20362. [DOI] [PubMed] [Google Scholar]
- 31.Kramer BW, Kallapur SG, Moss TJ, Nitsos I, Newnham JP, Jobe AH. Intra-amniotic LPS modulation of TLR signaling in lung and blood monocytes of fetal sheep. Innate Immun. 2009;15:101–7. doi: 10.1177/1753425908100455. [DOI] [PubMed] [Google Scholar]
- 32.Rindfleisch MS, Hasday JD, Taciak V, Broderick K, Viscardi RM. Potential role of interleukin-1 in the development of bronchopulmonary dysplasia. J Interferon Cytokine Res. 1996;16:365–73. doi: 10.1089/jir.1996.16.365. [DOI] [PubMed] [Google Scholar]
- 33.Bry K, Whitsett JA, Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol. 2007;36:32–42. doi: 10.1165/rcmb.2006-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Backstrom E, Hogmalm A, Lappalainen U, Bry K. Developmental stage is a major determinant of lung injury in a murine model of bronchopulmonary dysplasia. Pediatr Res. 2011;69:312–18. doi: 10.1203/PDR.0b013e31820bcb2a. [DOI] [PubMed] [Google Scholar]
- 35.Romero R, Garite TJ. Twenty percent of very preterm neonates (23–32 weeks of gestation) are born with bacteremia caused by genital mycoplasmas. Am J Obstet Gynecol. 2008;198:1–3. doi: 10.1016/j.ajog.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 36.Grattard F, Soleihac B, De Barbeyrac B, Bebear C, Seffert P, Pozzetto B. Epidemiologic and molecular investigations of genital mycoplasmas from women and neonates at delivery. Pediatr Infect Dis J. 1995;14:853–8. doi: 10.1097/00006454-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Goldenberg RL, Andrews WW, Goepfert AR, et al. The Alabama Preterm Birth Study: umbilical cord blood Ureaplasma urealyticum and Mycoplasma hominis cultures in very preterm newborn infants. Am J Obstet Gynecol. 2008;198:43.e1–5. doi: 10.1016/j.ajog.2007.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viscardi RM, Hashmi N, Gross GW, Sun CC, Rodriguez A, Fairchild KD. Incidence of invasive Ureaplasma in VLBW infants: relationship to severe intraventricular hemorrhage. J Perinatol. 2008;28:759–65. doi: 10.1038/jp.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clifford V, Tebruegge M, Everest N, Curtis N. Ureaplasma: pathogen or passenger in neonatal meningitis? Pediatr Infect Dis J. 2010;29:60–4. doi: 10.1097/INF.0b013e3181b21016. [DOI] [PubMed] [Google Scholar]
- 40*.Viscardi RM, Hasday JD. Role of Ureaplasma species in neonatal chronic lung disease: epidemiologic and experimental evidence. Pediatr Res. 2009;65:84R–90R . doi: 10.1203/PDR.0b013e31819dc2f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novy MJ, Duffy L, Axthelm MK, et al. Ureaplasma parvum or Mycoplasma hominis as sole pathogens cause chorioamnionitis, preterm delivery, and fetal pneumonia in rhesus macaques. Reprod Sci. 2009;16:56–70. doi: 10.1177/1933719108325508. [DOI] [PubMed] [Google Scholar]
- 42.Hee L. Likelihood ratios for the prediction of preterm delivery with biomarkers. Acta Obstet Gynecol Scand. 2011 May 13; doi: 10.1111/j.1600-0412.2011.01187.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 43.Estrada-Gutierrez G, Gomez-Lopez N, Zaga-Clavellina V, et al. Interaction between pathogenic bacteria and intrauterine leukocytes triggers alternative molecular signaling cascades leading to labor in women. Infect Immun. 2010;78:4792–9. doi: 10.1128/IAI.00522-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schelonka RL, Katz B, Waites KB, Benjamin DK., Jr Critical appraisal of the role of Ureaplasma in the development of bronchopulmonary dysplasia with metaanalytic techniques. Pediatr Infect Dis J. 2005;24:1033–9. doi: 10.1097/01.inf.0000190632.31565.83. [DOI] [PubMed] [Google Scholar]
- 45.Yoder BA, Coalson JJ, Winter VT, Siler-Khodr T, Duffy LB, Cassell GH. Effects of antenatal colonization with Ureaplasma urealyticum on pulmonary disease in the immature baboon. Pediatr Res. 2003;54:797–807. doi: 10.1203/01.PDR.0000091284.84322.16. [DOI] [PubMed] [Google Scholar]
- 46.Kasper DC, Mechtler TP, Bohm J, et al. In utero exposure to Ureaplasma spp. is associated with increased rate of bronchopulmonary dysplasia and intraventricular hemorrhage in preterm infants. J Perinat Med. 2011;39:331–6. doi: 10.1515/jpm.2011.022. [DOI] [PubMed] [Google Scholar]
- 47.Okogbule-Wonodi AC, Gross GW, Sun CC, et al. Necrotizing enterocolitis is associated with ureaplasma colonization in preterm infants. Pediatr Res. 2011;69:442–7. doi: 10.1203/PDR.0b013e3182111827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viscardi RM, Manimtim WM, Sun CCJ, Duffy L, Cassell GH. Lung pathology in premature infants with Ureaplasma urealyticum infection. Pediatr Devel Pathol. 2002;5:141–50. doi: 10.1007/s10024001-0134-y. [DOI] [PubMed] [Google Scholar]
- 49.Viscardi R, Manimtim W, He JR, et al. Disordered pulmonary myofibroblast distribution and elastin expression in preterm infants with Ureaplasma urealyticum pneumonitis. Pediatr Dev Pathol. 2006;9:143–51. doi: 10.2350/10-05-0112.1. [DOI] [PubMed] [Google Scholar]
- 50.Walsh WF, Butler J, Coalson J, Hensley D, Cassell GH, deLemos RA. A primate model of Ureaplasma urealyticum infection in the premature infant with hyaline membrane disease. Clin Infect Dis. 1993;17(Suppl 1):S158–62. doi: 10.1093/clinids/17.supplement_1.s158. [DOI] [PubMed] [Google Scholar]
- 51.Rudd PT, Cassell GH, Waites KB, Davis JK, Duffy LB. Ureaplasma urealyticum pneumonia: experimental production and demonstration of age-related susceptibility. Infect Immun. 1989;57:918–25. doi: 10.1128/iai.57.3.918-925.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crouse DT, Cassell GH, Waites KB, Foster JM, Cassady G. Hyeroxia potentiates Ureaplasma urealyticum pneumonia in newborn mice. Infect Immun. 1990;58:3487–93. doi: 10.1128/iai.58.11.3487-3493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viscardi RM, Kaplan J, Lovchik JC, et al. Characterization of a murine model of Ureaplasma urealyticum pneumonia. Infect Immun. 2002;70:5721–9. doi: 10.1128/IAI.70.10.5721-5729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viscardi RM, Atamas SP, Luzina IG, et al. Antenatal Ureaplasma urealyticum respiratory tract infection stimulates proinflammatory, profibrotic responses in the preterm baboon lung. Pediatr Res. 2006;60:141–6. doi: 10.1203/01.pdr.0000228322.73777.05. [DOI] [PubMed] [Google Scholar]
- 55.Normann E, Lacaze-Masmonteil T, Eaton F, Schwendimann L, Gressens P, Thebaud B. A novel mouse model of Ureaplasma-induced perinatal inflammation: effects on lung and brain injury. Pediatr Res. 2009;65:430–6. doi: 10.1203/PDR.0b013e31819984ce. [DOI] [PubMed] [Google Scholar]
- 56.Collins JJ, Kallapur SG, Knox CL, et al. Inflammation in fetal sheep from intra-amniotic injection of Ureaplasma parvum. Am J Physiol Lung Cell Mol Physiol. 2010;299:L852–60. doi: 10.1152/ajplung.00183.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polglase GR, Dalton RG, Nitsos I, et al. Pulmonary vascular and alveolar development in preterm lambs chronically colonized with Ureaplasma parvum. Am J Physiol Lung Cell Mol Physiol. 2010;299:L232–41. doi: 10.1152/ajplung.00369.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muglia LJ, Katz M. The enigma of spontaneous preterm birth. N Engl J Med. 2010;362:529–35. doi: 10.1056/NEJMra0904308. [DOI] [PubMed] [Google Scholar]
- 59.Cao L, Wang J, Tseu I, Luo D, Post M. Maternal exposure to endotoxin delays alveolarization during postnatal rat lung development. Am J Physiol Lung Cell Mol Physiol. 2009;296:L726–37. doi: 10.1152/ajplung.90405.2008. [DOI] [PubMed] [Google Scholar]
- 60*.Backstrom E, Lappalainen U, Bry K. Maternal IL-1beta production prevents lung injury in a mouse model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2010;42:149–60. doi: 10.1165/rcmb.2008-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beeton ML, Maxwell NC, Davies PL, et al. Role of pulmonary infection in the development of chronic lung disease of prematurity. Eur Respir J. 2011;37:1424–30. doi: 10.1183/09031936.00037810. [DOI] [PubMed] [Google Scholar]
- 62.Van Marter LJ, Dammann O, Allred EN, et al. Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr. 2002;140:171–6. doi: 10.1067/mpd.2002.121381. [DOI] [PubMed] [Google Scholar]
- 63.Velten M, Heyob KM, Rogers LK, Welty SE. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J Appl Physiol. 2010;108:1347–56. doi: 10.1152/japplphysiol.01392.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watterberg KL, Gerdes JS, Cole CH, et al. Prophylaxis of early adrenal insufficiency to prevent bronchopulmonary dysplasia: a multicenter trial. Pediatrics. 2004;114:1649–57. doi: 10.1542/peds.2004-1159. [DOI] [PubMed] [Google Scholar]
- 65.Gras-Le Guen C, Denis C, Franco-Montoya ML, et al. Antenatal infection in the rabbit impairs post-natal growth and lung alveolarisation. Eur Respir J. 2008;32:1520–8. doi: 10.1183/09031936.00023708. [DOI] [PubMed] [Google Scholar]
- 66.Novy MJ, Sadowsky DW, Grigsby PL, Duffy LB, Waites KB. Maternal azithromcyin (AZI) therapy in ureaplasma intramniotic infection (IAI) prevents advanced fetal lung lesions in rhesus monkeys. Reprod Sci. 2008;15(Suppl):184A. [Google Scholar]
- 67.Hassan HE, Othman AA, Eddington ND, et al. Pharmacokinetics, safety, and biologic effects of azithromycin in extremely preterm infants at risk for Ureaplasma colonization and bronchopulmonary dysplasia. J Clin Pharmacol. 2010 Nov 23; doi: 10.1177/0091270010382021. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turner MA, Jacqz-Aigrain E, Kotecha S. Azithromycin, Ureaplasma and chronic lung disease of prematurity: a case study for neonatal drug development. Arch Dis Child. 2011 June 22; doi: 10.1136/adc.2010.195180. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 69.Tyson JE, Wright LL, Oh W, et al. Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med. 1999;340:1962–8. doi: 10.1056/NEJM199906243402505. [DOI] [PubMed] [Google Scholar]
- 70.Schmidt B, Roberts RS, Davis P, et al. Caffeine therapy for apnea of prematurity. N Engl J Med. 2006;354:2112–21. doi: 10.1056/NEJMoa054065. [DOI] [PubMed] [Google Scholar]
- 71.Morris BH, Oh W, Tyson JE, et al. Aggressive vs. conservative phototherapy for infants with extremely low birth weight. N Engl J Med. 2008;359:1885–96. doi: 10.1056/NEJMoa0803024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72*.Laughon MM, Smith PB, Bose C. Prevention of bronchopulmonary dysplasia. Semin Fetal Neonatal Med. 2009;14:374–82. doi: 10.1016/j.siny.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Genc MR, Onderdonk A. Endogenous bacterial flora in pregnant women and the influence of maternal genetic variation. BJOG. 2011;118:154–63. doi: 10.1111/j.1471-0528.2010.02772.x. [DOI] [PubMed] [Google Scholar]
- 74.Kerk J, Dordelmann M, Bartels DB, et al. Multiplex measurement of cytokine/receptor gene polymorphisms and interaction between interleukin-10 (-1082) genotype and chorioamnionitis in extreme preterm delivery. J Soc Gynecol Invest. 2006;13:350–6. doi: 10.1016/j.jsgi.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 75.Hallman M, Marttila R, Pertile R, Ojaniemi M, Haataja R. Genes and environment in common neonatal lung disease. Neonatology. 2007;91:298–302. doi: 10.1159/000101345. [DOI] [PubMed] [Google Scholar]
- 76*.Lavoie PM, Dube MP. Genetics of bronchopulmonary dysplasia in the age of genomics. Curr Opin Pediatr. 2010;22:134–8. doi: 10.1097/MOP.0b013e328336eb85. [DOI] [PMC free article] [PubMed] [Google Scholar]