

Abstract
Despite the apparent appropriateness of left ventricular (LV) remodeling following myocardial infarction (MI), it poses an independent risk factor for development of heart failure. There is a paucity of studies into the molecular mechanisms of LV remodeling in large animal species. We took an unbiased molecular approach to identify candidate transcription factors (TFs) mediating the genetic reprogramming involved in post-MI LV remodeling in swine. Left ventricular tissue was collected from remote, non-infarcted myocardium, 3 weeks after MI-induction or sham-surgery. Microarray analysis identified 285 upregulated and 278 downregulated genes (FDR < 0.05). Of these differentially expressed genes, the promoter regions of the human homologs were searched for common TF binding sites (TFBS). Eighteen TFBS were overrepresented >two-fold (p < 0.01) in upregulated and 13 in downregulated genes. Left ventricular nuclear protein extracts were assayed for DNA-binding activity by protein/DNA array. Out of 345 DNA probes, 30 showed signal intensity changes >two-fold. Five TFs were identified in both TFBS and protein/DNA array analyses, which showed matching changes for COUP-TFII and glucocorticoid receptor (GR) only. Treatment of swine with the GR antagonist mifepristone after MI reduced the post-MI increase in LV mass, but LV dilation remained unaffected. Thus, using an unbiased approach to study post-MI LV remodeling in a physiologically relevant large animal model, we identified COUP-TFII and GR as potential key mediators of post-MI remodeling.
Electronic supplementary material
The online version of this article (doi:10.1007/s00395-011-0229-1) contains supplementary material, which is available to authorized users.
Keywords: Hypertrophy, Myocardial infarction, Transcription factors, Microarray, Animal models of human diseases, Systolic dysfunction
Introduction
Heart failure is currently the only major cardiovascular syndrome of which the prevalence is steadily increasing. For a significant part this is the result of improved survival of patients encountering an acute myocardial infarction (MI) in conjunction with an ageing population [59]. Loss of viable myocardium elicits a cascade of compensatory mechanisms, including neurohumoral activation, fluid retention and left ventricular (LV) remodeling, in an attempt to maintain normal pump function [27, 54]. However, despite its apparent appropriateness, post-MI remodeling—consisting of LV hypertrophy and dilation—constitutes an independent risk factor for the development of heart failure [63]. Mechanisms that have been implicated in the pathology of post-MI remodeling and heart failure include myofilament dysfunction [48, 57], alterations in excitation–contraction coupling [23] and mitochondrial function [25], decreased expression of atrophy mediators [5], inflammatory responses [3, 14, 31] and alterations in extracellular matrix [19, 47] and cell–cell interactions [32].
The molecular pathways underlying LV remodeling and its progression toward heart failure remain incompletely understood. The majority of studies into the molecular mechanisms of LV remodeling has been reductionistic in nature and has principally been performed in mice. These studies have yielded a wealth of information regarding the role of individual genes and proteins in LV remodeling [38]. However, there are marked differences in cardiac physiology between rodents and large mammals [10, 21]. Nevertheless, there is a paucity of studies into the molecular mechanisms of cardiac remodeling in large animal species mainly because the use of large animal models in molecular ‘–omics’ studies was limited by a lack of suitable tools together with incomplete annotation of the genome, transcriptome and proteome. However, porcine gene-expression arrays in conjunction with improved annotation have recently become available [6], allowing analysis of the molecular alterations in LV remodeling in pigs.
To identify candidate transcription factors (TFs) that mediate the genetic reprogramming involved in LV remodeling in the pig, we took an unbiased molecular approach (Fig. 1). First, global changes in gene expression were studied by microarray analysis of the remote non-infarcted LV tissue early after induction of MI. To identify the responsible transcription factors, these data were subsequently combined with transcription factor binding site (TFBS) analysis as well as with protein–DNA arrays using LV nuclear protein extracts. Rationale for this approach was that the coordinate upregulation and downregulation of a high number of different genes is likely achieved through altered function of a limited set of TFs. This integrated approach led to the identification of COUP-TFII and the glucocorticoid receptor as potential mediators of post-MI LV remodeling.
Fig. 1.
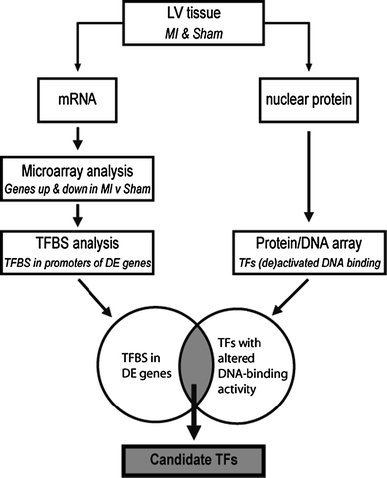
Workflow of our transcriptional genomics approach. RNA and nuclear proteins were isolated from LV tissue. The RNA was used in microarray analysis followed by TFBS analysis of differentially expressed genes. Nuclear protein extracts were used for protein/DNA array analysis to identify TFs that were increased or decreased in DNA-binding activity in post-MI hearts. Candidate TFs were identified by combining these two data sets. LV left ventricular, MI myocardial infarction, TFBS transcription factor binding site, DE differentially expressed, TF transcription factor
Methods
Experimental animals
Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and with approval of the Erasmus Medical Center Animal Care Committee. 51 pre-adolescent (2–3 months old) Yorkshire × Landrace swine (22.0 ± 0.4 kg) of either sex were used; males had been neutered.
Surgery
The animals were sedated with ketamine (20 mg/kg, intramuscularly (IM)) and midazolam (0.5 mg/kg, IM), anesthetized with thiopental (10 mg/kg, intravenously (IV)), intubated and ventilated with O2/N2 (1/2 v/v) enriched with 0.1–1% (vol/vol) isoflurane [51, 57]. Following a thoracotomy through the fourth left intercostal space, the heart was exposed via a small pericardial incision, the left circumflex artery (LCx) was dissected and a suture was placed around it. Subsequently, the LCx was permanently ligated in 31 swine designated to the MI group (producing a transmural infarction in the lateral wall encompassing 20–25% of the left ventricle [58]), while the suture was removed in the 20 swine designated to the sham group [11, 57, 58]. Then the pericardium and the chest were closed and animals were allowed to recover, receiving analgesia (0.3 mg buprenorphine IM) for 2 days and antibiotic prophylaxis (25 mg/kg amoxicillin and 5 mg/kg gentamycin IV) for 5 days.
Echocardiography and hemodynamic measurements
Three weeks after surgery, the animals were sedated with ketamine (20 mg/kg, IM) and midazolam (0.5 mg/kg, IM) and underwent echocardiography for the determination of LV end-diastolic cross-sectional area (EDA) and end-systolic cross-sectional area (ESA) [11]. 2D ejection fraction was calculated as 100 × (EDA − ESA)/EDA %. After induction of anesthesia with pentobarbital (10–15 mg/kg per h, IV) swine were instrumented for measurement of mean arterial pressure, heart rate, cardiac output, rate of rise in LV pressure at 40 mm Hg (LV dP/dtP40), time constant of relaxation (tau) and LV end diastolic pressure [11, 57, 58]. After completion of all measurements, the animals underwent a sternotomy and the heart was arrested and immediately excised. The left ventricle was divided into subendocardial and subepicardial tissue from the anterior, lateral, posterior and septal area of the LV wall and cardiac tissue was snap-frozen in liquid nitrogen (i.e., within 3–5 min of excising the heart) and stored at −80°C. For protein and RNA analysis, subendocardial tissue from the anterior wall was used, which is the remote non-infarcted area in MI animals.
Total RNA isolation
RNA was extracted from LV tissue from eight MI and eight sham animals (four males and four females per group). The frozen tissue samples were pulverized with a mortar and pestle and TRI reagent (Invitrogen, Carlsbad, CA, USA) was added immediately to the powder. The RNA was isolated according to manufacturer’s instructions, and cleaned with RNeasy clean-up kit (Qiagen, Valencia, CA, USA). Purity and quality of isolated RNA were assessed by RNA 6000 Nano assay on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). All samples showed a RNA integrity number >8.
Microarray analysis
Sixteen microarrays, eight for sham and eight for post-MI hearts, were run in three independent batches. From each heart, RNA (3 μg) was used for synthesis of biotinylated cRNA. Labeled cRNA was hybridized to the GeneChip Porcine Genome Array (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s instructions. The Affymetrix QC reports showed high quality of the samples and arrays, indicated by percentage of present calls, noise, background, and by a 3′:5′signal ratio for glyceraldehyde-3-phosphate dehydrogenase mRNA of <1.4.
Raw intensity values of all samples were normalized in R (http://www.r-project.org). The data were corrected for variability across batches and arrays by quantile normalization according to experimental group, followed by robust microarray average normalization, using the Affy package developed by Bioconductor [15]. The normalized expression values were loaded into Partek genomic suite (Partek Inc, St. Louis, MO, USA) and multivariate principal component analysis was performed [45]. All the microarray data have been submitted to the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database (GEO) (http://www.ncbi.nlm.nih.gov/geo) with GEO accession number GSE27962.
The class comparison tool of BRB-ArrayTools software (http://linus.nci.nih.gov/BRB-ArrayTools.html) was used for generation of lists of differentially expressed genes [49]. Genes were considered significantly different between MI and sham at a false discovery rate (FDR) <0.05. As the annotation of the porcine genechip is far from complete, the annotation of ANEXdb [6] was used. This open source application aligns Affymetrix porcine GeneChip target sequences to the Iowa Porcine Assembly, which is an assembly of all publicly available porcine-expressed consensus sequences. This was subsequently aligned to the NCBI RefSeq RNA database, to yield the homologous human RefSeq IDs.
Transcription factor binding site analysis
The genomic sequences of the human homologs of the differentially expressed genes were selected, and regions between position 500 upstream and 100 downstream of the transcription start site were searched for putative transcription factor binding sites (TFBS) using F-match implemented in the Explain Analysis System (Biobase GmbH, Wolfenbüttel, Germany) [29, 30]. The rationale for searching differentially expressed genes for common TFBS is the assumption that co-expressed genes are coordinately regulated by a limited set of TFs. Promoter regions of the differentially expressed genes were scanned for so-called positional weight matrices [29], constructed from collections of known binding sites for a given TF. The TFBS were searched using the entire vertebrate non-redundant set of transcription factors matrix from the TRANSFAC database [64]. To reduce false positive results in TFBS analysis, the choice of an appropriate control data set is of paramount importance. To identify TFBS that cause the increased expression of the upregulated genes, the promoters of the upregulated genes were taken as input set, while the promoters of the downregulated genes were taken as background set. For the identification of overrepresented TFBSs in downregulated genes, the promoters of downregulated were used as input set and the promoters of upregulated genes as background set. The number of binding sites in the promoter region of upregulated and downregulated genes per 600 basepairs for each TF is indicated. Overrepresentation was determined using a one-tailed Fisher exact probability test and considered significant when more than two-fold and at p < 0.01.
Protein/DNA array
With the protein/DNA array (Combo array, Panomics/Affymetrix, Milan, Italy), nuclear protein extracts were semi-quantitatively assayed by DNA binding activity for 345 TFs. Nuclear protein extracts were prepared from LV tissue as previously described [33]. Protein/DNA array analysis was performed according to the manufacturer’s instructions. Briefly, 5 μg of nuclear protein of a pool of four MI extracts and a pool of four sham extracts was used for binding to a mix of biotin-labeled TF-specific DNA probes in solution. Subsequently, all unbound DNA probes were washed away. The TF-bound probes were denatured and then hybridized to the array membrane. After addition of steptavidin–HRP, signals were generated by enhanced chemiluminescence (ECL) and exposure to Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ, USA). Various exposure times were used to obtain signals over a large dynamic range. Signal intensities were quantified using a Bio-Rad calibrated GS-800 scanner and Quantity One software (Bio-Rad, Hercules, CA, USA). Only non-saturated signals were used for further analysis. TF binding activity was considered significant when at least a two-fold signal difference between MI and sham was present.
Ingenuity pathway analysis
Pathway analysis (Ingenuity Systems, Redwood City, CA, USA) was performed to detect the biological functions and molecular networks of the differentially expressed genes, using the human Refseq IDs as input. Biological groups were identified with which the genes are significantly associated (p < 0.001). Interconnectivity of the genes was visualized by the molecular networks constructed by the program. These networks are constructed by connecting as many genes as possible, also using unchanged hub molecules. For linking TFs from the protein/DNA array to genes identified in the microarray analysis, the TFs were used as input for pathway analysis. In the build function and with the grow tool, the genes were linked to the TFs if they are known from the literature to be either expressed/transcribed by, or have an experimentally validated binding site for the TFs.
Statistics
Data are presented as mean ± SEM. Differences between two groups were analyzed by unpaired Student’s t-test. Differences between three groups were analyzed by one-way ANOVA followed by Student Newman-Keuls post-hoc test using GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA, USA) and p < 0.05 (two tailed) was considered to be statistically significant.
Results
LV remodeling and dysfunction after MI
Three weeks after MI-induction or sham surgery, LV eccentric hypertrophy was evident from a 15% higher LV to body weight ratio and a 70% larger LV end-diastolic lumen area in MI animals (Table 1). LV remodeling was associated with a maintained stroke volume and cardiac output, but the 2D-ejection fraction was lower in MI (37 ± 3%) than in sham (67 ± 3%) swine (p < 0.05) due to the increase in LV end-diastolic lumen area. Left ventricular systolic dysfunction was also reflected in the 20% lower LV dP/dtP40, while perturbed LV relaxation was suggested by the 20% higher time constant of relaxation tau. Finally, the elevations in LV end-diastolic pressure in conjunction with right ventricular hypertrophy suggest the presence of LV backward failure.
Table 1.
Anatomic, echocardiographic and hemodynamic data
| Sham | MI | |
|---|---|---|
| Anatomic data | (n = 20) | (n = 24) |
| Body weight (kg) | 30 ± 1 | 32 ± 1 |
| RVW/BW (g/kg) | 0.99 ± 0.05 | 1.35 ± 0.06*** |
| LVW/BW (g/kg) | 2.87 ± 0.09 | 3.38 ± 0.06*** |
| Echocardiographic data | (n = 9) | (n = 13) |
| LV EDA (mm2) | 921 ± 47 | 1,551 ± 157** |
| LV ESA (mm2) | 306 ± 34 | 985 ± 128*** |
| 2D-ejection fraction (%) | 67 ± 3 | 37 ± 3*** |
| Hemodynamic data | (n = 18) | (n = 20) |
| MAP (mm Hg) | 93 ± 5 | 94 ± 4 |
| Heart rate (bpm) | 115 ± 4 | 116 ± 7 |
| Cardiac output (l/min) | 3.4 ± 0.2 | 3.3 ± 0.2 |
| Stroke volume (ml) | 30 ± 2 | 30 ± 2 |
| LV dP/dtP40 (mm Hg/s) | 1743 ± 88 | 1,411 ± 51** |
| Tau (ms) | 36 ± 2 | 43 ± 2* |
| LV EDP (mm Hg) | 7.7 ± 0.8 | 13.0 ± 1.4** |
Values are mean ± SEM
BW body weight, RVW and LVW, right and left ventricle weight, EDA and ESA end-diastolic and end-systolic cross-sectional area, MAP mean aortic pressure, LV dP/dt P40 rate of rise in LV pressure at 40 mm Hg, EDP end-diastolic pressure
* p < 0.05; ** p < 0.01; *** p < 0.001 versus sham
Gene expression analysis
Global differences in gene expression in the remote non-infarcted part of the LV were determined by microarray analysis. In principal component analysis 33.7% of the variation in the data sets was explained by the first two components, with principal component 1 clearly separating MI from sham animals (Fig. 2a). There was no apparent clustering based on gender, which may be explained by the use of pre-adolescent pigs and neutered males. In MI swine, 285 genes were upregulated and 278 were downregulated (FDR < 0.05) compared to myocardium from sham swine.
Fig. 2.
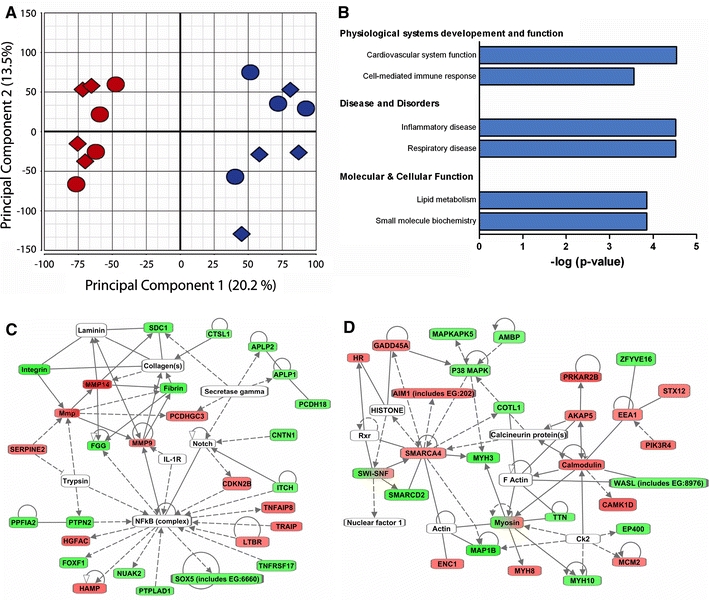
Differential gene expression. a Principal component analysis of the data from the individual microarrays shows separation into two groups, with the sham (blue) and post-MI (red) samples forming separate clusters. Females (diamonds) and males (circles) do not form separate clusters. b Clustering of the differentially expressed genes into biological groups was performed with Ingenuity pathway analysis (p < 0.001). c, d The top two networks of differentially expressed genes. Rectangles in green and red represent genes downregulated and upregulated after MI compared with sham. Dual-colored rectangles depict a group of genes, some of which are upregulated and some are downregulated after MI. White rectangles are hub molecules, of which the expression is not altered, but that generally have a large number of connections with the genes. Uninterrupted and dashed lines indicate physical and indirect interactions between molecules, respectively
To test whether differentially expressed genes clustered into ‘biological process’ groups, Ingenuity pathway analysis was used (Fig 2b). Besides ‘cardiovascular system development and function’, genes involved in ‘cell-mediated immune response’ and ‘inflammatory disease genes’ were significantly overrepresented among differentially expressed genes. These findings are in accordance with the concept that inflammation plays an important role in post-MI LV remodeling [40]. In addition, ‘lipid metabolism’ was significantly overrepresented, which may reflect the changes in energy metabolism associated with LV hypertrophy [66]. The genes that attribute to the subgroup clustering in Fig. 2b are listed in supplemental Table 1.
Ingenuity pathway analysis was also used to visualize relationships between the differentially expressed genes. Figure 2c displays the network with the highest number of genes. In this network NF-κB is a central hub molecule whose expression is not changed at the mRNA level, but which is known to be activated after MI in murine models [28] and in the failing human heart [65]. Calcineurin is a central hub molecule in the network with the second highest number of differentially expressed genes (Fig. 2d), and its role in cardiac hypertrophy in murine models has been well established [36]. In this network the ‘Calmodulin–A kinase anchor protein 5 (AKAP5)–protein kinase A regulatory subunit 2B (PRKAR2B)–axis’ is present, which was upregulated after MI. The latter observations are consistent with previous data from our laboratory, showing perturbations in PKA signaling in post-MI hearts [57].
Transcription factor binding site analysis
To identify which TFs are involved in the coordinate regulation of expression of differentially expressed genes, we scanned promoter regions of the homologous human genes for overrepresentation of transcription factor binding sites (TFBS). Eighteen TFBS were significantly overrepresented in promoter regions of the upregulated genes, while 13 were overrepresented in the downregulated genes (Table 2). These included a number of TFs involved in cell differentiation and proliferation (e.g., serum response factor (SRF) and BTB and CNC homology 1 (BACH1)), and a number of nuclear receptors (e.g., aryl hydrocarbon receptor (AhR), glucocorticoid receptor (GR), chicken ovalbumin upstream promoter transcription factor II COUP-TFII). For members of the FOX family of TFs, a different binding site was overrepresented in the promoters of upregulated and downregulated genes. Transcription factor binding sites for the nuclear factor of activated T-cells (NFAT) family of TFs were overrepresented in the downregulated genes. For each TFBS, the site was found on average in the proximal promoter region of 10–30% of the differentially expressed genes, whereas 37% of upregulated genes contained a site for COUP-TFII (Table 2).
Table 2.
Transcription factor binding sites overrepresented in upregulated or downregulated genes
| Matrix name | Transcription factor | Up/down | Frequency (sites/600 bp) | p value | |
|---|---|---|---|---|---|
| Up genes | Down genes | ||||
| V$HOXA3_01 | HOXA3/4 | 5.6 | 0.11 | 0.02 | 0.001 |
| V$SPIB_01 | SPIB | 4.7 | 0.12 | 0.03 | 0.001 |
| V$PAX4_01 | PAX4 | 4.4 | 0.11 | 0.03 | 0.002 |
| V$PAX3_B | PAX3 | 4.2 | 0.11 | 0.03 | 0.003 |
| V$SRF_Q5 | SRF | 4.1 | 0.12 | 0.03 | 0.003 |
| V$GR_Q6 | GR (NR3C1) | 3.2 | 0.13 | 0.04 | 0.006 |
| V$TBX5_02 | TBX5 | 3.1 | 0.16 | 0.05 | 0.002 |
| V$SMAD_Q6 | SMAD1-8 | 2.8 | 0.18 | 0.06 | 0.002 |
| V$HNF4_01 | HNF4gamma,HNF-4,HNF-4α | 2.8 | 0.12 | 0.04 | 0.01 |
| V$FOXD3_01 | FOXC1/D1/D3/F1/F2/H1/I1/J1/J2FOXL1/O1/O3/O4 | 2.8 | 0.19 | 0.07 | 0.002 |
| V$AR_03 | AR | 2.7 | 0.13 | 0.05 | 0.009 |
| V$MYC_Q2 | c-Myc,N-Myc,Max | 2.4 | 0.28 | 0.13 | 0.002 |
| V$HNF3B_01 | HNF-3beta | 2.3 | 0.17 | 0.08 | 0.008 |
| V$SOX10_Q6 | Sox2/4-6/9-12/18/20 | 2.3 | 0.20 | 0.09 | 0.005 |
| V$ARNT_02 | Arnt | 2.3 | 0.27 | 0.12 | 0.005 |
| V$ARP1_01 | COUP-TFI/II (NR2F1/2) | 2.2 | 0.37 | 0.17 | 0.0003 |
| V$E47_02 | MRF2,MASH-1,Myf5/6,MyoD,Myogenin, HTF4γ,E47 | 2.1 | 0.28 | 0.13 | 0.002 |
| V$NFAT_Q6 | NFATC1-4 | −4.8 | 0.02 | 0.11 | 0.002 |
| V$AHRARNT_02 | AhR,ARNT | −3.1 | 0.03 | 0.11 | 0.009 |
| V$TCF11MAFG_0 | BACH1/2,MAF,MAFB/E/G/K,NFE2,NFE2L1-3 | −3.1 | 0.03 | 0.11 | 0.009 |
| V$OLF1_01 | EBF1/3 | −3.1 | 0.03 | 0.11 | 0.009 |
| V$YY1_02 | YY1 | −2.7 | 0.7 | 0.16 | 0.005 |
| V$XFD3_01 | EP300,MEIS1,RRN3, SMAD2,TGIF1 | −2.7 | 0.06 | 0.15 | 0.005 |
| V$STAT3STAT3_Q3 | STAT3 | −2.7 | 0.07 | 0.19 | 0.002 |
| V$FAC1_01 | BPTF | −2.6 | 0.07 | 0.17 | 0.003 |
| V$CMYB_01 | MYB,MYBL2 | −2.4 | 0.06 | 0.15 | 0.009 |
| V$FREAC3_01 | FOXC1/D1/D3/F1/F2/H1/I1/J1/J2FOXL1/O1/O3/O4 | −2.4 | 0.07 | 0.17 | 0.007 |
| V$PAX8_01 | PAX8 | −2.4 | 0.07 | 0.16 | 0.007 |
| V$ATF1_Q6 | ATF1-4,ATF7,CREB1, CREM | −2.1 | 0.1 | 0.21 | 0.007 |
| V$HELIOSA_02 | IKZF1 | −2.1 | 0.12 | 0.24 | 0.006 |
Matrix name TFBS matrix from TRANSFAC database, Transcription factor TFs that can bind to the matrix element, Up/down factor overrepresentation of the TFBS in upregulated versus downregulated genes (+ sign) or in downregulated versus upregulated genes (− sign)
Protein/DNA array
To identify TFs with altered activity in remodeled myocardium, we performed protein/DNA array analysis of pooled LV nuclear extracts from eight post-MI hearts versus eight sham swine. Signals above background were detected for 186 out of the 345 probes in the array. DNA-binding activity of 10 TFs was found to be at least two-fold higher, while binding activity of 20 TFs was at least two-fold less, in remodeled myocardium of post-MI hearts compared to sham. Table 3 shows the top five of affected TFs (see Supplemental Table 2 for the full list). Two TFs were active only in one protein pool (Ikaros family zinc finger 1 (IKZF1) in MI and retinoic acid receptor (RAR) in sham), but signal intensity just surpassed the detection limit of the array.
Table 3.
Top 5 transcription factors showing increased or decreased DNA-binding activity in nuclear extracts from MI myocardium
| Probe name | Transcription factor | Ratio MI/sham |
|---|---|---|
| LyF | IKZF1 | ∞a |
| AIC/CBF | COUP-TFI/II (NR2F1/2) | 9.8 |
| AF-1 | COUP-TFII (NR2F2) | 4.6 |
| PEPCK PR | GR (NR3C1) | 2.5 |
| ACP BP | HIVEP1c | 2.5 |
| RAR(DR5) | RAR | −∞b |
| ORE | NFAT5 | −9.9 |
| HNF-3 | FOXA1/2/3 | −7.4 |
| NF-Y | NF-Y | −6.7 |
| MEF-1 | MyoDd | −6.5 |
Ingenuity pathway analysis was performed to link the TFs identified from the protein/DNA array to the differentially expressed genes from the microarray analysis (Fig. 3). The two TFs with the highest number of connections to the genes were CCAAT/enhancer-binding protein alpha (C/EBPα) and GR (also known as NR3C1). Most of the identified TFs had few or no links to the differentially expressed genes.
Fig. 3.
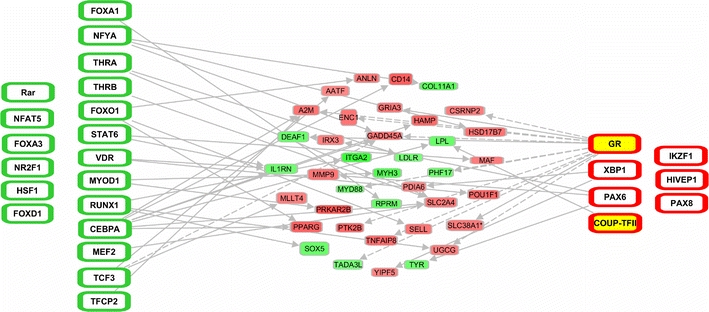
Linking of TFs identified by protein/DNA array to differentially expressed genes. TFs that show less (green outline) or more (red outline) DNA-binding activity in post-MI LV tissue are depicted at the far left and right and are connected with upregulated (red) and downregulated (green) genes depicted in the center. TFs in yellow show matching changes in both protein/DNA array and TFBS analysis. A line was drawn between a TF and a gene if it was known from the literature that the TF can cause the expression of the gene. Uninterrupted and dashed lines indicate physical and indirect interactions between molecules
Five TFs were identified in both the in silico TFBS analysis and the protein/DNA array. Paired box protein 8 (PAX8) and IKZF1 were overrepresented in the down-regulated genes after MI, while their DNA-binding activity was increased in remodeled myocardium, which could be explained if these TFs were repressors of gene expression. However, PAX8 is considered an activator of expression [41] and IKZF1 can act both as activator and repressor [16]. The transcriptional activator myogenic differentiation (MyoD) [55] showed decreased DNA-binding activity, while a TFBS, to which MyoD could bind was overrepresented in upregulated genes. For two TFs corresponding data were found in TFBS and protein/DNA array analysis, i.e. COUP-TFII (also known as NR2F2) and GR. As connections with the microarray data set were more abundant for GR than for COUP-TFII and since a pharmacological antagonist of COUP-TFII is, to our knowledge, not currently available we selected GR for validation of our transcriptional genomics approach.
Pharmacological blockade of GR
To test if GR is a critical mediator of LV remodeling, the GR antagonist mifepristone [2] (Mifegyne, Nordic Pharma BV, Baarn, the Netherlands) was administered to a subset of seven MI pigs starting 1 day after the MI. The animals received a daily dose of 200 mg p.o. for the first 3 days of treatment, and 100 mg/kg p.o. thereafter until the day of sacrifice. Mifepristone attenuated LV hypertrophy by 65% as reflected in the lower LVW/BW ratio measured at 3 weeks after MI (Fig. 4). However, mifepristone treatment of MI pigs had no effect on LV end-diastolic dimensions and did not improve 2D-ejection fraction or LVdP/dtP40 compared to untreated MI pigs (Fig. 4). In addition, mifepristone had no significant effect on stroke volume, cardiac output, tau, LV end-diastolic pressure, or right ventricular hypertrophy (see Supplemental Table 3).
Fig. 4.
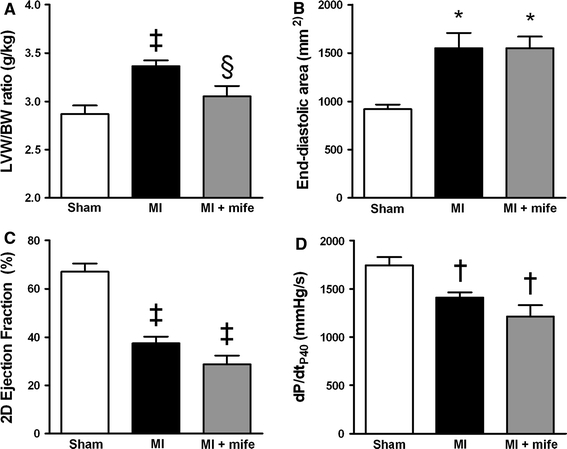
Effect of GR antagonist mifepristone on cardiac remodeling and function following MI. Sham (n = 20), MI (n = 24) and MI + mifepristone (n = 7) animals were studied. a LVW/BW ratios. b LV-dilation as indicated by LV end-diastolic area. c LV 2D-Ejection fraction. d LV dP/dtP40. *P < 0.05, † P < 0.01, ‡ P < 0.001 vs. sham, § P < 0.05 vs. MI. Measurements are mean ± SEM
Discussion
The present study is the first integrative unbiased investigation into the transcriptional control of post-MI LV hypertrophic remodeling in a large animal model, combining microarray data with TFBS data-mining and nuclear protein/DNA array data. Our approach identified two potential key players in LV remodeling, i.e., COUP-TFII and GR. Subsequent GR blockade revealed a critical role of this nuclear receptor in post-MI LV hypertrophy in swine, although it failed to blunt post-MI LV dilation.
In silico scanning of microarray data for TFBS in promoter regions of genes, identified a number of TFBS that were significantly overrepresented in either upregulated or downregulated genes. A number of these TFs have already been implicated in murine models of cardiac hypertrophy. SRF is part of the fetal gene program initiated after hypertrophy, and its transcriptional activity is higher in neonatal cardiomyocytes treated with hypertrophic stimuli [7]. Furthermore, SRF overexpression in a transgenic mouse model caused hypertrophy and cardiomyopathy [67]. BACH1 has been implicated in cardiac hypertrophy as knock-out mice display less hypertrophy following pressure-overload [35]. In our analysis BACH1 was overrepresented in downregulated genes, which is in line with its function as a transcriptional repressor [9]. Finally, AhR is considered to be a negative regulator of cardiac hypertrophy, as AhR−/− mice develop significant cardiac hypertrophy [56], and TFBS analysis showed that binding sites for AhR were overrepresented in downregulated genes after MI. Hannenhalli et al. [20] recently identified TFBS overrepresented in differentially expressed genes in human end-stage systolic heart failure. In that study, an important role for the family of FOX transcription factors was found, a number of which were also identified in our data set (see Table 2). Except for the FOX family of TFs, their dataset [20] shows remarkable little overlap with ours, which might be explained by the distinction between early LV hypertrophy versus end-stage heart failure, as well as the older age of the human patients compared with the swine used in this study and the usage of medication in the human patient population.
Protein/DNA array analysis simultaneously assays the DNA-binding activity of a large set of TFs and has previously been applied to cultured cardiomyocytes [8, 60], but not to studies on TF activity in LV remodeling. Of the TFs identified in the protein/DNA array that were not confirmed by the TFBS analysis, RAR has been implicated in hypertrophy, although its role is controversial.The RAR was found to suppress the α-adrenergic receptor-dependent hypertrophy in isolated cardiomyocytes [68], whereas overexpression of RAR in the heart caused dilated cardiomyopathy in mice [4]. Furthermore, a recent report suggests that retinoic acid induces a cardiac phenotype that is more compatible with physiological hypertrophy [13].
Overrepresentation of the TFBS for the NFATc1-4 family in downregulated genes was unexpected, as NFATs are considered transcriptional activators and NFATc1-4 are activated in cardiac hypertrophy, at least in rodents [36]. Indeed, NFAT transcriptional activity was reported to be increased in mice 3 weeks after MI [22]. This discrepancy might be explained by NFAT5 binding to the TFBS of NFATc1-4. In contrast to NFATc1-4, NFAT5 does not have its own entry in the TRANSFAC database and would therefore not be identified in the TFBS analysis. NFAT5 binds to the 5′-TGGAAA(C/A/T)A(T/A)-3′ motif, which contains the NFATc1-4 cognate element 5′-(T/A/C)GGAA(A/G)-3′ [37]. In line with this notion, using protein/DNA array, we found NFAT5 DNA-binding activity to be reduced in MI hearts. Similarly, NFAT5 DNA-binding activity is decreased in pacing-induced heart failure in dogs [53]. In contrast to NFATc1-4, NFAT5 is not activated by calcineurin-mediated dephosphorylation [37]. NFAT5 could play a role in cardiomyocyte survival, as expression of a dominant negative protein in neonatal cardiomyocytes decreased cell viability [24]. Interestingly, no known NFAT5 target genes were identified among the differentially expressed genes in our microarray analysis. Further studies into the role of NFAT5 in cardiac remodeling are warranted.
For two TFs, matching data were found in TFBS and protein/DNA array analysis, i.e., COUP-TFII and GR. COUP-TFs are involved in suppression of genes involved in fatty acid oxidation after pressure overload induced hypertrophy [46]. We found COUP-TFII DNA-binding activity to be increased in post-MI hearts and COUP-TFII binding sites to be overrepresented in promoter regions of upregulated genes. As COUP-TFs are mainly considered as repressors of gene expression [44], our data could be interpreted to suggest that the increase in COUP-TF binding activity in post-MI hearts represents a compensatory reaction, possibly as a response to the elevated expression of target genes. Unfortunately no inhibitors of COUP-TF are, to our knowledge, available, so its contribution to LV remodeling could not be validated pharmacologically in vivo. The second TF identified by our integrative approach was GR, which was extensively linked to the microarray data (Fig. 3). We found that the increase in cardiac mass was blunted by GR blockade with mifepristone. These findings indicate that glucocorticoids via the GR contribute to post-infarct hypertrophy of the remote myocardium.
Glucocorticoids have been prescribed as anti-inflammatory agents and a large body of data has accrued on its adverse effects on the heart. It was first recognized that the GR agonist dexamethasone, given as treatment for chronic lung disease in pre-term children, can result in cardiac hypertrophy [62]. In a large population-based study it was shown that patients using glucocorticoids were at increased risk of developing cardiovascular disease and a dose-dependent increase in risk for development of heart failure was observed [61]. Hyperproduction of cortisol in Cushing’s syndrome is associated with cardiac hypertrophy, which is reversed upon correction of hypercortisolism [43]. A role for local cardiac glucocorticoid production in mediating LV hypertrophy in response to hemodynamic overload was recently suggested by Ohtani et al. [42], who reported that GR expression was upregulated in pressure-overload hypertrophied LV of rats. Furthermore, cardio-specific elevation of glucocorticoid production augmented the hypertrophy response to pressure overload in mice [42]. Taken together these studies indicate that pharmacologically [61, 62], pathologically [43], or genetically [42] induced hypercortisolism is able to produce or augment hypertrophy. However, these studies do not answer the question whether glucocorticoid signaling contributes to LV hypertrophy under conditions of eucortisolism.
In mice with a recent MI, blocking GR with mifepristone was recently shown to significantly increase angiogenesis [50]. This increased angiogenesis was associated with improved infarct healing and a thicker scar area, which likely contributed to the improved 2D ejection fraction at 1–4 weeks after MI [34]. Unfortunately, the authors did not report on LV end-diastolic dimensions or LV weights. In contrast, we show here that GR contributes to post-MI LV hypertrophy, as mifepristone attenuated LV hypertrophy in MI pigs, while post-infarct LV dilatation and 2D ejection fraction as well as LV systolic and diastolic dysfunction were not affected. Thus, treatment with mifepristone specifically blunted the post-MI increase in LV mass, but failed to reverse the LV dilation and dysfunction. Although this observation is not readily explained, it could be speculated that GR blockade may have led to a pro-inflammatory state with a NF-κB mediated increase in activity of metalloproteinases (MMPs) in the remote area of the LV. Increased activity of MMPs has been implicated in post-MI LV remodeling [52] and the pro-hypertrophic calcineurin/NFAT pathway leads to upregulation of MMPs [47]. Whether this mechanism was indeed operative should be the subject of future studies. Mifepristone was used to validate our transcriptional genomics approach. Besides a GR antagonist, mifepristone also acts as a progesterone receptor antagonist [2]. In our study mifepristone was used to treat young prepubescent animals of either sex. As plasma levels of progesterone are very low before puberty [12], blocking of the progesterone receptor will have had little contribution to the attenuated hypertrophic response. Although mifepristone decreased LV hypertrophy there was a trend towards decreased ejection fraction and dP/dtP40. Hence, based on our data therapeutic application of GR inhibition in post-infarct patients would appear premature.
Pigs have a clear advantage over rodent models in resembling humans more closely in terms of heart rates and cardiac contractility [26], and autonomic control thereof [10, 21], as well as myofilament protein composition [10, 21] and function [18], and calcium homeostasis [17]. Notwithstanding its translational power, an inherent drawback of the pig as a model organism is that its genome has not been fully sequenced yet. This leads to methodological challenges such as poor microarray annotation and inability to perform TFBS analysis. With the ANEXdb the annotation of the porcine microarray has been vastly improved [6]. The output is the human homolog of the porcine gene, which can then be used for TFBS analysis or network analysis. The fact that human instead of porcine genes had to be used for the TFBS analysis made experimental validation of the results even more important. The protein/DNA array directly measures the DNA-binding activity of TFs, but an inherent limitation of the protein/DNA array is that only a selection of TFs is analyzed. A drawback of the TFBS analysis is that it cannot distinguish between TFs that bind to similar motifs, which leads to uncertainty as to which TF actually binds to the TFBS. By combining the two data sets, we circumvented these limitations and improved the reliability of the identification of TFs. Overcoming these experimental challenges is rewarded by gaining insight into cardiac remodeling in a large animal model with even better translational potential than murine models.
In conclusion, we showed that an unbiased ‘-omics’ approach is feasible in a large laboratory animal. Using this approach in which we combined microarray with in silico TFBS analysis and protein/DNA array analysis to study post-MI LV remodeling, we identified COUP-TFII and GR as potential key mediators in post-MI remodeling. Subsequent blockade of GR in vivo, demonstrated, for the first time, that GR contributes to the increase in LV mass after MI.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Peter van der Spek is gratefully acknowledged for providing the infrastructure for bioinformatical analysis and Justine K. Peeters for discussions on microarray analysis. This work was supported by grants from the Netherlands Heart Foundation (NHS2005B234 & NHS2000T042).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Brady JP, Kantorow M, Sax CM, Donovan DM, Piatigorsky J. Murine transcription factor alpha A-crystallin binding protein I. Complete sequence, gene structure, expression, and functional inhibition via antisense RNA. J Biol Chem. 1995;270:1221–1229. doi: 10.1074/jbc.270.3.1221. [DOI] [PubMed] [Google Scholar]
- 2.Cadepond F, Ulmann A, Baulieu EE. RU486 (mifepristone): mechanisms of action and clinical uses. Annu Rev Med. 1997;48:129–156. doi: 10.1146/annurev.med.48.1.129. [DOI] [PubMed] [Google Scholar]
- 3.Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, Frey N. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol. 2010;105:301–313. doi: 10.1007/s00395-009-0046-y. [DOI] [PubMed] [Google Scholar]
- 4.Colbert MC, Hall DG, Kimball TR, Witt SA, Lorenz JN, Kirby ML, Hewett TE, Klevitsky R, Robbins J. Cardiac compartment-specific overexpression of a modified retinoic acid receptor produces dilated cardiomyopathy and congestive heart failure in transgenic mice. J Clin Invest. 1997;100:1958–1968. doi: 10.1172/JCI119727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conraads VM, Vrints CJ, Rodrigus IE, Hoymans VY, Van Craenenbroeck EM, Bosmans J, Claeys MJ, Van Herck P, Linke A, Schuler G, Adams V. Depressed expression of MuRF1 and MAFbx in areas remote of recent myocardial infarction: a mechanism contributing to myocardial remodeling? Basic Res Cardiol. 2010;105:219–226. doi: 10.1007/s00395-009-0068-5. [DOI] [PubMed] [Google Scholar]
- 6.Couture O, Callenberg K, Koul N, Pandit S, Younes R, Hu ZL, Dekkers J, Reecy J, Honavar V, Tuggle C. ANEXdb: an integrated animal ANnotation and microarray EXpression database. Mamm Genome. 2009;20:768–777. doi: 10.1007/s00335-009-9234-1. [DOI] [PubMed] [Google Scholar]
- 7.Davis FJ, Gupta M, Camoretti-Mercado B, Schwartz RJ, Gupta MP. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J Biol Chem. 2003;278:20047–20058. doi: 10.1074/jbc.M209998200. [DOI] [PubMed] [Google Scholar]
- 8.Davis FJ, Pillai JB, Gupta M, Gupta MP. Concurrent opposite effects of trichostatin A, an inhibitor of histone deacetylases, on expression of alpha-MHC and cardiac tubulins: implication for gain in cardiac muscle contractility. Am J Physiol Heart Circ Physiol. 2005;288:H1477–H1490. doi: 10.1152/ajpheart.00789.2004. [DOI] [PubMed] [Google Scholar]
- 9.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 10.Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail. 2009;2:262–271. doi: 10.1161/CIRCHEARTFAILURE.108.814459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duncker DJ, Boontje NM, Merkus D, Versteilen A, Krysiak J, Mearini G, El Armouche A, de Beer VJ, Lamers JM, Carrier L, Walker LA, Linke WA, Stienen GJ, van der Velden J. Prevention of myofilament dysfunction by beta-blocker therapy in postinfarct remodeling. Circ Heart Fail. 2009;2:233–242. doi: 10.1161/CIRCHEARTFAILURE.108.806125. [DOI] [PubMed] [Google Scholar]
- 12.Elsaesser F, Parvizi N, Ellendorff F. Steroid feedback on luteinizing hormone secretion during sexual maturation in the pig. J Endocrinol. 1978;78:329–342. doi: 10.1677/joe.0.0780329. [DOI] [PubMed] [Google Scholar]
- 13.Freire CM, Azevedo PS, Minicucci MF, Oliveira SJ, Martinez PF, Novo R, Chiuso-Minicucci F, Matsubara BB, Matsubara LS, Okoshi K, Novelli EL, Zornoff LA, Paiva SA. Influence of different doses of retinoic acid on cardiac remodeling. Nutrition. 2011;27:824–828. doi: 10.1016/j.nut.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Garlie JB, Hamid T, Gu Y, Ismahil MA, Chandrasekar B, Prabhu SD (2011) Tumor necrosis factor receptor 2 signaling limits beta-adrenergic receptor-mediated cardiac hypertrophy in vivo. Basic Res Cardiol [ePub ahead of print]. doi:10.1007/s00395-011-0196-6 [DOI] [PubMed]
- 15.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Georgopoulos K. Haematopoietic cell-fate decisions, chromatin regulation and ikaros. Nat Rev Immunol. 2002;2:162–174. doi: 10.1038/nri747. [DOI] [PubMed] [Google Scholar]
- 17.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamdani N, de Waard MC, Messer AE, Boontje NM, Kooij V, van Dijk SJ, Versteilen A, Lamberts R, Merkus D, Dos Remedios C, Duncker DJ, Borbely A, Papp Z, Paulus W, Stienen GJ, Marston SB, van der Velden J. Myofilament dysfunction in cardiac disease from mice to men. J Muscle Res Cell Motil. 2008;29:189–201. doi: 10.1007/s10974-008-9160-y. [DOI] [PubMed] [Google Scholar]
- 19.Hammoud L, Lu X, Lei M, Feng Q. Deficiency in TIMP-3 increases cardiac rupture and mortality post-myocardial infarction via EGFR signaling: beneficial effects of cetuximab. Basic Res Cardiol. 2011;106:459–471. doi: 10.1007/s00395-010-0147-7. [DOI] [PubMed] [Google Scholar]
- 20.Hannenhalli S, Putt ME, Gilmore JM, Wang J, Parmacek MS, Epstein JA, Morrisey EE, Margulies KB, Cappola TP. Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation. 2006;114:1269–1276. doi: 10.1161/CIRCULATIONAHA.106.632430. [DOI] [PubMed] [Google Scholar]
- 21.Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- 22.Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H, Wollert KC. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci USA. 2005;102:1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heusch G, Schulz R. A radical view on the contractile machinery in human heart failure. J Am Coll Cardiol. 2011;57:310–312. doi: 10.1016/j.jacc.2010.06.057. [DOI] [PubMed] [Google Scholar]
- 24.Ito T, Fujio Y, Takahashi K, Azuma J. Degradation of NFAT5, a transcriptional regulator of osmotic stress-related genes, is a critical event for doxorubicin-induced cytotoxicity in cardiac myocytes. J Biol Chem. 2007;282:1152–1160. doi: 10.1074/jbc.M609547200. [DOI] [PubMed] [Google Scholar]
- 25.Javadov S, Rajapurohitam V, Kilic A, Hunter JC, Zeidan A, Said Faruq N, Escobales N, Karmazyn M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: the effect of NHE-1 inhibition. Basic Res Cardiol. 2011;106:99–109. doi: 10.1007/s00395-010-0122-3. [DOI] [PubMed] [Google Scholar]
- 26.Kass DA, Hare JM, Georgakopoulos D. Murine cardiac function: a cautionary tail. Circ Res. 1998;82:519–522. doi: 10.1161/01.res.82.4.519. [DOI] [PubMed] [Google Scholar]
- 27.Katz AM. The “modern” view of heart failure: how did we get here? Circ Heart Fail. 2008;1:63–71. doi: 10.1161/CIRCHEARTFAILURE.108.772756. [DOI] [PubMed] [Google Scholar]
- 28.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–H1344. doi: 10.1152/ajpheart.01175.2005. [DOI] [PubMed] [Google Scholar]
- 29.Kel A, Voss N, Jauregui R, Kel-Margoulis O, Wingender E. Beyond microarrays: find key transcription factors controlling signal transduction pathways. BMC Bioinformatics. 2006;7(Suppl 2):S13. doi: 10.1186/1471-2105-7-S2-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kel A, Voss N, Valeev T, Stegmaier P, Kel-Margoulis O, Wingender E. ExPlain: finding upstream drug targets in disease gene regulatory networks. SAR QSAR Environ Res. 2008;19:481–494. doi: 10.1080/10629360802083806. [DOI] [PubMed] [Google Scholar]
- 31.Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther. 2010;127:295–314. doi: 10.1016/j.pharmthera.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Krusche CA, Holthofer B, Hofe V, van de Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res Cardiol. 2011;106:617–633. doi: 10.1007/s00395-011-0175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuster DW, Merkus D, Jorna HJ, Dekkers DH, Duncker DJ, Verhoeven AJ. Nuclear protein extraction from frozen porcine myocardium. J Physiol Biochem. 2011;67:165–173. doi: 10.1007/s13105-010-0059-x. [DOI] [PubMed] [Google Scholar]
- 34.McSweeney SJ, Hadoke PW, Kozak AM, Small GR, Khaled H, Walker BR, Gray GA. Improved heart function follows enhanced inflammatory cell recruitment and angiogenesis in 11betaHSD1-deficient mice post-MI. Cardiovasc Res. 2010;88:159–167. doi: 10.1093/cvr/cvq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mito S, Ozono R, Oshima T, Yano Y, Watari Y, Yamamoto Y, Brydun A, Igarashi K, Yoshizumi M. Myocardial protection against pressure overload in mice lacking Bach1, a transcriptional repressor of heme oxygenase-1. Hypertension. 2008;51:1570–1577. doi: 10.1161/HYPERTENSIONAHA.107.102566. [DOI] [PubMed] [Google Scholar]
- 36.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morancho B, Minguillon J, Molkentin JD, Lopez-Rodriguez C, Aramburu J. Analysis of the transcriptional activity of endogenous NFAT5 in primary cells using transgenic NFAT-luciferase reporter mice. BMC Mol Biol. 2008;9:13. doi: 10.1186/1471-2199-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 39.Neuhold LA, Wold B. HLH forced dimers: tethering MyoD to E47 generates a dominant positive myogenic factor insulated from negative regulation by Id. Cell. 1993;74:1033–1042. doi: 10.1016/0092-8674(93)90725-6. [DOI] [PubMed] [Google Scholar]
- 40.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 41.Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol. 1999;19:2051–2060. doi: 10.1128/mcb.19.3.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohtani T, Mano T, Hikoso S, Sakata Y, Nishio M, Takeda Y, Otsu K, Miwa T, Masuyama T, Hori M, Yamamoto K. Cardiac steroidogenesis and glucocorticoid in the development of cardiac hypertrophy during the progression to heart failure. J Hypertens. 2009;27:1074–1083. doi: 10.1097/HJH.0b013e328326cb04. [DOI] [PubMed] [Google Scholar]
- 43.Pereira AM, Delgado V, Romijn JA, Smit JW, Bax JJ, Feelders RA. Cardiac dysfunction is reversed upon successful treatment of Cushing’s syndrome. Eur J Endocrinol. 2010;162:331–340. doi: 10.1530/EJE-09-0621. [DOI] [PubMed] [Google Scholar]
- 44.Pereira FA, Tsai MJ, Tsai SY. COUP-TF orphan nuclear receptors in development and differentiation. Cell Mol Life Sci. 2000;57:1388–1398. doi: 10.1007/PL00000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ringner M. What is principal component analysis? Nat Biotechnol. 2008;26:303–304. doi: 10.1038/nbt0308-303. [DOI] [PubMed] [Google Scholar]
- 46.Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA. 1997;94:6438–6443. doi: 10.1073/pnas.94.12.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saygili E, Rana OR, Meyer C, Gemein C, Andrzejewski MG, Ludwig A, Weber C, Schotten U, Kruttgen A, Weis J, Schwinger RH, Mischke K, Rassaf T, Kelm M, Schauerte P. The angiotensin-calcineurin-NFAT pathway mediates stretch-induced up-regulation of matrix metalloproteinases-2/-9 in atrial myocytes. Basic Res Cardiol. 2009;104:435–448. doi: 10.1007/s00395-008-0772-6. [DOI] [PubMed] [Google Scholar]
- 48.Schoenauer R, Emmert MY, Felley A, Ehler E, Brokopp C, Weber B, Nemir M, Faggian GG, Pedrazzini T, Falk V, Hoerstrup SP, Agarkova I. EH-myomesin splice isoform is a novel marker for dilated cardiomyopathy. Basic Res Cardiol. 2011;106:233–247. doi: 10.1007/s00395-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–17. [PMC free article] [PubMed] [Google Scholar]
- 50.Small GR, Hadoke PW, Sharif I, Dover AR, Armour D, Kenyon CJ, Gray GA, Walker BR. Preventing local regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci U S A. 2005;102:12165–12170. doi: 10.1073/pnas.0500641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sorop O, Merkus D, de Beer VJ, Houweling B, Pistea A, McFalls EO, Boomsma F, van Beusekom HM, van der Giessen WJ, VanBavel E, Duncker DJ. Functional and structural adaptations of coronary microvessels distal to a chronic coronary artery stenosis. Circ Res. 2008;102:795–803. doi: 10.1161/CIRCRESAHA.108.172528. [DOI] [PubMed] [Google Scholar]
- 52.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 53.Srivastava S, Chandrasekar B, Bhatnagar A, Prabhu SD. Lipid peroxidation-derived aldehydes and oxidative stress in the failing heart: role of aldose reductase. Am J Physiol Heart Circ Physiol. 2002;283:H2612–H2619. doi: 10.1152/ajpheart.00592.2002. [DOI] [PubMed] [Google Scholar]
- 54.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 55.Tapscott SJ. The circuitry of a master switch: MyoD and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- 56.Thackaberry EA, Gabaldon DM, Walker MK, Smith SM. Aryl hydrocarbon receptor null mice develop cardiac hypertrophy and increased hypoxia-inducible factor-1alpha in the absence of cardiac hypoxia. Cardiovasc Toxicol. 2002;2:263–274. doi: 10.1385/ct:2:4:263. [DOI] [PubMed] [Google Scholar]
- 57.van der Velden J, Merkus D, Klarenbeek BR, James AT, Boontje NM, Dekkers DH, Stienen GJ, Lamers JM, Duncker DJ. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004;95:e85–e95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- 58.van Kats JP, Duncker DJ, Haitsma DB, Schuijt MP, Niebuur R, Stubenitsky R, Boomsma F, Schalekamp MA, Verdouw PD, Danser AH. Angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade prevent cardiac remodeling in pigs after myocardial infarction: role of tissue angiotensin II. Circulation. 2000;102:1556–1563. doi: 10.1161/01.cir.102.13.1556. [DOI] [PubMed] [Google Scholar]
- 59.Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D’Agostino RB, Levy D, Kannel WB, Vasan RS. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–2062. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Venkatesan B, Valente AJ, Prabhu SD, Shanmugam P, Delafontaine P, Chandrasekar B. EMMPRIN activates multiple transcription factors in cardiomyocytes, and induces interleukin-18 expression via Rac1-dependent PI3 K/Akt/IKK/NF-kappaB andMKK7/JNK/AP-1 signaling. J Mol Cell Cardiol. 2010;49:655–663. doi: 10.1016/j.yjmcc.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med. 2004;141:764–770. doi: 10.7326/0003-4819-141-10-200411160-00007. [DOI] [PubMed] [Google Scholar]
- 62.Werner JC, Sicard RE, Hansen TW, Solomon E, Cowett RM, Oh W. Hypertrophic cardiomyopathy associated with dexamethasone therapy for bronchopulmonary dysplasia. J Pediatr. 1992;120:286–291. doi: 10.1016/s0022-3476(05)80446-9. [DOI] [PubMed] [Google Scholar]
- 63.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 64.Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I, Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–103. doi: 10.1161/01.cir.98.2.100. [DOI] [PubMed] [Google Scholar]
- 66.Zhang J, Duncker DJ, Ya X, Zhang Y, Pavek T, Wei H, Merkle H, Ugurbil K, From AH, Bache RJ (1995) Effect of left ventricular hypertrophy secondary to chronic pressure overload on transmural myocardial 2-deoxyglucose uptake. A 31P NMR spectroscopic study. Circulation 92:1274–1283. doi:10.1161/01.CIR.92.5.1274 [DOI] [PubMed]
- 67.Zhang X, Azhar G, Chai J, Sheridan P, Nagano K, Brown T, Yang J, Khrapko K, Borras AM, Lawitts J, Misra RP, Wei JY. Cardiomyopathy in transgenic mice with cardiac-specific overexpression of serum response factor. Am J Physiol Heart Circ Physiol. 2001;280:H1782–H1792. doi: 10.1152/ajpheart.2001.280.4.H1782. [DOI] [PubMed] [Google Scholar]
- 68.Zhou MD, Sucov HM, Evans RM, Chien KR. Retinoid-dependent pathways suppress myocardial cell hypertrophy. Proc Natl Acad Sci U S A. 1995;92:7391–7395. doi: 10.1073/pnas.92.16.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.