

Abstract
Ischemic stroke triggers a massive, although transient, glutamate efflux and excessive activation of NMDA receptors (NMDARs), possibly leading to neuronal death. However, multiple clinical trials with NMDA antagonists failed to improve, or even worsened, stroke outcome. Recent findings of a persistent post-stroke decline in NMDAR density, which plays a pivotal role in plasticity and memory formation, suggest that NMDAR stimulation, rather than inhibition, may prove beneficial in the subacute period after stroke.
Aim
This study aims to examine the effect of the NMDAR partial agonist d-cycloserine (DCS) on long-term structural, functional and behavioral outcomes in rats subjected to transient middle cerebral artery occlusion, an animal model of ischemic stroke.
Materials & methods
Rats (n = 36) that were subjected to 90 min of middle cerebral artery occlusion were given a single injection of DCS (10 mg/kg) or vehicle (phosphate-buffered saline) 24 h after occlusion and followed up for 30 days. MRI (structural and functional) was used to measure infarction, atrophy and cortical activation due to electrical forepaw stimulation. Memory function was assessed on days 7, 21 and 30 postocclusion using the novel object recognition test. A total of 20 nonischemic controls were included for comparison.
Results
DCS treatment resulted in significant improvement of somatosensory and cognitive function relative to vehicle treatment. By day 30, cognitive performance of the DCS-treated animals was indistinguishable from nonischemic controls, while vehicle-treated animals demonstrated a stable memory deficit. DCS had no significant effect on infarction or atrophy.
Conclusion
These results support a beneficial role for NMDAR stimulation during the recovery period after stroke, most likely due to enhanced neuroplasticity rather than neuroprotection.
Keywords: cognition, d-cycloserine, fMRI, infarction, NMDA receptor, stroke
Stroke is a leading cause of mortality and morbidity in the industrialized world. More than 795,000 people each year suffer from stroke in the USA alone, where it is the third leading cause of death and is associated with tens of billions of US dollars in costs [1,2,101]. The majority of strokes affect the middle cerebral artery (MCA) and reperfuse spontaneously [3]. Survival is often associated with long-term somatosensory, cognitive and motor deficits [4,5]. Treatment options for stroke victims are extremely limited. The only drug currently approved for treatment of stroke is tissue plasminogen activator, which has a very short therapeutic window (3 h) and is contraindicated in hemorrhagic stroke, greatly limiting the percentage of treatable subjects [6-10]. All Phase III clinical trials of putative neuroprotective agents aimed at improving neurological outcome in stroke survivors have failed to date [11,12].
Ischemia triggers a large, transient increase in excitatory amino acid transmitter efflux in the brain of experimental animals, as well as human subjects [13,14]. Glutamate activation of the NMDA receptor (NMDAR), which is a ligand-gated ion (calcium and sodium) channel, results in channel opening and ion influx into the cell. It has been suggested that this process, which causes cell death in neuronal culture, also mediates delayed excitotoxic neuronal death following brain ischemia [15], although the concept is not universally accepted [16]. Support for the involvement of NMDAR activation in neuronal death following ischemic brain injury has come from numerous studies showing that NMDAR antagonists reduce cell death and improve outcome in animal models of stroke, although NMDAR antagonists appear to be most efficacious when administered prior to or immediately after the insult and lose efficacy if administered more than 30–60 min postinjury (Table 1) [17,18]. The positive results from animal models led to the development and clinical testing of several NMDA antagonists in stroke and traumatic brain injury. Unfortunately, all of the NMDAR antagonists studied so far, including competitive, noncompetitive and partial (glycine site) antagonists, have failed to show efficacy in large controlled clinical trials. In some of these trials, NMDAR antagonists even worsened clinical outcome [11,19-22]. Importantly, the administration of NMDAR antagonist in clinical trials was initiated considerably later (mostly after a 6–24 h delay) and lasted longer (3–7 days) than in the animal studies (Table 1).
Table 1. Representative paradigms and outcomes of NMDA antagonists in animal models and human stroke.
| Study (year) | Species, model |
NMDA antagonist |
Time, duration | Follow- up |
Outcome | Ref. |
|---|---|---|---|---|---|---|
| Bielenberg and Beck (1991) |
Rat, MCAO | MK801 phencyclidine |
−30 min, 8, 16, 24 h (pre- and post- treatment) |
2 days | 50–55% reduction in infarction volume |
[61] |
| Izumi et al. (1991) | Rat, MCAO | Mg2+ | 1 h | 48 h | 44% reduction in infarction volume |
[62] |
| Chen et al. (1991) | Rat, MCAO | d-CPP-ene | −15 min, 1 h (pre- and post-treatment) |
6 h | 75% reduction in infarction volume (pretreatment), 13% reduction (post-treatment) |
[63] |
| Bullock et al. (1990) | Cat, MCAO | d-CPP-ene | −15 min | 6 h | 65% reduction in infarction volume |
[64] |
| Dezsi et al. (1994) | Cat, MCAO | MK801 | 30 min | 6 days | No reduction in infarction size | [65] |
| Yam et al. (2000) | Cat, MCAO | MK801 | −15 min | 6 h | No reduction of axonal injury | [66] |
| Reggiani et al. (2001) | Rat, MCAO | GV150526 | −5 min, 6 h (pre- and post-treatment) |
1 day | 84% reduction in infarction volume (pretreatment), 48% reduction (post-treatment) |
[67] |
| Ohtani et al. (2003) | Rat, MCAO | SM-31900 | 5, 30, 60 min | 1 day | 37% reduction in infarction volume |
[68] |
| Oda et al. (2007) | Mouse, MCAO | SM-31900 | −30 min, −10 min | 1 day | 46.4% reduction in infarction volume |
[69] |
| Albers et al. (1995) | Human, stroke | Dextrorphan | 12, 24, 48 h | 30 days | NIHSS no benefit, increased AEs with high dose |
[70] |
| Dyker and Lees (1999) | Human, stroke | Remacemide hydrochloride |
12 h, 3, 6 days | 4 weeks | Barthel index, transient benefit | [71] |
| Davis et al. (2000) | Human, stroke | Selfotel | 6 h | 3 months | NIHSS, no benefit, increased mortality |
[72] |
| Albers et al. (2001) | Human, stroke | Aptiganel | 6 h | 90 days | Rankin index, no benefit | [73] |
| Lees et al. (2001) | Human, stroke | AR-R15896AR | 24 h | 4 weeks | Barthel index, no benefit | [74] |
| Haley et al. (2005) | Human, stroke | Gavestinel | 6 h | 3 months | Barthel index, no benefit | [75] |
AE: Adverse effect; MCAO: Middle cerebral artery occlusion; NIHSS: NIH Stroke Scale.
The failure of NMDA antagonists in clinical trials coupled with animal data on the crucial role of NMDAR activation in neuroplasticity [23-25] led us (and others) to conclude that delayed and prolonged blockade of NMDARs might be harmful rather than beneficial in the aftermath of ischemic and traumatic brain injury [22,26,27]. Furthermore, work by us and others has shown that as soon as 1 h after ischemia, NMDAR availability is drastically reduced, remaining abnormally low for weeks after the insult [28-30], thus providing a likely explanation for the long-lasting cognitive and neurological deficits often observed in stroke victims.
To test this hypothesis, we have examined the effect of delayed stimulation, rather than inhibition, of NMDARs with the partial agonist d-cycloserine (DCS) [31] on functional, long-term outcomes in rats with occlusion of the MCA occlusion (MCAO) rats, a common animal model of ischemic stroke [32].
Materials & methods
Experimental design Adult female Sprague–Dawley rats (n = 56, 220–250 g, Taconic Farms, NY, USA) were kept under controlled light and dark conditions and given food and water ad libitum. The experiments were approved by the institutional animal care and use committee. Rats were initially randomized into three groups: two groups of nonischemic controls (intact, n = 11; and sham surgery, n = 9) and an ischemia group (MCAO, n = 36). Following stroke confirmation, the MCAO group was further randomized to receive DCS or vehicle (phosphate-buffered saline [PBS]).
Surgery
Middle cerebral artery occlusion was performed using an adaptation of published methods [28,32] on 36 rats. Animals were anesthetized with isofluorane (0.75–1.0%) mixed in O2-enriched air flow during the surgery. Body temperature was continuously monitored with a rectal probe and maintained at 37.0°C with a heating pad. A midline incision was made and the left common carotid artery, external carotid artery and internal carotid artery were exposed under an operating microscope. A 4-0 silicone rubber-coated monofilament (Doccol Corp., CA, USA) was inserted through the external carotid artery into the lumen of the internal carotid artery (~18–19 mm) until resistance was encountered, ensuring that the intraluminal suture had blocked the origin of the MCA. The incision was tightly closed with a 0-6 silk suture, leaving 1 cm of the monofilament protruding so it could be withdrawn to allow reperfusion. The silicone rubber-coated monofilament was allowed to remain in place for 90 min and then retracted so as to allow reperfusion of the ischemic region. Two nonischemic control groups (sham and intact) were also used; sham surgery (n = 9) consisted of anesthesia, incision and exposure of the arteries. Intact animals (n = 11) did not receive any intervention. Five rats died within 24 h of surgery, prior to randomization to treatment.
Postsurgical monitoring
Rats were examined for spontaneous locomotion, posture, forelimb placing, body weight and general appearance in the first few hours after surgery and daily thereafter. Animals showing signs of morbidity (e.g., severe weight loss) were euthanized.
Structural MRI
MRI scans were performed 24 h after MCAO to confirm infarction before randomization to drug treatment. MCAO animals and nonischemic controls were anesthetized with 3% isofluorane and 50 mg/kg intraperitoneally (ip.) of methohexital sodium and given glycopyrolate 0.04 mg/kg ip., then positioned in a custom-made cradle ensuring stabilization of the head and positioning in the center of the magnet where the main magnetic field is most homogeneous. The animals were continuously monitored with magnetic resonance (MR)-compatible optical physiological sensors including pulse oximetry, respiratory rate and body temperature. Anesthesia was maintained with 1.5–2% isoflurane with O2 gas mixture. MR images were acquired using a superconducting 9.4 T/210 horizontal bore magnet (Bruker BioSpin, AVANCE, MA, USA) with gradient strength of 950 mT/m equipped with an actively shielded 11.6-cm gradient set capable of providing 12 G/cm (Bruker). A birdcage coil (inner diameter: 72 mm) was used to transmit and a 30-mm diameter custom-made surface radio-frequency coil was used to receive the MR signal. Images were acquired in the axial plane using a T2-weighted (T2W) rapid acquisition refocusing echoes (RARE) spin echo sequence. The acquisition parameters were: repetition time = 2500 ms; echo time = 9.8 ms; slice thickness = 0.7 mm; slice gap = 0.1 mm; matrix size = 256 × 256; field of view = 3.00 cm; number of averages = 4; number of slices = 29; RARE factor = 8; spatial resolution = 0.117 mm; and total experiment time = 5 min 20 s.
Structural MRI analysis
The area of infarction and cross-sectional area of both hemispheres were measured using NIH Image routines from serial MRI slices covering the whole forebrain, summed and multiplied by the slice thickness and gap to provide a volume estimate. Percentage infarction was calculated as 100 × (infarct volume/volume of contralateral hemisphere). Percentage edema was calculated as 100 × ([ipsilateral hemisphere volume − contralateral hemisphere volume]/contralateral hemisphere volume). Atrophy (tissue loss) was calculated as percentage atrophy = 100 × ([contralateral hemisphere volume − ipsilateral hemisphere volume]/contralateral hemisphere volume).
Drug treatment
d-cycloserine (Sigma Aldrich Co., MO, USA; 10 mg/ml) was freshly prepared in PBS (pH 7.4). A total of 24 h after surgery, following confirmation of infarction by T2W MRI, rats were randomized to treatment with DCS (10 mg/kg in volume 1 ml/kg; n = 16) or PBS vehicle (1 ml/kg) given ip. (n = 15).
Functional MRI
Functional MRI (fMRI) studies were performed 30 or more days after MCAO. Rats were orally intubated under 3% isoflurane and methohexital sodium (50 mg/kg ip.) anesthesia and given glycopyrrolate (0.04 mg/kg ip.). After intubation, rats were placed on mechanical ventilation (Harvard Apparatus, Inspira ASV Inc., MA, USA) under 1.5–2% isofluorane with a 1:1 air:O2 gas mixture throughout the experiment. The femoral vein and artery were catheterized for α-chloralose and fluid administration, arterial blood pressure monitoring and collection of blood gas measurements during the study. All rats were maintained within the normal range of blood gas values and pH (pCO2 = 37 ± 3 mmHg; pO2 = 150 ± 20 mmHg; pH = 7.40 ± 0.05; cBaseEcf = −0.7 ± 1.8 mmol/l [33]. Intravenous bicarbonate was given when needed to correct for metabolic acidosis. Body temperature was measured with a rectal probe and maintained at approximately 37°C with a heating pad. All vital signs were continuously monitored during the experiment (SAM PC monitor™, SA Instruments, Inc., NY, USA). Rats were secured in a custommade stereotaxic head holder. Needle electrodes for electrical stimulation were inserted under the skin of each forepaw, between digits 2 and 3 and between 4 and 5 and the anesthetic regimen was switched from isoflurane to α-chloralose (Sigma Aldrich). α-chloralose was administered first as an intravenous bolus of 40 mg/kg over a 10-15 min time period followed by a continuous infusion of 25 mg/kg/h. Two to three trials of forepaw stimulation were administered to ensure an adequate depth of anesthesia (i.e., if the forepaw stimulation did not induce changes in mean arterial blood pressure, heart rate or the respiratory waveforms, the anesthetic depth was considered adequate).
A single-shot echo planar imaging spin-echo sequence was used for fMRI. The preprocessing setup included localized shimming using a fastmap sequence and adjustments to echo spacing. Customized pre-emphasis settings were used to correct for the Eddy current distorted gradient ramp in the readout direction. The following parameters were used: repetition time = 1500 ms; echo time = 30 ms; effective bandwidth = 227,272 Hz; field of view = 2.56 × 2.56 cm2; matrix = 64 × 64 with a resulting in-plane resolution of 400 μm; number of axial slices = 8; slice thickness = 1.4 mm; slice gap = 0.15 mm; and scan time = 3 s. To identify the anatomical location of the functional activation maps, higher-resolution T2W spin-echo images were also acquired with identical spatial geometry using a RARE pulse sequence.
Somatosensory stimulation was performed as previously described [34]. Briefly, pulse train forepaw stimulations were generated using a current stimulator (Isolated Pulse Stimulator 2100™, A-M Systems, WA, USA). Negative short rectangular current pulses of 0.3 ms duration were applied at a frequency of 3 Hz and a 2 mA current to either right or left forepaw. The paradigm consisted of 23 scans acquired during rest, ten scans acquired during stimulation (total stimulation time = 30 s) and followed by a poststimulation rest period of 30 scans (total acquisition time = 63 × 3 s = 3.15 min) [34]. Animals were allowed to rest for 5 min between stimulations. Complete datasets were obtained from 14 MCAO rats (seven treated with vehicle and seven treated with DCS) and 13 nonischemic controls.
fMRI data analysis
For fMRI data ana lysis we used STIMULATE V6.01 (Center for Magnetic Resonance Research [CMRR], University of Minnesota, Minneapolis, MN, USA) software. After background masking, the intensity time-course of each pixel during the scan was cross-correlated with a boxcar template according to the known stimulation profile. Correlation coefficients (r) of ≥0.3, corresponding to a p-value of 0.01, were used as thresholds to calculate the activation maps and detect the areas with a statistically significant blood oxygen level-dependent (BOLD) signal. A four-neighbor 2D cluster clustering was performed to remove scattered false activation. Two to four scans from each animal were analyzed. Time courses from all activated pixels in the somatosensory region were recorded. Both the number of pixels (nominally, each pixel accounts for a volume of 0.4 × 0.4 × 1.5 = 0.24 mm3) and the average time course of the statistically significantly active pixels (pixels with r ≥0.3) within the region of interest were extracted for each trial, and used to represent the spatial and temporal profile of the BOLD response, respectively. Each time course was then normalized to its mean value during the prestimulation baseline and the peak value was extracted to assess the response magnitude for each stimulation trial. All calculated and color-coded functional activity maps were overlaid on echo-planar images and anatomical locations of activated pixels were identified by comparing with corresponding high-resolution T2W MRIs.
Novel object recognition test
Memory function was assessed in nonischemic controls (n = 9) MCAO plus PBS (n = 15) and MCAO plus DCS (n = 16) rats using published methodology [35,36]. Briefly, on day 1, habituation was performed by placing each rat in a separate testing cage (a plastic box of 72 × 47 × 34 cm) for 1 h. On the following day (familiarization), rats were placed in the same cage with two identical objects. The cumulative time spent by the rat exploring the two objects was recorded during a 5-min interval. A total of 4 h later, the animals were reintroduced into the same cage for a 5-min test, where one of the two identical objects was replaced by a novel object. To offset location bias, the novel object was placed at the location of the old object, which the rat spent less time exploring (less than 45%) during the familiarization phase. The time (out of the 5-min total) spent exploring each of the objects was recorded. The outcome measure was percentage of time spent exploring the novel object during the testing phase, whereby normal healthy rodents will spend relatively more time exploring a novel object than a familiar (i.e., ‘memorized’) object. The proportion of time spent exploring the novel object during the testing phase was compared with the proportion of time spent exploring the object at the same location during the familiarization phase.
The test was administered before surgery and again at 7, 21 and 30 days after MCAO, using new pairs of objects for each time point.
Statistical analysis
The effects of surgery and drug treatment on object recognition task performance were analyzed by two-way ana lysis of variance (ANOVA) with repeated measures, with time as the repeated measure. fMRI data were analyzed by three-way ANOVA (by surgery, drug treatment and side). Significant ANOVAs were followed by a post hoc multiple comparisons test (Fisher’s protected least significant difference) using StatView software. p-values less than 0.05 were considered significant.
Results
Stroke induction
Stroke was induced in 36 animals subjected to transient (90-min) occlusion of the MCA, five of which died within the first 24 h after surgery.
Stroke validation: MRI & neurological examination
T2-weighted MRI performed 24 h after MCAO demonstrated unilateral hyperintensity in the MCA territory involving the striatum and some adjacent cortical regions. Cortical hyperintensity was noted most frequently in the parietal cortex and piriform cortex (Figure 1a). At this time point, most of the brains also showed edema, evidenced by a pronounced midline shift (Figure 1a). The MCAO procedure also resulted in impairment of placing of the contralateral (right) forelimb in all animals.
Figure 1. MRI confirmation of infarction and edema 24 h after middle cerebral artery occlusion.
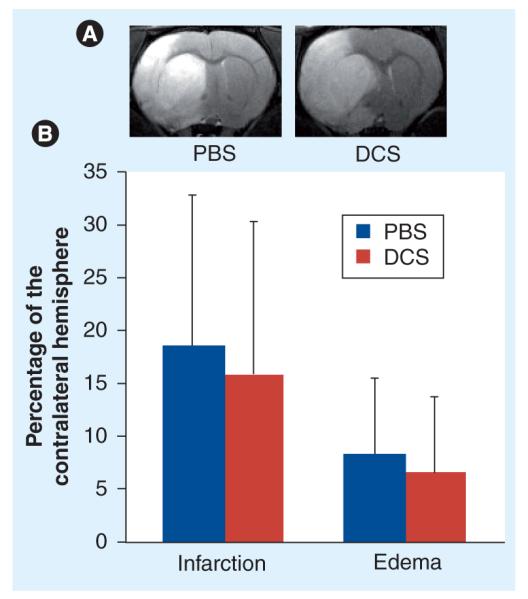
(A) Pretreatment infarction and edema in two animals later randomized to PBS or DCS treatment. Note the high signal intensity in these T2-weighted images, delineating infarction in the striatum and overlying cortex, and the midline shift and swelling of the infarcted hemisphere characteristic of edema. (B) Bars represent means and standard errors of infarction and edema expressed as percentage of the contralateral hemisphere. The area of infarction and cross-sectional area of both hemispheres were measured using NIH Image routines from serial MRI slices covering the whole forebrain, summed and multiplied by the slice thickness and gap to provide a volume estimate. Percentage infarction was calculated as 100 × (infarct volume/volume of contralateral hemisphere). Percentage edema was calculated as 100 × ([ipsilateral hemisphere volume − contralateral hemisphere volume]/contralateral hemisphere volume). There was no statistically significant difference between the animals randomized to receive vehicle (n = 7) or DCS (n = 7) in percentage infarction or percentage edema.
DCS: d-cycloserine; PBS: Phosphate-buffered saline.
Rats subjected to sham surgery and intact rats had no MRI findings or neurological deficits.
Animals randomized to receive PBS or DCS had similar pretreatment infarction and edema volumes (Figure 1b).
Somatosensory function
Figure 2 shows the BOLD signal changes in response to forepaw stimulation in nonischemic controls and MCAO animals treated with vehicle (MCAO + PBS) or DCS (MCAO + DCS) imaged 30 days or more after the surgery. The drug and vehicle were administered once only, 24 h after MCAO. As expected, nonischemic controls showed similar somatosensory activation in both hemispheres (top row of Figure 2), while transient focal ischemia followed by vehicle treatment resulted in a significant attenuation of the BOLD response in the somatosensory cortex ipsilateral to the lesion (Figure 2), which, with the threshold used in our experiment (r >0.3), was absent in the majority of the animals in this group (6/7). Ipsilateral activation deficits were evident in MCAO plus PBS animals with large infarcts (striatal + cortical infarcts; second row of Figure 2), as well as small infarcts (striatal infarct only; third row of Figure 2). Animals treated with DCS demonstrated remarkable preservation of brain activation in the ipsilateral hemisphere, which was present in six out of seven MCA plus DCS rats (Figure 2). Ipsilateral activation was evident even in animals with very large infarcts involving the primary somatosensory cortex, although in such animals, the BOLD signal change was shifted towards the secondary somatosensory cortex or the cingulate (fourth row of Figure 2). The mean amplitude of the BOLD signal changed to a similar extent in the right and left somatosensory cortices of nonischemic control rats in response to forepaw stimulation (5.9 ± 0.85% and 5.8 ± 0.87%, respectively) (Figure 3). The BOLD signal change was significantly reduced in the ipsilateral hemisphere of MCAO plus PBS rats (1.2 ± 0.9%; p < 0.001 relative to controls), but was in the control range in the contralateral hemisphere (6.5 ± 1.99%) (Figure 3).
Figure 2. Effect of ischemia and d-cycloserine on blood oxygen level-dependent activation maps 30 days after middle cerebral artery occlusion.
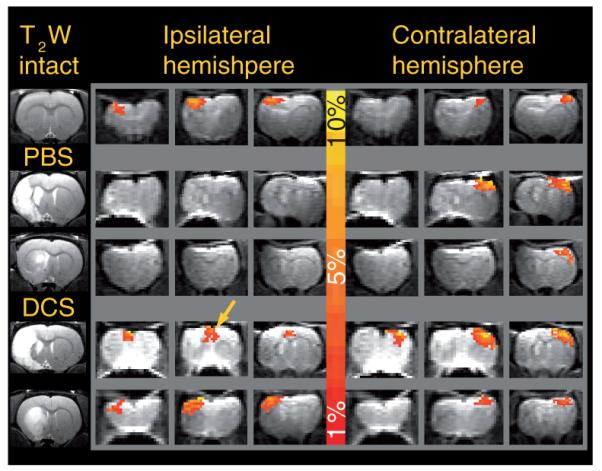
Structural T2-weighted coronal images at the level of maximal infarction are shown in the leftmost column, with a nonischemic control in the top row (intact), middle cerebral artery occlusion (MCAO) plus PBS (small and large infarct) in the middle rows and MCAO plus DCS (small and large infarct) in the bottom rows. The color bar corresponds to a positive blood oxygen level-dependent signal increase from 1% (red) to 10% (yellow). Serial coronal echo planar imaging images from the ipsilateral and contralateral hemisphere are shown to the left and right of the color bar, respectively. Note the symmetrical blood oxygen level-dependent signal intensity change produced in the nonischemic control, the absence of ipsilateral activation in the MCAO plus PBS animals and the presence of activation in the somatosensory cortex in a DCS-treated animal with a striatal infarct. Activation shifted towards the midline can be seen in the MCAO plus DCS animals with a large cortical infarct (arrow).
DCS: d-cycloserine; PBS: Phosphate-buffered saline; T2W: T2-weighted.
Figure 3. Effect of ischemia and d-cycloserine on percentage change in blood oxygen level-dependent signal amplitude 30 days after middle cerebral artery occlusion.
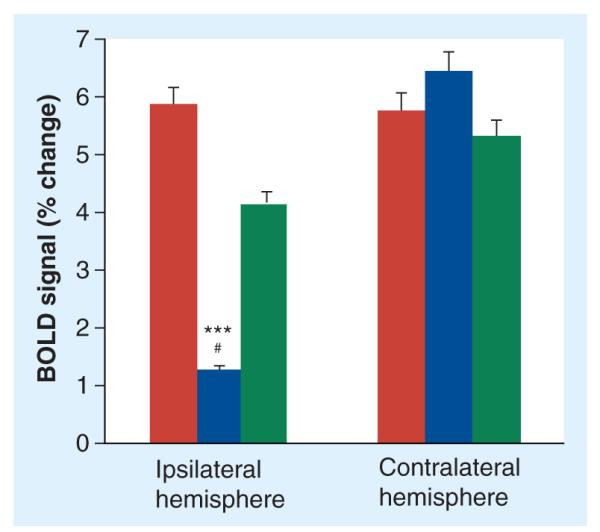
Bars represent means and standard errors of the percentage change in BOLD signal amplitude following right and left forepaw stimulation in nonischemic controls (n = 13, red bars), middle cerebral artery occlusion (MCAO) plus PBS (n = 7, blue bars) and MCAO plus DCS (n = 7, green bars). A three-way analysis of variance of percentage change in the amplitude of the BOLD signal revealed significant effects of surgery, side and drug treatment (p < 0.05). Post hoc ana lysis of the differences between individual groups and sides revealed significance for MCAO plus PBS compared with nonischemic controls in the ipsilateral hemisphere (***p < 0.001) and a trend for MCAO plus PBS compared with MCAO plus DCS in the ipsilateral hemisphere (#p = 0.08). The MCAO plus DCS and nonischemic controls were not significantly different from each other (p > 0.2) in the ipsilateral hemisphere. There were no effects of ischemia or drug treatment on the BOLD signal intensity in the contralateral hemisphere.
BOLD: Blood oxygen level-dependent; DCS: d-cycloserine; PBS: Phosphate-buffered saline.
The mean amplitude of the BOLD signal change in MCAO plus DCS rats was largely preserved and not significantly different from controls (4.2 ± 1.2% in the ipsilateral and 5.4 ± 1.26% in the contralateral somatosensory cortex). A three-way ANOVA (surgery, side and treatment) of percentage change in the amplitude of the BOLD signal revealed a significant effect of treatment (p < 0.05). The DCS and nonischemic control group were not significantly different from each other (p > 0.2) following post hoc ana lysis. There were no treatment effects on the BOLD signal intensity in the somatosensory cortex contralateral to the infarct.
Cognitive (memory) function
Animals tested in the object recognition task before surgery and randomization to treatment spent an increased proportion of their exploration time exploring a novel object (57.03 ± 4.30%) relative to a familiar object in the same location during a 5-min testing period. MCAO animals receiving PBS showed a persistent deficit in this task, with no preference for the novel object at 7, 21 and 30 days after surgery (36.21 ± 4.60%, 40.76 ± 3.87% and 33.01 ± 6.34%, respectively) (Figure 4), which was not significantly different from the percentage of time spent at the same location during the familiarization session. Nonischemic controls performed consistently at all time points, showing a similar preference for the novel object upon repeated testing with new object pairs (Figure 4). MCAO plus DCS rats demonstrated an apparent deficit on day 7, with progressive improvement at later time points, such that by day 30, they were similar to the nonischemic control group (Figure 4). Two-way ANOVA with repeated measures showed a significant difference between the MCAO plus PBS group and the non-ischemic control group, as well as the MCAO plus DCS group (p < 0.0001 and p < 0.05, respectively). The MCAO plus DCS and nonischemic control groups were not significantly different from each other at any time point.
Figure 4. Effect of d-cycloserine on novel object recognition task performance after middle cerebral artery occlusion.

Bars represent means and standard errors of the percentage of time spent by rats investigating a new object in the presence of a familiar one during a 5-min testing period. Rats were tested on day 0 (presurgery), and at 7, 21 and 30 days post-middle cerebral artery occlusion (MCAO) with new pairs of objects on each day. Note the initial (presurgical) adequate performance (preference of novel object) in all three groups, the consistent performance of the intact/sham group, the persistent deficit in the MCAO plus PBS rats and the gradual improvement to presurgical performance levels of the MCAO plus DCS group. Two-way analysis of variance with repeated measures revealed significant main effects for surgery and drug and a significant drug by time (repeated measure) interaction. Post hoc comparison of the individual groups and days revealed significance for MCAO plus PBS compared with nonischemic controls (***p < 0.0001), significance for MCAO plus PBS compared with nonischemic controls (**p < 0.001) and significance for MCAO plus PBS compared with MCAO plus DCS (*p < 0.05). The DCS and intact groups were not significantly different from each other at any time point.
DCS: d-cycloserine; PBS: Phosphate-buffered saline.
Atrophy
As expected, MCAO animals exhibited ventricular enlargement and tissue loss in the ipsilateral hemisphere. DCS treatment did not have a significant effect on late infarct size or tissue loss, measured from structural MRI scans performed 30 days or more after the surgery in six animal/groups (Figure 5).
Figure 5. No effect of d-cycloserine on long-term infarction and atrophy after middle cerebral artery occlusion.
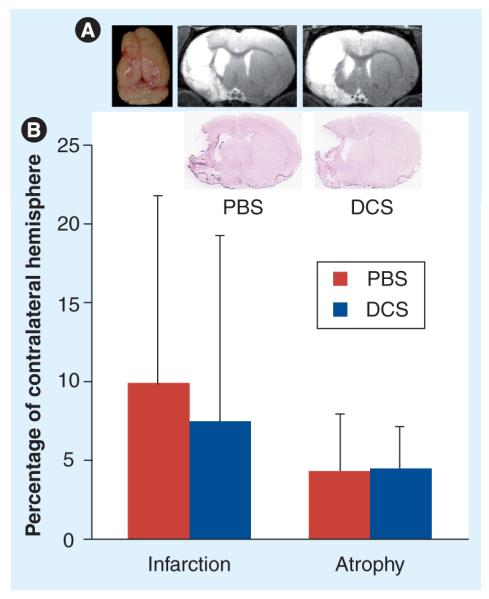
(A) Long-term tissue loss and infarction. From left, photograph (dorsal view) of a middle cerebral artery occlusion (MCAO) plus PBS brain showing shrinkage of the ipsilateral hemisphere, followed by MRI images of coronal slices and matching histological sections showing infarction, ipsilateral ventricular enlargement and tissue loss in the ipsilateral hemisphere in a MCAO plus PBS- and MCAO plus DCS-treated rat. (B) Results of a morphometric ana lysis of infarct volume and tissue loss perfomed on T2-weighted MRI images from six MCAO plus PBS and six MCAO plus DCS rats, with percentage infarction = 100 × (infarct volume/contralateral hemisphere volume) and percentage atrophy = 100 × ([contralateral hemisphere − ipsilateral hemisphere]/contralateral hemisphere). Bars represent means and standard errors of the infarction and atrophy expressed as percentage of contralateral hemisphere.
DCS: d-cycloserine; PBS: Phosphate-buffered saline.
Discussion
Using a common model of transient (90-min) focal ischemia that results in reliable infarction but minimal mortality in rats [37-39], we show here that stimulation of NMDARs with the partial agonist DCS improves long-term functional outcome. The functional improvement was not due to a reduction in infarction or tissue loss, since pretreatment infarct volume and subsequent tissue loss were similar in DCS- and vehicle-treated animals.
A total of 24 h after MCAO, prior to randomization, all rats included in the study showed signs of infarction and edema in the ipsilateral hemisphere, expressed as an increased signal intensity in the T2W MR images and increased volume of the ipsilateral hemisphere. As expected in this model as well as in human stroke, the extent of infarction and edema was quite variable, ranging from small, exclusively striatal infarcts to large infarcts with significant cortical involvement [40,41]. However, quantitative assessment of the infarction and edema confirmed that the drug- and vehicle-treatment groups were well matched for these two parameters.
Electrical forepaw stimulation resulted in a significant activation of the somatosensory cortex contralateral to the stimulated paw in intact and sham-operated animals, as previously demonstrated [34,42]. MCAO drastically reduced activation in the infarcted hemisphere, even when the infarct was restricted to the striatum, so that the functionally compromised region extended beyond the area of infarction, as previously reported in the literature [43]. Treatment with DCS preserved activation of the ipsilateral somatosensory cortex in animals with striatal and cortical infarcts not involving the primary somatosensory cortex. In animals with infarcts involving the somatosensory cortex treated with DCS, the activation shifted either towards the secondary somatosensory cortex or towards the cingulate, as reported in MCAO animals with spontaneous recovery [44,45].
d-cycloserine treatment had an additional beneficial effect on cognitive outcome, which has been previously demonstrated in rodent models of traumatic brain injury [46,47]. Using the novel object recognition task [35], we were able to test the animals several times (days 7, 21 and 30 after stroke), allowing an insight into the time course of the treatment effect. While the nonischemic control rats exhibited significant preference for the novel object at all time points and the vehicle-treated ischemic rats showed a stable deficit, DCS-treated rats had a reduced preference for the novel object, indicative of a memory deficit, in the earliest test (7 days post-MCAO) which improved gradually over time. A total of 30 days after ischemia, DCS-treated animals were no longer different from nonischemic controls. This progressive functional improvement following a single delayed administration of DCS was similar to the one we observed in head injured mice given a single NMDA or DCS injection 24 h after injury [27,47].
Measurement of the infarct and hemispheric volumes of T2W images obtained just prior to termination revealed no differences between the groups in the size of the infarct or the extent of tissue loss, although absolute infarct volume was reduced over time [48]. This finding suggests that the mechanisms underlying the effects of DCS are not related to prevention of early neuronal death and infarction, but rather to stimulation of neuronal plasticity and reorganization, which are thought to be the major mechanisms underlying spontaneous recovery from stroke [44,49,50]. A similar dissociation between changes in infarct size and functional outcome has been noted in the past in humans, as well as experimental animals [51,52].
Partial or complete spontaneous resolution of neurological deficits is well documented in the first few weeks or months after stroke [53], although the extent and rate of recovery can vary greatly, even among patients with identical clinical severity in the acute phase. While various explanations for these variations have been proposed, including reabsorption of perilesional edema and variability in the perfusion territory, it is clear that neural plasticity plays a major role in the recovery process [44,49,50] and is the likely target of DCS.
To elaborate, full and partial agonists of NMDAR, including NMDA, DCS and the naturally occurring amino acid d-serine, promote cell migration during development and improve memory function in adult animals [27,46,54,55]. DCS was also demonstrated to promote neuroplasticity in humans [56]. Recently, our group has shown that a single administration of 10 mg/kg DCS 24 h after closed head injury improved memory function as well as neurological deficits in mice, restored long-term potentiation and increased levels of brain-derived neurotrophic factor. Furthermore, we showed the beneficial effects of DCS were blocked by coadministration of the NMDAR antagonist MK801 and that there was no additional benefit derived from multiple administrations [47,57].
To our knowledge, this is the first study demonstrating beneficial effects of NMDAR stimulation in focal ischemic stroke. From the theoretical standpoint, these results support a re-evaluation of the role of glutamate in stroke pathology, emphasizing the importance of considering the dynamic nature of changes in glutamate transmission after stroke and trauma [27,28] in the selection of appropriate treatment. From the clinical standpoint, the well-established safety profile of DCS in humans [56,58,59] and the fact that it is already approved for human use as an antimicrobial agent [60] may facilitate the translation of these findings to the clinical domain, although further experiments are needed to establish the optimal dose and optimal dosing regimen for DCS in stroke.
Conclusion
Our results support a beneficial role for NMDAR stimulation during the recovery period after stroke, most likely due to enhanced neuroplasticity rather than neuroprotection.
Future perspective
Despite decades of intensive preclinical research and the identification of several promising interventions in rodents, treatment of human stroke, a major cause of chronic disability in the developed world, is still an elusive target. The short therapeutic window and possibility of complications with thrombolytic agents – the only drug intervention currently approved for stroke – effectively exclude large numbers of stroke victims who do not meet the criteria for such treatment. However, all pivotal clinical trials of drugs acting on brain targets have failed to date. Critical analysis of the many successful preclinical studies that led to resounding failures in large, controlled clinical stroke trials highlights the overwhelming use of populations (young males), treatment windows (immediately before or 15–30 min after ischemia), end points (infarct volume) and follow-up periods (24–48 h) that are grossly at odds with the clinical trial situation, as the majority of stroke victims are old and female, cannot be treated within less than 6–12 h at best and are expected to show improvements in functional, rather then radiological, outcomes, which will last at least 3 months. The studies described earlier demonstrate a beneficial effect of DCS in female rats, using a clinically relevant (24-h) therapeutic window, clinically relevant functional assessment tools and long follow-up (>30 days). Combined with the fact that DCS is already approved for human use in other indications, these findings are supportive of facile and relatively fast clinical development of DCS or other glutamatergic agonists for stroke treatment in the near future.
Executive summary.
Glutamate levels & ischemia
-
■
Focal ischemia (stroke) is defined as an interruption of blood supply in a cerebral artery, resulting in oxygen and glucose deprivation of the affected vessel territory.
-
■
Ischemia is followed within minutes by a large, although transient, increase in excitatory amino acid (glutamate and aspartate) transmitter efflux.
-
■
A transient increase in extracellular levels of glutamate was confirmed by microdialysis in animal models, as well as in human stroke and traumatic brain injury.
-
■
Brain infarction in the affected vessel territory is evident 24-48 h after ischemia.
-
■
Spontaneous partial recovery of sensory, motor and cognitive function is common among stroke victims, although many suffer from permanent deficits, some of which indicate dysfunction of brain regions that are remote from the ischemic core (diaschisis).
Glutamate & excitotoxicity
-
■
Exposure of neuronal cultures to high concentrations of glutamate or NMDA results in a dose- and time-dependent neuronal loss, referred to as excitotoxic neuronal death.
-
■
Excitotoxicity can be blocked by competitive and noncompetitive NMDA antagonists, as well as by indirect (glycine site) antagonists.
-
■
Excitotoxicity was accepted as the major mediator of infarction and neurological deficits in ischemic and traumatic brain injury.
Glutamate (NMDA) antagonists in animal models of stroke
-
■
A large number of competitive, noncompetitive and glycine site NMDA antagonists were examined in animal models of stroke in several species. The majority of these experiments reduced the size of infarction 24-48 h after ischemia, the most common end point employed.
-
■
A common characteristic of the various NMDA antagonists employed in animal models was a limited therapeutic window, whereby significantly improved outcome was achieved only when the drugs were administered either prior to the onset of ischemia or shortly (less than 1 h) after ischemia.
Glutamate (NMDA) antagonists in clinical trials of stroke
-
■
Several competitive, noncompetitive and glycine site NMDA antagonists were examined in randomized, double-blind, placebo-controlled clinical trials.
-
■
All NMDA antagonists employed in clinical trials failed to produce significant improvements in neurological function (measured by common scales such as the Rankin index, NIH Stroke Scale or Barthel index) and mortality.
-
■
In a few cases, mortality and neurological deficits were actually increased in the drug arm of the trials, which had to be abandoned upon the recommendation of the safety review board.
-
■
Importantly, there was no overlap between the clinical trials and the animal experiments in the time between the onset of stroke and drug administration. The drugs were invariably administered later, and often for a longer duration, in the clinical trials.
NMDA receptors & ischemia
-
■
Work from several groups has shown loss of functional NMDA receptors and NMDAR gene expression within a few hours of ischemia.
-
■
NMDA receptor loss is long lasting (at least 2 months) and observed in regions outside the infarct area, including the contralateral hemisphere, in a rat model of stroke.
-
■
The widespread and persistent loss of NMDA receptor function correlates well with the time course and nature of neurological and cognitive deficits in stroke and offers an explanation for the limited therapeutic window of NMDA antagonists.
NMDA agonists in stroke?
-
■
NMDA receptor activation is important in brain plasticity, learning and memory formation and neuronal survival.
-
■
Administration of the indirect (glycine site) agonist d-cycloserine (DCS) 24 h after stroke onset in rats resulted in long-term (>1 month) improvement of neurological and memory function, with no effect on infarct volume.
Future perspective
-
■
The well-established safety profile of DCS in humans supports facile development of this drug for the treatment of human ischemic stroke, although further experiments are needed to establish the dose and optimal dosing regimen for DCS in this indication.
Acknowledgements
The authors thank Y Mei for expert technical assistance.
Footnotes
Financial & competing interests disclosure
A Biegon is supported in part by NIH RO1NS050285. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate insti tutional review board approval or have followed the princi ples outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investi gations involving human subjects, informed consent has been obtained from the participants involved.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■ of considerable interest
- 1.Carandang R, Seshadri S, Beiser A. Trends in incidence, lifetime risk, severity, and 30-day mortality of stroke over the past 50 years. JAMA. 2006;296:2939–2946. doi: 10.1001/jama.296.24.2939. [DOI] [PubMed] [Google Scholar]
- 2.Rosamond W, Flegal K, Furie K, et al. Heart disease and stroke statistics 2008 update a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:E25–E146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 3.Wang KM, Kyoung KP, Yong SK, et al. Atherothrombotic middle cerebral artery territory infarction: topographic diversity with common occurrence of concomitant small cortical and subcortical infarcts. Stroke. 2000;31:2055–2061. doi: 10.1161/01.str.31.9.2055. [DOI] [PubMed] [Google Scholar]
- 4.Caplan L, Schmahmann J, Kase C, et al. Caudate infarcts. Arch. Neurol. 1990;47:133–143. doi: 10.1001/archneur.1990.00530020029011. [DOI] [PubMed] [Google Scholar]
- 5.Phipps L. Assessment of neurologic deficits in stroke: acute-care and rehabilitation implications. Nurs. Clin. North Am. 1991;26:957–970. [PubMed] [Google Scholar]
- 6.Albers GW, Clark WM, Madden KP, Hamilton SA. ATLANTIS trial: results for patients treated within 3 hours of stroke onset. Alteplase Thrombolysis for Acute Noninterventional Therapy in Ischemic Stroke (ATLANTIS) Stroke. 2002;33:493–496. doi: 10.1161/hs0202.102599. [DOI] [PubMed] [Google Scholar]
- 7.Mitka M. Tensions remain over tPA for stroke. JAMA. 2003;289:1363–1364. doi: 10.1001/jama.289.11.1363. [DOI] [PubMed] [Google Scholar]
- 8.Novak BL, Force RW. Detriments of tPA for acute stroke in routine clinical practice. J. Fam. Pract. 2003;52(2):95–96. [PubMed] [Google Scholar]
- 9.Hacke W, Kaste M, Bluhmki E. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 10.Wahlgren N, Ahmed N, Davalos A. Thrombolysis with alteplase 3 to 4.5 hours after acute ischaemic stroke (SITS-ISTR): an observational study. Lancet. 2008;372:1303–1309. doi: 10.1016/S0140-6736(08)61339-2. [DOI] [PubMed] [Google Scholar]
- 11.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginsberg MD. Current status of neuroprotection for cerebral ischemia: synoptic overview. Stroke. 2009;40:S111–S114. doi: 10.1161/STROKEAHA.108.528877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 1984;43:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. □ First article to document the increased efflux of excitatory amino acids induced by brain ischemia.
- 14.Davalos A, Castillo J, Serena J, Noya M. Duration of glutamate release after acute ischemic stroke. Stroke. 1997;28:708–710. doi: 10.1161/01.str.28.4.708. [DOI] [PubMed] [Google Scholar]
- 15.Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 16.Obrenovitch TP, Urenjak J, Zilkha E, Jay TM. Excitotoxicity in neurological disorders - the glutamate paradox. Int. J. Dev. Neurosci. 2000;18:281–287. doi: 10.1016/s0736-5748(99)00096-9. [DOI] [PubMed] [Google Scholar]
- 17.Rod MR, Auer RN. Pre- and post-ischemic administration of dizocilpine (MK-801) reduces cerebral necrosis in the rat. Can. J. Neurol. Sci. 1989;16:340–344. doi: 10.1017/s031716710002919x. □ Early demonstration of neuroprotective action of NMDA receptor (NMDAR) antagonist administration before or soon after brain ischemia.
- 18.Chen M, Bullock R, Graham DI, Frey P, Lowe D, McCulloch J. Evaluation of a competitive NMDA antagonist (d-CPPene) in feline focal cerebral ischemia. Ann. Neurol. 1991;30:62–70. doi: 10.1002/ana.410300112. [DOI] [PubMed] [Google Scholar]
- 19.Lees KR. Cerestat and other NMDA antagonists in ischemic stroke. Neurology. 1997;49:S66–S69. doi: 10.1212/wnl.49.5_suppl_4.s66. [DOI] [PubMed] [Google Scholar]
- 20.Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M, GAIN International Investigators Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomized controlled trial. Lancet. 2000;355:1949–1954. doi: 10.1016/s0140-6736(00)02326-6. [DOI] [PubMed] [Google Scholar]
- 21.Sacco RL, DeRosa JT, Haley EC, Jr, Levin B, Ordronneau P, Phillips SJ. Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA. 2001;28:1719–1728. doi: 10.1001/jama.285.13.1719. [DOI] [PubMed] [Google Scholar]
- 22.Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Curr. Mol. Med. 2004;4(2):131–136. doi: 10.2174/1566524043479248. □□ Critical ana lysis of the results of clinical trials employing NMDAR antagonists in stroke.
- 23.Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. □ Demonstration of the role of NMDAR activation in learning and memory formation.
- 24.Kossel AH, Williams CV, Schweizer M, Kater SB. Afferent innervation influences the development of dendritic branches and spines via both activity-dependent and non-activity-dependent mechanisms. J. Neurosci. 1997;17:6314–6324. doi: 10.1523/JNEUROSCI.17-16-06314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albensi BC. The NMDA receptor/ion channel complex: a drug target for modulating synaptic plasticity and excitotoxicity. Curr. Pharm. Des. 2007;13(31):3185–3194. doi: 10.2174/138161207782341321. [DOI] [PubMed] [Google Scholar]
- 26.Obviagle B, Kidwell CS, Satrkama S, Saver JL. Neuroprotective agents for the treatment of acute ischemic stroke. Curr. Neurol. Neuroci. Rep. 2003;3:9–20. doi: 10.1007/s11910-003-0031-z. [DOI] [PubMed] [Google Scholar]
- 27.Biegon A, Fry PA, Padan CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-d-aspartate receptors after closed head injury in mice: implications for treatment of neurological and cognitive deficits. Proc. Natl Acad. Sci. USA. 2004;101(14):5117–5122. doi: 10.1073/pnas.0305741101. □ First detailed description of NMDAR dynamics after traumatic brain injury, also demonstrating beneficial effects of delayed activation with a full agonist (i.e., NMDA).
- 28.Dhawan J, Benveniste H, Nawrocky M, Smith SD, Biegon A. Transient focal ischemia results in persistent and widespread neuroinflammation and loss of glutamate NMDA receptors. Neuroimage. 2010;51:599–605. doi: 10.1016/j.neuroimage.2010.02.073. □ Demonstrates long-term loss of NMDAR following transient focal ischemia.
- 29.Friedman LK, Belayev L, Alfonso OF, Ginsberg MD. Distribution of glutamate and proenkephalin messenger RNAs following transient focal cerebral ischemia. Neuroscience. 2000;95(3):841–857. doi: 10.1016/s0306-4522(99)00452-2. [DOI] [PubMed] [Google Scholar]
- 30.Ogawa N, Haba K, Mizukawa K, Asanuma M, Hirata H, Mori A. Loss of N-methyl-d-aspartate (NMDA) receptor binding in rat hippocampal areas at the chronic stage after transient forebrain ischemia: histological and NMDA receptor binding studies. Neurochem. Res. 1991;16(5):519–524. doi: 10.1007/BF00974869. □ Early demonstration of postischemic loss of NMDAR.
- 31.Watson GB, Bolanowski MA, Baganoff MP, Deppeler CL, Lanthorn TH. d-cycloserine acts as a partial agonist at the glycine modulatory site of the NMDA receptor expressed Xenopus oocytes. Brain Res. 1990;510:158–160. doi: 10.1016/0006-8993(90)90745-w. [DOI] [PubMed] [Google Scholar]
- 32.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniotomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 33.Hyder F, Rothman DL, Mason GF, Rangarajan A, Behar KL, Shulman RG. Oxidative glucose metabolism in rat brain during single forepaw stimulation: a spatially localized 1H[13C] nuclear magnetic resonance study. J. Cereb. Blood Flow Metab. 1997;17:1040–1047. doi: 10.1097/00004647-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 34.Luo Z, Mei Y, Smith SD, et al. The effect of intravenous lidocane on brain activation during non-noxious and acute noxious stimulation of the forepaw: a functional magnetic resonance imaging study in the rat. Anesth. Analg. 2009;108(1):334–344. doi: 10.1213/ane.0b013e31818e0d34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- 36.Hirshler Y, Polat U, Biegon A. Intracranial electrode implantation produces regional neuroinflammation and memory deficits in rats. Exp. Neurol. 2010;222:42–45. doi: 10.1016/j.expneurol.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Memezawa H, Minamisawa H, Smith ML, Siesjo BK. Ischemic penumbra in a model of reversible middle cerebral artery occlusion in the rat. Exp. Brain Res. 1992;89:67–78. doi: 10.1007/BF00229002. [DOI] [PubMed] [Google Scholar]
- 38.Memezawa H, Smith ML, Siesjo BK. Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke. 1992;23:552–559. doi: 10.1161/01.str.23.4.552. [DOI] [PubMed] [Google Scholar]
- 39.Spratt NJ, Fernandez J, Chen M, et al. Modification of the method of thread manufacture improves stroke induction rate and reduces mortality after thread-occlusion of the middle cerebral artery in young or aged rats. J. Neurosci. Methods. 2006;155:285–290. doi: 10.1016/j.jneumeth.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 40.Haefelin TN, Kastrup A, de Crespigny A, et al. Serial MRI after transient focal cerebral ischemia in rats dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke. 2000;31:1965–1973. doi: 10.1161/01.str.31.8.1965. [DOI] [PubMed] [Google Scholar]
- 41.Weber R, Ramos-Cabrer P, Hoehn M. Present status of magnetic resonance imaging and spectroscopy in animal stroke models. J. Cereb. Blood Flow Metab. 2006;26:591–604. doi: 10.1038/sj.jcbfm.9600241. [DOI] [PubMed] [Google Scholar]
- 42.Keilholz SD, Silva AC, Raman M, Merkle H, Koretsky AP. Functional MRI of the rodent somatosensory pathway using multislice echo planar imaging. Magn. Reson. Med. 2004;52:89–99. doi: 10.1002/mrm.20114. [DOI] [PubMed] [Google Scholar]
- 43.Weber R, Ramos-Cabrer P, Justicia C, et al. Early prediction of functional recovery after experimental stroke: functional magnetic resonance imaging, electrophysiology, and behavioral testing in rats. J. Neurosci. 2008;28(5):1022–1029. doi: 10.1523/JNEUROSCI.4147-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dijkhuizen RM, Singhal AB, Mandeville JB, et al. Correlation between brain reorganization, ischemic damage, and neurologic status after transient focal cerebral ischemia in rats: a functional magnetic resonance imaging study. J. Neurosci. 2003;23(2):510–517. doi: 10.1523/JNEUROSCI.23-02-00510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fishell G, Goldman JE. A silver lining to stroke: does ischemia generate new cortical interneurons? Nat. Neurosci. 2010;13(2):145–148. doi: 10.1038/nn0210-145. [DOI] [PubMed] [Google Scholar]
- 46.Temple MD, Hamm RJ. Chronic, post-injury administration of d-cycloserine, an NMDA partial agonist, enhances cognitive performance following experimental brain injury. Brain Res. 1996;741:246–251. doi: 10.1016/s0006-8993(96)00940-7. □ First demonstration of beneficial effects of d-cycloserine in experimental brain injury.
- 47.Yaka R, Biegon A, Grigoriadis N, et al. d-cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury. FASEB J. 2007;21:2033–2041. doi: 10.1096/fj.06-7856com. [DOI] [PubMed] [Google Scholar]
- 48.Henrich-Noack P, Baldauf K, Reiser G, Reymann KG. Pattern of time-dependent reduction of histologically determined infarct volume after focal ischaemia in mice. Neurosci. Lett. 2008;432:141–145. doi: 10.1016/j.neulet.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 49.Rossini PM. Brain redundancy: responsivity or plasticity? Ann. Neurol. 2001;48:128–129. [PubMed] [Google Scholar]
- 50.Rossini PM, Calautti C, Pauri F, Baron JC. Post-stroke plastic reorganization in the brain. Lancet Neurol. 2003;2:493–502. doi: 10.1016/s1474-4422(03)00485-x. [DOI] [PubMed] [Google Scholar]
- 51.Ji S, Kronenberg G, Balkaya M, et al. Acute neuroprotection by pioglitazone after mild brain ischemia without effect on long-term outcome. Exp. Neurol. 2009;216:321–328. doi: 10.1016/j.expneurol.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Mark VW, Taub E, Perkins C, Gauthier L, Uswatte G. MRI infarction load and CI therapy outcomes for chronic post-stroke hemiparesis. Restor. Neurol. Neurosci. 2008;26:13–33. [PubMed] [Google Scholar]
- 53.Twitchell TE. The restoration of motor function following hemiplegia in man. Brain. 1951;74:443–480. doi: 10.1093/brain/74.4.443. [DOI] [PubMed] [Google Scholar]
- 54.Mothet JP, Rouaud E, Sinet PM, et al. A critical role for the glial-derived neuromodulator d-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell. 2006;5:267–274. doi: 10.1111/j.1474-9726.2006.00216.x. [DOI] [PubMed] [Google Scholar]
- 55.Kim PM. Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc. Natl Acad. Sci. USA. 2005;102:2105–2110. doi: 10.1073/pnas.0409723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nitsche MA, Jaussi W, Liebetanz D, Lang N, Tergau F, Paulus W. Consolidation of human motor cortical neuroplasticity by d-cycloserine. Neuropsychopharmacology. 2004;29:1573–1578. doi: 10.1038/sj.npp.1300517. [DOI] [PubMed] [Google Scholar]
- 57.Adeleye A, Shohami E, Nachman D, et al. d-cycloserine improves functional outcome after traumatic brain injury with wide therapeutic window. Eur. J. Pharmacol. 2010;629:25–30. doi: 10.1016/j.ejphar.2009.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laake K, Oeksengaard AR. d-cycloserine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2002;2:CD003153. doi: 10.1002/14651858.CD003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duncan EJ, Szilagyi S, Schwartz MP, et al. Effects of d-cycloserine on negative symptoms in schizophrenia. Schizophr. Res. 2004;1:239–248. doi: 10.1016/j.schres.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 60.Heifets LB. Antimycobacterial drugs. Semin. Respir. Infect. 1994;9:84–103. [PubMed] [Google Scholar]
- 61.Bielenberg GM, Beck T. The effects of dizocilpine (MK-801), phencyclidine, and nimodipine on infarct size 48 h after middle cerebral artery occlusion in the rat. Brain Res. 1991;552:338–342. doi: 10.1016/0006-8993(91)90101-z. [DOI] [PubMed] [Google Scholar]
- 62.Izumi Y, Roussel S, Pinard E, Seylaz J. Reduction of infarct volume by magnesium after middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 1991;11(6):1025–1030. doi: 10.1038/jcbfm.1991.170. [DOI] [PubMed] [Google Scholar]
- 63.Chen M, Bullock R, Graham DI, Frey P, Lowe D, McCulloch J. Evaluation of a competitive NMDA antagonist (d-CPPene) in feline focal cerebral ischemia. Ann. Neurol. 1991;30:62–70. doi: 10.1002/ana.410300112. [DOI] [PubMed] [Google Scholar]
- 64.Bullock R, Graham DI, Chen MH, Lowe D, McCulloch J. Focal cerebral ischemia in the cat: pretreatment with a competitive NMDA receptor antagonist, d-CPP-ene. J. Cereb. Blood Flow Metab. 1990;10(5):668–674. doi: 10.1038/jcbfm.1990.120. [DOI] [PubMed] [Google Scholar]
- 65.Dezsi L, Greenberg JH, Sladky J, Araki N, Hamar J, Reivich M. Prolonged effects of MK-801 in the cat during focal cerebral ischemia and recovery: Survival, EEG activity and histopathology. J. Neurol. Sci. 1994;121(1):110–120. doi: 10.1016/0022-510x(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 66.Yam PS, Dunn LT, Graham DI, Dewar D, McCulloch J. NMDA receptor blockade fails to alter axonal injury in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2000;20:772–779. doi: 10.1097/00004647-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Reggiani A, Pietra C, Arban R, et al. The neuroprotective activity of the glycine receptor antagonist GV150526: an in vivo study by magnetic resonance imaging. Eur. J. Pharm. 2001;419(2-3):147–153. doi: 10.1016/s0014-2999(01)00948-7. [DOI] [PubMed] [Google Scholar]
- 68.Ohtani K, Tanaka H, Ohno Y. SM-31900, a novel NMDA receptor glycine-binding site antagonist, reduces infarct volume induced by permanent middle cerebral artery occlusion in spontaneously hypertensive rats. Neurochem. Int. 2003;42(5):375–384. doi: 10.1016/s0197-0186(02)00137-7. [DOI] [PubMed] [Google Scholar]
- 69.Oda M, Kure S, Sugawara T, et al. Direct correlation between ischemic injury and extracellular glycine concentration in mice with genetically altered activities of the glycine cleavage multienzyme system. Stroke. 2007;38:2157–2164. doi: 10.1161/STROKEAHA.106.477026. [DOI] [PubMed] [Google Scholar]
- 70.Albers GW, Atkinson RP, Kelley RE, Rosenbaum DM. Safety, tolerability, and pharmacokinetics of the N-methyl-d-aspartate antagonist dextrorphan in patients with acute stroke. Stroke. 1995;26:254–258. doi: 10.1161/01.str.26.2.254. [DOI] [PubMed] [Google Scholar]
- 71.Dyker AG, Lees KR. Remacemide hydrochloride: a double-blind, placebo-controlled, safety and tolerability study in patients with acute ischemic stroke. Stroke. 1999;30:1796–1801. doi: 10.1161/01.str.30.9.1796. [DOI] [PubMed] [Google Scholar]
- 72.Davis SM, Lees KR, Albers GW, et al. Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke. 2000;31:347–354. doi: 10.1161/01.str.31.2.347. [DOI] [PubMed] [Google Scholar]
- 73.Albers GW, Goldstein LB, Hall D, Lesko LM. Aptiganel hydrochloride in acute ischemic stroke: a randomized controlled trial. JAMA. 2001;286:2673–2682. doi: 10.1001/jama.286.21.2673. [DOI] [PubMed] [Google Scholar]
- 74.Lees KR, Dyker AG, Sharma A, Ford GA, Ardron ME, Grosset DG. Tolerability of the low-affinity, use-dependent NMDA antagonist AR-R15896AR in stroke patients: a dose-ranging study. Stroke. 2001;32:466–472. doi: 10.1161/01.str.32.2.466. [DOI] [PubMed] [Google Scholar]
- 75.Haley EC, Jr, Thompson JL, Levin B, et al. GAIN Americas and GAIN International Investigators. Gavestinel does not improve outcome after acute intracerebral hemorrhage: an ana lysis from the GAIN International and GAIN Americas studies. Stroke. 2005;36(5):1006–1010. doi: 10.1161/01.STR.0000163053.77982.8d. [DOI] [PubMed] [Google Scholar]
- Website ASA – American Stroke Association [Accessed 13 June 2011]; www.strokeassociation.org.