

Abstract
EGFR serves as an important therapeutic target because of its over-expression in many cancers. In this study, we investigated a peptide-based therapy aimed at blocking intracellular protein-protein interactions during EGFR signaling and evaluated a targetable lipid carrier system that can deliver peptides to intracellular targets in human cancer cells. EEEEpYFELV (EV), a nonapeptide mimicking the Y845 site of EGFR which is responsible for STAT5b phosphorylation, was designed to block EGFR downstream signaling. EV was loaded onto LPH nanoparticles that are comprised of a membrane/core structure including a surface-grafted polyethylene glycol (PEG) used to evade the reticuloendothelial system (RES) and anisamide (AA) for targeting the sigma receptor over-expressed in H460 human lung cancer cells. EV formulated with PEGylated and targeted LPH (LPH-PEG-AA) was taken up by the tumor cells and trafficked to the cytoplasm with high efficiency. Using this approach, EV acted as a dominant negative inhibitor of STAT5b phosphorylation, arrested cell proliferation, and induced massive apoptosis. Intravenous administration of EV loaded in LPH-PEG-AA led to efficient EV peptide delivery to the tumor in a xenograft mouse model, and multiple injections inhibited tumor growth in a dose-dependent manner. Our findings offer proof-of-concept for an intracellular peptide-mediated cancer therapy that is delivered by carefully designed nanoparticles.
Keywords: liposome, nanoparticle, intracellular peptide, drug delivery, PEGylation
Introduction
Most biopharmaceutical drugs act by binding to a protein target, resulting in a change in the protein’s behavior. Protein interactions crucially mediate many signaling pathways controlling cellular proliferation and apoptosis; blocking such interactions in an intracellular compartment holds great therapeutic potential for the development of anti-cancer drugs [1, 2]. Small molecules, however, have difficulty targeting these interactions because of the nature of the protein interface, which is normally extensive, shallow and hydrophobic [3]. Moreover, antibody therapeutics, albeit popular candidates because of advantages including high affinity and specificity, also demonstrate serious disadvantages, such as unfavorable immunogenicity, low drug penetration and hypersensitivity reactions [4]. Peptide drugs are an excellent compromise, as they moderate extensive, specific and highly affinitive interactions with proteins. In addition, peptide drugs are smaller than antibodies and are thus more readily deliverable into cells.
In order for a peptide to be an effective therapeutic, however, the peptide must either mask the active site of the target protein or distort the target sufficiently in order to suppress its biological activity [5]. Most importantly, the peptide must gain access to the intracellular compartment. To address this issue, we have developed strategies for the delivery of a peptide drug into the intracellular site of the target protein.
Although the protein transduction domain of a cell penetrating peptide can bring a peptide cargo into cells, the activity is non-specific and is accompanied by unexpected side effects [6, 7]. A few polymeric conjugates or nano-size carrier systems have been developed to enable the delivery of venom toxin or cell-lysis peptide [8–10]. In addition, polymer and gold nanoparticles have been used for intracellular peptide delivery [11–15]. Despite these advances, there have been no reports documenting a successful peptide drug delivery and subsequent inhibition of an intracellular signaling event in an animal model
In previous studies, we have developed effective LPD nanoparticle carriers for the intracellular delivery of negatively charged cargo such as oligonucleotides, including siRNA [16, 17]. These nanoparticles are especially modifiable; particle membranes can be coated with PEGylated targeting ligands, such as anisamide, that bind with the sigma receptor. In this paper, we demonstrate the use of a new carrier system (LPH), modified from LPD, for targeted, intracellular delivery of a peptide drug to block an important signaling protein in NCI-H460 human lung cancer cells.
Signal transducer and activator of transcription (STAT) proteins are latent cytoplasmic transcription factor proteins that regulate cellular processes such as proliferation, differentiation, and survival [18]. In particular, STAT5b, which is a crucial factor for controlling cell survival, cell cycle arrest, and cell death, can be activated by the phosphorylated Y845 kinase domain of EGFR in EGF stimulated tumor cells [19]. STAT5b activation is usually dependent on tyrosine phosphorylation. After reciprocal phosphotyrosine (pTyr)-SH2 interaction, STAT5b dimerizes, translocates to the nucleus, and begins controlling downstream gene transcription [20–22]. Also, loss of STAT5 can affect Bcl-2 and Bcl-xL expression with subsequent loss of mitochondrial membrane potential, followed by apoptosis through the Apaf-1/caspase-9 pathway [23, 24]. Thus, the inhibition of DNA binding STAT5b can induce apoptosis and arrest cell proliferation.
We selected a negatively charged peptide, EEEEpYFELV (EV peptide), that mimics the sequence of the Y845 kinase domain of EGFR. This fragment is likely to inhibit the phosphorylation of STAT5b by competitive interaction with EGFR. This paper will show the successful delivery of EV peptide by nanoparticles to block the phosphorylation of STAT5b and to induce apoptosis in tumor cells.
Materials and Methods
Materials
NCI-H460 human lung cancer cells were obtained from ATCC. Cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (Gibco, Invitrogen Corp., CA), 100 units/ml penicillin, and 100 μg/mL streptomycin (Gibco). NCI-H460 cells were shown to be sigma receptor positive by immunostaining and Western blot (data not shown). siRNAs for STAT5b were purchased from Thermo Scientific Dharmacon (siRNA SMART pool® reagent). Primary antibodies against p-STAT5b (mouse monoclonal), STAT5b (mouse monoclonal) and AIF were purchased from Santa Cruz Biotechnologies (Santa Cruz biotechnology Inc., CA). Caspase-3 antibody for IHC was obtained from Calbiochem (EMD chemical Inc. Germany), and alexa-488, used for synthesizing fluorescence peptides, was purchased from Invitrogen (Invitrogen Corp., CA). Phosphatase inhibitors (phosphatase inhibitor cocktail 1 and cocktail 2 including proteinase inhibitors) were obtained from Sigma (Sigma Aldrich Corp. MO). Therapeutic phosphorylated EV peptide (EEEEpYFELV) and control EE scrambled peptide (EpYELFEEVE) were synthesized commercially with ca. 90% purity (Peptide 2.0 Corp. VA).
Preparation and Characterization of LPH nanoparticle with EV peptide
Liposome-protamine-heparin (LPH) nanoparticles were prepared as follows. Briefly, small unilamellar liposomes consisting of DOTAP and cholesterol (molar ratio = 1:1) were prepared by thin film hydration followed by membrane extrusion. To prepare the cores of EV or EE peptide/LMWH and protamine, protamine (2 mg/mL) and a mixture of peptide (1 mg/mL) and LMWH (2.5 mg/mL) of weight ratio 1:1 were mixed in a 1.5 mL tube. The complex was allowed to stand at room temperature for 10 min, then the complex size and zeta potential were analyzed. The complex prepared with the determined optimal ratio of peptide and LMWH and protamine was mixed with 80 μL of DOTAP/cholesterol liposomes (total lipid concentration = 6.5 mM). Again, the particle size and zeta potential of the resulting particles were analyzed. In order to determine the EV peptide loading efficiency in the LPH nanoparticles, nanoparticles were applied on a Sepharose CL-2B (Amersham Biosciences, Uppsala, Sweden) column using distilled water as eluent. The optimal ratio of the LPH formulation was determined by the results from particle size, zeta potential and in vitro delivery efficiency. Non-targeted LPH and targeted LPH were prepared by incubating the LPH solution (238 μL) with a 10% micellar solution of either DSPE-PEG2000 or DSPE-PEG2000-anisamide (10 mg/mL), respectively, at 50 °C for 10 min. After incubation, the particles were allowed to stand at room temperature for 10 min, then the distribution of particle size of the samples was measured using a submicron particle sizer (NICOMP particle sizing systems, Autodilute PAT Model 370, Santa Barbra, CA) in the NICOMP mode. The polydispersity index was also checked to evaluate distribution characteristics. The zeta potential of the samples diluted in 1 mM KCl was determined by the Zeta Plus zeta potential analyzer (Brookhaven Instruments Corporation, Holtsville, NY). STAT5 siRNA was formulated in LPD-PEG-AA nanoparticles according to published procedure [17]
The H460 cell viability and cell growth changed by EV peptide
The MTT assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) acts as an indirect measure of H460 cell viability. The assay is based on the ability of viable cells to reduce MTT to insoluble colored formazan crystals. H460 cells were plated in 96-well plates (1×104) one day before treatment. After culture with or without LPH formulations (LPH-PEG-AA or LPH-PEG with EV or EE), cells were washed twice with PBS buffer and were incubated in 10 μl PBS buffer with MTT (0.5 mg/ml) for 4 hrs at 37°C. The supernatant was discarded, and cells in each well were lysed with 100 μl of dimethyl sulfoxide (DMSO) and were incubated for 20 min at room temperature. The colorimetric assay was measured at 570 nm on a Micro plate reader. After LPH formulation treatment, H460 cell growth was monitored by cell counting for 2 days. Optical microscopy was used to determine the proportion of live cells after tryphan blue staining.
Cell uptake and Western Blot
NCI-H460 cells were maintained with hEGF (human epidermal growth factor) in RPMI media (Gibco, Invitrogen Corp., CA). In order to identify the extent of LPH nanoparticle cellular uptake, Alexa-488 was conjugated to the amine group of the EV peptide. Alexa488 conjugated EV peptide was loaded into LPH, PEGylated LPH or PEGylated LPH with an anisamide targeting moiety, then H460 cells were treated with the various particle solutions for 4 h. PBS or free EV peptide served as an additional control treatment. After treatment, cells were exposed to trypsin-EDTA, then were collected to observe fluorescence peptide uptake. In order to quantify the difference in cellular uptake between LPH-PEG and LPH-PEG-AA, cells were treated with a mixture of either LPH-PEG or LPH-PEG-AA (2 μM), EV and Alexa-488 labeled EV for 4 h. PBS or a mixture of EV and Alexa-488 labeled EV peptide were used as controls. Samples were analyzed by flow cytometry (BD, RUO special order system, CA).
Protein levels were estimated by first acquiring whole cell extracts using cell extraction reagents (proteoextract® Native Membrane Protein Extraction Kit, Merck) supplemented with phosphatase inhibitor cocktails. The inhibition of p-STAT5b in whole cell lysate was then determined. Whole cell lysates containing the same amount of total protein were incubated with different concentrations of EV peptide with excess ATP for 1 hour. All incubated samples were then subjected to SDS-PAGE, transferred to a membrane, then exposed to anti-stat5a/b-phosphotyrosine (Y694/Y699, 5G4, Santa Cruz) and anti-stat5a/b (5G2, Santa Cruz), using the conventional Western Blotting method. The inhibition effect of p- STAT5b in the intact cell by EV or EE peptide formulated with LPH-PEG or LPH-PEG-AA was then determined. H460 cells were incubated with these formulations for 12 h with hEGF. Equal amounts of cell lysate were run on a 10% SDS-polyacrylamide gel and were transferred onto PVDF (Polyvinylidene difluoride) membrane. The detection was performed by Westernblot ™ 625 western blot kit with bright Qdot substrate. This signal was detected by the Alphaimager™ (Alpha Innotech, CA).
Flow Cytometry assay for detection of apoptosis and the arrest of cell cycle
Discrimination of apoptotic cellular subpopulations was done by individually staining groups of treated cells with Annexin V-FITC and PI. For each staining, 1 × 105 cells per sample were washed once with ice-cold PBS buffer and once with binding buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2). Collected samples were suspended in 50 μL of calcium binding buffer and 3 μL of Annexin V-FITC (0.5 mg/mL) was added to each sample. All samples were incubated in darkness for 20 min at room temperature (25 °C) to ensure adequate Annexin V binding. After washing with PBS buffer, PI (5 mg/mL) was added to the cells suspended in 500 μL of calcium binding buffer, and samples were immediately analyzed using a BD FACS Canto™ flow cytometer (BD, RUO special order system, CA). A 20-mW blue laser (488-nm beam emission) served as the excitation source for Annexin V-FITC (FL1) and PI (FL2). Green fluorescence of Annexin V was collected with a 530-nm band pass filter and red fluorescence of PI was collected with a 610-nm band pass filter. A total of 10,000 cells were analyzed per sample. The propidium iodide (PI) flow cytometric assay has been widely used for the evaluation of apoptosis and cell cycle analysis. We collected cells after treating EE or EV peptide formulated with LPH-PEG or LPH-PEG-AA for 24 h and resuspended the cell pellet in 500 μL of PBS. Cells were then fixed with 4.5 mL of 70% (v/v) cold ethanol to the cell suspension and were kept for 1–2 d at −20 °C. Supernatant (ethanol solution) was removed by centrifugation at 400g for 5 min. Cells were washed in 5 mL of PBS and centrifuged at 400g for 5 min. The cells were then treated with 0.1% Triton-X 100 for 5 min and then centrifuged and resuspended in 100 μL of PBS. The cells were stained with 100 μg/mL propidium iodide (PI) (Sigma Chemical Co., St. Louis, U.S.A.) solution prepared with 1 mg/mL RNase (Sigma). The samples were counted for positive staining by FACS Canto (BD, RUO special order system, CA). The cell cycle distribution was calculated from the resultant DNA histograms using Cell FIT software, based on a rectangular S phase model, and each sample was analyzed using at least 10,000 events corrected for debris and aggregate populations. The results were expressed as the percentage of cells in subG0, G0/G1, S and G2/M phase of the cell cycle.
Cell imaging by confocal microscopy
After 12 hours of treatment with alexa 488-conjugated EV peptide, H-460 cells were fixed with 2% paraformaldehyde and nuclei were counterstained with DAPI (Sigma). Confocal images were acquired with an SP2 Laser Scanning Confocal Microscope (Leica, Bannockburn, IL).
Pull-down assay for peptide activity on STAT5b protein
STAT5b antibodies were conjugated with biotin molecules using an NHS-PEG(4)-biotin labeling kit (EZ-Link NHS-PEG-Biotin kit, Pierce Co. IL). H460 cells were treated for 12 h with various concentrations of Alexa® 488 conjugated fluorescence–EE control and EV peptides in order to observe peptide interaction with STAT5b proteins. Cell extract samples, possibly including free EV peptide, free STAT5b or STAT5b bound with EV peptide, were incubated with biotinylated STAT5b antibodies for 10 min. All biotinylated STAT5b antibodies were captured by streptavidin conjugated magnetic beads immobilized in by an induced magnetic field column in a by a μMACS Column. Both the μMACS Column and the streptavidin magnetic beads were purchased from Miltenyi biotechnology. Before loading samples to the μMACS Column, the column was equilibrated with protein equilibrium buffer. After sample loading, the column was washed with PBS buffer, and then 500 μL buffer was added to elute the samples.
Tissue distribution and tumor uptake study
Mice with small tumors (0.5 to 0.8 cm in diameter) were given i.v. injections of peptides in different formulations, including LPH-PEG and LPH-PEG-AA (160 μg/kg). Free peptide was injected in a control mouse group. After 4 h, treated mice were sacrificed; tissue samples were collected, and subsequently imaged by the IVIS™ Imaging System (Xenogen Imaging Technologies, Alameda, CA). The excised tissues were homogenized in lysis buffer containing phosphatase inhibitors in order to quantify the accumulated doses. After centrifugation at 14,000 rpm for 10 minutes, 100 μL of supernatant was collected, and fluorescent–EV peptide was extracted and quantified with an ELISA reader.
Tumor growth retardation study and immunohistology analysis
Female nude mice 5–6 weeks of age were purchased from the NCI. All animal work was permitted by the UNC IACUC committee. NCI-H460 xenograft tumor- bearing mice (size 40–50 mm2) were produced on the 6th d after intracutaneous injections of 5 × 106 cells in the back of nude mouse. To quantify the accumulated doses, the excised tissues were homogenized in lysis buffer (based on organ weight) with phosphatase inhibitors and incubated at 65 °C for 10 min. One hundred μL of supernatant was collected after centrifugation at 14,000 rpm for 10 min. Fluorescent EV peptide was extracted in the supernatant and quantified using an ELISA reader. The accumulated dose in each organ was calculated from a standard curve obtained by spiking known amounts of free fluorescence peptide. EE peptide or EV peptide in different formulations (0.36 mg/kg, every other day) was given to each group for 20 days. Tumor growth in the treated mice was monitored thereafter and at the end of our experiment, all mice were sacrificed by cervical dislocation. Tumor tissues from mice were collected for immunohistology study. Samples were fixed in 10% formaldehyde solution for 2 h. A dilution (1:200) of anti-AIF or anti-caspase3 rabbit serum was produced by a previously described method and incubated for 2 h at 25°C. HRP-labeled secondary antibody (Santa Cruz biotechnology Inc., CA) was used at a dilution of 1:500 for 20 min. The sections were co-stained with H&E stain and observed with a microscope (Nikon Eclipse, Ti-U, Nikon instrument Inc. TX). In order to observe the EV peptide in vivo, tumor tissue was collected and frozen for further histology analysis 4 h after the injection of EV peptide.
Statistical analysis
Data was expressed as mean ± SEM and was analyzed using Microsoft Excel (office 2007) and Sigma plot Ver. 10 software. Statistical analysis was performed using a one-way Analysis of Variation (ANOVA) followed by Dunnett-test for differences among treatment groups (Keyplot ver 2.0 software). Values of P<0.05 were considered significant.
Results
The inhibitory effect of EV peptide in cell lysate
The effect of EEEEpYFELV peptide (EV) on STAT5b phosphorylation was measured in an H460 cell-free system. As depicted in Figure 1a, EV peptide treatment resulted in strong inhibition of STAT5b phosphorylation. In comparison, treatment against the Y845 site of EGFR (EEKEpYHAE) resulted in less inhibition of STAT5b phosphorylation. Since the EV peptide had a stronger inhibitory effect on STAT5b phosphorylation, it was chosen for further study. Figure 1b shows that STAT5b phosphorylation was inhibited by EV peptide in a dose dependent manner. The IC50 of EV peptide was determined to be about 2.5 μM after measuring the inhibition level of the phosphorylation of STAT5b with IMAGE J free software.
Figure 1.
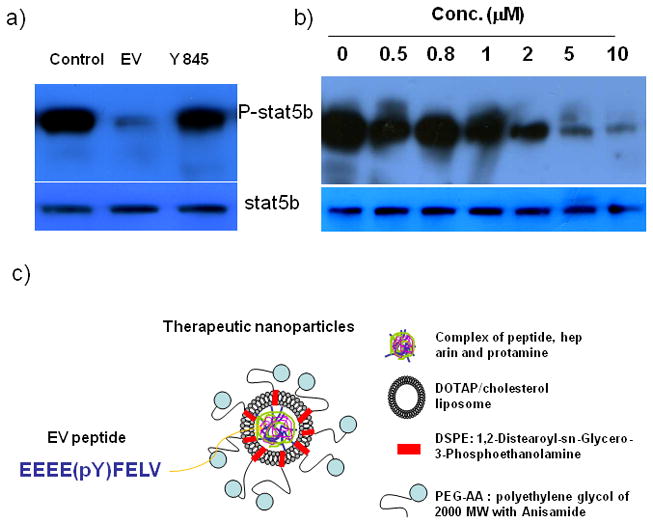
The inhibition effect of STAT5b phosphorylation (a) by the Y845 of EGFR and EV peptide and (b) by EV peptide with different concentrations in H460 cell lysate. And, schematic of tumor targetable nanoparticles (c).
Formulation of EV peptide with LPH nanoparticles
The condition for nanoparticle core formation was optimized with respect to the amount of both protamine and cationic liposomes (supplementary data I). A high amount of protamine was necessary for a tight core formation with peptide and low molecular weight heparin (LMWH), resulting in small, negatively-charged cores (ca. 170–180 nm) and a net negative charge (− 5~10 mV). Moreover, an appropriate amount (6.5 mM) of DOTAP/cholesterol liposomes was necessary for the final LPH preparation, with a diameter of less than 150 nm and a net positive charge (ca. 50–55 mV) [16–17]. After incubation with DSPE- PEG2000 or DSPE-PEG2000-AA, the final nanoparticles measured 130–150 nm in diameter and possessed a slightly positive or neutral charge with 70–75% EV peptide encapsulated.
Uptake of EV peptide by H460 cells
EV peptide labeled with Alexa®488 was formulated in different nanoparticles, then nanoparticle uptake in H460 cells was studied by fluorometry. As shown in Figure 2a, H460 cells treated with EV peptide formulated in LPH-PEG-AA had a 1.5 times higher fluorescent intensity than that of LPH-PEG. Flow cytometry results clearly show that LPH modified with PEG or PEG-AA efficiently delivers EV peptide compared to the free peptide (supplementary data II). We also examined the subcellular distribution of the EV peptide in H460 cells by confocal microscopy. As can be seen in Fig. 2b, fluorescent EV peptide was found in the cytoplasm of the cells, and not in the nuclei.
Figure 2.
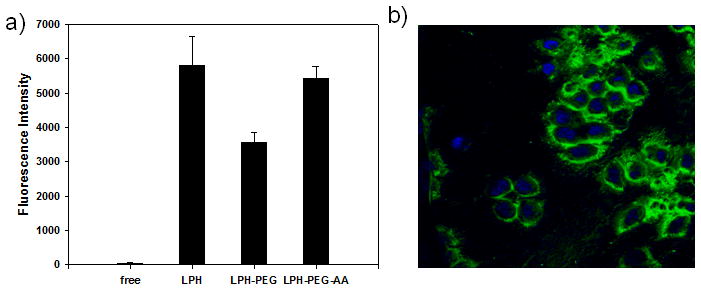
Uptake of Alexa488-labeled EV peptide in different formulations by H460 cells after 4 h incubation was measured by ELISA (a). Intracellular accumulation of fluorescence labeled EV peptide (green) formulated with LPH-PEG-AA observed by confocal microscopy. Nuclei were stained with DAPI (blue) (630×) (b).
Cytotoxicity and inhibition of cell growth of EV peptide
Having found that EV peptide could be delivered intracellularly by LPH-PEG-AA, we examined the effect of EV peptide on cell growth and survival. Cells treated with EV peptide formulated in LPH-PEG-AA showed significant death; viability was reduced by approximately 45%, 48 h after treatment (Figure 3a), while EV peptide formulated in LPH-PEG was only slightly decreased. For the control EE peptide, no significant decrease in viability was observed when the peptide was formulated in LPH-PEG. Some cell death was observed, however, when EE was formulated in the targeted LPH-PEG-AA; viability was about 75% after 48 h. These results suggested that EE peptide also had a minor toxic effect when it was delivered intracellularly. The cell growth curve was measured by counting cell survival numbers after peptide treatment. The result was similar to that of the cell viability measurement (Figure 3b). Cells treated with EV peptide formulated in LPH-PEG-AA showed the greatest growth arrest compared to the PBS treatment control. All other groups, including free EV or EE peptide, did not show a significant difference between each other.
Figure 3.
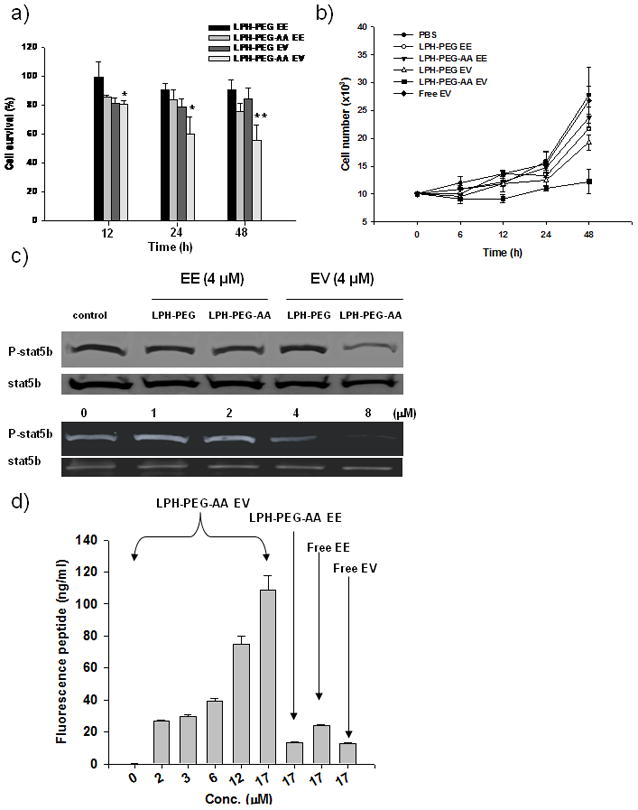
Cell viability at different incubation times after treatment of EV or EE peptide formulated in LPH-PEG or LPH-PEG-AA. Viability was measured by the MTT assay (P value: ** < 0.01, * < 0.05) (a). And, cell growth profile was observed by cell stain counting (b). Western blot analysis for p-STAT5b in the intact H460 cells after treating with EV or EE peptide (4 μM) formulated in LPH-PEG or LPH-PEG-AA confirmed the inhibition effect of STAT5b phosphorylation (upper panel). And, dose-response of STAT5b phosphorylation in the intact H460 cells after treating with EV peptide formulated in LPH-PEG-AA in different concentrations was observed by western blot analysis (lower panel). STAT5b serves as a loading control (c). Pull down assay for EV peptide and STAT5b was performed. Biotinylated STAT5b antibody was incubated with extracts of cells treated with fluorescence labeled EV peptide in different concentrations (from 1.5 to 17 μM). Fluorescence labeled free EE, EV and EE formulated with LPH-PEG-AA were used as the control (d). Mean ± SEM (n=3~5).
In order to elucidate the mechanism of cell growth inhibition by EV peptide, we examined the status of phosphorylated STAT5b (p-STAT5b) in H460 cells. As shown in Figure 3c, only cells treated with AA-targeted EV peptide showed a partial inhibition of p-STAT5b; no inhibition was seen with EV peptide delivered by untargeted formulation or by EE peptide delivered by either targeted or untargeted formulation. The lower panel of Figure 3c shows a dose-response inhibition of STAT5b phosphorylation by EV peptide delivered by LPD-PEG-AA; the apparent IC50 was approximately 4 μM. These results indicate that EV peptide specifically inhibited STAT5b phosphorylation after entering cells, analogous to results shown in the cell free system (Figure 1a).
The extent of EV peptide and STATb binding was also determined. Alexa-labeled EV peptide was delivered to intact H460 cells using LPH-PEG-AA. The amount of EV peptides bound to STAT5b was detected with biotinylated anti-STAT5b and the immune complex was eluted with a streptavidin bead column. As shown in Figure 3d, EV peptide binding with STAT5b increased as the peptide concentration increased. The control EE peptide showed only a small binding effect with STAT5b even at the highest concentration tested (17 μM). The result strongly supports the notion that EV, not EE, peptide binds specifically with STAT5b protein in intact cells.
Apoptosis induction and cell cycle control by EV peptide
After treating cells for 48 hours with either EV or EE peptide in different formulations, cells treated with EV peptide formulated with LPH-PEG-AA (Figure 4a) became smaller and more round. In the early stage of EV peptide treatment, cell retraction and plasma membrane blebbing was also observed (photo not shown). Small and fragmented nuclei were observed in cells treated with EV peptide using DAPI staining, indicating increased apoptosis. As shown in Figure 4b, H460 cells underwent extensive apoptosis (70–80%) after treatment of EV peptide formulated in LPH-PEG-AA for 48 h. Only 17% and 11%, of the cells treated with EE peptide delivered with LPH-PEG or LPH-PEG-AA, respectively, were induced for apoptosis. Thus, cell cytotoxicity was both peptide and delivery system dependent. Results also indicate that EV peptide addition can lead to cell cycle arrest. After treatment with formulated EV peptide, cells in the subG0 and G1 part of the cell cycle increased, with a concomitant decrease in G2/M (Figure 4c). It is not clear whether or not cell cycle arrest and apoptosis were a direct consequence of decreased p-STAT5b.
Figure 4.
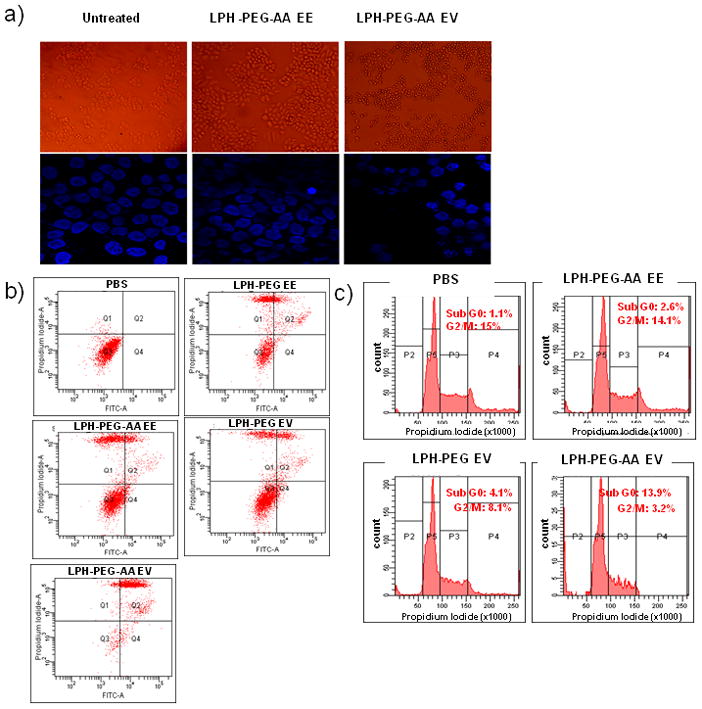
Change of cell morphology after treating with EV peptide formulated with LPH-PEG-AA for 48 h (top panel) and DAPI staining for the observation of fragmented DNA at 24 h after EV peptide treatment (bottom panel) (a). Flow cytometry for detection apoptotic events induced by EV peptide formulated with LPH-PEG or LPH-PEG-AA performed. PBS, EE peptide formulated with LPH-PEG or LPH-PEG-AA as the control treated for apoptosis study. Apoptosis was detected by annexin V (x-axis) and propidium iodide (PI, y-axis) (b). Cell cycle arrest of PBS treated H460 cells and those treated with EE peptide formulated with LPH-PEG-AA, EV peptide formulated with LPH-PEG or LPH-PEG-AA was observed by FACS analysis. Percentage of cells in sub G0/G1, G0/G1, S and G2/M were determined after staining with propidium iodide (PI) (c).
Tissue distribution of EV peptide after intravenous injection
Before testing the therapeutic effect of EV peptide in vivo, we evaluated the delivery of EV peptide to the tumor. As shown in Figure 5a, free EV peptide and EV peptide delivered by LPH or LPH-PEG mainly accumulated in the liver; little or no fluorescence was found in the tumor 4 h after injection (25, 33). However, EV peptide delivered by LPH-PEG-AA showed a high level of fluorescence intensity in the tumor and a comparatively minor intensity in the liver. All other organs did not show accumulation of the peptide, except the kidney, where some of the injected free peptide was found. Thus, we concluded that EV peptide could be delivered efficiently to the tumor by the targeted nanoparticle formulation.
Figure 5.
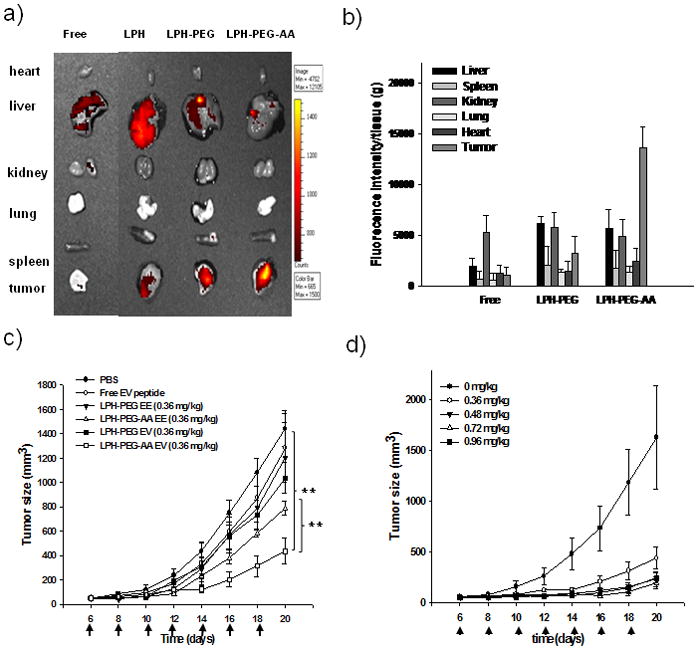
Distribution of fluorescence labeled EV peptide formulated in LPH in major organs (heart, lung, spleen, kidney, liver and tumor) was imaged using a Xenogen IVIS imaging system The LPH was prepared by coating DOTAP liposome on the complexes of EV peptide, heparin and protamine. LPH-PEG or LPH-PEG-AA was prepared by the inserting of DSPE-PEG or DSPE-PEG-AA on LPH nanoparticles. Free peptide was injected as the control. (a). Tissue distribution of EV peptide in mouse organs after intravenously administration of Alexa-488 labeled peptide formulated with LPH, LPH-PEG or LPH-PEG-AA was quantified (b). Tumor growth retardation effect of EV (0.36 mg/kg) or EE (0.36 mg/kg) peptide formulated with LPH-PEG or LPH-PEG-AA in H460 tumor model (b) and dosage effect of EV peptide formulated LPH-PEG-AA in H460 tumor model (d) was evaluated after intravenous injection every other d. Doses were 0.36, 0.48, 0.72 and 0.96 mg/kg of EV peptide formulated with LPH-PEG-AA.. (P value: * < 0.05, ** < 0.001), Mean ± SEM (n=5).
We examined whether EV peptide also exhibited anti-tumor effects after systematic delivery to xenograph tumors. A growth in tumor size over time in different treatment groups can be seen in Figure 5c. Tumor growth in mice treated with free EV peptide, EV peptide formulated in LPH-PEG, or EE peptide formulated in either LPH-PEG or LPH-PEG-AA all showed no significant differences from the PBS control. The only statistically significant difference in the growth rate was found in the group injected with EV peptide formulated in LPH-PEG-AA. Increasing the injection dose brought about an enhanced therapeutic effect (Figure 5d). Injection with EV peptide at 0.48 mg/kg resulted in the maximal therapeutic effect; further increase in the injection dose did not bring about any improved effect.
Immunohistological analysis of the tumor
We examined tumors after 7 injections of either EV or EE peptide in different formulations. Tumor sections were immunostained for the caspase-3 and AIF apoptotic markers. The AIF staining was strongly observed in the tumor tissues of mice treated with LPH-PEG-AA formulated EV peptide, but not in other groups (Figure 6, upper panel). This indicated that caspase independent apoptosis pathway [26] was induced by EV peptide. The same tumor samples were also stained for caspase-3 activation. Again, tumors in mice injected with LPH-PEG-AA containing EV peptide showed strong caspase-3 reactivity, but not in tumors of other groups (Figure 6, lower panel). These results indicated that both caspase dependent and independent apoptosis pathways in the tumor cells were activated by EV peptide delivered by the LPH-PEG-AA formulation. Examination of the tumor sections showed that the peptide was delivered to the interior of the tumor cells, but the distribution within the tumor was uneven (Figure 6, lower panel f). Most of the peptides were delivered to the cells in the outer layer of the tumor; only a minor amount was present in the middle of the tumor. Indeed, apoptosis was localized to the outer layer of the tumor (Figure 6, upper panel e).
Figure 6.
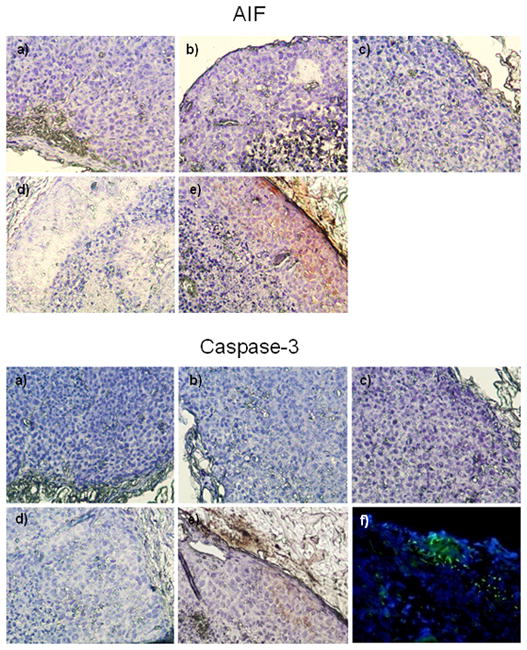
Localization of AIF and caspase-3 by immunohistochemistry in the tumor sections of mouse treated with EV or EE peptide formulated with LPH-PEG or LPH-PEG-AA. Sections from mice treated with PBS (a), EE peptide with LPH-PEG (b), EE peptide with LPH-PEG-AA (c), EV peptide with LPH-PEG (d), EV peptide with LPH-PEG-AA (e) were stained with AIF or caspase-3 antibody (brown staining). In figure f of lower panel, tumor tissues of mice intravenously injected with 0.36 mg/kg of Fluorescence-conjugated EV peptide (green) formulated in LPH-PEG-AA were sectioned and microscopically examined for fluorescence. 40×
Inhibiting other STAT proteins by EV peptide
A western blot analysis was also performed to determine the relationship between other anti-STAT proteins and EV. We found that EV peptide could indeed have a negative effect on the phosphorylation of STAT5a, as well as STAT5b, as shown by the western blot analysis of the cell extract incubated with formulated EV peptide (Supplementary III). Phosphorylation of other STAT proteins, such as STAT3, was not affected. This result indicates that EV peptide is specific for the phosphorylation site of STAT5 proteins in EGF–induced H460 cells.
Discussion
Clinical peptide drugs target extracellular or cell surface receptors. In spite of continuous efforts, delivery of peptide to an intracellular target site remains a challenge. Although conjugation with cell penetrating peptide or protein transduction domain can enhance cell penetration [27–29], the procedure is highly non-specific, and therefore not ideal for targeted therapies.
In the beginning of this study, we explored several peptide sequences containing EEEEpY, which had an inhibitory effect on p-STAT5b (data not shown) and found that EEEEpYFELV (EV) had the strongest activity in blocking p-STAT5b. EV peptide inhibited STATb phosphorylation far better than the Y845 sequence (Figure 1a). This result is not surprising; the natural Y845 domain may assume a favorable conformation only when the sequence is constrained by the context flanking sequences [30–31]. Although EV was active in inhibiting the phosphorylation of STAT5b, the peptide affinity was not particularly high (IC50 = 2.5 μM). It is possible that structural analogs of EV can offer higher affinity with STAT5b. Nevertheless, EV is likely to competitively inhibit activated EGFR.
In this study, we have developed a nanoparticle carrier system (LPH) for the delivery of macromolecular therapeutics in vivo by modification of the previously developed LPD system. LPD particles can be modified with surface grafted PEG chains containing ligand, such as anisamide, attached at the distal end [16, 32]. The current delivery system worked remarkably well for introducing EV peptide into cells. The apparent IC50 (4 μM) (Figure 3c) for p-STAT5b inhibition in intact cells was almost the same as that (2.5 μM) (Figure 1b) in the cell free system. This result indicates that the intracellular concentration of the peptide can be as high as the extracellular concentration without causing unreasonable apoptosis. Our creation of a highly effective, targeted delivery system with such low cytotoxicity is unprecedented. The result highly suggests that our nanoparticle platform will be clinically valuable as a safe, selective therapy. In addition, the carboxyl groups of EV peptide glutamic acids may be protonated in the low pH of endosomes, dissociating the nanoparticle core complex of EV peptide and macromolecules, and accelerating peptide release to the cytosol.
The LPH-PEG-AA formulation also delivered EV peptide efficiently to the tumor in a xenograft model (Figure 5a and 5b). EV peptide delivery was highly targetable, as particle uptake in the tumor was much higher than observed uptake in the liver and spleen. EV particles demonstrated a remarkable ability to evade the reticuloendothelial system of the LPH-PEG-AA nanoparticles while still maintaining a long circulation effect [33]. The nanoparticles contained a high density of grafted PEG chains on the surface; it is likely that these chains formed a dense brush to shield the cationic charges and resist opsonization by serum proteins. In a related formation, i.e. LPD, it was shown that highly PEGylated nanoparticles effectively evade uptake by liver Kupffer cells [34]. Although LPH nanoparticles contained a high density of the grafted PEG, the PEG chains will likely shed from the nanoparticles once injected into the blood [35]. PEG shedding has also been documented as an important part of peptide endosomolytic release in tumor cells [36]. The detailed relationship between PEG shedding and PK and pharmacodynamics of the nanoparticle formulation is the subject of on-going studies.
Cells treated with EV peptide displayed significant apoptosis (Fig. 4). Such phenotype should be similar to cells in which the target protein STAT5b is down-regulated by RNA interference. We compared the effect of peptide and siRNA treatments on the apoptotic status in the treated cells. Western blot analysis of H460 cells shown in Supplementary data IV indicates that EV peptide had induced more down-regulation of pSTAT5b than the corresponding siRNA. Such difference in the pSTAT5b status was reflected in the degree of apoptosis induced by each agent as measured by flow cytometry. Cells treated by PBS, siRNA and EV peptide showed 1.1 ± 0.1 %, 17.6 ± 2.3 % and 39.0 ± 5.7 % apoptosis, respectively, (p = 0.03 comparing the two treated groups, n=3). These results are consistent with the observation by others using a STAT5 knock-down model [37–38].
However, it is interesting to note that significant cell cycle arrest (Figure 4c) and cellular apoptosis (Figure 4a and b) occurred when p-STAT5b was inhibited to only 50–60% (Supplementary data V) at the effective dose applied to intact H460 cells. A plausible explanation is that EV inhibited the downstream signaling of another element, other than STAT5b, that led to the observed cellular phenotypes. Indeed, we found that EV peptide effectively interacted with, and inhibited the phosphorylation of another STAT protein, STAT5a (Supplementary data III). In addition, anionic EV peptide may have interactions with the positive charged domain of various intracellular proteins.
By blocking these crucial cellular signals, EV peptide was a potent inhibitor of tumor growth in vitro (Figure 3a and b) and its effect was nicely translated to the anti-tumor activity in a xenograft model of human lung cancer (Figure 5c). The in vivo toxicity of LPH formulations was evaluated by the analysis of serological or hematological parameters in CD-1 mice after injection of LPH formulations (data not shown). We found that six every other day injections of 0.36 mg/kg EV peptide formulated in LPH do not induce the toxicological effects in vivo. Moreover, based on the negative results of antibody probing for EV peptide and LPH core, we believe that injection of the EV peptide formulation does not induce an immune response.
In the immunohistochemical analysis for apoptosis in the tumor of the xenograft model, we observed markers for both caspase dependent (caspase-3) and independent (AIF) apoptotic pathways in animals injected with EV peptide formulated in LPH-PEG-AA. It is known that STAT 5b phosphorylation activates bcl-2 and bcl-xl, both of which are important anti-apoptosis factors [39–40]. Inhibition of STAT5b phosphorylation by the delivered EV peptide may lead to inhibition of bcl-2 and bcl-xl, which in turn activates the pro-apoptotic bak and/or bax, as well as apoptosis sensitivity [41]. Interestingly, we found that apoptosis was frequently induced in the exterior of the tumor and decreased towards the tumor middle. Indeed, when the distribution of Alexa-labeled EV peptide was examined, many more peptides were delivered to the exterior region of the tumor than the interior.
Conclusions
We have identified a nonapeptide (EV) which mimics the Y845 kinase domain of activated EGFR. Although the peptide showed only a modest affinity to STAT5b, PEGylated and targeted nanoparticle formulations were able to able to deliver sufficient EV amounts to inhibit the phosphorylation of STAT5b in intact cells. Only a partial inhibition of p-STAT5b was sufficient to induce both cell cycle arrest and cellular apoptosis. The same PEGylated and targeted formulation also effectively delivered EV peptide to the tumor cells in a xenograft model, leading to tumor growth inhibition. The current study serves as a proof-of-concept demonstration for delivering intracellular peptide as a therapeutic agent. It is our hope that the study will encourage new targeting strategies to block different protein/protein interactions via peptide delivery.
Supplementary Material
Acknowledgments
The work was supported by NIH grant CA129835. SKK was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-357-2008-1-E00066). Special thank goes to Kimberley Hoenerhoff and Michael Foote for manuscript preparation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gibbs JB. Mechanism-based target identification and drug discovery in cancer research. Science. 2000;287:1969–1973. doi: 10.1126/science.287.5460.1969. [DOI] [PubMed] [Google Scholar]
- 2.Gibbs JB, Oliff A. Pharmaceutical research in molecular oncology. Cell. 1994;79:193–198. doi: 10.1016/0092-8674(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 3.Poondra RR, Kumar NN, Bijian K, Prakesch M, Campagna-Slater V, Reayi A, Reddy PT, Choudhry A, Barnes ML, Leek DM, Daroszewska M, Lougheed C, Xu B, Schapira M, Alaoui-Jamali MA, Arya P. Discovery of Indoline-Based, Natural-Product-like Compounds as Probes of Focal Adhesion Kinase Signaling Pathways. J Comb Chem. 1994;11:303–309. doi: 10.1021/cc8001525. [DOI] [PubMed] [Google Scholar]
- 4.Chester KA, Hawkins RE. Clinical issues in antibody design. Trends Biotechnol. 1995;13:294–300. doi: 10.1016/S0167-7799(00)88968-4. [DOI] [PubMed] [Google Scholar]
- 5.Ladner RC, Sato AK, Gorzelany J, de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9:525–529. doi: 10.1016/S1359-6446(04)03104-6. [DOI] [PubMed] [Google Scholar]
- 6.Brugnano J, Ward BC, Panitch A. Cell penetrating peptides can exert biological activity: a review. BioMol Concepts. 2010;1:109–116. doi: 10.1515/bmc.2010.016. [DOI] [PubMed] [Google Scholar]
- 7.Zorko M, Langel Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Advanced Drug Delivery Reviews. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, Arbeit JM, Wickline SA, Schlesinger PH. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest. 2009;119:2830–42. doi: 10.1172/JCI38842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Cui F, Shi K, Cun D, Wang R. Design of high payload PLGA nanoparticles containing melittin/sodium dodecyl sulfate complex by the hydrophobic ion-pairing technique. Drug Dev Ind Pharm. 2009;35(8):959–68. doi: 10.1080/03639040902718039. [DOI] [PubMed] [Google Scholar]
- 10.Schluep T, Gunawan P, Ma L, Jensen GS, Duringer J, Hinton S, Richter W, Hwang J. Polymeric Tubulysin-Peptide Nanoparticles with Potent Antitumor Activity. Clin Cancer Res. 2009;181:181–9. doi: 10.1158/1078-0432.CCR-08-1848. [DOI] [PubMed] [Google Scholar]
- 11.Wei JS, Zeng HB, Liu SQ, Wang XG, Tay EH, Yang YY. Temperature and pH-sensitive core-shell nanoparticles self-assembled from poly(N-isopropylacrylamide-co-acrylic acid-co-cholesteryl acrylate) for intracellular delivery of anticancer drugs. Front Biosci. 2005;10:3058–67. doi: 10.2741/1762. [DOI] [PubMed] [Google Scholar]
- 12.Tkachenko A, Xie H, Franzen S, Feldheim DL. Assembly and characterization of biomolecule–gold nanoparticle conjugates and their use in intracellular imaging. Methods Mol Biol. 2005;303:85–99. doi: 10.1385/1-59259-901-X:085. [DOI] [PubMed] [Google Scholar]
- 13.Panyam J, Labhasetwar V. Targeting intracellular targets. Curr Drug Deliv. 2004;1:235–247. doi: 10.2174/1567201043334768. [DOI] [PubMed] [Google Scholar]
- 14.Duvall CL, Convertine AJ, Benoit DS, Hoffman AS, Stayton PS. Intracellular delivery of a proapoptotic peptide via conjugation to a RAFT synthesized endosomolytic polymer. Mol Pharm. 2010;7:468–476. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shenoy D, Fu W, Li J, Crasto C, Jones G, DiMarzio C, Sridhar S, Amiji M. Surface functionalization of gold nanoparticles using hetero-bifunctional poly(ethylene glycol) spacer for intracellular tracking and delivery. Int J Nanomedicine. 2006;1:51–57. doi: 10.2147/nano.2006.1.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li SD, Huang L. Surface-modified LPD nanoparticles for tumor targeting. Ann NY Acad Sci. 2006;1082:1–8. doi: 10.1196/annals.1348.001. [DOI] [PubMed] [Google Scholar]
- 17.Li SD, Huang L. Targeted delivery of antisense oligodeoxynucleotide and small interference RNA into lung cancer cells. Mol Pharm. 2006;3:579–88. doi: 10.1021/mp060039w. [DOI] [PubMed] [Google Scholar]
- 18.Johnson FM, Saigal B, Tran H, Donato NJ. Abrogation of signal transducer and activator of transcription 3 reactivation after Src kinase inhibition results in synergistic antitumor effects. Clin Cancer Res. 2007;17:4233. doi: 10.1158/1078-0432.CCR-06-2981. [DOI] [PubMed] [Google Scholar]
- 19.Kloth MT, Laughlin KK, Biscardi JS, Boerner JL, Parsons SJ, Silva CM. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J Biol Chem. 2003;278:1671–1679. doi: 10.1074/jbc.M207289200. [DOI] [PubMed] [Google Scholar]
- 20.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 21.Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2:536–50. doi: 10.1039/b606246f. [DOI] [PubMed] [Google Scholar]
- 22.Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, Rui H, Nevalainen MT. Inhibition of Transcription Factor STAT5 Induces Cell Death of Human Prostate Cancer Cells. J Biol Chem. 2003;278:27287–92. doi: 10.1074/jbc.M304307200. [DOI] [PubMed] [Google Scholar]
- 23.Shelburne CP, McCoy ME, Piekorz R, Sexl V, Roh KH, Jacobs-Helber SM, Gillespie SR, Bailey DP, Mirmonsef P, Mann MN, Kashyap M, Wright HV, Chong HJ, Bouton LA, Barnstein B, Ramirez CD, Bunting KD, Sawyer S, Lantz CS, Ryan JJ. STAT5 expression is critical for mast cell development and survival. Blood. 2003;102:1290–1297. doi: 10.1182/blood-2002-11-3490. [DOI] [PubMed] [Google Scholar]
- 24.Debierre-Grockiego F. Anti-apoptotic role of STAT5 in haematopoietic cells and in the pathogenesis of malignancies. Apoptosis. 2004;9:717–728. doi: 10.1023/B:APPT.0000045785.65546.a2. [DOI] [PubMed] [Google Scholar]
- 25.Li SD, Chen YC, Hackett MJ, Huang L. Tumor-targeted Delivery of siRNA by Self-assembled Nanoparticles. Mol Ther. 2008;16:163–169. doi: 10.1038/sj.mt.6300323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? Journal of Cell Science. 2002;115:4727–4234. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 27.Wadia JS, Dowdy SF. Protein transduction technology. Curr Opin Biotechnol. 1994;13:52–56. doi: 10.1016/s0958-1669(02)00284-7. [DOI] [PubMed] [Google Scholar]
- 28.Gariépy J, Kawamura K. Vectorial delivery of macromolecules into cells using peptide-based vehicles. Trends Biotechnol. 2000;19:21–26. doi: 10.1016/s0167-7799(00)01520-1. [DOI] [PubMed] [Google Scholar]
- 29.Morris MC, Chaloin L, Heitz F, Divita G. Translocating peptides and proteins and their use for gene delivery. Curr Opin Biotechnol. 2000;11:461–466. doi: 10.1016/s0958-1669(00)00128-2. [DOI] [PubMed] [Google Scholar]
- 30.de Mol NJ, Catalina MI, Dekker FJ, Fischer MJ, Heck AJ, Liskamp RM. Protein Flexibility and Ligand Rigidity: A Thermodynamic and Kinetic Study of ITAMBased Ligand Binding to Syk Tandem SH2. Chem Bio Chem. 2005;6:2261–2270. doi: 10.1002/cbic.200500141. [DOI] [PubMed] [Google Scholar]
- 31.Dekker FJ, Mol NJ, de Fischer MJE, Liskamp RMJ. Amino Propynyl Benzoic Acid Building Block in Rigid Spacers of Divalent Ligands Binding to the Syk SH2 Domains with Equally High Affinity as the Natural Ligand. Bioorg Med Chem Lett. 2003;13:1241–1244. doi: 10.1016/s0960-894x(03)00117-3. [DOI] [PubMed] [Google Scholar]
- 32.Needham D, Kim DH. PEG-covered lipid surfaces: bilayers and monolayers. Colloid Surf B: Biointerfaces. 2000;18:183–95. doi: 10.1016/s0927-7765(99)00147-2. [DOI] [PubMed] [Google Scholar]
- 33.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145(3):178–81. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: Role of the supported bilayer. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2009;1788:2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tseng YC, Huang L. Self-assembled lipid nanomedicines for siRNA tumor targeting. J Biomed Nanotechnol. 2009 Aug;5(4):351–63. doi: 10.1166/jbn.2009.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Huang L. In Vivo Delivery of RNAi with Lipid-Based Nanoparticles Annual Review of Biomedical Engineering. 13 doi: 10.1146/annurev-bioeng-071910-124709. (Volume publication date August 2011) [DOI] [PubMed] [Google Scholar]
- 37.Liang QC, Xiong H, Zhao ZW, Jia D, Li WX, Qin HZ, Deng JP, Gao L, Zhang H, Gao GD. Inhibition of transcription factor STAT5b suppresses proliferation, induces G1 cell cycle arrest and reduces tumor cell invasion in human glioblastoma multiforme cells. Cancer Letters. 2009;273:164–171. doi: 10.1016/j.canlet.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Xiong H, Su WY, Liang QC, Zhang ZG, Chen HM, Du W, Chen YX, Fang JY. Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in uman colorectal cancer cells. Laboratory Investigation. 2009;89:717–725. doi: 10.1038/labinvest.2009.11. [DOI] [PubMed] [Google Scholar]
- 39.Matte I, Lane D, Côté E, Asselin AE, Fortier LC, Asselin C, Piché A. Anti-apoptotic proteins Bcl-2/Bcl-XL inhibit Clostridium difficile toxin A-induced cell death in human epithelial cells. Infect Immun. 2009;77:5400–5410. doi: 10.1128/IAI.00485-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 41.Can J, John CR. Yeast and apoptosis. Nat Rev Mol Cell Bio. 2002;3:453–459. doi: 10.1038/nrm832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.