

Abstract
K201 has previously been shown to reduce diastolic contractions in vivo during β-adrenergic stimulation and elevated extracellular calcium concentration ([Ca2+]o). The present study characterised the effect of K201 on electrically stimulated and spontaneous diastolic sarcoplasmic reticulum (SR)-mediated Ca2+ release and contractile events in isolated rat cardiomyocytes during β-adrenergic stimulation and elevated [Ca2+]o. Parallel experiments using confocal microscopy examined spontaneous diastolic Ca2+ release events at an enhanced spatiotemporal resolution. 1.0 μmol/L K201 in the presence of 150 nmol/L isoproterenol (ISO) and 4.75 mmol/L [Ca2+]o significantly decreased the amplitude of diastolic contractions to ~16% of control levels. The stimulated free Ca2+ transient amplitude was significantly reduced, but stimulated cell shortening was not significantly altered. When intracellular buffering was taken into account, K201 led to an increase in action potential-induced SR Ca2+ release. Myofilament sensitivity to Ca2+ was not changed by K201. Confocal microscopy revealed diastolic events composed of multiple Ca2+ waves (2–3) originating at various points along the cardiomyocyte length during each diastolic period. 1.0 μmol/L K201 significantly reduced the (a) frequency of diastolic events and (b) initiation points/diastolic interval in the remaining diastolic events to 61% and 71% of control levels respectively. 1.0 μmol/L K201 can reduce the probability of spontaneous diastolic Ca2+ release and their associated contractions which may limit the propensity for the contractile dysfunction observed in vivo.
Electronic supplementary material
The online version of this article (doi:10.1007/s00395-011-0218-4) contains supplementary material, which is available to authorized users.
Keywords: Calcium, K201, JTV-519, Sarcoplasmic reticulum, Calcium waves
Introduction
Sarcoplasmic reticulum (SR)-mediated Ca2+ handling (which includes Ca2+ uptake via the sarcoplasmic/endoplasmic Ca2+ ATPase pump, SERCA, and Ca2+ release via the ryanodine receptor, RyR2) is altered in almost all models of heart failure and may lead to abnormal myocardial relaxation and filling during diastole [5, 6, 15, 22]. While many studies have correlated the consequences of altered SERCA-mediated Ca2+ uptake and diastolic dysfunction (DD) [1, 20], few have investigated the consequences of abnormal diastolic SR Ca2+ release on whole heart contractile function. To address this issue, a recent study by the authors demonstrated that while in vivo administration of a β-adrenergic agonist or an elevated extracellular calcium concentration ([Ca2+]o) separately did not produce any significant alteration of diastolic function in rat hearts, co-administration of a β-adrenergic agonist and an elevated [Ca2+]o led to an acute onset of DD [13]. A β-adrenergic agonist was used in this in vivo model to stimulate a PKA-mediated increase in SR Ca2+ load. When combined with an elevated [Ca2+]o, DD manifest as diastolic contractions, a raised end-diastolic pressure (EDP) but no alteration of ejection fraction [13].
K201 (JTV-519), a 1,4 benzothiazepine derivative structurally distinct from diltiazem, was originally discovered when screening compounds to prevent sudden cardiac cell death in a rat Langendorff model of myofibrillar over-contraction [12]. The drug has been suggested to improve contractile function in heart failure via a sole inhibitory effect on diastolic RyR2-mediated SR Ca2+ release [27, 29]. However, it is clear from the literature that K201 exhibits effects on additional cellular targets in a species-specific manner [3]. In guinea pig cardiomyocytes, K201 inhibits sodium (I Na) [16] and potassium currents (I K1, I Kr) [16, 17] and prolongs action potential duration [17], yet the effect on these targets has not been determined in alternative species. Investigations of the effect on L-type calcium current (I Ca) have yielded contrasting results with inhibition in the rat and guinea pig [11, 14], whilst no effect in the rabbit [18]. Inhibition of SERCA activity by K201 in rabbit cardiomyocytes has been demonstrated previously, but the effect in rat is unknown [18]. In the above rat model of DD, K201 limits diastolic contractures and reduces EDP [14] in vivo; however, the cellular mechanisms underlying these phenomena are unknown. It is possible that the observed benefit of K201 in vivo could originate from the drugs ability to (a) inhibit abnormal diastolic Ca2+ release events leading to improved contractile performance or (b) alteration of myofilament Ca2+ sensitivity. To test these hypotheses and given K201’s species specificity, this study was designed to characterise the effect of K201 on: (a) the stimulated Ca2+ transient and cell shortening, (b) spontaneous Ca2+ release and the corresponding spontaneous contractile event, (c) myofilament sensitivity to Ca2+ and (d) SR function, in isolated rat cardiomyocytes exposed to an elevated [Ca2+]o and a commonly used β-adrenergic agonist–isoproterenol (ISO).
Materials and methods
Adult rat ventricular cardiomyocyte isolation
Adult male Wistar rats (200–300 g, 8–9 weeks in age, n = 23) were euthanased using a schedule one procedure in accordance with the UK Animals (Scientific Procedures) Act 1986. Hearts were removed and perfused retrogradely at 5 mL min−1 (37°C) with a modified isolation Krebs-Henseleit (KH) solution for 4 min. The composition of the isolation KH was (in mmol/L): NaCl (120), KCl (5.4), HEPES (20), Na H2PO4 (0.52), MgCl26H2O (3.5), taurine (20), creatine (10), glucose (11.1), pH 7.4 with NaOH. This was followed by perfusion with KH containing 1 mg mL−1 collagenase (type I, Worthington Biochemical, Lakewood, NJ, USA), and 0.1 mg ml−1 protease (type XIV, Sigma-Aldrich, UK). After ~6.0 min, enzyme was removed and the heart perfused with nominally Ca2+-free isolation KH solution containing 1% BSA (Sigma-Aldrich, UK) for a further 6.0 min. The left ventricular free wall was then cut into strips and mixed to yield a single cell suspension. Cells were maintained in either Ca2+-free isolation KH solution or 1.0 mmol/L Ca2+ (via stepwise increments) until use.
Field stimulation of intact cardiomyocytes with simultaneous whole cell epi-fluorescence and shortening measurements
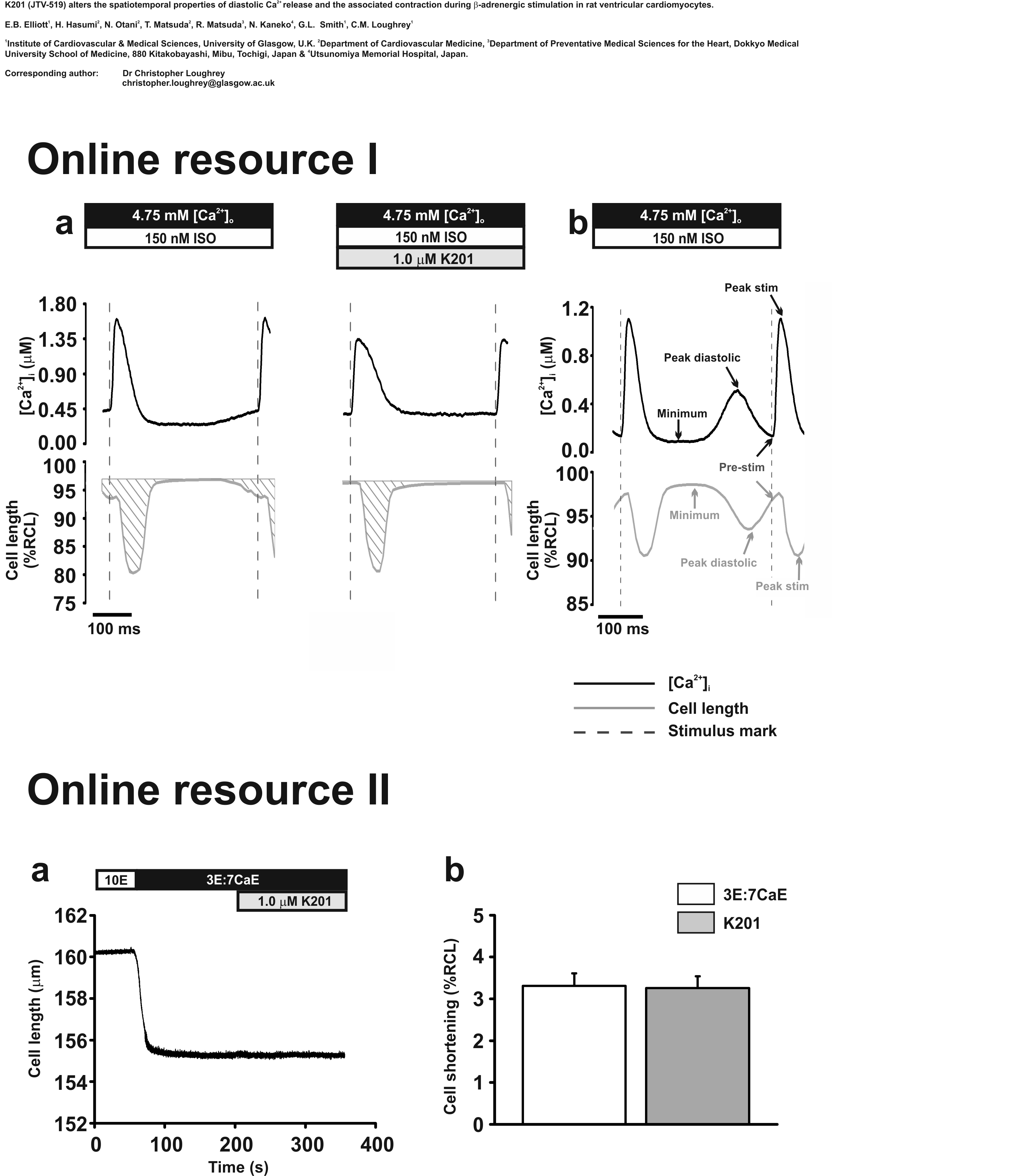
Intact cardiomyocytes in modified isolation KH solution (above) were loaded with a Ca2+ sensitive fluorophore (5.0 μmol/L Fura-4F AM, Invitrogen, UK) by incubation for ~10 min. The incubation medium was removed and the cells re-suspended in a second modified KH solution with the following composition (in mmol/L): NaCl (140), KCl (4.0), MgCl2 (1.0), HEPES (5.0), glucose (11.1), pH 7.4 with NaOH with 1.25 mmol/L [Ca2+] added. Cells were incubated for a further 30 min to ensure complete de-esterification. Cardiomyocytes were allowed to settle on a coverslip, placed on a bath (Cell Microcontrols, Norfolk, VA, USA) and super-fused with the same modified KH solution at 37°C. Cells were field stimulated with 2 ms duration voltage pulses delivered through parallel platinum wires (stimulation voltage set to 1.5 times the threshold). K201 (a gift from Aetas Pharma Co. Ltd., Japan) was prepared at a stock concentration of 1.0 mmol/L in 10% DMSO; the final concentration of K201 in the perfusate was 1.0 μmol/L. A parallel set of vehicle (DMSO) time control experiments were performed; all data obtained in K201 were normalised to these timed controls. The Fura-4F fluorescence (340 and 380 nm excitation; R 340/380nm) was measured using a spinning wheel spectrophotometer (Cairn Research Ltd., UK; sampling rate of 500 Hz) whilst cellular shortening was measured using a video edge detection system (IonOptix, Milton, MA, USA; sampling rate of 200 Hz). Data were analysed offline. Fura-4F fluorescence ratio was converted to intracellular [Ca2+] ([Ca2+]i) as previously described [21]. Mean [Ca2+]i and cell shortening signals were obtained by averaging 12 steady state transients (Origin; Online resource I). The mean minimum [Ca2+]i was measured from a single point at which [Ca2+]i was at its lowest during the diastolic period [see online resource I(b)]. Diastolic Ca2+ event amplitude, the degree of spontaneous Ca2+ release, was calculated from the minimum [Ca2+]i to the peak diastolic [Ca2+]i reached during the diastolic period. Diastolic Ca2+ events do not necessarily return to the minimum [Ca2+]i before the next action-potential stimulation. The [Ca2+]i immediately preceding the point of stimulation is therefore termed as the pre-stimulus [Ca2+]i (pre-stim). Pre-stim [Ca2+]i was subtracted from the stimulated [Ca2+]i transient peak to determine stimulated Ca2+ transient amplitude. Shortening data were expressed as percentage of resting cell length (% RCL) where resting cell length was taken at the quiescent cell length prior to commencement of field stimulation. Cell shortening parameters were measured as described above for [Ca2+]i and shown in Online resource Ib.
Myofilament Ca2+ sensitivity measurements in intact cells
Intact cardiomyocytes in modified KH solution were Fura-4F AM loaded and field stimulated as above. Fluorescence and cell shortening were assessed in three sub-sets of experiments during perfusion with KH solution containing: (a) a range of [Ca2+]o (0.5–1.8 mmol/L), (b) 1.8 mmol/L [Ca2+]o in the presence of K201 (0.3, 1.0 and 3.0 μmol/L) and (c) 1.8 mmol/L [Ca2+]o in the presence of diltiazem (4.0 and 8.0 μmol/L, Sigma-Aldrich, UK). Data obtained in KH solution with a [Ca2+]o of 1.8 mmol/L were obtained for each sub-set of experiments (to account for any variation between cell populations) and served as respective control groups. Fura-4F fluorescence was converted to [Ca2+]i as above and cell shortening amplitudes were expressed as a percentage of the shortening amplitude at a [Ca2+]o of 1.8 mmol/L. An additional set of experiments in permeabilised cells directly assessed the effect of K201 on the myofilaments (Online resource II).
Simultaneous field stimulation of intact cardiomyocytes with confocal imaging
Intact cardiomyocytes in a modified KH solution were loaded with Fluo-3AM and superfused with modified KH (composition as above). Confocal line-scan images of field stimulated cardiomyocytes were recorded using a Radiance 2000 confocal system (BioRad, UK). Fluo-3 was excited at 488 nm (Kr laser) and measured >515 nm using epifluorescence optics of an inverted microscope with a 60X/1.2 NA water-immersion objective lens. Fluorescence was acquired in line-scan mode at 2 ms line−1; pixel dimension was 0.3 μm (512 pixels/scan; zoom = 1.4) [2]. The scanning laser line was orientated parallel with the long axis of the cell and placed approximately equidistant between the outer edge of the cell and the nucleus/nuclei to ensure the nuclear area was not included in the scan line. Exact timing of electrical stimulation was marked in the confocal image by activating a light-emitting diode above the cell bath for 2 ms (i.e. duration of one line-scan), 8 ms before electrical stimulation. Fluorescence data were expressed as a ratio of the quiescent fluorescence (F/F 0). F/F 0 measurements and the number of initiation points/diastolic interval were calculated from 15 s periods of line scan trace.
Data analysis and statistical procedures
Data were expressed as mean ± SEM. For Ca2+ transient amplitude and Ca2+ wave parameters, comparisons were performed by applying the paired Student’s t test. ANOVA statistics with either a Tukey (Ca2+ transient and shortening parameters) or Dunnett (myofilament sensitivity) post-test were used in cases of multiple comparisons. Differences were considered significant when P < 0.05.
Results
Measurement of spontaneous diastolic events in isolated ventricular cardiomyocytes
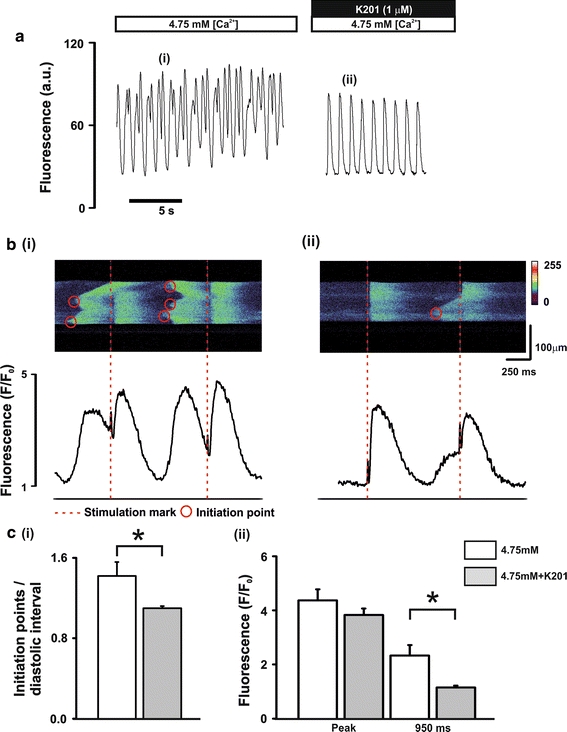
To gain an insight into the cellular mechanisms which contribute to the beneficial effect of K201 in a previously published pharmacologically induced in vivo rat model of diastolic dysfunction [14] isolated rat cells were perfused with a modified KH solution containing the [Ca2+]o and additions shown in Fig. 1a. Cells were field stimulated at 2.0 Hz to obtain a sufficient length of time between stimulations in which to characterise diastolic release events during an elevated [Ca2+]o and β-adrenergic stimulation. When cells were perfused with 4.75 mmol/L [Ca2+]o + 150 nmol/L isoproterenol (referred to hereafter as “ISO”), the amplitude of diastolic rises of [Ca2+]i was markedly increased compared to 4.75 mmol/L [Ca2+]o alone (referred to hereafter as “4.75”) [73.5 ± 4.8 vs. 483 ± 103 nmol/L; 4.75 vs. ISO: n = 8, P < 0.05; Fig. 1b(i vs. ii) top panel and c(i)]. These diastolic Ca2+ events consistently occurred in parallel with diastolic contractile events (Fig. 1b(i vs. ii) bottom panel and d(i)) and were similar to the diastolic rises of intra-ventricular pressure observed in vivo using a similar protocol [14]. When compared to DMSO vehicle time controls, perfusion with 4.75 mmol/L [Ca2+]o + 150 nmol/L isoproterenol + 1.0 μmol/L K201 for 4 min (referred to hereafter as “K201”) resulted in a cell-to-cell variable response on diastolic Ca2+ events. K201 significantly reduced the magnitude of diastolic Ca2+ events in all cells tested (100%) and, in ~50% cells, these were completely abolished (Fig. 1b(iii)). The mean response to K201 was to significantly reduce both the amplitudes of diastolic Ca2+ events [483 ± 103 vs. 154 ± 46 nmol/L; ISO vs. K201: n = 8, P < 0.05; Fig. 1c(i)] and diastolic contractile events [4.90 ± 0.73–0.77 ± 0.33% RCL; ISO vs. K201: n = 8, P < 0.05; Fig. 1d(i)].
Fig. 1.
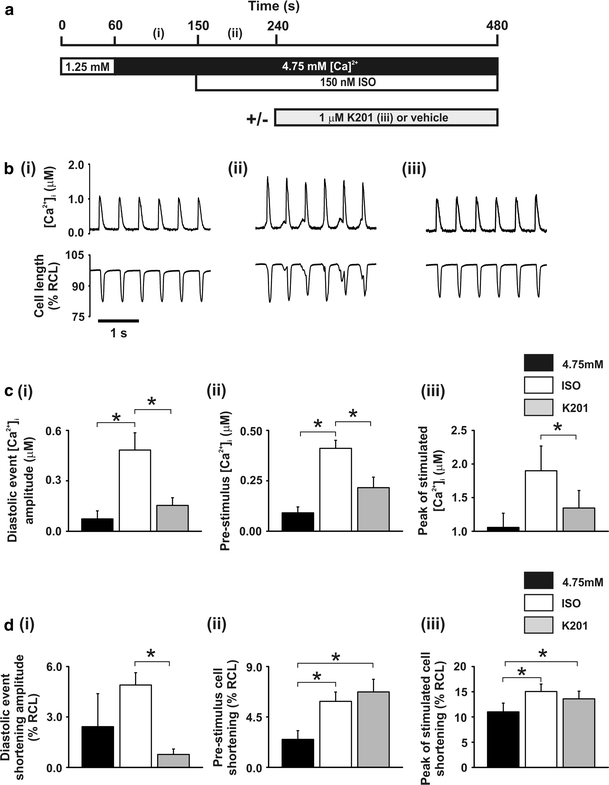
Effect of 1.0 μmol/L K201 on [Ca2+]i and cell shortening a perfusion protocol. b records of [Ca2+]i (upper) and cell length (lower) at points denoted in a. c mean ± SEM values of for (i) diastolic event free [Ca2+]i amplitude, (ii) pre-stimulus free [Ca2+]i and (iii) peak free [Ca2+]i of stimulated transient. d mean ± SEM values of cell shortening for (i) diastolic event amplitude, (ii) pre-stimulus and (iii) peak of stimulated transient. Black bars, 4.75: 4.75 mmol/L [Ca2+]o; White bars, ISO: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO; Grey bars, K201: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO + 1.0 μmol/L K201; n = 8, *P < 0.05
Application of 150 nmol/L ISO resulted in a significant increase in the mean minimum [Ca2+]i compared to 4.75 mmol/L [Ca2+]o alone (60.9 ± 1.7 vs. 159 ± 33.0 nmol/L; 4.75 vs. ISO: n = 8, P < 0.05; data not shown). Subsequent perfusion with K201 did not significantly alter the mean minimum [Ca2+]i (159 ± 33.0 vs. 120 ± 24.9 nmol/L; ISO vs. K201: n = 8, P > 0.05; data not shown).
Measurement of the peak stimulated event in isolated ventricular cardiomyocytes
The mean peak of the stimulated [Ca2+]i transient was significantly reduced by K201 [1,900 ± 367 vs. 1,350 ± 260 nmol/L; ISO vs. K201: n = 8, P < 0.05; Fig. 1c(iii)] as was the mean pre-stimulus [Ca2+]i [411 ± 40 vs. 216 ± 52 nmol/L; ISO vs. K201: n = 8, P < 0.05; Fig. 1c(ii)]. In contrast, there was no significant alteration in either the mean peak of the stimulated cell shortening [15.04 ± 1.49 vs. 13.60 ± 1.52% RCL; ISO vs. K201; n = 8, P > 0.05; Fig. 1d(iii)], mean pre-stimulus cell shortening [(5.89 ± 0.84 vs. 6.74 ± 1.12% RCL; ISO vs. K201; n = 8, P > 0.05; Fig. 1d(ii)] or the mean minimum cell shortening (3.23 ± 0.47 vs. 3.43 ± 0.47% RCL; ISO vs. K201; n = 8, P > 0.05; data not shown) following application of K201.
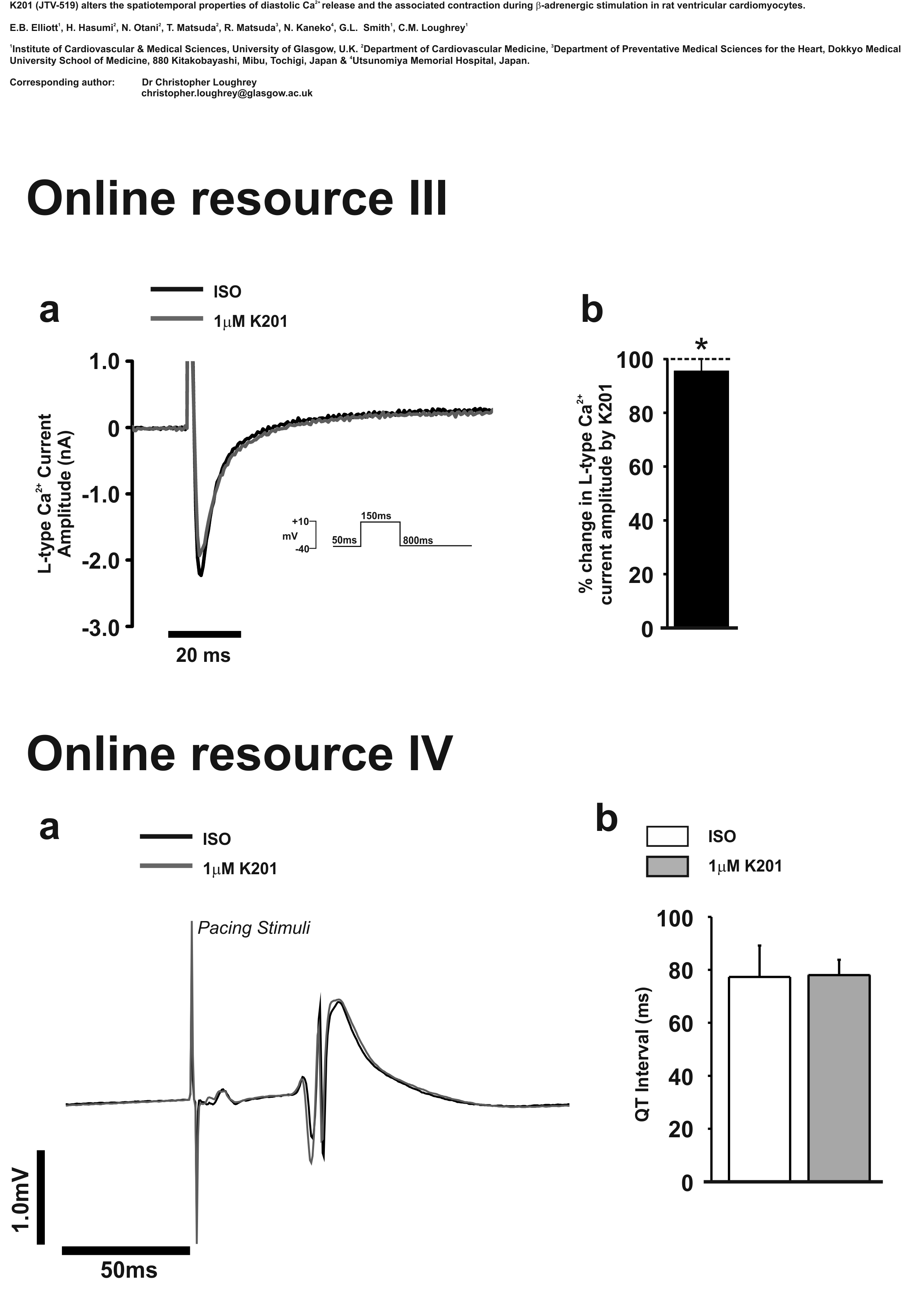
Separate voltage clamp measurements on cells under identical conditions (including the presence of an elevated [Ca2+]o) showed only a small decrease in L-type Ca2+ channel amplitude in response to 1.0 μmol/L K201 (95.6 ± 4.4% of timed control level—see Online resource III).
The effect of K201 on the overall electrical activity of ventricular cells was assessed from ECG measurements on isolated Langendorff perfused whole hearts (see Online resource IV). These revealed no significant differences in QT interval at a fixed stimulus rate suggesting no major change in action potential duration in rat hearts exposed to 1.0 μmol/L K201.
Effects of K201 on myofilament Ca2+ sensitivity
An assessment of the ability of K201 to alter myofilament Ca2+ sensitivity was made as this was a previously proposed mechanism to explain the in vivo improvement of function in the rat [14]. β-adrenergic stimulation alters the Ca2+ sensitivity of the myofilaments and an elevated Ca2+ concentration (i.e. 4.75 mmol/L [Ca2+]o) leads to the production of diastolic contractile events (which can affect the amplitude of the subsequent Ca2+ transient) therefore, these experiments were performed in the absence of β-adrenergic stimulation and at lower [Ca2+]o to ensure that diastolic Ca2+ events did not occur. Simultaneous measurements of [Ca2+]i and cell shortening were used to establish the relationship between Ca2+ transient parameters and cell shortening amplitudes in response to varying [Ca2+]o. Cells were field stimulated and perfused with modified KH solutions containing a range of [Ca2+]o from 1.8 to 0.5 mmol/L. As shown in Fig. 2a, reducing [Ca2+]o from 1.8 mmol/L (control) to 0.5 mmol/L resulted in a significant decrease in Ca2+ transient peak [Ca2+]i [617 ± 25 vs. 180 ± 6 nmol/L; 1.8 (control) vs. 0.5 mmol/L [Ca2+]o: n = 14, P < 0.05] and minimum [Ca2+]i [140 ± 4 vs. 81 ± 2 nmol/L; 1.8 (control) vs. 0.5 mmol/L [Ca2+]o: n = 14, P < 0.05]. This was accompanied by an ~90% reduction in cell shortening amplitude [100.0 vs. 9.28 ± 3.57%; 1.8 mmol/L (control) vs. 0.5 mmol/L [Ca2+]o: n = 14, P < 0.05].
Fig. 2.
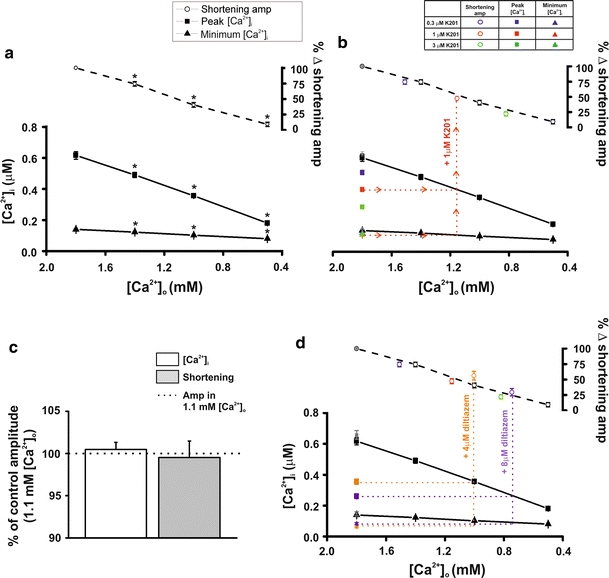
Effect of 1.0 μmol/L K201 on myofilament Ca2+ sensitivity a relationship between free Ca2+ transient parameters (peak and minimum) and cell shortening amplitudes (% shortening) in response to varying external [Ca2+] (n = 14, *P < 0.05 vs.1.8 mmol/L for each). b as in a with additional points obtained in 1.8 mmol/L external [Ca2+] and K201 (0.3, 1.0 and 3.0 μmol/L; n = 24, P < 0.05 for shortening amplitudes at all concentrations vs. 1.8 mmol/L, P < 0.05 for peak [Ca2+]i at 1 and 3 μmol/L K201 vs. 1.8 mmol/L). c mean ± SEM values for percentage change in amplitude of free [Ca2+]i and shortening amplitudes in 1.0 μmol/L K201 expressed relative to control in 1.1 mmol/L external Ca2+ (100%; n = 7). d as in a with additional points obtained in 1.8 mmol/L external [Ca2+] and diltiazem (4.0 and 8.0 μmol/L n = 8, P < 0.05 for shortening amplitudes and peak [Ca2+]i at both concentrations vs. 1.8 mmol/L and for minimum [Ca2+]i at 8.0 μmol/L diltiazem vs. 1.8 mmol/L)
In a second set of experiments, the effect of a range of concentrations of K201 (0.3–3.0 μmol/L) on Ca2+ transient parameters and cell shortening amplitudes was examined at a constant [Ca2+]o (1.8 mmol/L [Ca2+]i). Perfusion with K201 (0.3–3 μmol/L) led to a dose-dependent decrease in Ca2+ transient peak and minimum [Ca2+]i and cell shortening amplitude (Fig. 2b). Addition of 1.0 μmol/L K201 (red symbols) significantly reduced Ca2+ transient peak [Ca2+]i [640 ± 16 vs. 409 ± 13 nmol/L; 1.8 mmol/L [Ca2+]o (control, grey symbols) vs. 1.8 mmol/L [Ca2+]o + 1.0 μmol/L K201: n = 24, P < 0.05]. Similarly, 1.0 μmol/L K201 reduced cell shortening amplitude by ~53% [100.0 vs. 47.25 ± 3.99%; 1.8 mmol/L [Ca2+]o (control, grey symbol) vs. 1.8 mmol/L [Ca2+]o + 1.0 μmol/L K201: n = 24, P < 0.05 vs. control].
To determine the effect of K201 on the relationship between Ca2+ transient parameters and cell shortening amplitude shown in Fig. 2a, Ca2+ transient amplitudes at each K201 concentration were matched to those measured in Ca2+ alone by extrapolation of the K201 Ca2+ transient peak and minimum points (example for 1.0 μmol/L K201 is demonstrated by horizontal red dotted lines, Fig. 2b). Experimentally derived cell shortening amplitudes obtained in 1.8 mmol/L [Ca2+]o and each concentration of K201 were then plotted at the corresponding [Ca2+]o value (e.g. for 1.0 μmol/L K201, vertical red dotted line). As seen in Fig. 2b, the relationship between Ca2+ transient parameters and cell shortening in K201 is the same as that for varying [Ca2+]o alone. For example, the mean Ca2+ transient amplitude in 1.0 μmol/L K201 (297 ± 16 nmol/L) predicts a cell shortening amplitude of 49.1% of control which was confirmed by the experimentally measured value of 47.2 ± 4.0% in 1.0 μmol/L K201.
Using the relationship between Ca2+ transient parameters and cell shortening amplitude derived when varying [Ca2+]o alone (Fig. 2a), it was observed that a Ca2+ transient amplitude equivalent to that of 1.0 μmol/L K201 in 1.8 mmol/L [Ca2+]o (297 ± 16 nmol/L) was produced by perfusion with [Ca2+]o of 1.1 mmol/L (without K201). To verify the above negative result of K201 on myofilament Ca2+ sensitivity, these two solutions were sequentially perfused onto a separate sub-set of cells to directly assess the effect on cell shortening at equivalent Ca2+ transient amplitudes. Figure 2c shows mean data for [Ca2+]i and cell shortening obtained in response to 1.0 μmol/L K201 in 1.8 mmol/L Ca2+ expressed relative to that at 1.1 mmol/L [Ca2+]o alone (100%, without K201; dashed line). The mean data show that at equivalent Ca2+ transient amplitudes, K201 does not significantly alter cell shortening (transient amplitude: 100.48 ± 0.85% of control; shortening amplitude: 99.53 ± 1.95% of control; n = 7, P > 0.05 for each).
To expand this finding, a third set of experiments were performed utilising diltiazem as a tool to alter Ca2+ transient parameters to a similar degree as reducing [Ca2+]o alone. Diltiazem (4.0 and 8.0 μmol/L) caused a dose-dependent decrease in transient peak and minimum [Ca2+]i (Fig. 2d). Equivalent Ca2+ transient amplitudes in diltiazem were matched to the relationship derived in Ca2+ alone (in Fig. 2a) and cell shortening amplitudes were plotted as before. At equivalent Ca2+ transient amplitudes, cell shortening in the presence of diltiazem was not significantly different from the levels predicted by altering [Ca2+]o alone.
Effects of K201 on SR function
Following 1.5 min of 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO then 4.0 min perfusion with or without 1.0 μmol/L K201, rapid application of 10.0 mmol/L caffeine was used to asses SR Ca2+ content (Fig. 3a(i)). Perfusion with K201 resulted in no significant alteration of SR free Ca2+ content as determined by the mean amplitude of the caffeine-induced Ca2+ transient [1,310 ± 230 vs. 1,400 ± 260 nmol/L; 4.75 mmol/L ISO vs. K201; n = 8, P > 0.05; Fig. 3a(ii)]. The rate of decline of the caffeine-induced transients is a measure of the rate of sarcolemmal Ca2+ extrusion, these rates were not altered following K201 (1.35 ± 0.15 vs. 1.21 ± 0.15 μmol/L s−1; ISO vs. K201; n = 8, P > 0.05; data not shown).
Fig. 3.
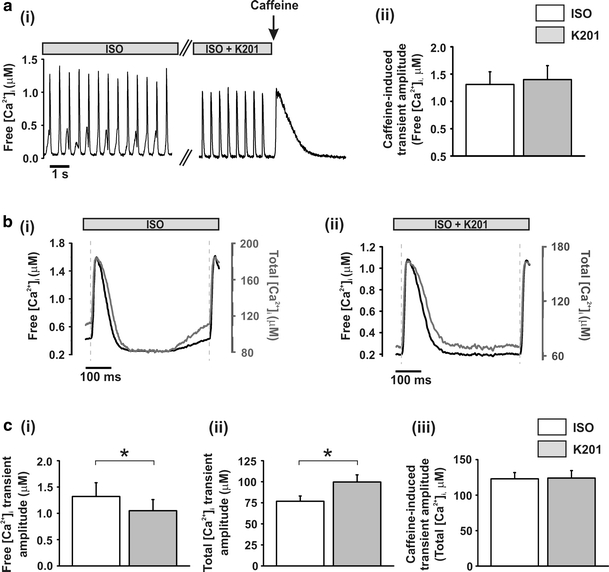
Effect of 1.0 μmol/L K201 on Ca2+ transient amplitude and SR Ca2+ content a (i) typical records of free [Ca2+]i in a cell perfused with 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO then 1 μmol/L K201 with subsequent caffeine application (arrow); (ii) mean ± S.E.M. values of caffeine-induced free Ca2+ transient amplitude (n = 8). b Average transient in free Ca2+ (black trace) and total Ca2+ (grey trace) in the presence of (i) ISO: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO and (ii) ISO + K201: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO + 1.0 μmol/L K201; dashed lines denote stimulus mark. c mean ± SEM values of (i) free Ca2+ transient amplitude (n = 11) and (ii) total Ca2+ transient amplitude (n = 11) both measured from the peak Ca2+ minus pre-stim Ca2+ of their respective averaged traces; (iii) caffeine-induced total Ca2+ transient amplitude. (n = 8). White bars, ISO: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO, Grey bars, K201: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO + 1.0 μmol/L K201; *P < 0.05
Given the dramatic reduction in the amplitude of diastolic Ca2+ release events by K201, the [Ca2+]i immediately preceding the stimulus (pre-stim) was lower after K201 (see Fig. 1c(ii)). The consequences of this alteration in diastolic Ca2+ level at the point of action potential-induced Ca2+ release must be considered when assessing the effect of K201 on the Ca2+ transient amplitude. To allow for varying degrees of intracellular buffering of SR release under these two conditions, mean traces were converted to total cellular [Ca2+] using previously reported intracellular buffer concentrations and affinities (Fig. 3b(i, ii), [8]). The Ca2+ transient amplitude was measured by subtracting the pre-stim from the peak of the stimulated transient for the average signals converted to free [Ca2+] and total [Ca2+]. This analysis revealed that whilst K201 produced a significant reduction in the amplitude of the free Ca2+ transient [1.32 ± 0.26 vs. 1.05 ± 0.21 μmol/L; ISO vs. K201: n = 11, P < 0.05; Fig. 3c(i)] there was a significant increase in the amplitude of the total Ca2+ transient [76.8 ± 6.3 vs. 99.8 ± 8.5 μmol/L; ISO vs. K201: n = 11, P < 0.05; Fig. 3c(ii)]. Conversion of the caffeine-induced Ca2+ transient to total [Ca2+]i showed no significant difference between SR content in ISO and K201 (Fig. 3c(iii)).
Characterisation of the stimulated Ca2+ transients shown in Fig. 1 revealed that whilst 1.0 μmol/L K201 did not alter the maximum rate of rise of the stimulated Ca2+ transient it significantly decreased both the maximum rate of fall [4.3 × 10−5 ± 1.1 × 10−5 vs. 2.4 × 10−5 ± 6.4 × 10−6 M s−1; ISO vs. K201: n = 8, P < 0.05; Online resource V(a)] and the rate of fall at 500 nmol/L [Ca2+]i [1.8 × 10−5 ± 2.2 × 10−6 vs. 1.1 × 10−5 ± 1.3 × 10−6 M s−1; ISO vs. K201: n = 8, P < 0.05; Online resource V (b and c), respectively]. Separate direct measurements of the effect of 1.0 μmol/L K201 on SERCA activity showed no significant inhibition of the rate constant of decay of free [Ca2+]i [index of SERCA function in a permeabilised cell assay as described in Online resource Materials, Online resource V(d)]. While the effect of K201 was not apparent at 1.0 μmol/L, at the higher concentration of 10.0 μmol/L, the rate constant of decay was significantly reduced to 73.9 ± 2.5% [Online resource V(e), n = 4, P < 0.05].
Confocal measurements of diastolic Ca2+ events during β-adrenergic stimulation
To visualise the effect of K201 on individual diastolic Ca2+ release events at an enhanced spatiotemporal resolution, a set of experiments was performed using confocal imaging of rat cardiomyocytes perfused with the same [Ca2+]o and additions as in Fig. 1 (Fig. 4a). Line scan confocal imaging revealed a synchronous rise of [Ca2+]i along the length of the scan line at each stimulation point (Fig. 4b; red dashed lines denote stimulation mark). Upon perfusion with 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO, spontaneous diastolic Ca2+ events were visualised in the line scan (Fig. 4b(ii) vs. b(i) top panels) and corresponding fluorescence intensity profiles (Fig. 4b(ii) vs. b(i) bottom panels). Diastolic Ca2+ events were initiated at discrete points along the scan line (Fig. 4b(ii) top panel) and then propagated along the cell length identifying these diastolic events as being the result of multiple Ca2+ waves. Initiation points of the Ca2+ waves were identified on the basis of evidence from the line scan image of propagation away from a separate site. The number of initiation points which led to Ca2+ waves was markedly increased when ISO was added to the perfusing solution and typically resulted in multiple waves in the diastolic interval (Fig. 4b(ii) top panel). Application of 1.0 μmol/L K201 caused a significant reduction in the frequency of diastolic events (12.45 ± 1.36 vs. 7.55 ± 1.53 diastolic events/15 s period; ISO vs. K201: n = 11, P < 0.05—data not shown) and, as illustrated in Fig. 4b(iii) and c(i), a significant reduction in the initiation points/diastolic interval in the remaining diastolic Ca2+ release events [1.43 ± 0.09 vs. 1.02 ± 0.02; ISO vs. K201: n = 11, P < 0.05; Fig. 4c(i)]. This was accompanied by no significant change in stimulated transient peak fluorescence but a significant reduction in the fluorescence measured at 950 ms post stimulation compared to control [2.33 ± 0.20 vs. 1.48 ± 0.15 F/F 0; ISO vs. K201: n = 11, P < 0.05; Fig. 4c(ii)]. Measurements of Ca2+ spark events during the diastolic period revealed a significant decrease in Ca2+ spark amplitude (3.24 ± 0.18 vs. 2.84 ± 0.16; ISO vs. K201: n = 416 control sparks and n = 183 K201 sparks, P < 0.05) and a trend towards a decrease in Ca2+ spark frequency (see Online resource VI).
Fig. 4.
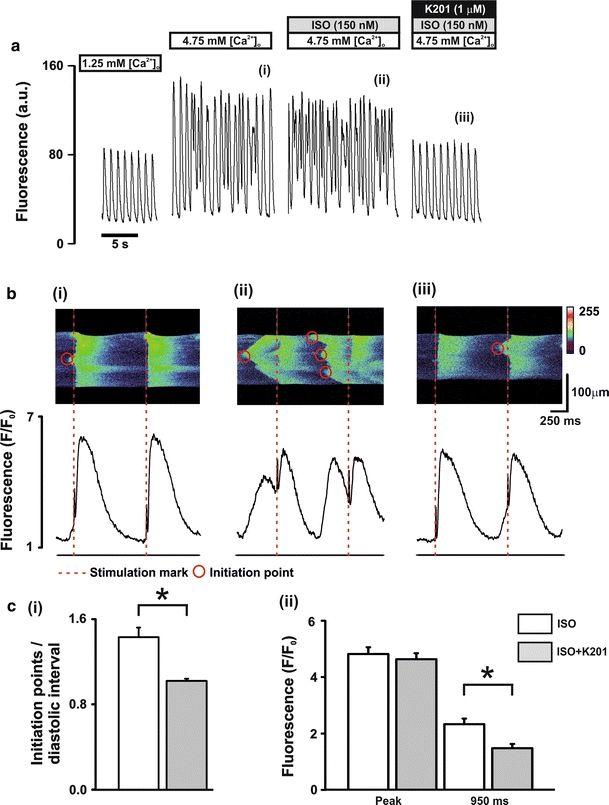
Characterisation of diastolic Ca2+ release events a perfusion protocol (upper) and typical records of Fluo-3 fluorescence (lower). b Typical line scan images (upper) and corresponding fluorescence profiles (lower) in the presence of (i) 4.75 mmol/L [Ca2+]o, (ii) 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO and (iii) 4.75 mmol/L[Ca2+]o + 150 nmol/L ISO + 1.0 μmol/L K201; dashed lines (red) represent stimulus mark, circles represent wave initiation points. c mean ± SEM values of (i) number of wave initiation points/diastolic interval and (ii) fluorescence signals measured at the peak and at 950 ms post stimulation. White bars, ISO: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO; Grey bars, ISO + K201: 4.75 mmol/L [Ca2+]o + 150 nmol/L ISO + 1.0 μmol/L K201; n = 11, *P < 0.05
Confocal measurements of diastolic Ca2+ events during elevated [Ca2+]o
A sub-set of cells exhibited a propensity to produce diastolic Ca2+ events when exposed to 4.75 mmol/L [Ca2+]o without ISO (Fig. 5a). Confocal images obtained from these cells showed a synchronous rise of [Ca2+]i at each stimulation point and multiple waves originating at various initiation points during the diastolic interval (Fig. 5b(i)). Application of 1.0 μmol/L K201 without ISO (referred to hereafter as 4.75 + K201) markedly reduced the frequency of these initiation points/diastolic interval [1.33 ± 0.13 vs. 1.03 ± 0.02; 4.75 vs. 4.75 + K201: n = 4, P < 0.05; Fig. 5b(ii), c(i)] along with a significant decrease in the diastolic fluorescence measured at 950 ms post stimulation compared to 4.75 mmol/L [Ca2+]o without K201 [2.33 ± 0.39 vs. 1.15 ± 0.07 F/F o; 4.75 vs. 4.75 + K201: n = 4, P < 0.05; Fig. 5c(ii)]. No significant change in stimulated transient peak fluorescence was observed.
Fig. 5.
Characterisation of diastolic Ca2+ release events in the absence of ISO a perfusion protocol (upper) and typical records of Fuo-3 fluorescence (lower). b typical line scan images (upper) and corresponding fluorescence profiles (lower) in the presence of (i) 4.75 mmol/L [Ca2+]o and (ii) 4.75 mmol/L [Ca2+]o + 1.0 μmol/L K201; dashed lines (red) represent stimulus mark, circles represent wave initiation points. c mean ± SEM values of (i) number of wave initiation points/diastolic interval and (ii) fluorescence signals measured at the peak and at 950 ms post stimulation. White bars, 4.75: 4.75 mmol/L [Ca2+]o; Grey bars, 4.75 + K201: 4.75 mmol/L [Ca2+]o + 1.0 μmol/L K201; n = 4, *P < 0.05
Confocal measurements of Ca2+ wave initiation points and diastolic Ca2+ event amplitude
To examine the relationship between the number of initiation points per diastolic event and corresponding diastolic event amplitude, the confocal images of a number of cells (n = 8) perfused with 4.75 mmol/L [Ca2+]o and ISO were examined as in Fig. 6a(i–iii) with respective fluorescence intensity profiles (Fig. 6b top–bottom). Number of initiation points (highlighted by red circles—Fig. 6a) per diastolic event was plotted against the amplitude of that event (Fig. 6c). The mean data showed a positive correlation between the two parameters with significant differences between 1, 2 and 3 initiation points (1.06 ± 0.1, 1.87 ± 0.1, 2.58 ± 0.26 F/F 0; P < 0.05; Fig. 6c), respectively.
Fig. 6.
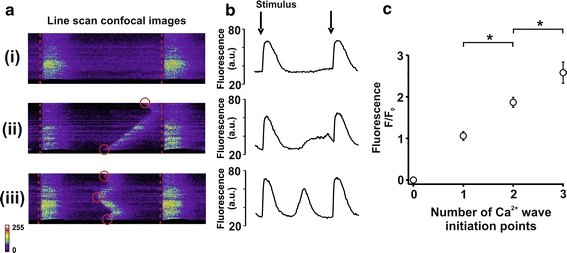
Characterisation of Ca2+ wave initiation points a (i–iv) typical line scan images with varying numbers of initiation points; red lines and circles denote stimulus mark and initiation points, respectively. b (top to bottom): fluorescence intensity profiles corresponding to the images in a; arrows represent the time of stimulation. c mean ± SEM data showing the relationship between diastolic Ca2+ event initiation points and diastolic Ca2+ event amplitude taken from 8 cells in Fig. 4 (1, 2 and 3 initiation points, n = 42, 30 and 6 diastolic Ca2+ events, respectively, *P < 0.05)
Discussion
We have provided for the first time a detailed characterisation of the effects of K201 on the stimulated Ca2+ transient, spontaneous diastolic SR mediated Ca2+ release and contractile events during application of β-adrenergic stimulation and an elevated [Ca2+]o in rat cardiomyocytes. Our main findings are: (a) K201 has no effect on stimulated cell shortening but significantly reduces spontaneous diastolic contractile events induced by elevated Ca2+ loading, (b) K201 produces a small but significant reduction in stimulated free Ca2+ transient amplitude and significantly reduces spontaneous Ca2+ release events with no alteration in SR Ca2+ content, (c) when intracellular buffering is taken into account, K201 produces a significant increase in total Ca2+ transient amplitude, (d) K201 had no effect on myofilament Ca2+ sensitivity thereby discounting this previously proposed mechanism to explain the in vivo improvement of function in the rat [14], (e) 1.0 μmol/L K201 does not inhibit SERCA in rat cardiomyocytes (which is in contrast to the effect in rabbit [18]) and (f) spontaneous diastolic Ca2+ events occurred in the form of multiple propagating Ca2+ waves (each with an initiating point) and that K201 acts to reduce both the event frequency and number of initiation points per diastolic event.
Comparison between the effect of K201 on diastolic contractions in vivo and in isolated cardiomyocytes
A previous study has demonstrated that in vivo application of a β-adrenergic agonist and an elevated [Ca2+]o led to the production of diastolic contractions resulting in a raised intra-ventricular pressure measured at the R wave [14]. This pharmacological intervention resulted in a model of severe acute diastolic dysfunction without a significant change in systolic peak pressure. Application of 1.0 μmol/L K201 in vivo reduced the occurrence of the diastolic contractures and significantly reduced the mean intra-ventricular pressure measured at the R wave with no significant alteration of the peak pressure. With the caveat that detailed comparison of contractile function between isolated cardiomyocyte and whole heart experiments is problematic, the aforementioned effect of K201 on the mechanical function of the whole rat heart demonstrates some similarities to shortening measurements in isolated rat cardiomyocytes performed in this study (Fig. 1). In the present study, β-adrenergic stimulation of ventricular cardiomyocytes perfused with an elevated [Ca2+]o also led to the production of spontaneous diastolic cellular contractions. The amplitude of the spontaneous diastolic contractures was significantly reduced by K201 (to ~16% of control) with no alteration of the stimulated shortening amplitude. One difference between the in vivo rat study and the isolated cardiomyocyte experiments is the response of the cells in the current study with elevated [Ca2+]o to ISO, which resulted in a positive ionotropy (Fig. 1d(iii)). A trend to positive inotropy also existed within the in vivo model (reflected by the increase in left ventricular pressure) after 25 min of an elevated [Ca2+]o and β-adrenergic stimulation but this did not reach significance. Reasons for this discrepancy include the fact that the single cardiomyocytes were (a) only exposed to β-adrenergic stimulation and an elevated [Ca2+]i for ~5.0 min and (b) field stimulated (paced) and therefore not affected by the changes in heart rate which accompany an in vivo response.
The relationship between the stimulated Ca2+ transient and cell shortening amplitude
The current data reveal for the first time the relationship between the effect of K201 on stimulated Ca2+ transient amplitude together with the degree of cell shortening. The results demonstrate that although 1.0 μmol/L K201 significantly reduced the stimulated free Ca2+ transient amplitude by 16.1% [P < 0.05; Fig. 3a(ii)], the stimulated shortening amplitude was not significantly affected. No data exist in the literature as to the effect of K201 on the myofilament sensitivity to [Ca2+]i. To investigate this, the relationship between Ca2+ transient amplitude and degree of cell shortening was determined by lowering [Ca2+]o to various levels (Fig. 2). This relationship was unaltered in the presence of a range of K201 concentrations or an alternative benzothiazepine derivative (diltiazem) thereby demonstrating that K201 does not alter the sensitivity of the myofilaments under these conditions. Further verification of this was obtained via direct myofilament measurements performed in permeabilised rat ventricular cardiomyocytes at a free [Ca2+]i of 840 nmol/L (Online resource II) and confirmed at 375 nmol/L (data not shown). The latter concentration is equivalent to the mean [Ca2+]i observed during perfusion of 1.0 μmol/L K201 during β-adrenergic stimulation and an elevated [Ca2+]o (359 ± 43.9; n = 8; Fig. 1). Thus, the absence of a negative inotropic effect of K201 may be based on the non-linear relationship between peak [Ca2+]i and shortening under conditions of Ca2+ overload. It is not clear why K201 did not alter mean minimum [Ca2+]i; this may reflect the lack of effect of K201 on sarcolemmal extrusion as previously reported [18].
The effect of K201 on the SR Ca2+ content
Previous studies have attributed the beneficial effect of K201 on diastolic cardiac function to the ability of K201 to ensure maximal binding of FKBP12.6 to the RyR2 thus reducing the open probability of RyR2 [26]. The resultant effect would be to prevent diastolic Ca2+ release and subsequently to reduce arrhythmias and diastolic contractions. Venetucci et al. [25] have demonstrated in rat cardiomyocytes that the sole inhibitory effect on RyR2 by tetracaine (a drug which decreases the open probability of RyR2) can lead to inhibition of diastolic Ca2+ events between depolarisations but leads to an increase in the amplitude of systolic Ca2+ transient and SR Ca2+ content [4, 25]. The current study observed significant inhibition of diastolic Ca2+ spark activity by 1.0 μmol/L K201 supporting the concept that K201 reduces the activity of RyR2 but no significant difference in SR content was observed. However, due to the cell-to-cell variability in the caffeine response it is acknowledged that the measurements in the current study may have been unable to discriminate a ~10–12% change in SR Ca2+ content. Despite this limitation, it should be recognised that K201 is not the only drug which demonstrates inhibition of RyR2 without a concomitant increase in SR Ca2+ content. Flecainide has been reported to reduce Ca2+ wave amplitude in permeabilised rat cardiomyocytes without alteration in the SR Ca2+ content or SERCA activity. This is thought to have been achieved by inhibition of RyR2 via an open state block; under these circumstances comparable leak is achieved via significantly higher spark rates despite a significantly reduced “spark mass” [7].
The effect of K201 on the Ca2+ transient amplitude
K201 has been shown to prevent diastolic dysfunction and improve systolic function during Ca2+ overload conditions in terminally failing human myocardial preparations [24]. In contrast to the current study, no direct measurements of the Ca2+ transient or [Ca2+]i were performed. In another previous study by the authors, on this occasion in rabbit cardiomyocytes, the amplitude of depolarisation-induced Ca2+ transients was reduced by 1.0 μmol/L K201 but this did not reach significance [18]. Cellular shortening measurements were not reported in this previous work. The current study permits further insight into the effects of K201 using a fluorophore (Fura 4F AM) with a lower affinity which is able to determine the systolic [Ca2+] transient with more accuracy [28]. These novel data suggest that the action potential-induced free Ca2+ transients demonstrate a small but significant reduction in amplitude in response to 1.0 μmol/L K201. These changes occurred in the absence of major changes in ventricular electrophysiology (ECG recordings; Online resource IV) or the activity of common Ca2+ handling proteins, e.g. SERCA or NCX (see Online Resource V). In particular, 1.0 μmol/L K201 caused only a small decrease (~5%) in I Ca amplitude in the presence of an elevated [Ca2+]o. This contrasts with a previous study which showed a reduction of ICa peak by 22% in rat cardiomyocytes [10], but is similar to the ~3% reduction observed in rabbit cardiomyocytes [18]. This slight reduction in I Ca (~5%) would not be expected to cause a significant (>5%) change in Ca2+ transient amplitude [9]. Superficially, it would appear difficult to reconcile the absence of the effects of K201 on SR Ca2+ content and I Ca with the reduction in the peak of the stimulated free Ca2+ transient. Particularly, since K201 was very effective in inhibiting spontaneous diastolic Ca2+ release which should result in more Ca2+ being available for release on the subsequent stimulus. The reconciliation of these data comes when allowance is made for the intracellular buffering which, as shown by previous studies [8], is highly nonlinear over the range of [Ca2+]i measured in this study (~75–2,000 nmol/L). Using the values of reported intracellular buffer concentrations and affinities, the free [Ca2+]i in this study were converted to total cellular [Ca2+]i. On this basis, the amplitude of the Ca2+ transient (peak-pre-stim) when expressed as total [Ca2+]i indicates that on addition of K201, the amount of Ca2+ release from the SR increased (to ~140% of control). This was not observed in terms of free [Ca2+]i because although more SR Ca2+ appeared to be released after K201, the cellular buffer value was higher resulting in a decreased peak systolic Ca2+ level. Increased Ca2+ released from the SR on abolition of spontaneous Ca2+ release is consistent with studies by Venetucci et al. [25] who also reported increased SR Ca2+ content on abolition of Ca2+ waves. This was not evident from this study, but the variability of the data would prevent increases of SR content of ~10% being detected; an increase in SR Ca2+ content as small as 10% would be sufficient to explain an increase of SR Ca2+ release of ~140% [23].
Other contributing factors which were not investigated include alteration in the intracellular [Na+]i. Future work using the voltage clamp technique and monitoring of intracellular [Na+] i may uncover to what degree K201 is able to alter the balance between Ca2+ influx and efflux and control for certain parameters which are not achievable with the field stimulation technique.
Confocal imaging reveals characteristics of diastolic Ca2+ events
SR mediated Ca2+ release occurs via clusters of ryanodine receptors. During diastole this occurs in the form of either localised (Ca2+ sparks) or propagating whole cell Ca2+ release (Ca2+ waves). Ca2+ waves are recognised as events which could potentially lead to delayed after depolarisations (DADs) and triggered arrhythmias. A DAD occurs when a transient inward current is produced upon extrusion of SR-mediated diastolic Ca2+ via NCX. The ability of this inward current to generate a DAD is, amongst other factors, dependent upon the size of the Ca2+ wave and the inward current generated [19]. While there is an established link between Ca2+ waves and electrical abnormalities, the link between Ca2+ waves and mechanical dysfunction is less clear. To ascertain the spatiotemporal properties of the spontaneous diastolic Ca2+ release events, confocal microscopy of ventricular cardiomyocytes was employed using a similar protocol to that in Fig. 1. The experiments revealed that the diastolic events were predominantly in the form of propagating Ca2+ waves (Figs. 4, 5), which were significantly reduced in frequency and amplitude by 1.0 μmol/L K201. Characterisation of these events revealed multiple initiating points, the number of which was reduced upon application of 1.0 μmol/L K201. Thus, whilst K201 acts to significantly reduce the probability of spontaneous diastolic events (by ~50%) it also acts to modulate the characteristics of the remaining events. The significance of the transition in the latter is illustrated in Fig. 6. Diastolic Ca2+ events with multiple initiation points (Fig. 6a(iii vs. ii)) tend towards being more synchronous in activation (similar to the stimulated Ca2+ transient (Fig. 6a (i)). Analysis of the whole cell fluorescence (Fig. 6b) reveals that the larger number of initiation points results in a larger amplitude of spontaneously released Ca2+ from the SR (Fig. 6b (bottom)) and hence may increase the propensity to generate a transient inward current. This is summarised in the mean data presented in Fig. 6c, which demonstrate that for every extra initiation point per diastolic Ca2+ event during a short diastolic interval, the amplitude of the global Ca2+ release increases by ~1.8 times. Conversely, a reduction in the number of these initiation points (Fig. 6b (middle)) would be expected to lead to a reduction in Ca2+ available for extrusion at any one time by NCX. An insight into the net effect of this response to K201 in a multi-cellular preparation can be seen in the averaged traces of [Ca2+]i and contraction (Online Resource Ia).
Synchronicity of initiation points in the ISO data set (Fig. 4) was measured using the time between the first and last initiation point in each diastolic interval. This value was then correlated with the Ca2+ spark frequency in that cell. In cells with low spark frequency (6.0 ± 1.1/100 μm/s), the initiation points were on average 120.4 ± 13.9 ms between the first and last wave in a diastolic interval. In cells with a much higher spark frequency (35.4 ± 1.1/100 μm/s), the initiation points were on average 69.4 ± 7.3 ms between the first and last wave in a diastolic interval. This proved to be a significant correlation (P < 0.05). In these two subgroups, Ca2+ spark amplitude was, however, not significantly different. Therefore, while the action of K201 to reduce Ca2+ spark frequency (albeit not significantly, see online resource VI) may lead to a reduction in the synchronicity of Ca2+ initiation points, the effect is not enough to explain the reduction in number of Ca2+ initiation points.
Is the effect of K201 on Ca2+ waves related to β-adrenergic antagonism?
It is possible that the effect of K201 to reduce diastolic Ca2+ release event amplitude and number of initiation points in this study is due to an ability of K201 to antagonise the effect of β-adrenergic stimulation. While the results in Fig. 5 cannot discount this hypothesis, it seems unlikely given that in the sub-set of cells where Ca2+ waves were produced in the absence of β-adrenergic stimulation, the effect of K201 to limit these Ca2+ waves was identical to that in the presence of β-adrenergic stimulation (Fig. 4).
Summary and conclusion
The anti-arrhythmic effect of K201 has been attributed to reducing the open probability of RyR2 thus preventing excessive diastolic SR mediated Ca2+ leak [26]. This study provides several new mechanistic insights which advance our knowledge of K201. For the first time, we have examined the effect of K201 on intracellular Ca2+ and contractile function of isolated rat cardiomyocytes and have shown the drug significantly inhibits diastolic Ca2+ release and their associated contractile events leading to an increased action potential-induced SR Ca2+ release. These effects occur in the absence of altered myofilament Ca2+ sensitivity. Our data also provide greater insight into the effect of K201 on the spatiotemporal properties of Ca2+ handling introducing the concept that a reduction in Ca2+ wave initiation points has the potential to be anti-arrhythmic due to the limitation of the amplitude of the Ca2+ wave and subsequent increase in transient inward current. This may therefore be a route through which both the deleterious arrhythmogenic and mechanical effects of Ca2+ waves can be limited.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Thanks to Aetas Pharma Co. Ltd., Tokyo, Japan for the gift of K201. This work was supported by the British Heart Foundation [grant number PG/05/130 to CML & GLS].
Conflict of interest
NK is employed in the position of Chairman by Aetas Pharma Co. Ltd (the company supplying K201). Aetas Pharma Co. Ltd. agree with this submission to Basic Research in Cardiology.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- [Ca2+]i
Intracellular calcium concentration
- [Ca2+]o
Extracellular calcium concentration
- DAD
Delayed after depolarisations
- DD
Diastolic dysfunction
- F/F0
Quiescent fluorescence
- ICa
L-type Ca2+ current
- KH
Krebs–Henseleit solution
- NCX
Sodium–calcium exchanger
- NE
Nor-epinephrine
- RCL
Resting cell length
- RyR2
Ryanodine receptor
- SERCA
Sarcoplasmic/endoplasmic Ca2+ ATPase pump
- SR
Sarcoplasmic reticulum
References
- 1.Andersson KB, Birkeland JAK, Finsen AV, Louch WE, Sjaastad I, Wang Y, Chen J, Molkentin JD, Chien KR, Sejersted OM, Christensen G. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol. 2009;47:180–187. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 2.Colotti G, Zamparelli C, Verzili D, Mella M, Loughrey CM, Smith GL, Chiancone E. The W105G and W99G sorcin mutants demonstrate the role of the D helix in the Ca(2+)-dependent interaction with annexin VII and the cardiac ryanodine receptor. Biochemistry. 2006;45:12519–12529. doi: 10.1021/bi060416a. [DOI] [PubMed] [Google Scholar]
- 3.Currie S, Elliott EB, Smith GL, Loughrey CM. Two candidates at the heart of dysfunction: The ryanodine receptor and calcium/calmodulin protein kinase II as potential targets for therapeutic intervention-An in vivo perspective. Pharmacol Ther. 2011;131(2):204–220. doi: 10.1016/j.pharmthera.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Eisner DA, Choi HS, Diaz ME, O’Neill SC, Trafford AW. Intergrative analysis of calcium cycling in cardiac muscle. Circ Res. 2000;87:1087–1094. doi: 10.1161/01.res.87.12.1087. [DOI] [PubMed] [Google Scholar]
- 5.Eisner DA, Isenberg G, Sipido KR. Normal and pathological excitation-contraction coupling in the heart: an overview. J Physiol (Lond) 2003;546:3–4. doi: 10.1113/jphysiol.2002.036756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heusch G. Diastolic heart failure: a misNOmer. Basic Res Cardiol. 2009;104:465–467. doi: 10.1007/s00395-009-0025-3. [DOI] [PubMed] [Google Scholar]
- 7.Hilliard FA, Steele DS, Laver D, Yang Z, Le Marchand SJ, Chopra N, Piston DW, Huke S, Knollmann BC. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48:293–301. doi: 10.1016/j.yjmcc.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hove-Madsen L, Bers DM. Passive Ca buffering and SR Ca uptake in permeabilized rabbit ventricular myocytes. Am J Physiol. 1993;264:C677–C686. doi: 10.1152/ajpcell.1993.264.3.C677. [DOI] [PubMed] [Google Scholar]
- 9.Hussain M, Orchard CH. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during beta-adrenergic stimulation. J Physiol. 1997;505(Pt 2):385–402. doi: 10.1111/j.1469-7793.1997.385bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inagaki K, Kihara Y, Hayashida W, Izumi T, Iwanaga Y, Yoneda T, Takeuchi Y, Suyama K, Muso E, Sasayama S. Anti-ischemic effect of a novel cardioprotective agent, JTV519, is mediated through specific activation of delta-isoform of protein kinase C in rat ventricular myocardium. Circulation. 2000;101:797–804. doi: 10.1161/01.cir.101.7.797. [DOI] [PubMed] [Google Scholar]
- 11.Inagaki K, Kihara Y, Izumi T, Sasayama S. The cardioprotective effects of a new 1, 4-benzothiazepine derivative, JTV519, on ischemia/reperfusion-induced Ca2+ overload in isolated rat hearts. Cardiovasc Drugs Ther. 2000;14:489–495. doi: 10.1023/A:1007884905461. [DOI] [PubMed] [Google Scholar]
- 12.Kaneko N. New 1, 4-benzothiazepine derivative, K201, demonstrates cardio-protective effects against sudden cardiac cell death and intracellular calcium blocking action. Drug Dev Res. 1994;33:429–438. doi: 10.1002/ddr.430330406. [DOI] [Google Scholar]
- 13.Kaneko N, Matsuda R, Nakajima T, Shinozaki T, Ohtani N, Oda K, Hasumi H, Shimamoto K. Norepinephrine-induced diastolic dysfunction with aortic valve opening under calcium-loading in rats. Drug Dev Res. 2006;67:511–518. doi: 10.1002/ddr.20109. [DOI] [Google Scholar]
- 14.Kaneko N, Matsuda R, Ohtani N, Nakajima T, Arikawa T, Suzuki H, Toyoda S, Kikuchi M, Hata Y, Abe S, Taguchi I, Shimamoto K. K201 improves norepinephrine-induced diastolic dysfunction with preserved ejection fraction. Drug Dev Res. 2006;67:852–861. doi: 10.1002/ddr.20153. [DOI] [Google Scholar]
- 15.Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- 16.Kimura J, Kawahara M, Sakai E, Yatabe J, Nakanishi H. Effects of a novel cardioprotective drug, JTV-519, on membrane currents of guinea pig ventricular myocytes. Jpn J Pharmacol. 1999;79:275–281. doi: 10.1254/jjp.79.275. [DOI] [PubMed] [Google Scholar]
- 17.Kiriyama K, Kiyosue T, Wang JC, Dohi K, Arita M. Effects of JTV-519, a novel anti-ischaemic drug, on the delayed rectifier K+ current in guinea-pig ventricular myocytes. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;361:646–653. doi: 10.1007/s002100000230. [DOI] [PubMed] [Google Scholar]
- 18.Loughrey CM, Otani N, Seidler T, Craig MA, Matsuda R, Kaneko N, Smith GL. K201 modulates excitation-contraction coupling and spontaneous Ca2+ release in normal adult rabbit ventricular cardiomyocytes. Cardiovasc Res. 2007;76:236–246. doi: 10.1016/j.cardiores.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Schlotthauer K, Bers DM. Sarcoplasmic Reticulum Ca2+ release causes myocyte depolarisation. Circ Res. 2000;87:774. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD, Kranias EG. Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc Res. 2002;56:248–259. doi: 10.1016/S0008-6363(02)00541-2. [DOI] [PubMed] [Google Scholar]
- 21.Seidler T, Loughrey CM, Zibrova D, Kettlewell S, Teucher N, Kogler H, Hasenfuss G, Smith GL. Overexpression of FK-506 binding protein 12.0 modulates excitation contraction coupling in adult rabbit ventricular cardiomyocytes. Circ Res. 2007;101:1020–1029. doi: 10.1161/CIRCRESAHA.107.154609. [DOI] [PubMed] [Google Scholar]
- 22.Sossalla S, Maurer U, Schotola H, Hartmann N, Didie M, Zimmermann WH, Jacobshagen C, Wagner S, Maier LS. Diastolic dysfunction and arrhythmias caused by overexpression of CaMIIδ can be reversed by inhibition of late Na+ current. Basic Res Cardiol. 2011;106:263–272. doi: 10.1007/s00395-010-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic calcium simultaneous measurements of sarcoplasmic reticulum and sarcolemmal calcium fluxes in rat ventricular myocytes. J Physiol. 2000;522.2:259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toischer K, Lehnart SE, Tenderich G, Milting H, Korfer R, Schmitto JD, Schondube FA, Kaneko N, Loughrey CM, Smith GL, Hasenfuss G, Seidler T. K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Res Cardiol. 2010;105:279–287. doi: 10.1007/s00395-009-0057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venetucci LA, Trafford AW, Diaz ME, O’Neill SC, Eisner DA. Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes. Circ Res. 2006;98:1299–1305. doi: 10.1161/01.RES.0000222000.35500.65. [DOI] [PubMed] [Google Scholar]
- 26.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–296. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 27.Wehrens XHT, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. PNAS. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wokosin DL, Loughrey CM, Smith GL. Characterization of a range of fura dyes with two-photon excitation. Biophys J. 2004;86:1726–1738. doi: 10.1016/S0006-3495(04)74241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Kohno M, Matsuzaki M. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477–484. doi: 10.1161/01.CIR.0000044917.74408.BE. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.