

Abstract
Familial hypertrophic cardiomyopathy (FHC) is an autosomal dominant disease, which in about 30% of the patients is caused by missense mutations in one allele of the β-myosin heavy chain (β-MHC) gene (MYH7). To address potential molecular mechanisms underlying the family-specific prognosis, we determined the relative expression of mutant versus wild-type MYH7-mRNA. We found a hitherto unknown mutation-dependent unequal expression of mutant to wild-type MYH7-mRNA, which is paralleled by similar unequal expression of β-MHC at the protein level. Relative abundance of mutated versus wild-type MYH7-mRNA was determined by a specific restriction digest approach and by real-time PCR (RT-qPCR). Fourteen samples from M. soleus and myocardium of 12 genotyped and clinically well-characterized FHC patients were analyzed. The fraction of mutated MYH7-mRNA in five patients with mutation R723G averaged to 66 and 68% of total MYH7-mRNA in soleus and myocardium, respectively. For mutations I736T, R719W and V606M, fractions of mutated MYH7-mRNA in M. soleus were 39, 57 and 29%, respectively. For all mutations, unequal abundance was similar at the protein level. Importantly, fractions of mutated transcripts were comparable among siblings, in younger relatives and unrelated carriers of the same mutation. Hence, the extent of unequal expression of mutated versus wild-type transcript and protein is characteristic for each mutation, implying cis-acting regulatory mechanisms. Bioinformatics suggest mRNA stability or splicing effectors to be affected by certain mutations. Intriguingly, we observed a correlation between disease expression and fraction of mutated mRNA and protein. This strongly suggests that mutation-specific allelic imbalance represents a new pathogenic factor for FHC.
Electronic supplementary material
The online version of this article (doi:10.1007/s00395-011-0205-9) contains supplementary material, which is available to authorized users.
Keywords: Hypertrophic cardiomyopathy, Allelic imbalance, Cardiac β-myosin heavy chain, Myosin missense mutation, mRNA quantification
Introduction
Familial hypertrophic cardiomyopathy (FHC), the most common genetic heart disease, is characterized by hypertrophy of the left ventricle and the inter-ventricular septum (IVS), in the absence of overt etiological factors [23]. The hypertrophy is often asymmetric and variable in severity [16]. The prevalence of hypertrophic cardiomyopathy is 1 in 500; it affects individuals at every age [22]. FHC is often associated with arrhythmias, syncope and progression to heart failure or sudden cardiac death even at young age [23].
The genetically heterogeneous disease is transmitted as an autosomal dominant trait [38]. Mutations in more than 18 genes, mostly encoding sarcomeric proteins, have been linked to this disease in humans (Human Gene Mutation Database at http://www.hgmd.cf.ac.uk/ac/index.php). To date, approximately 30% of all genotyped cases were found to be associated with missense mutations in the β-myosin heavy chain (β-MHC) gene (MYH7) with chromosomal localization 14q12 [34]. Notably, the human β-MHC isoform (UniProt ID P12883), besides being the principal ventricular myosin isoform, is also expressed in the slow skeletal muscle fibers, e.g., of musculus soleus [35].
Several mutations in the β-MHC are associated with early onset and high incidence of sudden cardiac death, while others have a less severe disease expression and outcome [3]. This is usually explained by the type and location of the mutations within the molecule and their different effects on structure and function of the encoded protein [32]. Furthermore, even among affected members of the same family, variability in the clinical manifestations of FHC has been observed, which points to additional risk factors [23].
A further mechanism for different clinical prognosis of FHC-related mutations, however, could be the relative level of mutated versus wild-type protein in patients with different mutations or even in patients with the same mutation. Nearly all FHC patients are heterozygous for the respective mutation, and both mutated and wild-type proteins are assumed to be co-dominantly expressed from the corresponding alleles. Equal proportions of the allelic messages of the MYH7 gene and thus of the encoded protein are assumed to be present in every muscle cell. Yet, a higher or lower expression of the two allelic transcripts and proteins resulting in a deviation from an expected 50-to-50 ratio, known as allelic imbalance, has been shown previously for expression of different genes in humans [8, 13, 48]. Thus, allelic imbalance could well be quite significant for disease expression in genetic disorders [29], as has been shown, e.g., for hyperkalemic periodic paralysis in horses [50].
Evidence for allelic imbalance in FHC arises from our previous work on functional effects of β-MHC missense mutations. Since model systems of FHC often do not reveal the same mutation effect as it may occur in humans [19, 25], we study these mutations in M. soleus fibers and ventricular biopsies of FHC patients. In muscle fibers from individuals who were essentially asymptomatic but carried β-MHC mutations G584R or V606M, respectively, we did not detect any effects on sarcomere function. The fraction of mutated β-MHC for mutations V606M and G584R was only 12 and 23% of the total β-MHC in the sarcomeres, respectively [26]. This raised several questions: (1) Is the deviation from the expected 50-to-50 ratio of wild-type versus mutated myosin at the protein level paralleled by similar changes at the mRNA level due to allelic imbalance? (2) Is an unequal ratio of mutant versus wild-type protein and mRNA a common feature in FHC caused by myosin mutations and how is it correlated to the severity of the disease?
In the present study, we addressed these questions by measuring the relative abundance of mutated and wild-type MYH7-mRNA (RefSeq accession NM_000257) as well as of the encoded protein for several different mutations in M. soleus and cardiac muscle biopsies of FHC patients with different disease severity. Wild-type and mutated MYH7-mRNAs are co-expressed in the same cells/muscle fibers and therefore are subjected to identical conditions. Thus, variations due to confounding factors such as environmental influences and drugs, of hormones, or differences in tissue sampling or experimental procedures could be minimized. For all five MYH7 missense mutations studied, we found a deviation from the usually expected equimolar ratio of wild-type versus mutated β-MHC. The deviation was very similar at the protein- and mRNA level and was identical in myocardial tissue and M. soleus. Most interestingly, the unequal abundance appears to be directly related to the particular missense mutation, because it is found to be essentially the same in all carriers of a given mutation, including relatives of different generations and unrelated individuals. Our results suggest that unequal allelic expression of β-MHC contributes to the complex phenotype of FHC.
Materials and methods
Patients and muscle biopsies
The present study was performed on muscle biopsies from individuals previously characterized clinically as FHC patients and genetically as carriers of point mutations in the β-MHC head domain. All patients were heterozygous for the mutations they carried. In Table 1, clinical details available to us and the age of all patients at the time of biopsy are given.
Table 1.
Fraction of mutated β-MHC transcript and protein for all patients studied and clinical data
| Mutationa | Patienta | Age at biopsy (years) | Fraction of mutated MYH7- mRNAb | Fraction of mutated β-myosin | NYHA class | ICD, HT or sudden death | Heart blocks | ABN Q | LVH ±ST | PWT (mm) | IVS (mm) | Classification of mutationc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fraction in M. soleus | ||||||||||||
| V606M [12, 45] | H5 [45] | ~30 | 28 ± 4 | 12 ± 6d | n.p. | benign | ||||||
| H6 [12] | >45 | n.d. | 13 ± 2 | n.p. | Malignant | |||||||
| G584R [2] | H2 | ~30 | n.d. | 23 ± 1d | n.p. | Benign | ||||||
| I736T [30] | H18 | 51 | 38 ± 4 | 43 ± 13 | II | − | − | + | + | 11 | 14 | Benign |
| H19 | 54 | 39 ± 4 | 33 ± 3 | II | − | LAHB | + | + | 10 | 18 | Benign | |
| H20 | 49 | 39 ± 5 | 42 ± 7 | II | − | − | + | + | 10 | 17 | Benign | |
| R719W [2] | H13 | 24 | 57 ± 5 | 54 ± 7 | I | ICD | BBB | − | − | 26 | n.p. | Malignant |
| R723G [11] | H27 | 54 | 66 ± 7 | 62 ± 6d | II | ICD, HT | BBB | − | + | 13 | 16 | Malignant |
| H28 | 61 | 62 ± 4 | 62 ± 7d | III | ICD, HT | BBB | − | + | 12 | 12 | Malignant | |
| H71 | 34 | 70 ± 8 | n.d. | I | − | − | + | + | 11 | 23 | Malignant | |
| H72 | 30 | 66 ± 4 | n.d. | I | ICD | − | − | + | 11 | 22 | Malignant | |
| Fraction in ventricular tissue | ||||||||||||
| R723G [11] | H27 left ventr. | 55 | 69 ± 9 | 63 ± 11e | II | ICD, HT | BBB | − | + | 13 | 16 | Malignant |
| H27 right ventr. | 55 | 69 ± 3 | n.d. | II | ICD, HT | BBB | − | + | 13 | 16 | Malignant | |
| H29 left ventr. | 53 | 67 ± 5 | n.d. | IV | HT | BBB | + | + | 10 | 12 | Malignant | |
Values expressed as mean ± SD
n.d. not determined, n.p. data not published, NYHA, New York Heart Association, ICD implantable cardioverter-defibrillator, LAHB left anterior hemiblock, BBB bundle-branch block bifascicular and left, ABN Q abnormal Q wave, LVH ± ST left ventricular hypertrophy with voltage criteria and/or repolarization changes, PWT posterior wall thickness (mm), for H13 this is maximal left ventricular wall thickness (mm), IVS interventricular septal thickness (mm)
aNumbers in square brackets represent references for family and clinical details
bmRNA data from restriction digest approach
cClassification is based on life expectancy (benign, associated with near normal life expectancy; malignant, those which significantly reduce life expectancy; intermediate, those with a moderate effect on life expectancy [2]) or on status of patient and family (for G584R)
dPreviously published values are shown here for comparison
ePreliminary data
For all patients, FHC features were evaluated on the basis of medical history, physical examination, two-dimensional and M-mode echocardiography, and 12-lead electrocardiogram (ECG). Criteria for FHC were hypertrophy of the left ventricular wall, interventricular septal thickness (IVS) ≥13 mm in the absence of other known causes of hypertrophy and major ECG abnormalities like negative T-waves, pathological Q-waves, or heart blocks. Clinical records and family history of disease-related deaths and age at death were used for assessment of life expectancy with a given mutation (Table 1).
Mutation R723G: We obtained M. soleus tissue from FHC patients of three families with this mutation [11]. Biopsies were from two brothers (H27, Family 26, II-5; and H28, Family 26, II-2) and from a younger patient of the same family (H71, Family 26, III-5). Both brothers had received an ICD after syncopal episodes; H71 at the time of biopsy was still NYHA class I. A soleus biopsy was obtained from a younger patient of a third, unrelated family (H72, Family 157, III-2) who had received an ICD. Left ventricular tissue was available from patient H27 and an unrelated female patient (H29, Family 11, III-8). Mutation R719W: We obtained soleus tissue from a British female (H13; [2]). After cardiac arrest and resuscitation, she had received an ICD. Mutation V606M: Soleus biopsies were available from a British individual (H5; [45]) and from an unrelated American individual (H6; [12]. Mutation I736T: We obtained soleus biopsies from three siblings of a Kyrgyz family (Caucasian origin; [30]), one male (H18, Family A, II-2) and two females (H19, Family A, II-4 and H20, Family A, II-7) with moderate left ventricular and septal hypertrophy (14, 18, 17 mm, for H18, H19 and H20, respectively) and pathological ECGs. Mutation G584R: The biopsy was from a young British male [45]. This patient’s tissue underwent relative quantification of mutated protein only. Controls: Soleus biopsies of several healthy individuals, i.e., volunteers without any known impairment of cardiac function. Detailed information on sample preparation is given in Online Resource and [15]. Inclusion criteria for FHC of the individual patients can be obtained from the respective references.
Written, informed consent was obtained from all individuals according to approved Ethics Committee protocols of Hannover Medical School (protocols no. 2228 and no. 2729), of Hospital Clinic, Barcelona and of St. Georges Hospital, London. The investigations conformed to the principles of the Declaration of Helsinki [46].
RNA isolation, synthesis of sscDNA and PCR
For mutations I736T, R719W and V606M, total RNA was isolated from approximately 3 mg pieces of frozen muscle using Trizol (GIBCO, Karlsruhe, Germany). Isolated RNA was reverse transcribed into cDNA (SuperScript First-Strand Synthesis System for RT-PCR, GIBCO, Karlsruhe, Germany). For mutation R723G, the RNA isolation from both myocardium and soleus muscle, and the cDNA synthesis steps were performed using cells-to-cDNA kit (Ambion, Darmstadt, Germany). For reverse transcription, an MYH7-specific primer RTR719W was used for all samples. Amplification of regions of cDNA encompassing the mutation site was performed using mutation-specific primers (Online Resource). For mutations R719W, I736T and V606M, the PCR reaction was performed by 1 min of initial denaturation at 94°C, 22–25 cycles of 30 s at 94°C, 3 min at 68°C and a final elongation for 3 min at 68°C. For mutation R723G, the reaction was performed using HotStarTaq DNA Polymerase (Qiagen, Hilden, Germany). Cycling included initial activation of the enzyme for 15 min at 95°C, 35 cycles of 95°C for 1 min, 68°C for 30 s, 72°C for 2.5 min and a final extension at 72°C for 5 min. Control reactions without mRNA, without reverse transcriptase and without cDNA template, were run during cDNA synthesis and PCR, respectively, to rule out contamination or genomic DNA as a source of positive signals.
Reconditioning PCR
To avoid errors from heteroduplex formation at high cycle numbers, we performed a reconditioning PCR [4, 41]. The PCR product of up to 35 cycles was used at a final dilution of 1:100 and was run in several parallel PCR reactions up to a low cycle number in the exponential range (Fig. 1b). Prior to the restriction digest, the products of the parallel reactions were pooled and precipitated overnight by addition of 3 M sodium acetate to a final concentration of 200 mM plus addition of three volumes of absolute alcohol. For precipitation, the sample was kept at −80°C.
Fig. 1.
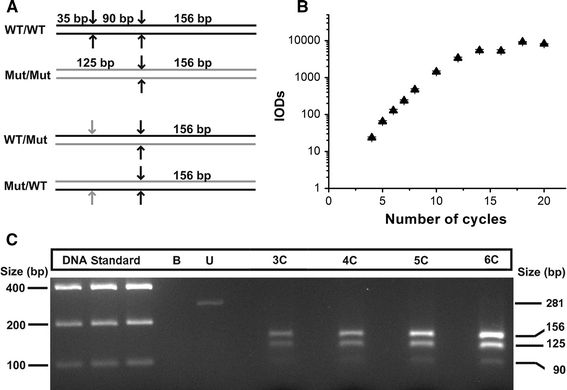
Experimental approach to minimize heteroduplex formation. a Schematic representation of restriction fragments produced by Nde II digestion of wild-type and mutated homoduplexes for mutation R723G (upper panels). Formation of heteroduplexes (lower two panels) generates one refractory restriction site (gray arrows), thus producing the 125-bp fragment from both strands of the heteroduplexes. b For reconditioning PCR, after 35 PCR cycles 1:100 dilutions of the product were subjected to another PCR and quantified after each cycle. The reaction was exponential until the seventh cycle. IOD, integrated optical density (SD from densitometric analysis at different exposure times). c 3.5% agarose gel with restriction digests of PCR products of a sample from the left ventricular lateral wall of a patient heterozygous for mutation R723G. The 35-bp fragment is not seen due to its small size. Cleavage of the reconditioning PCR products of three, four, five or six cycles (lanes 3C–6C) yielded mutated MYH7-mRNA fractions of 73, 82, 77 and 75%, respectively (on average 74 ± 8% in this muscle sample). There was no indication for heteroduplex-induced increase of mutated MYH7-mRNA at increasing cycle numbers. Lanes 1–3 12, 15 and 18 μl of equimolar DNA standard; B blank; U undigested PCR product
Restriction digests
To discriminate between products from wild-type versus mutated mRNA, the PCR products were digested with the following restriction enzymes, according to the manufacturers’ recommendations: Nde II (Promega, Mannheim, Germany) for R723G, MspI (New England Biolabs, Frankfurt, Germany) for R719W, Hpy8I (Fermentas, St. Leon-Rot, Germany) for I736T and Nla III (New England Biolabs, Frankfurt, Germany) for V606M. The enzymes were selected such that one cleavage site in the PCR product was specific for either the mutated or wild-type sequence (for detailed description, see Online Resource).
Allele-specific relative quantification of RNA transcripts
The DNA fragments generated by the restriction digests were separated on 2.5 or 3.5% EtBr-agarose gels, using an agarose for high-resolution separation of small DNA molecules (Metaphor agarose, Cambrex Bio Science, Rockland, USA; NuSieve agarose, Biozym, Oldendorf, Germany). Since the intensities of EtBr-stained DNA bands correlate with the total number of base pairs in the respective bands, for quantification of relative transcript levels we used the intensity of EtBr staining of the digested PCR products (for details and controls, see Online Resource). The integrated intensities of the individual bands were obtained by curve fitting of the intensity profiles of the lanes using the software Origin™ (Microcal™ Software, Northampton, USA). Because we aimed at a relative measure of transcripts from the two alleles in the same sample, normalization to a reference housekeeping gene was not required.
Control experiments for the relative mRNA quantifications with known plasmid mixtures
Reconstitution experiments were used to test the linearity and accuracy of the mRNA quantification method over essentially the whole range of variations in allele ratio. The wild-type and mutant MYH7 sequences that correspond to the PCR products of each mutation were cloned into plasmid pGA4 (Geneart, Regensburg, Germany) (R723G), vector pMX (I736T and V606M) or vector pMA-T (R719W) using the KpnI and SacI restriction sites. Several defined mixtures of the respective wild-type and mutated constructs ranging from 0 to 100% were applied to mutation-specific PCR and subsequently quantified as described for the patient samples.
Assessment of heteroduplex formation
The 125-bp fragment of the restriction digest of a 35-cycle PCR product with mutation R723G was used to estimate the fraction of heteroduplexes, which may form at high cycle numbers (Fig. 1a, lower panels). The 125-bp band was excised from the agarose gel and the DNA was purified using gel purification kit (Qiagen, Hilden, Germany). The fragments were cloned into a pAlli10 vector using the Alligator Cloning Kit (Trenzyme, Konstanz, Germany) and sequenced at GATC (Konstanz, Germany) in the forward as well as reverse orientations to determine the sequences of both strands.
Allele-specific real-time PCR (RT-qPCR) assay
As an alternative approach to determine relative levels of wild-type and mutated MYH7-mRNA in cardiac biopsies with mutation R723G (patient H27, left and right ventricular wall), allele-specific RT-qPCR was performed. The hybridization probes specific for the detection of either wild-type or R723G-mutant alleles of MYH7 were fluorescently labeled at the 5′-ends with VIC and 6-FAM, respectively, and with a non-fluorescent quencher at the 3’end (Assays-by-Design Service, Applied Biosystems, Foster City, CA) (Online Resource). Amplification was performed using identical primers for both alleles [47], excluding bias caused by unequal amplification efficacy. For experimental conditions and analysis, see Online Resource. Detection of the two alleles occurred in a single-tube analysis. Relative quantification was performed according to the delta-CT method [31].
Relative quantification of wild-type versus mutated protein
The relative abundance of wild-type and mutated β-MHC was determined by liquid chromatography/electrospray-ionization mass-spectrometry as described in detail previously [5]. Quantification is based on myosin extraction and generation of peptides using endoproteinase Lys-C (Roche, Mannheim, Germany). Peptides with mutant and wild-type sequences and an isotopically enriched (13C, 15N) leucine in each sequence (Coring System Diagnostix, Gernsheim, Germany) served as internal standards. Relative quantification of wild-type and mutated β-myosin was based on peak areas obtained by MS for the stable isotope-labeled and for the native peptides. Alternatively, for some samples the peptides generated by Lys-C were quantified using our previously published method of HPLC and MS coupled to capillary zone electrophoresis [26].
Statistics
Results are presented as mean ± SD. For each individual, mRNA and protein quantifications were based on at least three different muscle biopsy pieces. Quantification occurred by repeated PCR reactions [given as n independent experiments (assays) in results], each of which was analyzed in two to four lanes on each gel. For one patient with mutation V606M of American origin, only one sample of soleus muscle tissue was available for analysis. Statistical significance of deviations from a 50-to-50 ratio of mutant versus wild-type mRNA and protein, or differences between two samples, was tested using one-sample t test and two-sample unpaired t test, respectively. p values <0.05 were considered to be significant.
Results
Reconditioning PCR for relative quantification of wild-type and mutant MYH7-mRNA
To quantify the ratio of wild-type and mutant MYH7-mRNA in the samples of human FHC patients, we performed a highly specific reverse transcriptase-polymerase chain reaction followed by mutation-specific restriction analysis. The MYH7-mRNA was reverse transcribed using a MYH7-specific primer for all analyzed mutations. For the restriction analysis, cDNA encompassing the respective mutation site was amplified using mutation-specific primers (Online Resource). Mutant and wild-type fragments were subsequently cleaved by DNA-restriction enzymes specific for restriction sites either generated (I736T, c.2313T>C; V606M, c.1922G>A) or disrupted (R719W, c.2261C>T) by the respective mutations. Mutation R723G (c.2273C>G) does not lead to a changed restriction site. Here, a mutagenic forward primer was used to create an additional cleavage site for Nde II in the wild-type PCR product only (Online Resource). The mutation-specific DNA-restriction fragments were then quantified to determine the ratio of wild-type and mutated MYH7-mRNA in each sample.
To obtain sufficient PCR product for the subsequent restriction digest analysis, up to 35 cycles had been necessary. At such high cycle numbers, formation of heteroduplexes between wild-type and mutated strands in the PCR product is possible (Fig. 1a, lower panels). Sequence analysis of 105 cloned mutation-specific 125-bp fragments showed only two fragments to contain sequences that result from formation of heteroduplexes. Thus, we could assert with 95% confidence that the heteroduplexes formed at 35 cycles were <10%.
Yet, to minimize heteroduplex formation, we used a reconditioning PCR method for most quantifications [4, 41]. After 35 cycles, the PCR product was diluted 100-fold and submitted to about ten parallel PCR reactions. Starting from cycle 4, the reaction was stopped for one aliquot at each cycle. Aliquots were run on a gel and relatively quantified (Fig. 1b). The exponential range where the slope of the regression curve is close to two was limited to about the seventh cycle. Analysis of the ratio of mutation to wild-type specific restriction fragments in four subsequent PCR cycles from the exponential range revealed comparable ratios from cycles 3 to 6 (Fig. 1c). This indicates that our results are not influenced by a bias caused by heteroduplex formation, which would lead to an increase of the uncleaved 125-bp fragment (Fig. 1a, lower panels). For each mutation, the exponential range of the reconditioning PCR was determined and the number of necessary aliquots for pooling was set by the signal intensities needed for quantification on the gels (data not shown).
Relative quantification of mutant versus wild type in defined mixtures of plasmids reveals accuracy of method
To test the sensitivity of our method for mRNA quantification and to verify the analysis procedure, we examined whether the observed ratio of mutated versus wild-type fragments in reconstituted heterozygous samples reflects the selected input ratio. We generated standard vectors that contained the PCR fragments for each mutation and the respective wild-type DNA (Online Resource). Defined mixtures of mutated and wild-type vectors in the ratios 0:100, 20:80, 40:60, 50:50, 70:30 and 90:10 100:0 were subjected to reconditioning PCR. After restriction digestion of the PCR products, gels were loaded such that the bands were in the linear intensity range of EtBr fluorescence (Fig. 2a; see also Online Resource).
Fig. 2.
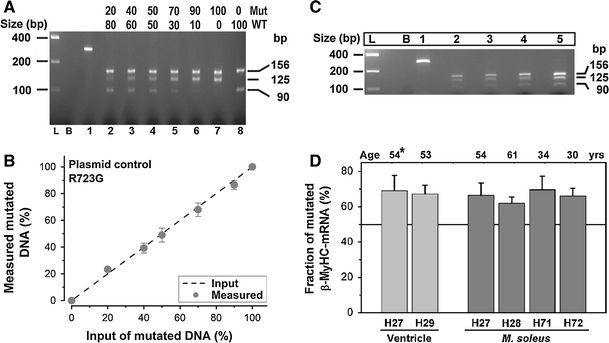
Relative quantification of R723G mutated and wild-type sequences in defined plasmid mixtures and biopsies. a Restriction fragments of the 281-bp PCR product generated from defined mixtures of R723G-mutant and wild-type plasmid (Lanes 2–8). Lane L, equimolar DNA standard; lane B, blank; lane 1, undigested product. The increasing ratio (indicated above lanes) of mutant versus wild-type sequence resulted in brighter bands of 125 bp and weaker bands of 90 bp. b Input versus experimentally determined mutation-specific DNA in plasmid mixtures (n = 3 assays). The dashed line indicates the expected values. c Representative gel with the undigested 281-bp PCR product (lane 1) and restriction fragments after reconditioning PCR for three, four, five and six cycles (lanes 2–5) from a M. soleus sample of patient H27. L, equimolar DNA standard; B, blank. d Fraction of mutated mRNA in the left ventricular wall (light columns) and M. soleus (dark columns) of patients from three unrelated families with mutation R723G. The age (years) of patients at M. soleus biopsy is indicated; *patient H27 was transplanted ≈ 1 year after soleus biopsy)
As shown exemplarily for mutation R723G, the fractions of mutant DNA obtained from the above mentioned mixtures of plasmids were 0, 23.3 ± 1.5, 39.3 ± 3.7, 49 ± 5.2, 68 ± 5.1, 86.5 ± 3.5 and 100%, respectively (Fig. 2b; n = 3). Thus, the mixed allelic templates were amplified and detected essentially in direct proportion to the stoichiometric fraction of each template over a wide range of mutant to wild-type template ratios. In addition, the absence of the mutation-specific 125-bp fragment in the 0:100 mixture (Fig. 2a; lane 8) confirms complete digestion at all restriction sites. Similar results were also obtained with plasmid mixtures for the other mutants I736T, V606M and R719W (Online Resource).
Unequal allelic expression of wild-type and mutated MYH7-mRNA
The established assays were used to quantify the relative expression levels of wild-type and mutated MYH7-mRNA isoforms in M. soleus biopsies from FHC patients with mutations R723G, I736T, R719W and V606M (Table 1; see also Online Resource).
For mutation R723G, the fraction of mutated transcript was determined from the ratio of IOD/bp values of the mutation-specific 125-bp fragment versus the 156-bp fragment. Analysis of four patients revealed fractions of mutated MYH7-mRNA of 62 ± 3.5% (H28, n = 4 assays), 66.1 ± 4.3% (H72, n = 4 assays), 66.4 ± 6.9% (H27, n = 5 assays) and 69.6 ± 7.7% (H71, n = 4 assays) of the total MYH7-mRNA (Fig. 2d; Table 1). The expression level of mutated MYH7-mRNA among all patients with this mutation available to us was not significantly different (p ≥ 0.05), but only for H28 it was slightly lower (p = 0.05). This indicates that the expression of the mutated MYH7-mRNA was very similar even among patients of different generations and from unrelated families.
For relative quantification of mutant and wild-type MYH7-mRNA with mutation I736T, M. soleus biopsies of three siblings were available to us. The fraction of mutated MYH7 transcript was calculated from the IOD/bp value of the mutation-specific 205-bp fragments and the wild-type specific 354-bp fragments (Online Resource). The fraction of mutated MYH7-mRNA was 37.6 ± 4.2% (n = 8 assays), 39.1 ± 4.1% (n = 9 assays) and 38.6 ± 5.3% (n = 9 assays) of total MYH7-mRNA for FHC patients H18, H19 and H20, respectively.
For mutation V606M, the fraction of mutated MYH7-mRNA in the biopsies was calculated based on the IOD/bp values of the mutant-specific 180-bp fragment versus the total IOD/bp value of wild-type specific 267-bp fragment plus the mutant-specific 180-bp fragment (Online Resource). This yielded on average 28 ± 6% MYH7-mRNA of total MYH7-mRNA with mutation V606M (n = 12 assays).
For the patient with mutation R719W, the fraction of mutated MYH7-mRNA was 57.1 ± 5.4% of the total MYH7-mRNA (n = 9 assays), indicating a small yet significant (p < 0.01) deviation from equal abundance of wild-type and mutated transcript (Online Resource). The fraction of mutated MYH7-mRNA was calculated based on the IOD/bp values of the mutation-specific 269-bp fragment versus the average of the two wild-type specific fragments of 146 and 123 bp.
Same deviation from the equimolar ratio in myocardium and slow skeletal muscle for mutation R723G
Samples of ventricular tissue of two members of two unrelated Spanish families with mutation R723G were available to us. One patient (H27) underwent heart transplantation about 1 year after the M. soleus biopsy was taken. The other myocardial sample was of a transplanted female patient (H29) of an unrelated family. In the left ventricular wall of H27 and H29, the fraction of mutated transcript was 69 ± 8.8% (n = 20 assays) and 67.2 ± 5% (n = 9 assays) of total MYH7-mRNA, respectively (Table 1). Hence, at least for R723G patients, the deviation from the equimolar ratio of wild-type versus mutant MYH7-mRNA and thus the fraction of mutated transcript is essentially the same in skeletal and ventricular biopsies (Fig. 2d).
Validation of allelic imbalance for mutation R723G by allele-specific real-time PCR (RT-qPCR)
To corroborate the results of the restriction digest approach, we performed allele-specific RT-qPCR from left and right ventricular biopsies of patient H27 with mutation R723G. Both alleles were analyzed in the same reaction using mutation or wild-type specific probes, respectively, which were labeled by two distinct dyes (Online Resource). Using the delta-CT method, we found on average 61.7 ± 8.9% (n = 8 assays) and 64 ± 13.3% (n = 18 samples in two assays) of mutated transcript in the left and right ventricular muscle, respectively. Both values are well consistent with data obtained on the same samples with the restriction digest approach (Fig. 3; [7]).
Fig. 3.
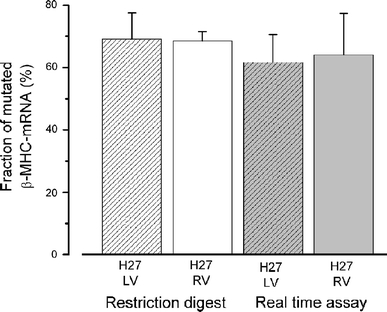
Allele-specific RT-qPCR for quantification of mutated versus wild-type MYH7-mRNA in myocardium. For RT-qPCR, mRNA from cardiac tissue samples was reverse transcribed using a MYH7-specific RT-primer. Subsequent comparative quantitative PCR was accomplished using identical primers and differently labeled mutation or wild-type specific probes in one reaction tube (Online Resource). RT-qPCR data are shown in comparison to data from restriction digest approach performed on the same cardiac samples from left ventricular (LV) anterior wall and right ventricular (RV) wall of H27
Corresponding deviations from the equimolar ratio for β-MHC mutations also at the protein level
The fraction of wild-type and mutated β-myosin was determined after myosin extraction and enzymatic cleavage using liquid chromatography/electrospray-ionization mass-spectrometry (LC/ESI-MS) or LC/MS in combination with capillary zone electrophoresis, which we previously developed [5, 26]. Figure 4 shows ESI-MS spectra of Lys-C digested native β-MHC peptides from a patient carrying mutation R723G and the corresponding synthetic stable-isotope labeled internal standard (IS) peptides. Equal amounts of mutated and wild-type IS-peptides yielded slightly different integrated peak areas (Fig. 4a, b). This most likely is due to different competition for ionization of IS-peptides with peptides of the Lys-C digest, and is taken into account for quantification (Fig. 4c, d).
Fig. 4.
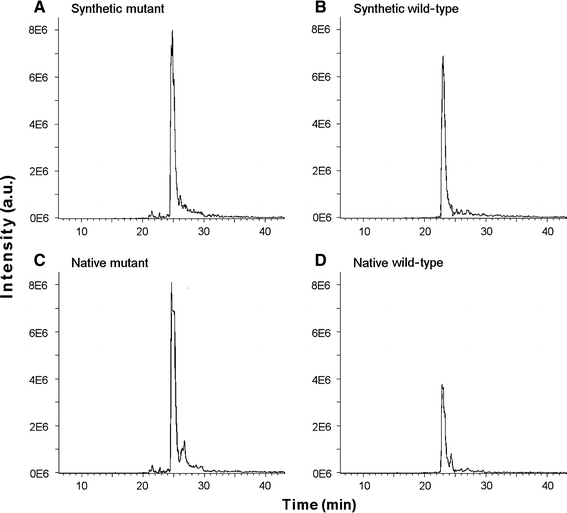
MS spectra of native and synthetic β-MHC wild-type peptides and of peptides with mutation R723G. The Lys-C digest fraction of β-myosin containing native wild-type and R723G-mutated peptides was analyzed by nanoLC–ESI-MS after adding equimolar amounts of stable isotope-labeled synthetic wild-type and mutant internal standard peptides. The traces represent ion signals of the fivefold charged synthetic mutant (a; m/z 764.1), synthetic wild-type (b; m/z 783.9), native mutant (c; m/z 763.1) and native wild-type (d; m/z 782.8) peptides. Note the quite large difference in peak areas between native wild-type and mutated peptides mainly representing the difference in abundance of the two peptides and thus of the two parent proteins in the muscle sample
β-MHC quantifications in M. soleus biopsies (Table 1) of two patients with mutation R723G (H27 and H28) yielded 61.6 ± 6% (n = 36 peptide samples) and 62.4 ± 7% (n = 31 peptide samples) mutated myosin of total β-MHC, respectively [5]. Preliminary β-MHC quantification of cardiac samples from patient H27 yielded essentially the same fraction of 62.7 ± 11% (n = 2 peptide samples) mutated myosin of total myosin as found in M. soleus. The fraction of mutated myosin in the soleus muscle biopsy of the patient with mutation R719W was 54 ± 7% (n = 4 muscle samples). The siblings with mutation I736T had 43 ± 13, 33 ± 3 and 42 ± 7% (n = 4–7 muscle samples) mutated myosin of total β-MHC in the sarcomeres of their M. soleus, respectively. The American patient with mutation V606M had 13 ± 2% (n = 3 peptide samples of 1 muscle sample) mutated myosin of total β-MHC. As previously published, the British patient with V606M had 12 ± 6% and the patient with mutation G584R had 23 ± 1% mutated myosin of total β-MHC, respectively [26].
Interestingly, the mRNA levels were similar to the corresponding protein levels for mutations R723G, R719G and I736T (Fig. 5), as determined by non-significant deviations (Student’s t test, p > 0.5). Note that the deviation between protein and mRNA quantification for one patient with mutation V606M might be because mRNA analysis for this specific patient was carried out on tissue initially frozen for histology. Nevertheless, both the fraction of mutated mRNA and protein for this mutation are significantly below 50%.
Fig. 5.
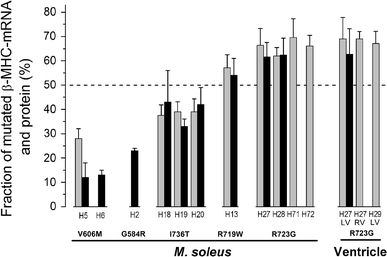
Fractions of mutated MYH7-mRNA and β-myosin for different FHC mutations. The fractions of mutated mRNA (gray bars) and protein (black bars) in M. soleus are summarized for mutations V606M, I736T, R719W and R723G. Additionally, the R723G-mutated mRNA fractions in myocardium and previously published protein levels of mutations G584R and V606M [26] are depicted. mRNA data shown here are from the restriction digest approach. All data are also listed in Table 1. All mutations significantly deviate from equal abundance of wild-type and mutated transcript with similar deviations also at the protein level. Similar deviations of mRNA and protein levels were found in M. soleus and myocardium for mutation R723G (LV, left ventricle; RV, right ventricle). Note the intra- and/or inter-familial similarity in mutated MYH7-mRNA/protein expression for I736T, R723G and V606M
Discussion
Levels of mutated versus wild-type MYH7-mRNA and protein deviate from an equimolar ratio
In general, both alleles of autosomal genes are assumed to be co-dominantly expressed. Thus, in a tissue sample carrying a heterozygous missense mutation, an equimolar ratio is expected for wild-type and mutated transcripts and encoded proteins. Yet, assessment of these ratios of five different FHC-causing β-myosin missense mutations revealed an allelic imbalance at the mRNA as well as at the protein level. The fraction of mutated MYH7-mRNA and β-myosin ranges from less than 20–70%. Most interestingly, these ratios seem to be characteristic for the respective mutations, as shown for mutations V606M, I736T and R723G. Further, they are identical for both myocardium and skeletal muscle at least with mutation R723G.
The finding that in all cases studied, the fraction of mutated protein was essentially the same as the mRNA in the same tissue indicates that at the translational level and upon incorporation of protein into the sarcomere, no large additional deviations arise, e.g., due to poor incorporation or degradation of mutant β-myosin protein. It is unlikely that missense mutations in MYH7 cause haploinsufficiency, although relative quantification cannot fully exclude this. It has been demonstrated that β-myosin with missense mutations in the head domain is incorporated into the sarcomeres of cardiac and slow-twitch skeletal muscles [5, 21, 26]. No alterations of sarcomeric structure, packing of myofibrils or assembly of the thick filaments were found [6, 17]. The mutations studied here rather act by inducing functional changes of the acto-myosin interactions and, therefore, of the whole sarcomere [15, 17, 36].
How may missense mutations affect steady-state mRNA levels?
The data suggest that the deviation from the equimolar ratio is an inherent property of each mutation itself. Allele-specific expression of genes is mostly regulated by cis- or trans-acting factors of either genetic or epigenetic origin. To assess whether allelic imbalance may be due to autosomal imprinting or is independent of parental origin [33], we analyzed the available family pedigrees of the different patients. Since mutations R723G and V606M were passed on through either the paternal or the maternal germ line, imprinting is highly unlikely. For mutation I736T, the genetic status of the parents is not known. The analysis of the molecular mechanisms of imprinting renders this probability rather unlikely. The altered efficiency of transcription due to methylation of cytosines in the CpG dinucleotide sequence provides an important mechanism of sequence-dependent imprinting of genes [14]. However, the two CpG-islands within the MYH7 gene (Accession number NG_007884) at positions 15,421 to 15,723 and 20,455 to 20,682 (http://www.ebi.ac.uk/Tools/emboss/cpgplot/) remain unaffected by each of the missense mutations examined here and no additional CpG-island is predicted to be created. However, it cannot fully be excluded that a linked non-coding variant in cis causes the allelic imbalance in individuals from apparently unrelated families. Haplotype analysis of the three Spanish kindreds with mutation R723G suggests a common founder mutation [11]. For the unrelated patients with mutation V606M, such analysis has not been reported.
Allele-specific expression of MYH7-mRNA due to single nucleotide substitutions could be caused by alteration of mRNA stability. One determinant of RNA-stability is its secondary structure that can affect, e.g., the accessibility to stability-regulating proteins or the recruitment into RNA-induced silencing complexes (RISCs) [20]. We bioinformatically examined the influence of each point mutation on the secondary structure of the MYH7-mRNA using the RNAfold program (http://www.rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). Severe changes in the secondary structure for mutations V606M (c.1922G>A) and R723G (c.2273C>G) (Fig. 6a) were identified, whereas mutations R719W (c.2261 C>T) and I736T (c.2313 T>C) did not alter the mRNA structure. These findings are a first hint that structural changes induced by mutations V606M and R723G may influence the MYH7-mRNA turnover and thereby lead to different protein levels.
Fig. 6.
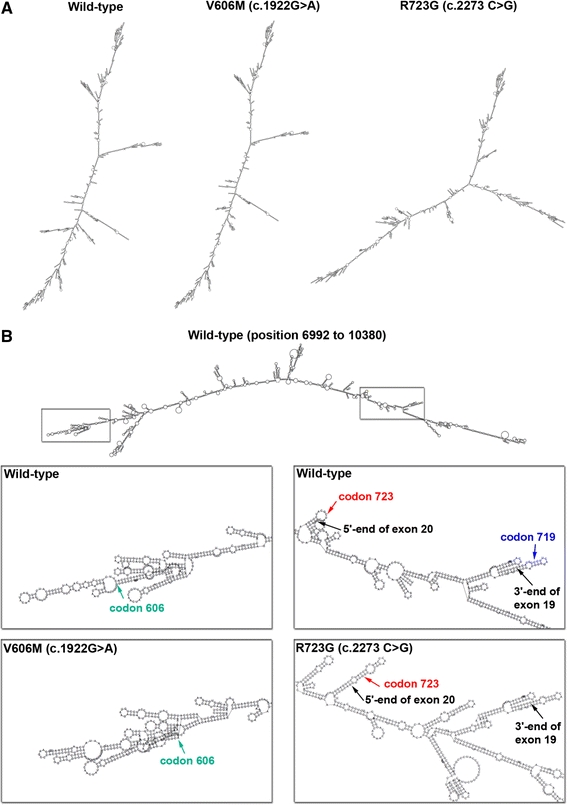
Secondary structure of MYH7 pre-mRNA and mature mRNA. The RNAfold program (minimum free energy) was used to compare the secondary structure of the MYH7 mRNA of mutated and wild-type isoforms. Mutations R719W and I736T generate identical structures as the wild-type sequence and are therefore not depicted. a Secondary mRNA structures of wild-type (NM_000257), mutation V606M (c.1922G>A) and mutation R723G (c.2273C>G). b Secondary structure of the pre-mRNA of wild-type exon 15 to exon 21 (position 6,992–10,380, NG_007884). Enlarged boxes: sites of structural alterations caused by mutations V606M (c.1922G>A, green arrow) and R723G (c.2273C>G, red arrow). Blue arrow in the wild-type sequence: location of mutation R719W that does not alter the pre-mRNA structure
Allelic imbalance could also be caused by differential splicing of the mutated allele. The splicing efficacy can be affected either by the secondary structure of the transcript, namely hairpin structures located close to exonic splicing enhancers (ESE) or exonic splicing silencers (ESS) [18], or by sequence motifs [9]. We thus examined the structure of the pre-mRNA from exon 15 to 21, the region including each analyzed mutation (Fig. 6b) and the effect of the mutations on splicing regulatory motifs using the Human Splicing Finder program (http://www.umd.be/HSF/) [9] and the ESE Finder program (http://www.rulai.cshl.edu/ESE; version 3.0).
Analysis of mutation V606M revealed the generation of novel hairpin structures in exon 16 (Fig. 6b) and the disruption of ESE motifs. Such effects have been reported to cause exon skipping [49]. Skipping of exon 16 in the MYH7-mRNA would cause a premature stop-codon at position 552 and thereby most likely lead to nonsense-mediated decay (NMD) of the mutated allele. In contrast, mutation I736T (c.2313 T>C) is predicted to strengthen an existing ESE at position 2,308–2,315 (variation +13.4%). This could lead to the activation of a cryptic splice site and thereby either to NMD or to the generation of mRNA that is not detected by our assay and encodes for a non-functional β-MHC.
Mutations R719W (c.2261C>T) and R723G (c.2273C>G) lead to increased mutated mRNA- and protein fractions. The mechanisms underlying the regulation of such hypermorphic mutations are poorly understood. Interestingly, both mutations are located in hairpin structures at intron–exon boundaries (Fig. 6b). Such mutations have been reported to influence splicing efficacy [18, 39]. Therefore, one could speculate that a more efficient splicing of the mutated as compared to the wild-type pre-mRNA may lead to the different mRNA and protein levels. Further studies will have to reveal whether changes in mRNA stability, differential splicing or a linked cis-acting variant could provide the underlying mechanisms of the observed allelic imbalance.
Does the abundance of mutated protein contribute to severity of the FHC phenotype?
Most interestingly, our results suggest that the abundance of mutated mRNA and mutated protein is correlated with the reported severity of the clinical outcome of FHC and the extent of functional changes at the sarcomeric level (Table 1). For nearly all of the mutations studied, a large fraction of mutated transcript and protein correspond to a severe long-term disease progression in the families and pronounced effects on the function of the contractile apparatus.
One very malignant mutation is R723G, where about 2/3 of total MYH7-mRNA and β-myosin are mutated. Patients with this mutation have a chance of only about 18% to reach the age of 50 years without a cardiac event and with NYHA class II or better. Mean survival in the three families studied by Enjuto et al. [11] was 51 years. Some patients with this mutation develop severe heart failure in their forties or fifties [11], which is generally found in a subgroup of FHC patients [40]. Patient H27 in our study, as a typical example, initially developed the classical clinical phenotype of FHC with diastolic dysfunction and septal hypertrophy. This phenotype then progressed into LV dilatation and systolic heart failure resulting in a heart transplant [7]. Interestingly, our own preliminary analysis of phosphorylation of sarcomeric proteins in cardiac tissue of patients H27 and H29 with mutation R723G showed significantly reduced phosphorylation, as commonly found in heart failure. The typically increased calcium sensitivity of cardiomyocytes from failing hearts [42], however, was not seen. Instead, calcium sensitivity was dominated by the mutation effect (own, unpublished observations). In studies on M. soleus fibers from patients H27 and H28, we previously observed a significant reduction in calcium sensitivity due to mutation R723G [15]. A similar reduced calcium sensitivity was also found in M. soleus fibers with mutation R719W (patient H13). Both mutations, R723G and R719W, also caused a pronounced increase in resistance to elastic deformation and substantial increase in force generation of the mutated myosin heads [15, 17, 36]. The large functional effects of mutation R719W at the sarcomeric level correspond to the fairly high expression level of 57% mutated MYH7-mRNA of total MYH7-mRNA, and the high risk of sudden death with this mutation [2]. Note that age-related penetrance of the disease plays an important role as well. In patients with mutation R723G, a malignant mutation appears benign at young age (patients H71 and H72), while mutation R719W is already very severe in young patients. Average life expectancy with mutation R719W is 38 years [2]. In muscle tissue with mutation I736T, only slightly more than 1/3 of total myosin was mutated and only small effects of the mutation on the contractile apparatus were observed [15, 36]. All three patients harboring mutation I736T showed mild clinical symptoms with a normal life expectancy [30]. Similarly, the individual with mutation G584R (23% of mutated protein) was essentially unaffected by the disease [2] and no functional changes were detected at the sarcomeric level.
For mutation V606M, the correlation between expression level and severity is less clear. The low abundance of mutated protein corresponds to the lack of functional changes in the muscle fibers and to the benign course of the disease in one of these individuals (H5) and in several families with this mutation [44, 45]. Survival at 50 years of age was 95%, which indicates nearly normal life expectancy. However, despite the low fraction of myosin with mutation V606M, the family of the other patient (H6) had a malignant form of the disease based on the fact that four out of eight HCM patients in the kindred studied died prematurely of sudden cardiac death [12]. It is unclear whether this is due to additional, as yet undetected etiological factors. Alternatively, since in patient H6 not all major candidate genes for mutations causing FHC have been analyzed, an additional mutation in another sarcomere gene contributing to disease severity cannot be excluded, although only up to 5% of all FHC patients are double or compound heterozygotes [34]. This holds also true for the other patients in our study.
Nevertheless, the correlation between life expectancy or severity of disease and expression level of mutated β-MHC is an additional aspect in FHC genotype–phenotype correlation and may represent a new prognostic parameter. In the past, mutations that result in a change in charge of the encoded amino acid have been correlated with a significantly shorter life expectancy [2, 44]. In later studies, it was suggested that the striking clinical and pathological heterogeneity is at least partially a function of the affected sarcomeric protein [10, 24]. For instance, families with cardiac troponin-T mutations appeared to have a particularly high risk of sudden cardiac death, while mutations in cardiac myosin-binding protein C are characterized by a benign course of the disease [27, 43]. Yet, the risk of disease-related complications for each patient varies also with modifying genes and lifestyle [1, 10, 37]. Our observation that the relative abundance of mutated and wild-type protein correlates with disease severity at least for mutations in the β-MHC could be a reason why in FHC no strong genotype–phenotype correlation has been established so far. Based on our functional studies on β-MHC mutations, we hypothesize that the mechanism responsible for the severe phenotype in patients with a large fraction of mutated β-myosin is a functional imbalance among individual cardiomyocytes because of unequal expression of the mutant protein from cell to cell [15]. Functional imbalances, e.g., in force generation, might represent a unifying mechanism triggering a common pathological pathway in FHC for different mutations, despite variable primary functional effects [28, 37]. Further studies on more FHC mutations, also in other sarcomeric proteins, are necessary to corroborate the correlation between disease severity and overall expression level of mutated protein.
In summary, relative quantification of wild-type and mutated MYH7 transcript and protein in muscle tissue of FHC patients shows that altered structure and function of the protein most likely are not the only determinants of its pathogenic effect. In addition, the sequence-specific unequal abundance of mutated and wild-type protein may contribute to the development of the complex phenotype in FHC. It would be of high clinical interest to further investigate (1) the mechanisms underlying the observed allelic imbalance and (2) whether the fraction of mutated transcript and protein is an indicator/determinant of prognosis and survival for a given mutation and family also for FHC mutations in other proteins.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We are indebted to the FHC patients and control individuals for their donation of muscle tissue. We thank C.E. and J.G. Seidman (Harvard Medical School, Boston, USA) for the initial methodological support, N. Bit-Avragim (Max-Delbrück-Centrum für Molekulare Medizin, Berlin, Germany) for help with patient recruitment, J. Meissner (Physiology, Hannover Medical School) for technical support and M. Ebeling (Hoffmann-La Roche, Basel, Switzerland) for bioinformatics input. M. Raida (Experimental Therapeutic Center, Singapore) is acknowledged for his contributions to protein quantification. The Clinics of Gastroenterology, Hepatology and Endocrinology and of Immunology and Rheumatology, Hannover Medical School, are acknowledged for making their ABI7000 available to us. The authors are grateful to F. Matinmehr, B. Piep, A. Lingk and A. Radocaj (all Physiology, Hannover Medical School) for help in establishing the methods, for excellent technical support and for help with data analysis, respectively. This work was supported by grants (KR 1187/5-3, 4 and KR 1187/19-1) of the Deutsche Forschungsgemeinschaft to TK.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19(1):104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 2.Anan R, Greve G, Thierfelder L, Watkins H, McKenna WJ, Solomon S, Vecchio C, Shono H, Nakao S, Tanaka H, et al. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest. 1994;93(1):280–285. doi: 10.1172/JCI116957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet. 2002;11(20):2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 4.Becker-Andre M, Hahlbrock K. Absolute mRNA quantification using the polymerase chain reaction (PCR). A novel approach by a PCR aided transcript titration assay (PATTY) Nucleic Acids Res. 1989;17(22):9437–9446. doi: 10.1093/nar/17.22.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker E, Navarro-Lopez F, Francino A, Brenner B, Kraft T. Quantification of mutant versus wild-type myosin in human muscle biopsies using nano-LC/ESI-MS. Anal Chem. 2007;79(24):9531–9538. doi: 10.1021/ac701711h. [DOI] [PubMed] [Google Scholar]
- 6.Becker KD, Gottshall KR, Hickey R, Perriard JC, Chien KR. Point mutations in human beta cardiac myosin heavy chain have differential effects on sarcomeric structure and assembly: an ATP binding site change disrupts both thick and thin filaments, whereas hypertrophic cardiomyopathy mutations display normal assembly. J Cell Biol. 1997;137(1):131–140. doi: 10.1083/jcb.137.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borchert B, Tripathi S, Francino A, Navarro-Lopez F, Kraft T. The left and right ventricle of a patient with a R723G mutation of the beta-myosin heavy chain and severe hypertrophic cardiomyopathy show no differences in the expression of myosin mRNA. Cardiol J. 2010;17(5):518–522. [PubMed] [Google Scholar]
- 8.Buckland PR. Allele-specific gene expression differences in humans. Hum Mol Genet. 2004;13((Spec No 2)):R255–R260. doi: 10.1093/hmg/ddh227. [DOI] [PubMed] [Google Scholar]
- 9.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363(9424):1881–1891. doi: 10.1016/S0140-6736(04)16358-7. [DOI] [PubMed] [Google Scholar]
- 11.Enjuto M, Francino A, Navarro-Lopez F, Viles D, Pare JC, Ballesta AM. Malignant hypertrophic cardiomyopathy caused by the Arg723Gly mutation in beta-myosin heavy chain gene. J Mol Cell Cardiol. 2000;32(12):2307–2313. doi: 10.1006/jmcc.2000.1260. [DOI] [PubMed] [Google Scholar]
- 12.Fananapazir L, Epstein ND. Genotype–phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation. 1994;89(1):22–32. doi: 10.1161/01.cir.89.1.22. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmeyer S, Assum G, Kaufmann D, Krone W. Unequal expression of NF1 alleles. Nat Genet. 1994;6(4):331. doi: 10.1038/ng0494-331. [DOI] [PubMed] [Google Scholar]
- 14.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 15.Kirschner SE, Becker E, Antognozzi M, Kubis HP, Francino A, Navarro-Lopez F, Bit-Avragim N, Perrot A, Mirrakhimov MM, Osterziel KJ, McKenna WJ, Brenner B, Kraft T. Hypertrophic cardiomyopathy-related beta-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. Am J Physiol Heart Circ Physiol. 2005;288(3):H1242–H1251. doi: 10.1152/ajpheart.00686.2004. [DOI] [PubMed] [Google Scholar]
- 16.Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26(7):1699–1708. doi: 10.1016/0735-1097(95)00390-8. [DOI] [PubMed] [Google Scholar]
- 17.Köhler J, Winkler G, Schulte I, Scholz T, McKenna W, Brenner B, Kraft T. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc Natl Acad Sci USA. 2002;99(6):3557–3562. doi: 10.1073/pnas.062415899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Zhou Y, Hu Z, Sun T, Denise A, Fu XD, Zhang Y. Regulation of splicing enhancer activities by RNA secondary structures. FEBS Lett. 2010;584(21):4401–4407. doi: 10.1016/j.febslet.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J Biol Chem. 2008;283(29):20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahen EM, Watson PY, Cottrell JW, Fedor MJ. mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS. 2010;8(2):e1000307. doi: 10.1371/journal.pbio.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malinchik S, Cuda G, Podolsky RJ, Horowits R. Isometric tension and mutant myosin heavy chain content in single skeletal myofibers from hypertrophic cardiomyopathy patients. J Mol Cell Cardiol. 1997;29(2):667–676. doi: 10.1006/jmcc.1996.0309. [DOI] [PubMed] [Google Scholar]
- 22.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary artery risk development in (Young) adults. Circulation. 1995;92(4):785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 23.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, 3rd, Spirito P, Ten Cate FJ, Wigle ED. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J. 2003;24(21):1965–1991. doi: 10.1016/s0195-668x(03)00479-2. [DOI] [PubMed] [Google Scholar]
- 24.McKenna WJ. The future in hypertrophic cardiomyopathy: important clues and potential advances from an understanding of the genotype phenotype relationship. Ital Heart J. 2000;1(1):17–20. [PubMed] [Google Scholar]
- 25.Neulen A, Stehle R, Pfitzer G. The cardiac troponin C mutation Leu29Gln found in a patient with hypertrophic cardiomyopathy does not alter contractile parameters in skinned murine myocardium. Basic Res Cardiol. 2009;104(6):751–760. doi: 10.1007/s00395-009-0038-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nier V, Schultz I, Brenner B, Forssmann W, Raida M. Variability in the ratio of mutant to wildtype myosin heavy chain present in the soleus muscle of patients with familial hypertrophic cardiomyopathy. A new approach for the quantification of mutant to wildtype protein. FEBS Lett. 1999;461(3):246–252. doi: 10.1016/s0014-5793(99)01433-7. [DOI] [PubMed] [Google Scholar]
- 27.Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338(18):1248–1257. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 28.Palmiter KA, Solaro RJ. Molecular mechanisms regulating the myofilament response to Ca2+: implications of mutations causal for familial hypertrophic cardiomyopathy. Basic Res Cardiol. 1997;92(Suppl 1):63–74. doi: 10.1007/BF00794070. [DOI] [PubMed] [Google Scholar]
- 29.Pastinen T, Hudson TJ. Cis-acting regulatory variation in the human genome. Science. 2004;306(5696):647–650. doi: 10.1126/science.1101659. [DOI] [PubMed] [Google Scholar]
- 30.Perrot A, Schmidt-Traub H, Hoffmann B, Prager M, Bit-Avragim N, Rudenko RI, Usupbaeva DA, Kabaeva Z, Imanov B, Mirrakhimov MM, Dietz R, Wycisk A, Tendera M, Gessner R, Osterziel KJ. Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Mol Med. 2005;83(6):468–477. doi: 10.1007/s00109-005-0635-7. [DOI] [PubMed] [Google Scholar]
- 31.Pfaffl MW (2006) Relative quantification. In: Dorak T (ed) Real time PCR, vol 2. International University Line, La Jolla, pp 63–82
- 32.Rayment I, Holden HM, Sellers JR, Fananapazir L, Epstein ND. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 1995;92(9):3864–3868. doi: 10.1073/pnas.92.9.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2(1):21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 34.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107(17):2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 35.Schiaffino S, Reggiani C. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol Rev. 1996;76(2):371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- 36.Seebohm B, Matinmehr F, Kohler J, Francino A, Navarro-Lopez F, Perrot A, Ozcelik C, McKenna WJ, Brenner B, Kraft T. Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys J. 2009;97(3):806–824. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seidman CE, Seidman JG. Molecular genetic studies of familial hypertrophic cardiomyopathy. Basic Res Cardiol. 1998;93(Suppl 3):13–16. doi: 10.1007/s003950050196. [DOI] [PubMed] [Google Scholar]
- 38.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104(4):557–567. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 39.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35(2):371–389. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spirito P, Maron BJ, Bonow RO, Epstein SE. Occurrence and significance of progressive left ventricular wall thinning and relative cavity dilatation in hypertrophic cardiomyopathy. Am J Cardiol. 1987;60(1):123–129. doi: 10.1016/0002-9149(87)90998-2. [DOI] [PubMed] [Google Scholar]
- 41.Thompson JR, Marcelino LA, Polz MF. Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res. 2002;30(9):2083–2088. doi: 10.1093/nar/30.9.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Velden J, Boontje NM, Papp Z, Klein LJ, Visser FC, de Jong JW, Owen VJ, Burton PB, Stienen GJ. Calcium sensitivity of force in human ventricular cardiomyocytes from donor and failing hearts. Basic Res Cardiol. 2002;97(Suppl 1):I118–I126. doi: 10.1007/s003950200040. [DOI] [PubMed] [Google Scholar]
- 43.Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332(16):1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 44.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326(17):1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 45.Watkins H, Thierfelder L, Anan R, Jarcho J, Matsumori A, McKenna W, Seidman JG, Seidman CE. Independent origin of identical beta cardiac myosin heavy-chain mutations in hypertrophic cardiomyopathy. Am J Hum Genet. 1993;53(6):1180–1185. [PMC free article] [PubMed] [Google Scholar]
- 46.WMA (1997) World Medical Association Declaration of Helsinki. Cardiov Res 35:2–3. doi:10.1016/S0008-6363(97)00109-0 [PubMed]
- 47.Wolstencroft EC, Hanlon K, Harries LW, Standen GR, Sternberg A, Ellard S. Development of a quantitative real-time polymerase chain reaction assay for the detection of the JAK2 V617F mutation. J Mol Diagn. 2007;9(1):42–46. doi: 10.2353/jmoldx.2007.060083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297(5584):1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 49.Zatkova A, Messiaen L, Vandenbroucke I, Wieser R, Fonatsch C, Krainer AR, Wimmer K. Disruption of exonic splicing enhancer elements is the principal cause of exon skipping associated with seven nonsense or missense alleles of NF1. Hum Mutat. 2004;24(6):491–501. doi: 10.1002/humu.20103. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Spier SJ, Beech J, Hoffman EP. Pathophysiology of sodium channelopathies: correlation of normal/mutant mRNA ratios with clinical phenotype in dominantly inherited periodic paralysis. Hum Mol Genet. 1994;3(9):1599–1603. doi: 10.1093/hmg/3.9.1599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.