

Abstract
We investigated the historical demography of Anopheles albimanus using mosquitoes from five countries and three different DNA regions, the mitochondrial cytochrome oxidase subunit I gene (COI), the single copy nuclear white gene and the ribosomal internal transcribed spacer two (ITS2). All the molecular markers supported the taxonomic status of a single species of An. albimanus. Furthermore, agreement between the COI and the white genes suggested a scenario of Pleistocene geographic fragmentation (i.e., population contraction) and subsequent range expansion across southern Central America.
Keywords: Anopheles albimanus, COI, white, ITS2, Pleistocene environmental changes, geographic fragmentation, population expansion
1. Introduction
Several phylogeographic studies have depicted a complex biogeographical history across the Isthmus of Panama and northern Colombia where significant genetic differentiation is seen at short distances, and waves of colonization, extinctions and re-invasions appear to be the main forces shaping the distribution of genetic diversity. Most of these studies have placed diversification events in a time frame earlier than the complete formation of the Isthmus of Panama (3 – 3.5 mya) (Bermingham and Martin, 1998; Zeh et al., 2003; Weigt et al., 2005; Miller et al., 2008), and therefore, much less is known about the impact of more recent environmental changes across this region during the Pleistocene (0.01 – 1.8 mya). Pleistocene climatic oscillations have had a profound effect on the genetic structure of vectors of pathogens, such as Anopheles mosquitoes, worldwide. For instance, in Southeast Asia, Anopheles dirus and Anopheles baimaii, two important malaria vectors, have expanded demographically due to changes in forest structure and climatic conditions in the Pleistocene, 172,202 years ago and 234,443 ya, respectively (O’Loughlin et al., 2008). Likewise, Anopheles darlingi, an important malaria vector in most of the Neotropics, has undergone Pleistocene population expansion in parts of South America likely due to changes in climatic conditions leading to forest fragmentation and refugia isolation, although in a much more recent time frame when compared with the Asian mosquitoes (25,312 ya) (Mirabello and Conn, 2006a,b). These and other studies have uncovered historical changes in demography that have ultimately shaped the geographical distribution and relative abundance of Anopheles species, and perhaps their capabilities to vector malarial parasites at a regional scale (Walton et al., 2000; Mirabello and Conn, 2008). Similar studies are scarce in southern Central America, despite the acknowledged geological and environmental complexity of this region during the Pleistocene (Crawford, 2003; Cortes-Oritz et al., 2003; Nettel et al., 2008; Miller et al., 2008).
Anopheles albimanus is a primary malaria vector throughout the northern Neotropics, yet its population history with respect to Pleistocene environmental changes has not been examined in depth. Furthermore, no accurate information exists on the geographical origin of An. albimanus, and its likely initial colonization path throughout the Americas. Recent research on An. albimanus uncovered considerable geographic structuring across the Isthmus of Panama and northern Colombia (Gutiérrez et al., 2009; Loaiza et al., 2010). Several divergent groups of mtDNA COI haplotypes were found co-occurring across this region, and were hypothesized to be the result of late Pleistocene geographic fragmentation and multiple re-introductions via demographic expansion. The population expansion, in central-eastern Panama and the Caribbean coast of Colombia, was believed to be due to climatic oscillation around 22,000 ya, and thus An. albimanus is not at mutation-drift equilibrium regionally (Gutiérrez et al., 2009; Loaiza et al., 2010). The co-occurrence of several maternal lineages of An. albimanus across Panama and northern Colombia may have important implications for malaria control, especially if they are differentially involved in malaria transmission. Nevertheless, three hypotheses remain to be tested: (1) Is the population divergence in recent studies of An. albimanus due to cryptic speciation?; (2) Is this divergence associated with Pleistocene environmental changes?; and (3) Is the population expansion of An. albimanus supported by a single copy nuclear gene?. To answer these questions, we combined the data from Gutiérrez et al. (2009) and Loaiza et al. (2010), with 123 additional mtDNA COI sequences from Nicaragua, eastern Panama and Ecuador, expanding the sampling scheme and augmenting the statistical power of the analyses. In addition, we sequenced a subset of individuals for the single copy nuclear white gene; because the white locus mutates more slowly than the COI gene, we used it to infer the initial colonization path of An. albimanus across southern Central America. Finally, we looked for fixed substitutional changes and length differences in the ribosomal DNA ITS2 marker as this may indicate the existence of cryptic species or restricted gene flow in An. albimanus (Collins and Paskewitz, 1996).
2. Materials and methods
2.1. Sample processing and laboratory procedures
Information on mosquito sampling, collection sites, laboratory procedures and GenBank accession numbers for the COI gene can be found in Gutiérrez et al. (2009) and Loaiza et al. (2010). Additional samples were obtained either as adults or in larval collections. The latter were reared to adulthood and processed as in Loaiza et al. (2010). A total of 612 sequences of the COI, all shortened to 776bp, were analyzed in this study (new COI sequence submission GenBank accession nos. HM030881 – HM030907). We sequenced the white gene for 175 individuals (HM042289 – HM042297) and the ITS2 for 173 (HM042298 – HM042301) from a subset of randomly selected samples (Table 1). Information on PCR-amplification conditions, and sequencing reactions for the ITS2 region and the white gene can be found in Linton et al. (2002) and Mirabello and Conn (2008), respectively.
Table 1.
Summary of diversity measures for Anopheles albimanus
| Gene marker | N | No. of COI haplotypes | K | Hd ± SD | π ± SD |
|---|---|---|---|---|---|
| COI | 612 | 191 (131) | 11.23 | 0.91 ± 0.008 | 0.014 ± 0.0003 |
|
| |||||
| NCRWP | 135 | 55 (30) | 6.76 | 0.95 ± 0.011 | 0.008 ± 0.0003 |
| CEPCO | 309 | 93 (65) | 3.41 | 0.79 ± 0.024 | 0.004 ± 0.0004 |
| PCOLE | 168 | 53 (36) | 7.42 | 0.94 ± 0.009 | 0.009 ± 0.0005 |
|
| |||||
| Gene marker | N | white alleles | K | Hd ± SD | π ± SD |
|
| |||||
| White | 175 | I - XVIII | 0.66 | 0.50 ± 0.001 | 0.0008 ± 0.0005 |
|
| |||||
| NCRWP | 56 | I, II, III, IV, V, XIII, XVIII | 0.57 | 0.39 ± 0.001 | 0.0007 ± 0.0001 |
| CEPCO | 61 | I, II, III, X, XI, XII, XIII, XIV, XV, XVI, XVII, | 0.12 | 0.12 ± 0.003 | 0.0001 ± 0.0002 |
| PCOLE | 58 | I, II, III, IV, VI, VII, VIII, IX | 1.06 | 0.73 ± 0.004 | 0.0014 ± 0.0002 |
|
| |||||
| Gene marker | N | ITS2 variants | K | Hd ± SD | π ± SD |
|
| |||||
| ITS2 | 173 | a, b | 0.52 | 0.54 ± 0.00 | 0.0011 ± 0.00 |
|
| |||||
| NCRWP | 56 | a - 2; 17 | 0.42 | 0.33 ± 0.00 | 0.0014 ± 0.00 |
| CEPCO | 61 | a - 2; 21 | 0.11 | 0.11 ± 0.00 | 0.0003 ± 0.00 |
| PCOLE | 56 | a - 2, b - 3; 0 | 0.03 | 0.04 ± 0.00 | 0.0001 ± 0.00 |
N = number of individuals of An. albimanus sequenced per gene marker and population deme as define by SAMOVA (see Fig. 1). The number in parentheses indicates the number of COI singletons. Bold letters are shared white alleles or ITS2 variants and plain letters are unique in that population deme. (a) and (b) are insertions in positions 281-283 (ATG) and 291-292 (AG) of the ITS2; these are followed by their frequencies shown by the number in italic. Underlined numbers are the number of individuals with multiple gene copies of the ITS2. K is the average number of nucleotide differences among sequences. Hd, haplotype diversity ± standard deviation; π, nucleotide diversity ± standard deviation.
2.2. Neutral expectation and genetic diversity
The program DNASP v4.50.02 (Rozas et al., 2003) was used to calculate Tajima’s D to determine whether or not the COI and the white gene sequences conformed to neutral expectations. Because the white gene is nuclear we allowed for recombination while testing for significant deviation from neutrality, thus accounting for overestimation of directional selection. We computed basic sequence statistics for all the markers in ARLEQUIN v3.11 (Excoffier et al., 2005) (Table 1).
2.3. Population divergence and historical demography
COI
We employed the spatial analysis of molecular variance (SAMOVA v1.0) (Dupanloup et al., 2002) to define aggregates of collection sites that are geographically homogeneous, but genetically differentiated from other similar aggregates. We ran SAMOVA from K = 2 to 30 and implemented 10,000 independent simulated annealing steps each starting from 100 random sets of initial conditions. The Mantel analysis was used to test for the isolation by distance (IBD) pattern, using a pairwise matrix of linearized genetic distances, estimated by ΦST, and the natural log-transformed geographic distance. Because An. albimanus has a coastal distribution we used the shortest geographic distances among populations along the shore instead of straight-line distances. The significance of the Mantel test was determined by n = 10,000 permutations using the IBD web service v3.15 (http://ibdws.sdsu.edu; Jensen et al., 2005). Genetic structure within population demes as defined by SAMOVA was further explored using the Mantel test as explained above. The program MDIV was used to distinguish between (1) a scenario of past isolation with no subsequent migration from (2) a scenario with no isolation, but contemporary gene flow or secondary contact among populations using MDIV@BioHPC (http://cbsuapps.tc.cornell.edu/mdiv.aspx; Nielsen and Wakeley, 2001). We ran MDIV under the HKY model, each simulation 6 ×106 times and assumed a 10% burn-in period with priors for Max T = 5 – 10, and Max M = 5 – 12. Estimates of θ, M and T were taken from the highest posterior probability. The net divergence DA (Nei, 1987), the between population distance minus the within population distance, was calculated using the Tamura and Nei model in MEGA v4.0 (Tamura et al., 2007). To assess the long-term stability of An. albimanus we used the mismatch distribution and the raggedness (r) statistic (Rogers and Harpending, 1992), both calculated in ARLEQUIN v3.11 (Excoffier et al., 2005). Neutrality tests of Fu’s FS (1997) and R2 (Ramos-Onsins and Rosas, 2002) were obtained in DNASP v4.50.02 (Rozas et al., 2003), and 10,000 coalescence simulations assessed significance. Dates of population expansion were estimated with the formula T = τ/2u (Rogers and Harpending, 1992) using 10 generations per year (Walton et al., 2000) and the mutation rate of Drosophila estimated at 1.2 ×10-8 substitutions per site per year (Powell et al., 1986). We examined changes in the effective population size through time implementing the Bayesian skyline plot (BSKP). The software BEAST v.1.4.2. (Drummond and Rambaut, 2007) was used to generate the BSKP with the SRD06 model of sequence evolution as suggested when analyzing mitochondrial protein coding genes. The analysis was implemented under a relaxed clock with rate for each branch drawn from a log normal distribution. The Markov Chain Monte Carlo algorithm was iterated for 10 × 107generations with a burn-in of 2 ×104 generations. We assessed the genealogical relationship among COI haplotypes using the statistical parsimony (SP), and the median-joining (MJ) network methods. These analyses were implemented in the programs TCS v1.12 (Clement et al., 2000) and NETWORK v4.2.0.1 (http://www.fluxus-engineering; Bandelt et al., 1999), respectively, and Anopheles triannulatus, in the same subgenus Nyssorhynchus, was used as the outgroup in the MJ network to provide directionality.
White
For the white gene data set we computed the SP network (Clement et al., 2000), the neutrality tests Fu’s FS (1997) and R2 (Ramos-Onsins and Rosas, 2002), the mismatch distribution (Rogers and Harpending, 1992) and the BSKP (Drummond and Rambaut, 2007). We used the Drosophila substitution rate of 0.004 – 0.008 per site per million years (Langley and Aquadro, 1987; Miyashita and Langley, 1988), 10 generations per year and 759bp of the white gene to calculate time of population growth or decline in the BSKP.
3. Results
3.1. Neutral expectation and genetic diversity
In total, 191 COI hapotypes and eighteen white alleles were recovered in this study (Table 1; Fig. 1). Both markers conformed to neutral expectations, as estimates of Tajima’s D were not significantly different from zero (Tajima’s D, COI = - 1.38, P > 0.05 and white D = - 0.59, P > 0.05). Two short insertions were found in the ITS2 region of An. albimanus. These insertions were located in positions 281-283 (ATG) and 291-292 (AG), respectively and were found in 9 (5.2%) of 173 individuals. Furthermore, one base pair ambiguity in position 234 (i.e., multiple copies of the ITS2) separated 38 (21.9%) individuals from Nicaragua (4), Costa Rica (13) and Panama (21) from the remaining 135, all of which had a single peak for that nucleotide and were distributed over the entire geographic area (Table 1).
Fig. 1.
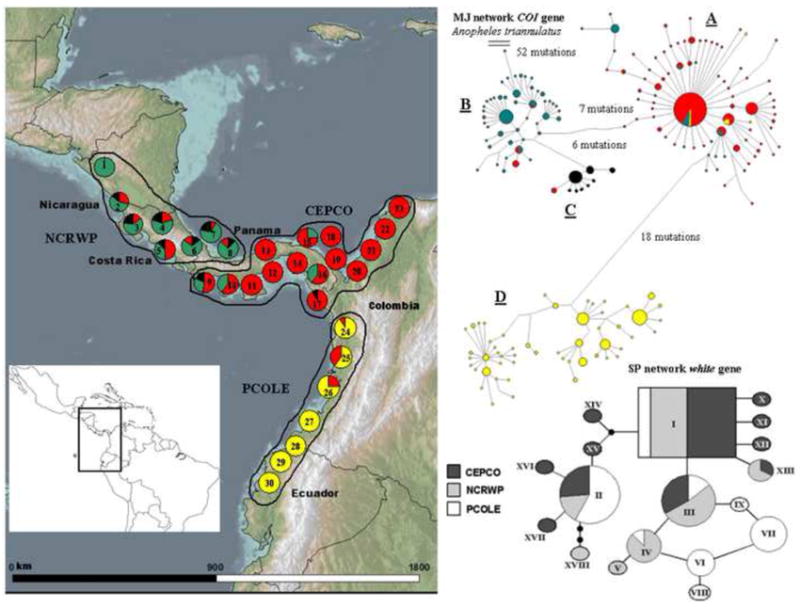
The map shows the geographic distribution of four COI haplogroups (A, red), (B, green), (C, black) and (D, yellow) from the MJ haplotype network of Anopheles albimanus, located in the upper right corner. The circled numbers correspond to 30 localities, positioned on the map according to the longitude and latitude of each site. Three population demes defined by SAMOVA are outlined: Nicaragua, Costa Rica and the Atlantic coast of western Panama (NCRWP = localities 1– 8); the Pacific coast of western Panama, central – eastern Panama and the Caribbean coast of Colombia (CEPCO = localities 9 – 23); and the Pacific coast of Colombia and Ecuador (PCOLE = localities 24 – 30). The statistical parsimony network of nine white gene alleles (I – IX) and their geographical distributions are shown in the lower right corner. The inset map depicts the geographic position of the study area.
3.2. Population divergence and historical demography
In agreement with previous studies the COI showed considerable population structure in An. albimanus. SAMOVA defined three population demes with the highest among group variation at 64.75% (FCT = 0.6475, P > 0.0001). These population demes were clearly segregated geographically indicating either the existence of specific barriers to gene flow or separate demographic origins. The Mantel analyses to test for IBD among all populations, and within each population deme were all statistically insignificant (all populations, R2 = 0.0152, P = 0.813; NCRWP, R2 = 0.0134, P = 0.931; CEPCO, R2 = 0.0612, P = 0.429; PCOLE, R2 = 0.0074, P = 0.701), suggesting no association between geographic and genetic distances overall, and shallow genetic structure within population demes. These results have to be interpreted cautiously though, as population demes may be better defined by current migration rather than by historical gene flow.
MDIV analysis supported secondary contact as a better model to explain the COI geographical pattern versus historical isolation without subsequent gene flow among population demes (Fig. S1 Supplementary data). The net nucleotide substitution per site between NCRWP and CEPCO (DA = 0.005 ± 0.002, 95% CI) placed the time of divergence between these population demes around 250,000 (285,000 ± 215,000) years ago in the late Pleistocene. Furthermore, divergence times between NCRWP vs. PCOLE (DA = 0.017 ± 0.004, 95% CI) and CEPCO vs. PCOLE (DA = 0.016 ± 0.003, 95% CI) were around 850,000 (975,000 ± 725,000) and 827,000 (952,000 ± 702,000) years ago, respectively, in the middle Pleistocene. Fu’s FS (1997) neutrality test was negative and highly significant in all of the population demes (NCRWP Fs = - 24.444, P < 0.0001; CEPCO Fs = - 25.719, P < 0.0001; PCOLE Fs = - 24.633, P < 0.0001), therefore rejecting the mutation-drift equilibrium assumption in An. albimanus and strongly favoring the interpretation of population expansion. Similarly, the R2 test (Ramos-Onsins and Rosas, 2002) was low, positive and significant (NCRWP R2 = 0.0628, P = 0.0053; CEPCO R2 = 0.0224, P = 0.0012; PCOLE R2 = 0.0634, P = 0.0032), thus reinforcing a scenario of population expansion. It is noteworthy that the mismatch distribution for the entire COI data statistically fit the model of sudden expansion, but showed three defined peaks reflecting the existence of three population demes, each of them expanding, albeit at different times (Fig. S3 Supplementary data).
The SP and the MJ networks further supported the population divergence defined by SAMOVA, and provided deeper resolution of the geographic structure of An. albimanus uncovering four different COI haplogroups (Table 1; Fig. 1; Fig. S4 Supplementary data). These four maternal lineages were separated by 6 – 18 mutational steps and segregated among the three geographical regions defined by SAMOVA. Haplogroups (B) and (C) co-occurred and were more prevalent in NCRWP; (A) was prevalent in CEPCO, and (D) was largely predominant in PCOLE (Fig. 1). The net nucleotide substitution per site among these haplogroups (DA = 0.008 – 0.026) placed the time of divergence between them around (400,000 – 1,300,000) years ago, in the late and middle Pleistocene. We re-calculated the mismatch and neutrality tests for each mitochondrial haplogroup separately, because the coalescence signal of the COI gene (i.e., the time since expansion) may be confounded by subsequent gene flow and more recent secondary contact among population demes. Both the Fu’s FS and the R2 tests rejected the mutation-drift equilibrium assumption in all the COI haplogroups, likely due to population expansion, and this was corroborated by the non-significant raggedness indexes and unimodal mismatch distributions (Fig. S5 Supplementary data). The expansion times for A (τ = 3.658), B (τ = 5.132), C (τ = 2.929) and D (τ = 4.765) were 19,641 (95% CI 9,324 – 39,641), 27,555 (95% CI 14,177 – 47,533), 15,727 (95% CI 6,324 – 35,641) and 25,585 (95% CI 10,077 – 46,533), respectively, all in the late Pleistocene. The BSKPs further supported significant population growth and showed an increase in effective population size in each of these four COI haplogroups, all within 150,000 – 550,000 years before the present (Fig. S2 Supplementary data).
In contrast to the COI gene, genetic diversity was lower for the white gene (Table 1). Eighteen alleles were joined by 1 – 3 nucleotide differences in a SP network (Fig. 1). These alleles were very closely related and not segregated geographically as were the four COI haplogroups. Instead, one ancestral allele (I) was present in all the sample locations, but more frequently in localities across NCRWP, and CEPCO, and almost fixed for those in Costa Rica and Panama. Other three alleles, of intermediate-frequency (II, III, and IV), were shared among regions whereas the rest were less frequent and more restricted geographically (Table 1; Fig. 1). It is noteworthy that seven out of ten white gene singletons were recovered exclusively from CEPCO, and this may suggest a recent pattern of regional population growth (Fig. 1). Furthermore, Fu’s FS (1997) neutrality test was negative and significant (Fs = - 7.65, P < 0.0001), and the R2 test (Ramos-Onsins and Rosas, 2002) was low, positive and highly significant (R2 = 0.0112, P = 0.0013). The expansion time for the white alleles based on τ = 2.55 from the mismatch distribution (Fig. S5 Supplementary data), was between 20,586 – 41,996 years ago, in the late Pleistocene. Nevertheless, the BSKP for the nuclear marker depicted a recent decrease in the effective population size of An. albimanus around 50,000 years before the present, although it was not statistically significant (Fig. S2 Supplementary data). Given the use of non-anopheline mutation rates in the present study, our estimates of the divergence and expansion times for the COI and the white genes have to be interpreted with caution as they may reflect only approximate values.
4. Discussion
The present study provides further evidence that late Pleistocene environmental changes had a profound effect on the demography and regional geographic structure of An. albimanus in the northern Neotropics. The COI findings appear to be the result of historical fragmentation leading to four COI haplogroups that were geographically isolated during the middle Pleistocene, but reconnected across southern Central America via more recent secondary contact and a common late Pleistocene expansion event. Although the mismatch distributions for NCRWP, CEPCO and PCOLE were not statistically different from a model of sudden population expansion, they were all visually bimodal, perhaps reflecting the admixing of COI haplogroups among different regions. In addition, MDIV estimated higher secondary contact between NCRWP and CEPCO, and this is supported by less divergence between haplogroups (A), (B) and (C) (6 – 13 mutations) than between (A, B, C) and (D) (18 – 25 mutations). Clearly, this pattern reflects the fact that haplogroup D is the most geographically restricted because none of its haplotypes were encountered outside PCOLE (Fig. 1).
Alternatively, the COI divergence may be due to directional selection in the mitochondrial genome, for instance, an advantageous mutation sweeping to fixation across southern Central America. However, selection is generally a locus specific force, whereas demographic changes affect the entire genome. The latter is more consistent with the evidence of population expansion in both the COI and white genes. Moreover, all the COI haplogroups showed an excess of low frequency haplotypes indicating a common pattern of recent population growth regardless of their geographical distributions. This would make a selective sweep unlikely because the same advantageous mutation would have to appear independently in each of NCRWP, CEPCO and PCOLE.
On the other hand, two short insertions, found in the ITS2 of An. albimanus, may support the presence of unidentified cryptic taxa, but very few individuals harbored these indels, and only one of these insertions was found exclusively in PCOLE (Table 1). Besides these indels, no fixed substitutional changes or length differences were encountered in the ITS2 sequences of 52, 59, and 52 individuals from NCRWP, CEPCO and PCOLE, respectively. Although based on our ITS2 analysis, we cannot reject substantial historical gene flow among populations of An. albimanus; we cannot entirely reject the existence of multiple taxa (i.e., cryptic speciation) either. For instance, the levels of divergence between our sympatrically occurring mtDNA lineages are far in excess of those observed between sister taxa in other Anopheles species (Foley et al., 2006). The restricted occurrence of intragenomic variability in the ITS2 of samples from NCRWP and CEPCO, and an exclusive insertion in three samples from PCOLE, may suggest past geographical fragmentation as proposed for the COI data, though the alternative hypothesis of recent, perhaps slight limits to gene flow seems equally likely (Bower et al., 2008).
Anopheles albimanus is believed to have originated in the Caribbean islands and then colonized the American continent, and a founder effect may have been a main factor shaping its continental population structure (Molina-Cruz et al., 2004). The colonization routes suggested by these authors hypothesize that An. albimanus invaded South America from northern Central America. In the present analysis, Anopheles triannulatus joined through 52 mutations to haplogroup B in the COI MJ network (Fig. 1) indicating that haplogroups (C) and (A) originated from (B), and then (D) originated from (A). This pattern is consistent with the scenario suggested by Molina-Cruz et al. (2004). The co-occurrence of two distinct COI haplogroups (B and C) in NCRWP supports this view and indicates that successful initial colonization of South America was achieved by two separate introductions of An. albimanus from Nicaragua into Colombia and Ecuador. Nevertheless, genetic diversity as measured by both genes does not decrease progressively from north to south, as would be expected under a founder effect and sequential bottleneck in the colonizing front. Instead genetic diversity is lower in CEPCO than in Nicaragua (NCRWP) and Ecuador (PCOLE) (Table 1).
Coalescent theory predicts that the most frequent and widely distributed gene variants are ancestral, because they have had more time to disperse (Uthicke and Benzie, 2003). In our data, both of these variants, the COI haplotype (A1) and the white allele (I), were prevalent across Panama, implying a possible Panamanian geographical origin for An. albimanus (Fig. 1; Fig. S4 Supplementary data). This view is unlikely, though, given the recent geological origin of the Isthmus of Panama, dated approximately 3 – 3.5 mya (Bermingham and Martin, 1998). Due to the non-equilibrium frequencies of variants in both genes and our incomplete sampling of the geographic range of An. albimanus, we do not have enough evidence to draw firm conclusions about its geographical origin and initial colonization path across southern Central America.
The observed geographic structure seems to be the result of population contraction of an ancestral and perhaps stable population of An. albimanus across the Isthmus of Panama (i.e., the four COI haplogroups diverged and expanded within the same time frame). This view is supported by lower diversity in the COI and white genes across Panama (CEPCO), which may indicate that An. albimanus went through a population bottleneck followed by a subsequent demographic expansion toward Nicaragua and Ecuador (Table 1). Previous work using four microsatellite loci and the mtDNA ND5 gene also indicated significantly reduced diversity for one locality in central Panama, thus supporting our hypothesis (Molina-Cruz et al., 2004). Our results from two independent molecular markers may represent two temporal pictures of the demography of An. albimanus in the study area, with the white gene showing a bottleneck and the COI depicting a subsequent demographic expansion, and this may be caused by different mutation rates, and effective population sizes in these genes. The bottleneck hypothesis may be supported by a drop of 35% precipitation and changes in vegetation and faunal structure in central - eastern Panama in the late Pleistocene (Piperno and Jones, 2003; Gonzales et al., 2006). Additional signals of Pleistocene geographic fragmentation across Panama have been demonstrated in howler monkeys, black mangroves and dirt frogs, and hypothesized to be caused by forest fragmentation, sea level changes, and climatic refugia, respectively (Crawford, 2003; Cortes-Oritz et al., 2003; Nettel et al., 2008).
5. Conclusions
Anopheles albimanus appears to be a single, albeit polymorphic, species that is not at mutation-drift equilibrium due to past geographic fragmentation and regional fluctuation in its effective population size. There is a strong geographic component in its genetic structure with three population demes and an admixture zone across eastern Panama and northern Colombia. The COI gene suggests a common pattern of historical isolation, subsequent haplotype mixing, and population expansion of four mtDNA COI lineages in the late Pleistocene. Data from the white gene does not reflect the high genetic diversity of the COI, but it is consistent with the scenario of late Pleistocene population expansion, thus supporting demographic phenomena as the cause of structure rather than a selective sweep. Finally, lower genetic diversity by both the COI and white genes across Panama suggests that An. albimanus populations contracted, then subsequently expanded toward Nicaragua and Ecuador.
Supplementary Material
Acknowledgments
Financial support was obtained partially from the Secretariat for Science, Technology and Innovation of Panama (SENACYT) through a research grant (COL08-066) awarded to Jose R. Loaiza. Additional financial support was provided by the Smithsonian Tropical Research Institute (STRI), National Institutes of Health (NIH) grant (AI) R0154139-02 to JEC, Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología (COLCIENCIAS) grant 1115-05-16879 to MCO, Comité para el Desarrollo de la Investigación (CODI) Universidad de Antioquia grant 8700-039 and E-01233, to MCO, and Natural Sciences and Engineering Research Council of Canada (NSERC). Research at the Institute of Parasitology is supported by a regroupement stratégique from Fonds Québécois de la recherche sur la nature et les technologies (FQRNT). We would like to acknowledge Dr. Efrain Beltran from the National Malaria Eradication Service (SNEM), in Ecuador, and Luis Guillermo Chaverri Sánchez from Instituto Nacional de Biodiversidad de Costa Rica (INBio) for technical assistance. We are grateful to Matthew Miller and Laura B. Geyer from STRI and to Martin Donnelly from the Liverpool School of Tropical Medicine for helpful discussions and comments on earlier versions of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Bermingham E, Martin AP. Comparative mtDNA phylogeography of Neotropical fresh water fishes: testing shared history to infer the evolutionary landscape of lower Central America. Mol Ecol. 1998;7:499–517. doi: 10.1046/j.1365-294x.1998.00358.x. [DOI] [PubMed] [Google Scholar]
- Bower JE, Dowton M, Cooper RD, Beebe NW. Intraspecific concerted evolution of the rDNA ITS1 in Anopheles farauti sensu stricto (Diptera: Culicidae) reveals recent patterns of population structure. J Mol Biol. 2008;67:397–411. doi: 10.1007/s00239-008-9161-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FH, Paskewitz SM. A review of the use of ribosomal DNA (rDNA) to differentiate among cryptic Anopheles species. Insect Mol Biol. 1996;5(1):1–9. doi: 10.1111/j.1365-2583.1996.tb00034.x. [DOI] [PubMed] [Google Scholar]
- Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- Cortes-Ortíz L, Bermingham E, Rico C, Rodríguez -Luna E, Sampaio I, Ruiz-Garcíae M. Molecular systematics and biogeography of the Neotropical monkey genus, Alouatta. Mol Phyl Evol. 2003;26:64–81. doi: 10.1016/s1055-7903(02)00308-1. [DOI] [PubMed] [Google Scholar]
- Crawford AJ. Huge populations and old species of Costa Rican and Panamanian dirt frogs inferred from mitochondrial and nuclear gene sequences. Mol Ecol. 2003;12:2525–2540. doi: 10.1046/j.1365-294x.2003.01910.x. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11:2571–81. doi: 10.1046/j.1365-294x.2002.01650.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schmeider S. Arlequin ver 3.0: An integrated software package for population genetic data analysis. EBO. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Foley DH, Wilkerson RC, Cooper RD, Volovsek ME, Bryan JH. A molecular phylogeny of Anopheles annulipes (Diptera: Culicidae) sensu lato: The most species-rich anopheline complex. Mol Phylogenet Evol. 2006;43:283–297. doi: 10.1016/j.ympev.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzáles C, Urrego LE, Martínez JI. Late quaternary vegetation and climate change in the Panama basin: Palynological evidence from marine cores ODP 677 and TR 163-38. PALAEO. 2006;234:62–80. [Google Scholar]
- Gutiérrez LA, Naranjo NJ, Cienfuegos AV, Muskus CE, Luckhart S, Conn JE, Correa MM. Population structure analyses and demographic history of the malaria vector Anopheles albimanus Wiedemann (Diptera: Culicidae) from the Caribbean and the Pacific regions of Colombia. Mal J. 2009;8:259. doi: 10.1186/1475-2875-8-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genetics. 2005;6:13. doi: 10.1186/1471-2156-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley CH, Aquadro CF. Restriction-map variation in natural populations of Drosophila melanogaster: white-locus region. Mol Biol Evol. 1987;151:651–663. doi: 10.1093/oxfordjournals.molbev.a040467. [DOI] [PubMed] [Google Scholar]
- Linton YM, Samanidou-Voyadjoglou A, Harbach RE. Ribosomal ITS2 sequence data for Anopheles maculipennis and An. messeae in northern Greece, with a critical assessment of previously published sequences. Insect Mol Biol. 2002;11(4):379–383. doi: 10.1046/j.1365-2583.2002.00338.x. [DOI] [PubMed] [Google Scholar]
- Loaiza JR, Scott ME, Bermingham E, Rovira R, Conn JE. Evidence for Pleistocene population divergence and expansion of Anopheles albimanus in Southern Central America. Am J Trop Med Hyg. 2010;82(1):156–164. doi: 10.4269/ajtmh.2010.09-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Bermingham E, Klicka J, Escalante P, Raposo do Amaral FS, Weir JT, Winker K. Out of Amazonia again and again: episodic crossing of the Andes promotes diversification in a lowland forest flycatcher. Proc R Soc B. 2008;275:1133–1142. doi: 10.1098/rspb.2008.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Conn JE. Molecular population genetics of the malaria vector Anopheles darlingi in Central and South America. Heredity. 2006a;96:311–321. doi: 10.1038/sj.hdy.6800805. [DOI] [PubMed] [Google Scholar]
- Mirabello L, Conn JE. Correction to Heredity (2006) 96, 311-321: Molecular population genetics of the malaria vector Anopheles darlingi in Central and South America. Heredity. 2006b;97:438. doi: 10.1038/sj.hdy.6800805. [DOI] [PubMed] [Google Scholar]
- Mirabello L, Conn JE. Population analysis using the nuclear white gene detects Pliocene/Pleistocene lineage divergence within Anopheles nuneztovari in South America. Med Vet Entomol. 2008;22:109–119. doi: 10.1111/j.1365-2915.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- Miyashita N, Langley CH. Molecular and phenotypic variation of the white locus region in Drosophila melanogaster. Genetics. 988;120:199–212. doi: 10.1093/genetics/120.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Cruz A, De Merida AM, Mills K, Rodríguez F, Schoua C, Yurrita MM, Molina E, Palmieri M, William CB. Gene flow among Anopheles albimanus populations in Central America, South America, and the Caribbean assessed by microsatellites and mitochondrial DNA. Am J Trop Med Hyg. 2004;71:350–359. [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary genetics. New York: Columbia University Press; 1987. [Google Scholar]
- Nettel A, Dodd RS, Afzal-Rafii Z, Tovilla-Hernandez C. Genetic diversity enhanced by ancient introgression and secondary contact in East Pacific black mangroves. Mol Ecol. 2008;17:2680–2690. doi: 10.1111/j.1365-294X.2008.03766.x. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: A Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loughlin SM, Okabayashi T, Honda M, Kitazoe Y, Kishino H, Somboon P, Sochantha S, Nambanya PK, Saikia VDEV, Walton C. Complex population history of two Anopheles dirus mosquito species in South Asia suggests the influence of Pleistocene climate change rather than human-mediated effects. J Evol Biol. 2008;21:1555–1569. doi: 10.1111/j.1420-9101.2008.01606.x. [DOI] [PubMed] [Google Scholar]
- Piperno DR, Jones JG. Paleoecological and archaeological implications of the Late Pleistocene Early Holocene record of vegetation and climate from the Pacific coastal plain of Panama. Quaternary Res. 2003;59:79–87. [Google Scholar]
- Powell JR, Caccone A, Amato GD, Yoon C. Rate of nucleotide substitution in Drosophila mitochondrial DNA and nuclear DNA are similar. Proc Natl Acad Sci USA. 1986;83:9090–9093. doi: 10.1073/pnas.83.23.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19:2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034. [DOI] [PubMed] [Google Scholar]
- Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;62:122–127. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sanchez-Del Rio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescence and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Walton C, Handley JM, Tun-Lin W. Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Mol Biol Evol. 2000;17:962–974. doi: 10.1093/oxfordjournals.molbev.a026377. [DOI] [PubMed] [Google Scholar]
- Uthicke S, Benzie JAH. Gene flow and population history in high dispersal marine invertebrates: mitochondrial DNA analysis of Holothuria nobilis (Echinodermata: Holoturoidea) populations from the Indo-Pacific. Mol Ecol. 2003;12:2635–2648. doi: 10.1046/j.1365-294x.2003.01954.x. [DOI] [PubMed] [Google Scholar]
- Weigt LA, Crawford AJ, Stanley Rand A, Ryan M. Biogeography of the tungara frog, Physalaemus pustulosus: a molecular perspective. Mol Ecol. 2005;14:3857–3876. doi: 10.1111/j.1365-294X.2005.02707.x. [DOI] [PubMed] [Google Scholar]
- Zeh JA, Zeh DW, Bonilla MM. Phylogeography of the harlequin beetle-riding pseudoscorpion and the rise of the Isthmus of Panama. Mol Ecol. 2003;12:2759–2769. doi: 10.1046/j.1365-294x.2003.01914.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.