

Abstract
This review describes the most commonly used rodent models and outcome measures in preclinical stroke research and discusses their strengths and limitations. Most models involve permanent or transient middle cerebral artery occlusion with therapeutic agents tested for their ability to reduce stroke-induced infarcts and improve neurological deficits. Many drugs have demonstrated preclinical efficacy but, other than thrombolytics, which restore blood flow, none have demonstrated efficacy in clinical trials. This failure to translate efficacy from bench to bedside is discussed alongside achievable steps to improve the ability of preclinical research to predict clinical efficacy: (i) Improvements in study quality and reporting. Study design must include randomization, blinding and predefined inclusion/exclusion criteria, and journal editors have the power to ensure statements on these and mortality data are included in preclinical publications. (ii) Negative and neutral studies must be published to enable preclinical meta-analyses and systematic reviews to more accurately predict drug efficacy in man. (iii) Preclinical groups should work within networks and agree on standardized procedures for assessing final infarct and functional outcome. This will improve research quality, timeliness and translational capacity. (iv) Greater uptake and improvements in non-invasive diagnostic imaging to detect and study potentially salvageable penumbral tissue, the target for acute neuroprotection. Drug effects on penumbra lifespan studied serially, followed by assessment of behavioural outcome and infarct within in the same animal group, will increase the power to detect drug efficacy preclinically. Similar progress in detecting drug efficacy clinically will follow from patient recruitment into acute stroke trials based on evidence of remaining penumbra.
LINKED ARTICLES
This article is part of a themed issue on Translational Neuropharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-4
Keywords: middle cerebral artery occlusion, penumbra, infarct, neuroprotection, MRI, neurological deficit, rodent
Stroke – some facts and figures
Stroke is a common and devastating disease. It is the second leading cause of death after coronary heart disease in developed countries (Donnan et al., 2008) and is the greatest cause of disability, leaving 50% of survivors permanently disabled. Identification of risk factors such as arterial hypertension, high cholesterol, diabetes and obesity has led to successful stroke prevention strategies (McArthur and Lees, 2010). Some progress has been made in effective rehabilitation following the acute stroke period (Langhorne et al., 2009; Kalra, 2010) with evidence for improvements in motor recovery associated with high intensity and repetitive task-specific practice (e.g. constraint-induced movement therapy). However, there is insufficient data at present to identify specific interventions that would be widely applicable. The area that has progressed least in the last 15 years, despite a significant preclinical and clinical research effort is identification of acute therapies that can limit stroke-induced brain damage and disability. There is still no clinically effective neuroprotective drug licensed for stroke.
Strokes are classified as haemorrhagic or occlusive/ischaemic, with the majority (80%) falling into the latter category (Heiss, 2010). For occlusive stroke, thrombolysis with recombinant tissue-type plasminogen activator (rt-PA), and care provided by specialized acute stroke units, has been proven to improve outcome (Donnan et al., 2008).
Rt-PA is directed at the occluding blood clot and by enhancing the endogenous formation of plasmin from plasminogen helps dissolve the clot by disruption of fibrin, thereby enabling restoration of blood flow to ischaemic tissues. However, it does not offer any direct protection to ischaemic tissues and in fact has neurotoxic potential within the parenchyma if not confined within the vasculature (Yepes et al., 2009). Since thrombolysis with rt-PA also carries a risk of symptomatic intracranial haemorrhage (∼2%), and probability of benefit declined rapidly with time beyond 4.5 post- stroke (NINDS & Stroke rt-PA Stroke Study Group, 1995; Hacke et al., 2008; Lees et al., 2010), guidelines recommend treatment restricted to the first 4.5 h following stroke onset (ESO, 2008; Del Zoppo et al., 2009). In England, Wales and Northern Ireland, only 3.8% of patients are reported to receive the treatment (Royal College of Physicians, 2010; National Sentinel Audit of Stroke). Similar figures are reported in the USA (∼2% of eligible patients) (Kleindorfer et al., 2008), leaving the majority of stroke patients with no access to effective drug treatments.
Targeting the ischaemic cascade
The last 30 years have seen significant progress in our understanding of the pathophysiology of ischaemic stroke and the identification of mechanisms that contribute to tissue damage. The ischaemic cascade, starting with a severe focal reduction in cerebral blood flow (CBF) and culminating in cell death and infarction, has many intervening and interlinking steps that have provided a number of potential drug targets (reviewed in Moskowitz et al., 2010). Pharmacological agents have been developed to block the major mediators of injury: high, toxic concentrations of extracellular glutamate, intracellular calcium and free radicals. Ischaemia causes widespread cellular depolarization, with Na+, K+, Ca2+ and Cl- fluxes through activated ionotropic receptors and gated ion channels, loss of ionic concentration gradients across cell membranes, uncontrolled neurotransmitter release and glutamate-induced excitotoxicity. Calcium dysregulation is the trigger for enzymatic destruction of the cell and its components with activation of proteases, lipases and nucleases. Activation of enzymes such as neuronal nitric oxide synthase, NADPH oxidase, cyclooxygenase and lipo-oxygenase generates free radicals that attack cellular membranes and dismantle key functions of the cell such as ATP production, protein synthesis and the sequestration and storage of intracellular calcium within intracellular organelles. Inflammation represents another key component of the ischaemic cascade with pro-inflammatory phospholipase A2 liberating arachidonic acid, a substrate for cyclooxygenase and lipo-oxygenase and precursor of prostaglandins and leukotrienes. The culmination of this complex, self-generating programme of events is either rapid necrotic cell death or delayed apoptotic cell death depending on the severity and duration of ischaemia. Drug targets include neurotransmitter receptors and ion channels, free radicals, cytokines and inflammatory mediators, enzymes and membranes. Drugs developed to block these targets are often referred to as ‘neuroprotective’. However, their role is not simply to protect the neurone (i.e. cell body, axon and nerve terminals) but rather the neurovascular unit: the neurone plus the supporting glial and vascular cells within its immediate environment, which includes astrocytes, pericytes, microglia, oligodendrocytes and the endothelial cells of microvessels (del Zoppo, 2010; Moskowitz et al., 2010). A number of drugs have demonstrated neuroprotective efficacy in preclinical studies where focal ischaemic insults produce reproducible infarcts and behavioural deficits (O'Collins et al., 2006). However, human stroke is a highly heterogeneous condition, and treatment targeted at a single mechanism in the ischaemic cascade is unlikely to be universally effective. Combination therapy or single drugs with multiple targets and actions are more likely to be effective.
Preclinical stroke research
Preclinical stroke research is carried out using in vitro and in vivo models. In vitro studies use neuronal or mixed cell cultures and organotypic slice preparations as model systems that recreate some of the consequences of a focal ischaemic insult. Cells and tissues are exposed to excitotoxic concentrations of glutamate, NMDA, alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), Kainate, hypoxia or oxygen glucose deprivation. Compounds are tested for their ability to reduce cell death and inhibit the targeted deleterious mechanism (Richard et al., 2010), and the most effective are then taken forward for in vivo evaluation.
Stroke is studied in animal models by permanently or transiently occluding a cerebral artery that causes a severe reduction in CBF in the territory of that artery. In other words, a focal ischaemic insult is induced in a defined region of brain tissue. This is different from a global ischaemic insult, where the entire brain or forebrain is exposed to a severe reduction in its blood supply (e.g. as a consequence of cardiac arrest, severe hypotension or peripheral haemorrhage, strangulation or drowning). Animal models of both global and focal ischaemia have been developed and reveal differences in the mechanisms of injury and patterns of brain damage (reviewed in Ginsberg and Busto, 1989; Traystman, 2003).
Models of focal cerebral ischaemia have been established in a number of species, most commonly in lysencephalic species such as rats and mice. Guidelines for drug development recommend that once efficacy is established in rodents, studies are carried out in gyrencephalic species such as cats, pigs and non-human primates before taking the drug through to studies in man (STAIR, 1999). The current review focuses specifically on rodent models of focal cerebral ischaemia. Rodents are the species of first choice for a number of reasons. It is generally considered ethically more acceptable to use rodents rather than higher mammals. Animal and maintenance costs are low, and the vascular anatomy is similar to man. Rodent neuroanatomy and the cascade of molecular mechanisms leading to ischaemic cell death are well characterized with additional information from stroke studies in transgenic mice where specific genes are overexpressed or knocked out/down. Rodent strains and transgenic mice developed to display comorbidities such as hypertension, atherosclerosis, diabetes and obesity are also available. Relevance to human stroke is supported by the knowledge that these mechanisms are similar between rodents and man, and that thrombolysis, the one treatment known to be efficacious in man, is equally efficacious in rodent focal ischaemia models (Back et al., 2007).
Selection of the model
A wide range of rodent focal cerebral ischaemia models are available for preclinical drug development and selection of the most pertinent model will depend on the class of drug under study and its perceived mechanism of action. For example, embolic stroke models are used to test new thrombolytic drugs, models of transient focal ischaemia are commonly used to test free radical scavengers and anti-inflammatory agents and the first glutamate antagonists to be developed were tested in models of permanent focal ischaemia. However, given the heterogeneity of human stroke, drugs should demonstrate preclinical efficacy in a range of different models and species before being considered for translation through to clinical trials.
Middle cerebral artery occlusion (MCAO)
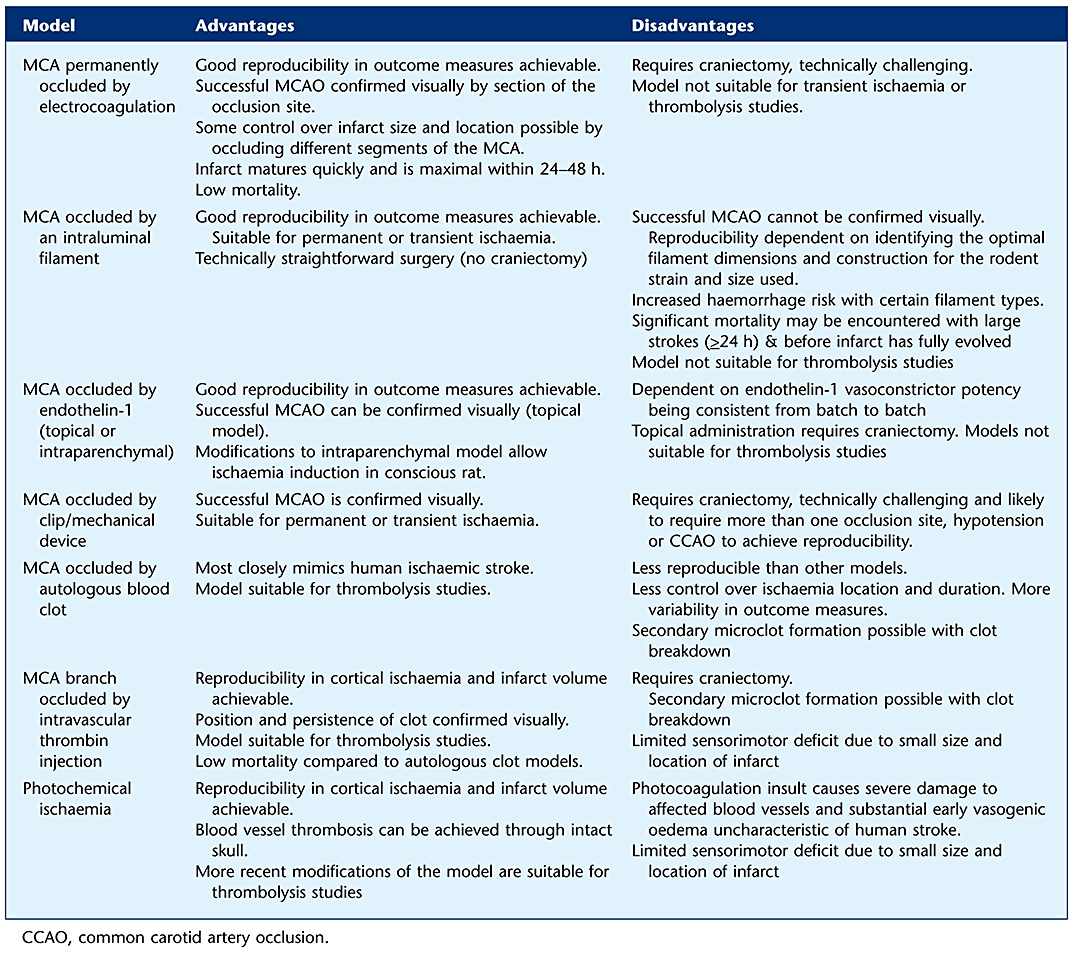
The middle cerebral artery (MCA) is the most commonly affected blood vessel in human occlusive/ischaemic stroke (Mohr et al., 1986) and is the artery most commonly targeted in rodent stroke models (see Figure 1). A range of MCAO models have been developed, and there are advantages and disadvantages associated with each approach (Table 1). Following surgical exposure of the blood vessel via a craniectomy, the MCA can be occluded in a number of ways: by electrocoagulation of the blood within it and destruction of the blood vessel per se (Tamura et al., 1981); mechanically, by applying an occluding device such as a clip or ligature (van Bruggen et al., 1999); or pharmacologically by applying a potent and prolonged vasoconstrictor such as endothelin-1 (Macrae et al., 1993). MCAO models that do not require a craniectomy include stereotaxic injection of endothelin-1 into parenchyma adjacent to the MCA that results in a longer-lasting vasoconstriction (Sharkey et al., 1993). Alternatively, the skull is left intact, and an intravascular approach via the carotid artery is used to advance a filament or embolus to the point where it blocks the origin of the MCA (Koizumi et al., 1986; Longa et al., 1989; Zhang et al., 1997).
Figure 1.
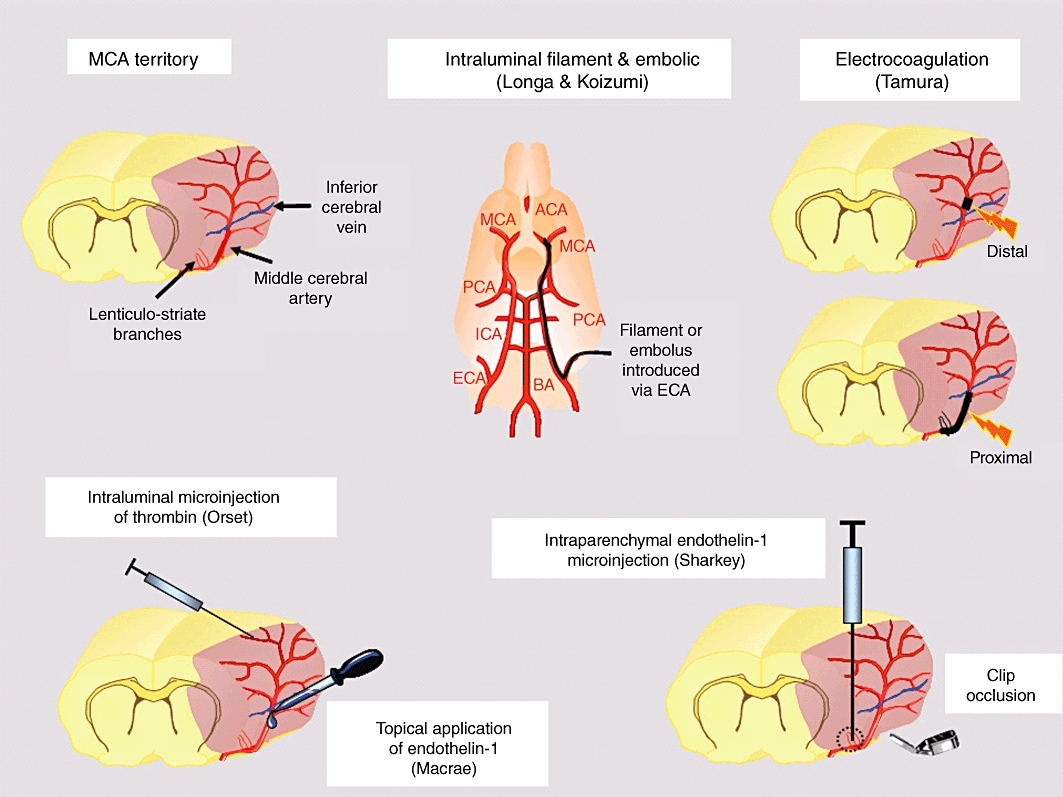
MCAO models in rats and mice. Pink shading on MCA territory diagram represents the tissues supplied by the MCA. Pink shading on the electrocoagulation diagrams represents ischaemia and demonstrates the ability to induce purely cortical ischaemia with a distal MCAO versus cortical and subcortical ischaemia with a proximal MCAO including the lenticulostriate branches.
Table 1.
Advantages and disadvantages of the different focal ischaemia models
 |
MCA occlusion by electrocoagulation or application of an occluding device
All the models within this category require a craniectomy and section of the dura mater to expose the MCA. Models that require a craniectomy are associated with low or absent mortality (Table 2) since the craniectomy prevents ischaemia-induced increases in intracranial pressure. This can be a significant advantage, particularly where large strokes are studied over a number of days. Oedema and brain swelling correlate with infarct size and the increases in brain volume associated with large MCA strokes over the first 24–48 h post-stroke would cause significant mortality if the skull was intact.
Table 2.
Published mortality data for different rodent focal ischaemia models
| MCAO model (duration of ischaemia) | Species, strain (m/f) | Craniectomy | Mortality (time period examined) | References |
|---|---|---|---|---|
| Proximal MCAO (permanent) | Rat, Wistar (m) | Yes | 0% (10 weeks) | (Yonemori et al., 1999) |
| Electrocoagulation | ||||
| Proximal MCAO (permanent) | Rat, SD (m) | No | 18.2% (24 h) | (Lu et al., 2009) |
| Intraluminal filament | Rat SD (m) | No | 42% (72 h) | (Schöller et al., 2004) |
| Rat SD (m) | No | 0% (24 h), 12.5–33%* (48 h) | (Aspey et al., 1998) | |
| Rat Wistar (m) | No | 75% (24 h) | (Aspey et al., 1998) | |
| Rat Fischer 344 (m) | No | 8% (24 h) | (Aspey et al., 1998) | |
| Distal MCAO (permanent) | Mouse, C57BL/6 (m) | Yes | 0% (6 h-8 days) | (Kuraoka, et al, 2009) |
| Electrocoagulation | ||||
| TMCAO (proximal, 60 min) | Rat SD (m) young, 3–4 months | Yes | 6.3% (24 h), 6.3% (28 days) | (Wang et al., 2003) |
| Ligature + BCCAO | ||||
| Rat SD (m) old, 22–24 months | Yes | 35% (24 h), 43.5% (28 days) | ||
| TMCAO Intraluminal filament (proximal, 60 min) | Mouse, SV129/J (m) | No | 13, 37 & 57% (4, 5 & 6 days, resp) | (Meisel et al., 2004) |
| (proximal, 45 min) | Mouse, 129S6SvEv (m) | No | 33% (72 h) | (Braun et al., 2007) |
| Thromboembolic (autologous clot) | Rat SD | No | 30–50% (24 h) | (Orset et al., 2010) |
| Rat SD (m) | No | 44% (14 days) | (Rasmussen et al., 2008) | |
| Thromboembolic (intra-arterial thrombin injection) | Mouse, Swiss (m) | Yes | <1% (24 h and 3 months) | (Orset et al., 2010) and (D. Vivien, pers. comm.) |
| Photochemical stroke | Mouse and rat | No | Low (not defined) | Witte (2010 |
| Endothelin-1 topical application to MCA | Rat SD (m) | Yes | <5% (4 h) | (IM. Macrae, unpublished) |
| Endothelin-1 injection into piriform cortex adjacent to proximal MCA | Rat SD (m) | # | 7% (24 h), thereafter 0% out to 3 months | J. Sharkey (pers. comm.) |
| 15% (4 h) | (Nikolova et al., 2009) |
Mortality levels encountered are dependent not only on the model employed but also the survival period and the level of experience and skills of the surgeon.
Dependent on suture used.
Small burr hole on dorsal surface of the skull for insertion of stereotaxic needle and/or guide cannula.
BCCAO, bilateral common carotid artery occlusion; f, female; m, male; SD, Sprague–Dawley; TMCAO, transient middle cerebral artery occlusion.
Following surgical exposure, the MCA is permanently occluded by coagulating the blood within it using an electric current passed through the tips of fine diathermy forceps (Tamura et al., 1981). The occluded portion of the artery is then cut confirming complete occlusion. Electrocoagulation models have been successfully adapted for use in larger species such as the cat (Bullock et al., 1990), miniature pig (Imai et al., 2006) and baboon (Yonas et al., 1981). Maintaining sterile conditions and careful post-stroke care of the wound is particularly important to avoid any infection associated with the surgery, which will increase ischaemic damage and confound the ability to determine drug-induced effects on infarct size and functional recovery. The main advantages of the model are good reproducibility in infarct size and functional deficit, low mortality, visual confirmation of successful MCAO and the ability to adapt the model to produce infarcts of different size and location. The main disadvantages are that the model induces permanent focal ischaemia and is not therefore suitable for investigation of thrombolytic agents or drugs designed to target the reperfusion phase following ischaemia. Inducing MCAO is technically demanding. Exposing the artery and applying electrocoagulation without rupturing the blood vessel or damaging the underlying cortex requires significant surgical skill. The surgery required for proximal MCA occlusion can also cause jaw alignment problems in rats requiring replacement of standard chow with soft diet and regular monitoring and tooth filing to avoid overgrowth.
Alternative MCA occlusion models use devices such as microaneurysm clips, hooks (used to lift the artery from the cortical surface until flow ceases), ligatures and in, larger species, inflatable cuffs to occlude the artery remotely (Shigeno et al., 1985; van Bruggen et al., 1999). These models have the advantage of control over the duration of ischaemia and allow subsequent reperfusion of ischaemic tissue. They provide visual confirmation of successful MCAO and reperfusion when the occluding device is removed. However, microaneurysm clips are too small to apply by hand in rats and have to be loaded into a special applicator for attachment to the MCA. Applying and removing the clips without damaging the artery is technically difficult, particularly when targeting the proximal MCA. Other disadvantages include greater variability in infarct size compared with electrocoagulation models, particularly when a single point on the MCA is occluded. Reproducibility is improved by occluding the artery at more than one site, combining MCAO with hypotension or ipsilateral common carotid artery occlusion, or using rat strains with poor collateral supply (e.g. the spontaneously hypertensive rat and the spontaneously hypertensive stroke-prone rat; see Coyle and Jokelainen, 1983) to increase the severity of the ischaemic insult. However, for considering neuroprotection studies, it is worth considering that steps such as these taken to improve reproducibility in infarct size are also likely to result in less potentially salvageable penumbral tissue being available for rescue.
Modifications to the model
The position and length of MCA occluded provides some control over the severity of the ischaemic insult, the amount of penumbral tissue available for rescue and the neurological deficit produced. Proximal occlusion of a long segment of the MCA including the lenticulostriate branches gives rise to a large consistent ischaemia and infarct in cortical and subcortical structures (see electrocoagulation, Figure 1) and a reproducible neurological deficit. However, the greater the volume of ischaemic tissue within the MCA territory, the less penumbral tissue will be available for the drug under test to rescue. Occlusion of a shorter (1–2 mm) segment of the MCA, distal to the inferior cerebral vein, will induce a smaller region of ischaemia and infarct, mainly confined to the cortex, and provide a larger volume of target penumbral tissue. However, the neurological deficit produced will be much milder and more difficult to detect.
MCA occlusion induced pharmacologically
The peptide endothelin-1 (ET-1) lends itself to cerebral ischaemia research because it induces profound and prolonged vasoconstriction of cerebral vessels (Asano et al., 1989; Robinson and McCulloch, 1990). ET-1 topically applied to the abluminal surface of the exposed MCA induces an ischaemic insult of sufficient severity and duration to produce a reproducible infarct (Figure 1). This model was developed in the rat (Macrae et al., 1993) and has been adapted for use in marmosets (Virley et al., 2004). Advantages include visual confirmation of ischaemia, with some control over the severity and duration by adjusting the ET-1 concentration, a gradual reperfusion and low mortality. Disadvantages include stability and potency issues relating to peptides that can increase variability. An alternative model, where ET-1 is administered by stereotaxic injection into piriform cortex immediately adjacent to the proximal MCA, is less demanding surgically and induces a more persistent ischaemia that topical application (Figure 1, Sharkey et al., 1993). This model is more useful for fast throughput screening of drugs and has been adapted for induction of stroke in the conscious animal by injecting ET-1 via a previously implanted guide cannula. If ET-1 is replaced with endothelin-3, it is possible to reverse vasoconstriction and downstream ischaemia with a second injection of an endothelin-A receptor antagonist (Henshall et al., 1999). Intraparenchymal injection of ET-1 has been adapted for anterior cerebral artery occlusion (Ward et al., 1998) and selective white matter ischaemia in the internal capsule (Frost et al., 2006; Lecrux et al., 2008).
MCA occlusion by intraluminal filament
The intraluminal filament (or suture) method is currently the most widely used model of focal ischaemia in rats and mice and is used to induce both permanent and transient ischaemia. No craniectomy is required as the occluding device, a flexible monofilament, is introduced directly into the internal carotid artery (or with modification, via the external carotid artery) and advanced until it blocks the origin of the MCA (Figure 1). The method was introduced by Koizumi et al. (1986) with the first of many modifications described by Longa et al. (1989). The advantages of this model are the ability to precisely control the duration of ischaemia and the fact it is technically easier to master than the craniectomy models. The disadvantages mainly relate to difficulties in reproducibility and mortality. Modifications to improve reproducibility include filament construction, coating and design of the tip and are covered in a recent review (Durukan and Tatlisumak, 2007). Filament diameter is carefully matched to a defined body weight range in rodents to ensure adequate occlusion of the origin of the MCA. Laser Doppler flowmetry probes, placed on the skull above the sensorimotor cortex, are commonly used to guide correct placement of the filament, to assess the severity of the drop in CBF and to avoid advancing the filament too far, thereby risking blood vessel puncture and haemorrhage. Filament insertion to block the origin of the MCA should induce cortical and subcortical ischaemia, but tissue infarction may be confined to subcortical structures with minimal cortical involvement if the rodent strain used has good collateral blood supply to the cortex (Coyle and Jokelainen, 1983; Duverger and Mackenzie, 1988). Increasing the duration of ischaemia, tandem occlusion of the carotid artery(s) or hypotension is used to increase cortical involvement. Since the filament is advanced along the internal carotid artery to reach the origin of the MCA, depending on filament construction, other arteries that branch off the internal carotid, proximal to the origin of the MCA, may also be occluded by the filament. Therefore, although this model is employed for MCA occlusion, other arteries, such as the anterior choroidal and hypothalamic arteries may also be blocked, leading to damage beyond MCA territory and corresponding associated deficits (e.g. hyperthermia associated with ischaemic damage to hypothalamus; Li et al., 1999).
For longitudinal studies, problems with morbidity and mortality are encountered when ischaemia is permanent or prolonged (e.g. ≥90 min). This is manifest within the first 24–48 h in animals with large MCA territory strokes due to brain swelling and increased intracranial pressure (Table 2).
Embolic MCA occlusion models
Embolic models fall into two main categories: (i) embolization induced by the introduction of blood clots or artificial emboli; (ii) localized chemically initiated thromboembolism. Thromboemboli cause most human strokes and therefore models that mimic this type of occlusion are useful for testing new thrombolytic agents. Blood drawn from the animal to form autologous clots of specific size and composition ex vivo are subsequently (e.g. 24 h later) introduced into the cerebral circulation via a cannula inserted and advanced along the internal carotid artery (using a similar approach to intraluminal filament insertion). Longa et al. (1989) first described the induction of thromboembolic stroke in the rat, with subsequent modifications published by Busch et al. (1997). Similar models have been established in mice (Zhang et al., 1997), rabbits (Lapchak et al., 2000), cats (Yamaguchi et al., 2000) and non-human primates (Watanabe et al., 1977; Kito et al., 2001). The advantages of embolic models include their clinical relevance, the fact that the surgery required is straightforward and does not involve a craniectomy. When the clot is correctly positioned, large infarcts can be induced, which include both cortical and sub-cortical structures, and give rise to pronounced behavioural deficits. The blood clot is broken down and reperfusion induced by administration of thrombolytics such as rt-PA. Meta-analysis of the preclinical literature demonstrates similarities with the clinical literature: rt-PA administered within the first 3–4 h post-stroke in rodents reduces infarct volume and improves neurobehavioural scores in rats, with an increased probability of haemorrhage (Perel et al., 2007). However, quality scores for the 113 studies identified were poor with evidence of publication bias. There are a number of pertinent disadvantages associated with embolic models. Intravascular introduction of emboli can result in multifocal ischaemia with significant variability in infarct size and location as well as early autolysis, depending on emboli composition (see Busch et al., 1997). Brain haemorrhage is frequent with embolic models, and mortality rates are generally much higher (e.g. ≥30% in rat) than for other models (Table 2).
Composition and stability of the embolus are important in preventing spontaneous clot disintegration. Thrombolytic breakdown of the blood clot and recanalization of the occluded artery is related to the amount of red cells in the emboli and inversely related to the volume of the emboli, fibrin content and density of the clots. (Overgaard et al., 1994). Since early in vitro studies demonstrated that the rat's fibrinolytic system was 10-fold less sensitive to rt-PA than the human system (Korninger and Collen, 1981), the majority of in vivo studies have used rt-PA at a 10 fold higher dose (10 mg·kg−1) than the human dose (0.9 mg·kg−1). Successful thrombolysis and reperfusion achieved with 10 mg·kg−1 rt-PA administered within 2–4 h of stroke reduced infarct size and improved survival and neurological deficits in rodent models. However, the higher dose may not be necessary or recommended for future preclinical research since a recent study, comparing the two doses administered 45 min after stroke in a rat embolic model, has demonstrated that the lower (human) dose was equally effective in inducing reperfusion (although slower in onset) and significantly reduced infarct size and oedema (Haelewyn et al., 2010). The 10 mg·kg−1 dose is no longer effective beyond 4 h post-stroke and increases infarct size and haemorrhagic transformation (Kano et al., 2000; Lapchak., 2010).
Artificial emboli
A variety of materials have been used to form non-clot emboli (see Durukan and Tatlisumak, 2007), the most common being microspheres of defined diameter. Suspensions of calibrated microspheres injected into the internal carotid artery produce microembolization and slowly evolving ischaemic lesions and a model in rabbits has been used for the study of stroke pharmacotherapy (Zivin et al., 1987). However, the utility of microspheres is more limited than clot-based emboli in that vessel occlusion is permanent with no potential for recanalization, either spontaneously or interventionally (Mayzel-Oreg et al., 2004).
Chemically initiated thromboembolism
Chemically initiated thromboembolism is induced photochemically by systemic injection of a photosensitive dye (e.g. Rose Bengal or erythrosin B) in combination with irradiation through the exposed skull with light of a specific wavelength (Watson et al., 1985, rats; Sugimori et al., 2004, mice). The reaction between the light and the intravascular dye generates oxygen radicals causing peroxidation of endothelial lipids and blood elements, thereby inducing platelet aggregation and thrombosis (Ginsberg and Busto, 1989). The model is minimally invasive, produces a reproducible cortical photothrombosis in both rats and mice and uses specialist laser equipment that provides precision over the exact location of ischaemia. Mortality associated with the model is low, and the size and depth of ischaemic damage can be controlled by adjusting the plasma concentration of the dye and the intensity and duration of light. Limitations of the model include its end-arterial occlusive nature that makes the lesion resistant to flow enhancement strategies. The rapid progression of ischaemic damage is associated with significant early cytotoxic and vasogenic oedema formation that is different from the situation in human stroke. Early versions of the model induced a severe insult with rapidly evolving ischaemic damage and no salvageable penumbral tissue, but more recent modifications, using a ring model and modified laser parameters, display ‘region-at-risk’ in the ring-encircled interior region (Wester et al., 1995; Hilger et al., 2004). Further developments include the use of optical fibres stereotaxically implanted or directed at the surgically exposed proximal MCA to produce photothrombosis in subcortical sites. These models demonstrate evidence of penumbra on magnetic resonance imaging (MRI) scans and reversal of ischaemia with rt-PA (Chen et al., 2007; Kuroiwa et al., 2009).
Recently, a new model of thromboembolic stroke has been developed in the mouse where in situ microinjection of thrombin into a branch of the MCA is used to trigger local clot formation (Figure 1; Orset et al., 2007; 2010). A craniectomy is required to expose the MCA branch and insertion of the tip of a thrombin-filled micropipette into the blood vessel. Thrombin injection induces the formation of a clot in situ, a persistent downstream ischaemia and a reproducible cortical infarct. Recanalization of the occluded branch is achievable using intravenous rt-PA (10 mg·kg−1 i.v. administered 20 min later) with restoration of cortical cerebral blood flow and reduced final infarct size. The advantages of this thromboembolic model include more precise control of the location of ischaemia and the ability to visually determine the permanency of vessel occlusion, and successful recanalization in thrombolysis studies. The size and location of cortical ischaemia and infarction is more reproducible than for the autologous clot models and mortality is minimal. Disadvantages include the requirement for a craniotomy, the possibility of spontaneous thrombus disruption and reperfusion with microclot formation (although this can be identified visually and built into exclusion criteria) and the lack of a robust neurological deficit, due to the small cortical lesion.
Outcome measures and endpoints
The major preclinical outcome measures for stroke pharmacotherapy studies are final infarct size (i.e. the amount of permanent brain damage produced by the stroke) and neurological deficit (generally sensorimotor deficits induced by MCAO). The latter is the more clinically relevant but also the more challenging to accurately assess in animal models as these deficits can rapidly resolve in rodent models. For flow enhancing and thrombolytic drugs, successful recanalization and the quality of reperfusion are also assessed using CBF techniques.
Infarct volumes are quantified from histologically stained brain sections (e.g. haematoxylin and eosin, cresyl violet or tetrazolium salts such as TTC) or non-invasively with T2-weighted MRI (Durukan and Tatlisumak, 2007; Sommer, 2010; see Figure 2). The infarct develops over a number of hours (permanent MCAO) or days (transient MCAO). Consequently, it is fundamentally important in drug efficacy studies that the time point at which the measurement is made is late enough for the infarct to have fully evolved. Otherwise, drugs that delay rather than stop the progression of ischaemic damage could be wrongly assessed as neuroprotective. Infarct area is first quantified on sufficient numbers of coronal sections throughout the MCA territory to allow accurate determination of infarct volume (e.g. eight slices described in Osborne et al., 1987 for proximal MCAO induced by electrocoagulation). However, since brain oedema also develops within and around the infarct over the first 2–3 days post-stroke, calculations should be corrected for the space-occupying effect of brain oedema, to avoid overestimation of infarct volume. Corrections for brain swelling are made using published formulae (e.g. Swanson et al., 1990). Infarct volume is assessed indirectly as the volume of the contralateral hemisphere minus the non-infarcted volume of the ipsilateral hemisphere and is based on the assumption that oedema develops almost exclusively within the infarct. However, there are limitations associated with this type of correction. Tissue sections examined at the peak of stroke-induced oedema clearly show brain swelling in both infarct and surrounding peri infarct tissue. Therefore, infarct volume correction using these methods may still be confounded by oedema (Dirnagl, 2010). Alternative approaches to correct for oedema involve identifying ischaemic damage on tissue sections and transcribing the information onto line diagrams of standard size from a stereotaxic atlases (Osborne et al., 1987).
Figure 2.
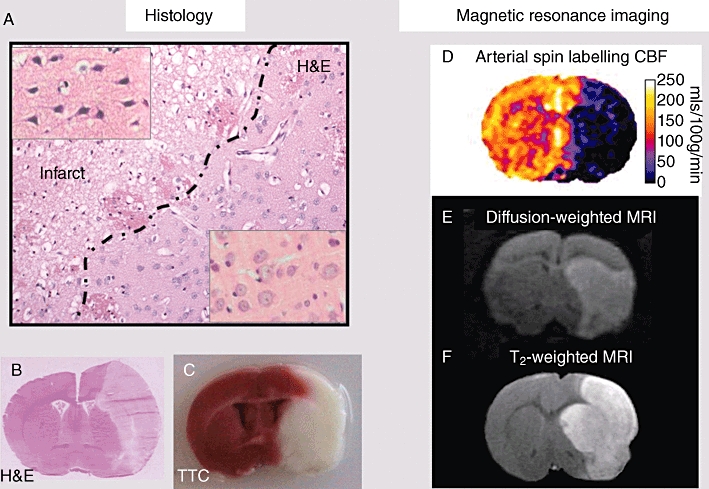
Histology and MRI measures of outcome. (A) Haematoxylin and eosin (H&E)-stained section illustrating the ability to discriminate between irreversibly damaged and morphologically normal tissue in perfusion-fixed paraffin-embedded tissue. With this level of tissue processing, irreversible ischaemic damage is distinguished from changes in neuronal morphology and background neurophil that becomes irregular and vacuolated as early as 4 h after permanent MCAO. Higher magnification inset panels show the characteristic morphology of dark, shrunken, pyknotic, ischaemically damaged neuronal cell bodies (top left), compared with the pale-stained, round morphology of normal neuronal cell bodies (bottom right). (B) By 24 h after permanent MCAO, a low-magnification image of a fresh frozen cryostat section stained with H&E will clearly display the boundary between infarct (area of palor on RHS of section) and non-ischaemic tissue. Note also the brain swelling evident in the ipsilateral hemisphere. (C) By 24 h after permanent MCAO, TTC staining of fresh tissue slices will also reveal a clear boundary between infarct (white) and non-ischaemic tissue (red). (D–F) These MRI scans are non-invasive and do not require administration of contrast agents. (D) Arterial spin labelling provides serial scans of cerebral blood flow to map the severity and location of ischaemia. (E) DWI provides serial scans of ischaemic injury (hyperintensity), which in the first hours after stroke may be reversible on reperfusion. (F) T2-weighted imaging identifies infarct (hyperintensity) from 24 h post stroke.
Infarct size can be expressed either as an absolute volume in mm3 or as a relative value (e.g. percentage of the contralateral hemisphere, or brain volume). Relative values allow more straightforward comparison between studies as absolute brain size will vary depending on animal sex and age. Tissue dehydration and processing also affects brain size with different techniques causing markedly different degrees of shrinkage of the tissue (e.g. greater in paraffin embedded that in frozen tissue). For chronic studies, where rodents survive >7 days post-stroke, the removal of dead tissue by macrophages and microglia results in the infarct being gradually replaced by a fluid-filled cyst. The difference between the volume of the remaining ipsilateral and the contralateral hemisphere provides an alternative measurement of tissue loss as a consequence of the stroke.
Diffusion-weighted MRI is non-invasive and is particularly useful for serial scanning in the acute stroke period (first 3–4 h post-stroke). Affected tissue appears hyperintense, allowing tracking of the evolution of ischaemic injury and the consequences of therapeutic intervention. However, acute hyperintensity on diffusion-weighted imaging (DWI) scans does not signify irreversible ischaemic damage as this abnormality can disappear on early reperfusion of ischaemic tissue. Following serial scanning, and anaesthetic withdrawal, animals can be recovered for behavioural assessment of neurological deficits at later time points and re-anaesthetized for assessment of final infarct with T2-weighted MRI (Ebisu et al., 2001).
Functional outcome represents an essential component of the preclinical testing of drugs targeting the acute (neuroprotection) and chronic (repair and recovery) stages of stroke. Testing functional outcome is extremely time-consuming and labour-intensive. It is still an evolving science with groups developing new and modifications of existing tests, with no consensus as yet on a prescribed battery of tests to determine stroke outcome. Not only are there many different tests being used, but there is also no consistency in the number and frequency of tests applied within a study. The current lack of consensus is a weakness in the field that needs to be addressed if we are to improve the ability of animal models to predict therapeutic efficacy in man. There has to be agreement amongst preclinical researchers and guidelines drawn up on the specific sensorimotor tests that offer the best opportunity to identify long-term deficits in rodents, the optimum (i.e. minimum) number required and the frequency with which the animals should be tested.
The range of behavioural tests developed for quantifying the severity of the sensorimotor deficit and the extent of subsequent recovery are covered in recent reviews (see Schallert, 2006; Metz, 2010). Specific tests, such as the Morris water maze, are used to determine cognitive deficits, which may arise when artery territories other than the MCA are affected, and more general neurological scoring systems have been developed to provide an overall score of the animal's condition. They range from simple 0–3 point scores based on forelimb flexion, resistance to lateral push and circling (Bederson et al., 1986) to more detailed (3–18 point or 21 point) scoring systems incorporating sensory and motor assessments (Garcia et al., 1995; Hunter et al., 2000). However, scoring systems have limited value and tend to be either fast to carry out but limited in ability to differentiate between different levels of deficit or time-consuming and over-detailed, with some of the component parts contributing little to the sensitivity of the method. Hence, each group tends to apply further modifications to the published systems to reduce the time required to score each animal or to increase sensitivity to pick up a deficit. Specific behavioural tests assessing skilled motor function appear more informative and clinically relevant. The most commonly used tests include sensorimotor asymmetry (sticky label or dot test), and fore/hindlimb use (e.g. cylinder test, ledged/tapered beam, horizontal ladder walking task, Figure 3). Animals are assessed for ability to undertake and complete the tasks (e.g. time taken and order in which sticky labels are removed from the affected and normal forelimbs), scored for preference of use of uninjured versus injured limb (e.g. cylinder test) or number of foot faults made in completing a motor task (e.g. tapered beam, ladder walking test). Assessment of functional outcome in rodents is challenging as neurological deficits are often not overt, even when the animal sustains a large infarct. Difficulties may be encountered in animal compliance to undertake the task, and the deficit may be short-lived, as rodents often exhibit learned compensatory behaviour that may be wrongly identified as recovery. However, chronic deficits post stroke have been identified with behavioural testing and used to assess the effects of therapy on functional outcome. For example, the whiskers reflex, rotameter test with amphetamine and bilateral asymmetry (sticky label) test continue to detect sensorimotor deficits out to 12 weeks from stroke onset (60 min ischaemia using the intraluminal thread model; Modo et al., 2000) and the sticky label and rotameter tests were sensitive enough to detect improvements in sensorimotor deficits following stem cell injection into the putamen at 4 weeks post-stroke (Stroemer et al., 2009).
Figure 3.

Behavioural tests in rodent stroke models. Illustrations of four commonly used sensorimotor tests. The cylinder test reveals forelimb preference when the animal rears to explore its environment by making forelimb contact with the cylinder walls. The horizontal ladder and tapered beam tests reveal foot faults as the animal traverses the ladder, which has irregularly spaced rungs, or the beam that gets gradually narrower as the animal approaches its home cage.
CBF measurement is useful for determining blood vessel recanalization in thrombolytic studies, identifying drug effects on stroke severity or quality of reperfusion and for confirming comparable severities of ischaemic insult between groups prior to drug administration. The main techniques employed in preclinical research are laser Doppler flowmetry and hydrogen clearance, which provide good temporal resolution, [14C] iodoantipyrine and [99 m Tc] HMPAO autoradiography, terminal techniques with good spatial resolution and MRI perfusion-weighted imaging (PWI) that provides spatial and temporal information on CBF. Laser Doppler flowmetry probes are placed on the surface of the skull or stereotaxically implanted into discrete neuroanatomical sites to provide a semiquantitative read out of change in blood flow over time, but data are generated in relative (flux), not absolute units with data restricted to a limited volume of tissue around the probe tip. Autoradiographic techniques provide fully quantitative ([14C]-iodoantipyrine) or semiquantitative ([99 m Tc] HMPAO) information on CBF throughout the brain but restricted to a single time point. Non-invasive MRI perfusion techniques such as arterial spin labelling require no exogenous contrast agent and can be calibrated to provide fully quantified serial maps of CBF (Figure 2D).
The importance of the ischaemic penumbra in preclinical and clinical stroke research
Occlusion of a cerebral artery causes a severe reduction in CBF within the brain territory the occluded artery normally supplies. However, because the territories of the major cerebral arteries are linked via collateral blood vessels (anastomoses), there is heterogeneity of tissue perfusion within the ischaemic region. Tissue furthest from collateral supply and exposed to the most severe reduction in CBF (<20% of baseline) is referred to as the ‘ischaemic core’ and becomes irreversibly damaged within minutes as ATP supplies are rapidly exhausted. Surrounding the ischaemic core is a border zone where collateral flow results in a less severe reduction in CBF. However, CBF levels are not sufficient to sustain normal function. This zone, referred to as the ‘ischaemic penumbra’, has preserved structural integrity but a limited lifespan (of hours), due to the inadequate supply of oxygen and glucose, limiting its capacity to generate ATP. It is referred to as ‘tissue at risk’ and ‘potentially salvageable’ as prompt restoration of CBF leads to penumbral tissue survival and recovery of function (Symon, 1987; Heiss, 2010). Without restoration of blood flow, penumbral tissue gradually becomes incorporated into the irreversibly damaged ischaemic core. Penumbral tissue, detected and followed over time using MRI perfusion/diffusion (PWI/DWI) mismatch, becomes gradually incorporated into the ischaemic core over a period of 4–6 h in rodent permanent MCAO models (Meng et al., 2004; McCabe et al., 2009). Penumbra therefore represents the therapeutic target for acute stroke therapies.
In the past, clinical trials were compromised by the lack of penumbra imaging, patients being recruited on the basis of time from stroke onset, rather than detection of potentially salvageable tissue. This combined with unrealistically long therapeutic time windows meant inclusion of patients with no remaining penumbra who were unlikely to benefit from acute stroke therapies, diminishing the power to identify effective drugs.
The amount of salvageable penumbra varies widely among acute stroke patients. Some have no remaining penumbra (or chance to benefit from treatment) even within a few hours of stroke, while others may have large amounts of penumbra persisting far longer than average. Consequently, some drugs that demonstrated preclinical efficacy but subsequently failed in clinical stroke trials may have had clinical efficacy that was missed due to the lack of appropriate patient selection (e.g. inclusion of patients with large strokes and no remaining penumbra, or small strokes). In clinical trials, using very simple brain scanning, the thrombolytic rt-PA reduced death and disability when given within 4.5 h of stroke, with probability of benefit declining rapidly with time thereafter (NINDS & Stroke rt-PA Stroke Study Group, 1995; Hacke et al., 2008; Lees et al., 2010). However, with more sophisticated imaging, targeting rt-PA treatment to those patients with persisting penumbra, the time frame for benefit could be potentially extended at least out to 6 h, since outcomes in the 3–6 h window with MRI selection did not differ from treatment within 3 h (Kohrmann et al., 2006).
PET imaging (CBF, CMRO2, OEF) is also used to detect penumbra, but low availability, high cost and poor spatial resolution limit its utility for routine clinical use and preclinical rodent studies. MRI perfusion/diffusion mismatch and CT perfusion imaging are being validated for penumbra detection in animal and clinical scanners and will be key in identifying effective acute stroke therapies in the future. MRI provides not only longitudinal data on survival of penumbra but also complementary multiparametric data on the evolution of stroke (Table 3). However, as for the induction of stroke, the use of anaesthesia to limit movement during scanning has to be acknowledged as a limitation of MRI-based stroke research in animals.
Table 3.
MRI techniques used in preclinical stroke research
| MRI technique | Potential use in preclinical stroke research |
|---|---|
| MR Angiography | Occlusion/patency status of the targeted blood vessel |
| Perfusion-weighted imaging PWI | Serial CBF maps reveal severity and location of ischaemia, and any spontaneous or induced reperfusion or drug-induced changes in CBF |
| Diffusion-weighted imaging DWI | Serial DWI and ADC maps reveal acute and evolving brain injury, brain swelling |
| PWI/DWI mismatch | Serial assessment of penumbra and its incorporation into ischaemic core over time. |
| Functional MRI (fMRI) combined with a stimulus such as forepaw stimulation | Serial fMRI studies combined with a specific stimulus will reveal ischaemia-induced loss of function in response to the stimulus, any compensatory changes or subsequent return of function |
| Contrast-enhanced T1-weighted imaging | Blood-brain barrier breakdown |
| T1, T2 and T2*-weighted imaging | Brain haemorrhage |
| Diffusion tensor imaging –DTI | White matter damage |
| T2-weighted imaging | Infarct, brain swelling |
ADC, apparent diffusion coefficient.
Additional confounding factors and general limitations of stroke studies in rodents
Generally, in neuroprotection studies, the drug under test is administered either before, or within the first 1–2 h post-stroke, in order to provide the best opportunity to identify efficacy. Although this provides a good first step in identifying promising drugs, there are clear limitations in relying on this type of study design to identify drugs/therapies that have the potential to translate successfully to the clinic. The available time window for delaying treatment should also be determined.
In the past, most drug efficacy studies were carried out on inbred young, healthy male animals expressing none of the comorbidities commonly associated with stroke. The use of such animals limits costs and helps control variability in outcome measures such as infarct size and sensorimotor deficits. The failure of drugs, tested only in healthy animals, to show efficacy in clinical trials has challenged the relevance of these stroke models and highlighted the need for additional preclinical efficacy studies in aged (i.e. ∼18 month old for normotensive rat), and female animals, and in animals with relevant comorbidities (i.e. hypertension, diabetes, obesity, infection, inflammation and atherosclerosis). However, it is important to recognize that in addition to cost, stroke severity, morbidity and mortality are likely to be increased in these models. The severity of the ischaemic insult should therefore be adjusted to a level where morbidity and mortality are within ethically acceptable levels. Equally important for stroke pharmacotherapy studies is the fact that the amount of penumbra is also likely to be less than in young, healthy animals. This will need to be incorporated into power analysis for sample size calculation so that studies are not underpowered to detect drug efficacy. A range of rodent disease models incorporating comorbidities and risk factors are available (see McColl et al., 2010): (i) hypertension: genetically determined hypertensive strains such as the spontaneously hypertensive (SHR), spontaneously hypertensive stroke-prone rat (SHRSP, also insulin resistant), and models of induced hypertension such as the DOCA salt rat; (ii) diabetes: models displaying diabetes/hyperglycaemia include the streptozotocin and biobreeding (BB) rat models and the non-obese diabetic (NOD) mouse models of type-1 diabetes, Zucker and Goto-Kakizaki rat models of type 2 diabetes and the fructose fed rat model of metabolic syndrome (insulin resistance, hyperinsulinemia and hypertension); (iii) obesity: diet-induced obesity can be generated with high-fat and high-sugar diets in Sprague–Dawley and Long Evans rats and C57B16/J mice. Models incorporating both diabetes and obesity include the Zucker diabetic fatty rat, the db/db (spontaneous mutation in leptin receptor gene) and ob/ob (spontaneous mutation in Lep gene encoding the protein leptin) mouse models; and (iv) atherosclerosis: genetically modified mouse models are most prevalent. The early foam cell/fatty streak stage of the disease can be induced in wild-type mice but requires aggressive and prolonged (4–5 months) dietary manipulation. Advanced atherosclerosis can be induced in genetically modified murine strains with the addition of dietary modification (e.g. Paigen or Western diets). The best characterized of these are the apolipoprotein E (apoE)−/− and low-density lipoprotein receptor (LDLR)−/− mouse models.
Irrespective of the model to be used, the stroke must be induced in as sterile an environment as possible. Although fairly resistant to infection, wound infections can occur in rodents, particularly in chronic studies. This can be avoided by keeping the operating area clean and as sterile as possible. Surgical sites should be shaved and disinfected, and instruments and occluding devices (e.g. intraluminal filaments, clips, etc.) sterilized prior to use. Surgery-related infections can lead to meningitis and significant recruitment of inflammatory cells into the brain, thereby influencing the severity of the ischaemic insult and increasing the final infarct size. Infections will also increase variability in outcome measures and confound interpretation of drug study results. Since inflammation is also a natural downstream consequence of focal ischaemia (Moskowitz et al., 2010, review), it is important to ensure that any inflammation identified in the brain is a consequence of the ischaemic insult and not an infectious agent introduced as a result of poor aseptic technique.
The lack of white matter in rodents compared to gyrencephalic species and man represents another important limitation. However, small ischaemic lesions can be induced in the larger white matter tracts (e.g. internal capsule) by stereotaxic injection of vasoconstrictor endothelin-1 (Frost et al., 2006; Lecrux et al., 2008). Quantification of white matter damage is achievable using immunohistochemistry (e.g. amyloid precursor protein accumulation at sites of injury, Gentleman et al., 1999; Imai et al., 2002) and MRI (e.g. diffusion tensor imaging, Sotak, 2002).
Finally, it is important to be aware that certain mouse strains display a high frequency of cerebrovascular anomalies leading to an incomplete circle of Willis (Kitagawa et al., 1998; Kelly et al., 2001). This will influence the severity of focal ischaemic insults by compromising collateral flow and represents a major source of variability in outcome measures.
Recommendations for the future
The failure to predict acute neuroprotective drug efficacy from preclinical data has led to the relevance of animal stroke models being questioned. There is some merit in this viewpoint. What are described in this review are NOT in fact rodent models of stroke; they are models of focal cerebral ischaemia. They model the consequences of an ischaemic insult but, in the main, do not recreate the background pathophysiology that would give rise to an endogenous stroke. Therefore, we have to accept the limitations inherent in these models and, follow early proof of efficacy studies in young healthy animals with more rigorous studies in models incorporating age and comorbidities. Only drugs that demonstrate efficacy in a range of these models should be considered for further investigation in larger gyrencephalic models and subsequent translation to clinical trials.
For drugs targeting the brain, ability to cross the intact blood–brain barrier is crucial and should be established in animals before the drug progresses through to human stroke trials.
Translational problems will be reduced with further improvement in preclinical study design and conduct. A number of substandard practices have been identified and recommendations published to improve the quality and reporting of preclinical stroke research (STAIR I, 1999 subsequently updated by Fisher et al., 2009; Macleod et al., 2009; Kilkenny et al., 2010): study design should include randomization and blinding to limit bias and sample size calculations to ensure studies are adequately powered to detect real differences between treatment and control groups. Oedema-corrected infarct size should be measured when the lesion has reached its maximum size and not before (i.e. when the infarct is still evolving). If significant mortality is encountered prior to this time point, the severity of the ischaemic insult should be reduced to limit morbidity and improve survival. Information on inclusion/exclusion criteria, excluded animals and mortality should be included in preclinical stroke publications. Despite the fact that the first STAIR recommendations have now been in press for more than a decade, issues relating to preclinical study design and quality are still evident in the literature. A recent systematic survey examined all cerebrovascular research studies published in the Journal of Cerebral Blood Flow and Metabolism in 2008. ‘Few studies reported a primary research hypothesis, statement of purpose or measures to safeguard internal validity (such as randomization, blinding, exclusion or inclusion criteria). Many studies lacked sufficient information regarding methods and results to form a reasonable judgment about their validity.’ (Vesterinen et al., 2010). The deficiencies identified are in no way exclusive to the Journal of Cerebral Blood Flow and Metabolism. They are evident throughout the neuroscience literature and are just as prevalent, if not more so, for other neurodegenerative diseases (Jucker, 2010; Macleod, 2011; Rooke et al., 2011). The survey by Vesterinen and colleagues exemplifies both the major problem and the achievable solution to the preclinical end of the translational roadblock in stroke research. There is an onus on researchers and manuscript referees to ensure that the basic principles of good study design and reporting are adhered to, to bring the preclinical literature up to the same standards as the clinical literature. Journal editors have the power to improve the standard of preclinical publications and should update instructions to authors to make specific statements on study design mandatory.
Journal editors also have the powers to reduce publication bias, another issue that has influenced our ability to predict drug efficacy from preclinical data. Reluctance to publish negative or neutral drug studies has resulted in overestimation of drug efficacy and has severely limited the ability of preclinical meta-analyses and systematic reviews to predict drug efficacy in man (Dirnagl and Macleod, 2009). However, this failing is now being addressed with some journal editors recognizing the importance of publishing properly conducted studies that fail to identify drug efficacy (e.g. a new Negative Results article type in the Journal of Cerebral Blood Flow and Metabolism, for data that fails to identify a difference between the experimental groups and/or reproduce published findings).
All the focal ischaemia models described require induction of anaesthesia and some surgery, both of which can give rise to variability in outcome in neuroprotection studies. Anaesthesia can affect blood pressure, blood gases and body temperature, all of which influence stroke severity and outcome. In addition, many anaesthetics (e.g. isoflurane, halothane, xenon, propofol, ketamine) have inherent neuroprotective characteristics (Kawaguchi et al., 2005; Traystman, 2010). Duration of anaesthesia should therefore be as short as possible. In studies where the whole experiment is carried out in the anaesthetized animal, physiological monitoring is recommended to ensure physiological parameters stay within normal limits. This will help to control variability in outcome and thereby increase the power to detect a therapeutic effect. Physiological monitoring can also be useful in detecting any drug-induced influences on these parameters that could either limit or enhance efficacy.
Using a similar format to multicentre clinical trials, networks of preclinical researchers establishing and using standardized techniques and confirming results across centres will achieve much more than individual groups competing for funds. A standardized set of tests for assessment of functional outcome from stroke in each model would be a priority to reach agreement on.
In summary, since the positive rt-PA NINDS trial in 1995, many neuroprotective drugs have been tested in >114 clinical trials (http://www.strokecenter.org/trials/), but none has shown clinical efficacy. This dismal lack of progress has led to a devastating reduction in acute stroke research and development programmes within the major pharmaceutical companies. Published guidelines provide the steps required to address the limitations in study design and reporting on the preclinical side and problems with patient selection for trials on the clinical side of the translational roadblock. Addressing these limitations is eminently achievable with the establishment of preclinical stroke networks, more widespread use of diagnostic imaging and better links between preclinical and clinical scientists. Progress will have significant cost implications, requiring greater numbers of animals (to include aged and comorbidities) and increased use of both preclinical and clinical imaging. However, identification of just one neuroprotective/neurorepair therapy with clinical efficacy for stroke would more than justify the costs, providing an alternative to rt-PA for the majority of patients who are not currently treated and reversing the roadblock by restoring confidence in the pharmaceutical industry for further drug development.
Acknowledgments
Research in the author's laboratory is financially supported by the Medical Research Council, Wellcome Trust, Chief Scientist Office and Neurosciences Foundation.
Glossary
Abbreviations
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- CBF
cerebral blood flow
- CMRO2
cerebral metabolic rate of oxygen
- DWI
diffusion-weighted imaging
- HMPAO
hexamethylpropyleneamine oxime
- MCAO
middle cerebral artery occlusion
- OEF
oxygen extraction fraction
- PET
positron emission tomography
- PWI
perfusion-weighted imaging
- rt-PA
recombinant tissue-type plasminogen activator
- TTC
triphenyltetrazolium chloride
Conflict of interest
There are no conflicts of interest.
References
- Aspey BS, Cohen S, Patel Y, Terruli M, Harrison MJ. Middle cerebral artery occlusion in the rat: consistent protocol for a model of stroke. Neuropathol Appl Neurobiol. 1998;24:487–497. doi: 10.1046/j.1365-2990.1998.00146.x. [DOI] [PubMed] [Google Scholar]
- Asano T, Ikegaki I, Suzuki Y, Satoh S-I, Shibuya M. Endothelin and the production of cerebral vasospasm. Biochem Biophys Res Comm. 1989;159:1345–1351. doi: 10.1016/0006-291x(89)92258-4. [DOI] [PubMed] [Google Scholar]
- Back T, Otto D, Kittner D, Schuler OG, Hennerici MG, Mennel H-D. Failure to improve the effect of thrombolysis by memantine in a rat embolic stroke model. Neurol Res. 2007;29:264–269. doi: 10.1179/174313206X154012. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Braun JS, Prass K, Dirnagl U, Meisel A, Meisel C. Protection from brain damage and bacterial infection in murine stroke by the novel caspase-inhibitor Q-VD-OPH. Exp Neurol. 2007;206:183–191. doi: 10.1016/j.expneurol.2007.03.032. [DOI] [PubMed] [Google Scholar]
- van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104:1613–1620. doi: 10.1172/JCI8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock R, McCulloch J, Graham DI, Lowe D, Chen MH, Teasdale GM. Focal ischemia in the cat: pre-treatment with a competitive NMDA receptor antagonist, D-CPP-ene. J Cereb Blood Flow Metab. 1990;10:668–674. doi: 10.1038/jcbfm.1990.120. [DOI] [PubMed] [Google Scholar]
- Busch E, Krüger K, Hossmann K-A. Improved model of thromboembolic stroke and rt-PA induced reperfusion in the rat. Brain Res. 1997;778:16–24. doi: 10.1016/s0006-8993(97)01008-1. [DOI] [PubMed] [Google Scholar]
- Chen F, Suzuki Y, Nagai N, Sun X, Wang H, Yu J, et al. Microplasmin and tissue plasminogen activator: comparison of therapeutic effects in rat stroke model at multiparametric MR imaging. Radiology. 2007;244:429–438. doi: 10.1148/radiol.2442061316. [DOI] [PubMed] [Google Scholar]
- Coyle P, Jokelainen PT. Differential outcome to middle cerebral artery occlusion in spontaneously hypertensive stroke-prone rats (SHRSP) and Wistar Kyoto (WKY) rats. Stroke. 1983;14:605–611. doi: 10.1161/01.str.14.4.605. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ. The neurovascular unit in the setting of stroke. J Int Med. 2010;267:156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Zoppo GJ, Saver JL, Jauch EC, Adams HP., Jr Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke. 2009;40:2945–2948. doi: 10.1161/STROKEAHA.109.192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U. Complexities, confounders and challenges in experimental stroke research: a checklist for researchers and reviewers. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 263–277. Springer Protocols Neuromethods 47. [Google Scholar]
- Dirnagl U, Macleod MR. Stroke research at a road block: the streets from adversity should be paved with meta-analysis and good laboratory practice. Br J Pharmacol. 2009;157:1154–1156. doi: 10.1111/j.1476-5381.2009.00211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Duverger D, MacKenzie ET. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, and age. J Cereb Blood Flow Metab. 1988;8:449–461. doi: 10.1038/jcbfm.1988.86. [DOI] [PubMed] [Google Scholar]
- Ebisu T, Katsuta K, Fujikawa A, Aoki I, Umeda M, Naruse S, et al. Early and delayed neuroprotective effects of FK506 on experimental focal ischemia quantitatively assessed by diffusion-weighted MRI. Magn Reson Imaging. 2001;19:153–160. doi: 10.1016/s0730-725x(01)00233-8. [DOI] [PubMed] [Google Scholar]
- European Stroke Organisation (ESO) Executive Committee; ESO Writing Committee. Guidelines for management of ischaemic stroke and transient ischaemic attack 2008. Cerebrovasc Dis. 2008;25:457–507. doi: 10.1159/000131083. [DOI] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. STAIR Group Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost SB, Barbay S, Mumert ML, Stowe AM, Nudo RJ. An animal model of capsular infarct: endothelin-1 injections in the rat. Behav Brain Res. 2006;169:206–211. doi: 10.1016/j.bbr.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, McKenzie JE, Royston MC, McIntosh TK, Graham DI. A comparison of manual and semi-automated methods in the assessment of axonal injury. Neuropathol Appl Neurobiol. 1999;25:41–47. doi: 10.1046/j.1365-2990.1999.00159.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Progress review. Stroke. 1989;20:1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- Haelewyn B, Risso J-J, Abraini JH. Human recombinant tissue-plasminogen activator (alteplase): why not use the ‘human’ dose for stroke studies in rats? J Cereb Blood Flow Metab. 2010;30:900–903. doi: 10.1038/jcbfm.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss W-D. The concept of the penumbra: can it be translated to stroke management? Int J Stroke. 2010;5:290–295. doi: 10.1111/j.1747-4949.2010.00444.x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Butcher SP, Sharkey J. A rat model of endothelin-3-induced middle cerebral artery occlusion with controlled reperfusion. Brain Res. 1999;843:105–111. doi: 10.1016/s0006-8993(99)01896-x. [DOI] [PubMed] [Google Scholar]
- Hilger T, Blunk JA, Hoehn M, Mies G, Wester P. Characterization of a novel chronic photothrombotic ring stroke model in rats by magnetic resonance imaging, biochemical imaging, and histology. J Cereb Blood Flow Metab. 2004;24:789–797. doi: 10.1097/01.WCB.0000123905.17746.DB. [DOI] [PubMed] [Google Scholar]
- Hunter AJ, Hatcher J, Nelson VP, Irving E, Hadingham SJ, Parsons AA. Functional assessments in mice and rats after focal stroke. Neuropharmacology. 2000;39:806–816. doi: 10.1016/s0028-3908(99)00262-2. [DOI] [PubMed] [Google Scholar]
- Imai H, McCulloch J, Graham DI, Masayasu H, Macrae IM. A new method for the quantitative assessment of axonal damage in focal cerebral ischaemia. J Cereb Blood Flow Metab. 2002;22:1080–1089. doi: 10.1097/00004647-200209000-00005. [DOI] [PubMed] [Google Scholar]
- Imai H, Konno K, Nakamura M, Shimizu T, Kubota C, Seki K, et al. A new model of focal cerebral ischaemia in the miniature pig. J Neurosurg. 2006;104:123–132. doi: 10.3171/ped.2006.104.2.123. [DOI] [PubMed] [Google Scholar]
- Jucker M. The benefits and limitations of animal models for translational research in nuerodegenerative diseases. Nat Med. 2010;16:1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- Kalra L. Stroke rehabilitation 2009: old chestnuts and new insights. Stroke. 2010;41:e88–e90. doi: 10.1161/STROKEAHA.109.572297. [DOI] [PubMed] [Google Scholar]
- Kano T, Katayama Y, Tejima E, Lo EH. Hemorrhagic transformation after fibrinolytic therapy with tissue plasminogen activator in a rat thromboembolic model of stroke. Brain Res. 2000;854:245–248. doi: 10.1016/s0006-8993(99)02276-3. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M, Furuya H, Patel PM. Neuroprotective effects of anesthetic agents. J Anesth. 2005;19:150–156. doi: 10.1007/s00540-005-0305-5. [DOI] [PubMed] [Google Scholar]
- Kelly S, McCulloch J, Horsburgh K. Minimal ischaemic neuronal damage and HSP70 expression in MF1 strain mice following bilateral common carotid artery occlusion. Brain Res. 2001;914:185–195. doi: 10.1016/s0006-8993(01)02801-3. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, et al. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab. 1998;18:570–579. doi: 10.1097/00004647-199805000-00012. [DOI] [PubMed] [Google Scholar]
- Kito G, Nishimura A, Susumu T, Nagata R, Kuge Y, Yokota C, et al. Experimental thromboembolic stroke in cynomolgus monkey. J Neurosci Methods. 2001;105:45–53. doi: 10.1016/s0165-0270(00)00351-4. [DOI] [PubMed] [Google Scholar]
- Kleindorfer D, Lindsell CJ, Brass L, Koroshetz W, Broderick JP. National US estimates of recombinant tissue plasminogen activator use: ICD-9 codes substantially underestimate. Stroke. 2008;39:924–928. doi: 10.1161/STROKEAHA.107.490375. [DOI] [PubMed] [Google Scholar]
- Kohrmann M, Juttler E, Fiebach JB, Huttner HB, Siebert S, Schwark C, et al. MRI versus CT-based thrombolysis treatment within and beyond the 3 h time window after stroke onset: a cohort study. Lancet Neurol. 2006;5:661–667. doi: 10.1016/S1474-4422(06)70499-9. [DOI] [PubMed] [Google Scholar]
- Koizumi J, Yoshida Y, Nakazawa T, Ooneda G. Experimental studies of ischemic brain edema: 1. A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn Stroke J. 1986;8:1–8. [Google Scholar]
- Korninger C, Collen D. Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb Haemost. 1981;46:561–565. [PubMed] [Google Scholar]
- Kuraoka M, Furuta T, Matsuwaki T, Omatsu T, Ishii Y, Kyuwa S, et al. Direct experimental occlusion of the distal middle cerebral artery induces high reproducibility of brain ischemia in mice. Exp Anim. 2009;58:19–29. doi: 10.1538/expanim.58.19. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Xi G, Hua Y, Nagaraja TN, Fenstermacher JD, Keep RF. Development of a rat model of photothrombotic ischemia and infarction within the caudoputamen. Stroke. 2009;40:248–253. doi: 10.1161/STROKEAHA.108.527853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhorne P, Coupar F, Pollock A. Motor recovery after stroke: a systematic review. Lancet Neurol. 2009;8:741–754. doi: 10.1016/S1474-4422(09)70150-4. [DOI] [PubMed] [Google Scholar]
- Lapchak PA. Translational stroke research using a rabbit embolic stroke model: a correlative analysis hypothesis for novel therapy development. Transl Stroke Res. 2010;1:96–107. doi: 10.1007/s12975-010-0018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Chapman DF, Zivin JA. Metalloproteinase inhibition reduces thrombolytic (tissue plasminigen activator)-induced hemorrhage after thromboembolic stroke. Stroke. 2000;31:3034–3039. doi: 10.1161/01.str.31.12.3034. [DOI] [PubMed] [Google Scholar]
- Lecrux C, McCabe C, Weir CJ, Gallagher L, Mullin J, Touzani O, et al. Effects of magnesium treatment in a model of internal capsule lesion in spontaneously hypertensive rats. Stroke. 2008;39:448–454. doi: 10.1161/STROKEAHA.107.492934. [DOI] [PubMed] [Google Scholar]
- Lees KR, Bluhmki E, von Kummer R, Brott TG, Toni D, Grotta JC, et al. Time to treatment with intravenous alteplase and outcome in stroke: an updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet. 2010;375:1695–1703. doi: 10.1016/S0140-6736(10)60491-6. [DOI] [PubMed] [Google Scholar]
- Li F, Omae T, Fisher M. Spontaneous hyperthermia and its mechanism in the intraluminal suture middle cerebral artery occlusion model of rats. Stroke. 1999;30:2464–2470. doi: 10.1161/01.str.30.11.2464. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Lu A, Clark JF, Broderick JP, Pyne-Geithman GJ, Wagner KR, Khatri P, et al. Mechanical reperfusion is associated with post-ischemic hemorrhage in rat brain. Exp Neurol. 2009;216:407–412. doi: 10.1016/j.expneurol.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur K, Lees KR. Advances in emerging therapies. Stroke. 2010;41:e67–e70. doi: 10.1161/STROKEAHA.109.571539. [DOI] [PubMed] [Google Scholar]
- McCabe C, Gallagher L, Gsell W, Graham D, Dominiczak AF, Macrae IM. Differences in the evolution of the ischaemic penumbra in the SHRSP and WKY rat. Stroke. 2009;40:3864–3868. doi: 10.1161/STROKEAHA.109.559021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl B, Howells D, Rothwell N Denes A. Modelling risk factors and confounding effects in stroke. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 93–120. Springer Protocols Neuromethods 47. [Google Scholar]
- Macleod MR. Study quality and publication bias in experimental stroke. 2011. International Stroke Conference presentation available at http://www.camarades.info.
- Macleod MR, Fisher M, O'Collins V, Sena ES, Dirnagl U, Bath PM, et al. Good laboratory practice: preventing introduction of bias at the bench. Stroke. 2009;40:e50–e52. doi: 10.1161/STROKEAHA.108.525386. [DOI] [PubMed] [Google Scholar]
- Macrae IM, Robinson MJ, Graham DI, Reid JL, McCulloch J. Endothelin induced reductions in cerebral blood flow: dose-dependency, time course and neuropathological consequences. J Cereb Blood Flow Metab. 1993;13:276–284. doi: 10.1038/jcbfm.1993.34. [DOI] [PubMed] [Google Scholar]
- Mayzel-Oreg O, Omae T, Kazemi M, Li F, Fisher M, Cohen Y, et al. Microsphere-induced embolic stroke: an MRI study. Magn Reson Med. 2004;51:1232–1238. doi: 10.1002/mrm.20100. [DOI] [PubMed] [Google Scholar]
- Meisel C, Prass K, Braum J, Victorov I, Wolf T, Megow D, et al. Preventive antibacterial treatment improves the general medical and neurological outcome in a mouse model of stroke. Stroke. 2004;35:2–6. doi: 10.1161/01.STR.0000109041.89959.4C. [DOI] [PubMed] [Google Scholar]
- Meng X, Fisher M, Shen Q, Sotak CH, Duong TQ. Characterizing the diffusion/perfusion mismatch in experimental focal cerebral ischemia. Ann Neurol. 2004;55:207–212. doi: 10.1002/ana.10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz G. Behavioural testing in rodent models of stroke. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 199–212. Springer Protocols Neuromethods 47. [Google Scholar]
- Modo M, Stroemer RP, Tang E, Veizovic T, Sowniski P, Hodges H. Neurological sequelae and long-term behavioral dysfunction in a rat model of stroke. J Neurosci Methods. 2000;194:99–109. doi: 10.1016/s0165-0270(00)00329-0. [DOI] [PubMed] [Google Scholar]
- Mohr JP, Gautier JC, Hier D, Stein RW. Middle cerebral artery. In: Barnett HJM, Stein BM, Mohr JP, Yatsu FM, editors. Stroke, Vol 1, Pathophysiology, Diagnosis and Management. New York: Churchill Livingstone; 1986. pp. 377–450. [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The Science of Stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova S, Moyanova S, Hughes S, Bellyou-Camilleri M, Lee TY, Bartha R. Endothelin-1 induced MCAO: dose dependency of cerebral blood flow. J Neurosci Methods. 2009;179:22–28. doi: 10.1016/j.jneumeth.2009.01.009. [DOI] [PubMed] [Google Scholar]
- O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Orset C, Macrez R, Young AR, Panthou D, Angles-Cano E, Maubert E, et al. Mouse model of in situ thromboembolic stroke and reperfusion. Stroke. 2007;38:2771–2778. doi: 10.1161/STROKEAHA.107.487520. [DOI] [PubMed] [Google Scholar]
- Orset C, Haelewyn B, Vivien K, Vivien D, Young AR. Rodent models of thromboembolic stroke. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 55–70. Springer Protocols Neuromethods 47. [Google Scholar]
- Osborne KA, Shigeno T, Balarsky AM, Ford I, McCulloch J, Teasdale GM, et al. Quantitative assessment of early brain damage in a rat model of focal cerebral ischaemia. J Neurol Neurosurg Psychiatry. 1987;50:402–410. doi: 10.1136/jnnp.50.4.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard K, Sereghy T, Perdersen H, Boysen G. Effect of delayed thrombolysis with rt-PA in a rat embolic stroke model. J Cereb Blood Flow Metab. 1994;14:472–477. doi: 10.1038/jcbfm.1994.58. [DOI] [PubMed] [Google Scholar]
- Perel P, Roberts I, Sena E, Wheble P, Briscoe C, Sandercock P, et al. Comparison of treatment effects between animal experiments and clinical trials: systematic review. BMJ. 2007;334:197. doi: 10.1136/bmj.39048.407928.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen RS, Overgaard K, Pakola S, Boysen G. Effects of microplasmin on recovery in a rat embolic stroke model. Neurol Res. 2008;30:75–81. doi: 10.1179/016164107X181860. [DOI] [PubMed] [Google Scholar]
- Richard MJ, Saleh TM, El Bahh B, Zidichouski JA. A novel method for inducing focal ischemia in vitro. Neurosci Methods. 2010;190:20–27. doi: 10.1016/j.jneumeth.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, McCulloch J. Contractile responses to endothelin in feline cortical vessels in situ. J Cereb Blood Flow Metab. 1990;10:285–289. doi: 10.1038/jcbfm.1990.46. [DOI] [PubMed] [Google Scholar]
- Rooke EDM, Vesterinen HM, Sena ES, Egan KJ, Macleod MR. Dopamine agonists in animal models of Parkinson's disease: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2011;17:313–320. doi: 10.1016/j.parkreldis.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Royal College of Physicians. National Sentinel Stroke Audit Organisational Audit 2010: Royal College of Physicians. 2010. http://www.rcplondon.ac.uk/sites/default/files/2010-stroke-public-report.pdf.
- Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006;3:497–504. doi: 10.1016/j.nurx.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöller K, Zausinger S, Baethmann A, Schmid-Elsaesser R. Neuroprotection in ischemic stroke–combination drug therapy and mild hypothermia in a rat model of permanent focal cerebral ischemia. Brain Res. 2004;1023:272–278. doi: 10.1016/j.brainres.2004.01.094. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Ritchie IM, Kelly PAT. Perivascular microapplication of endothelin-1: a new model of focal cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1993;13:865–871. doi: 10.1038/jcbfm.1993.108. [DOI] [PubMed] [Google Scholar]
- Shigeno T, Teasdale GM, McCulloch J, Graham DI. Recirculation model following MCA occlusion in rats. Cerebral blood flow, cerebrovascular permeability and brain edema. J Neurosurg. 1985;63:272–277. doi: 10.3171/jns.1985.63.2.0272. [DOI] [PubMed] [Google Scholar]
- Sommer C. Histology and infarct volume determination. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 213–226. Springer Protocols Neuromethods 47. [Google Scholar]
- Sotak CH. The role of diffusion tensor imaging in the evaluation of ischemic brain injury – a review. NMR Biomed. 2002;15:561–569. doi: 10.1002/nbm.786. [DOI] [PubMed] [Google Scholar]
- Stroemer P, Patel S, Hope A, Oliveira C, Pollock K, Sinden J. The neural stem cell line CTX0E03 promotes behavioral recovery and endogenous neurogenesis after experimental stroke in a dose-dependent fashion. Neurorehabil Neural Repair. 2009;23:895–909. doi: 10.1177/1545968309335978. [DOI] [PubMed] [Google Scholar]
- Stroke Therapy Academic Industry Roundtable (STAIR) Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- Sugimori H, Yao H, Ooboshi H, Ibayashi S, Iida M. Krypton laser-induced photothrombotic distal middle cerebral artery occlusion without craniectomy in mice. Brain Res, Brain Res Protoc. 2004;13:189–196. doi: 10.1016/j.brainresprot.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Symon L. Recovery of brain function after ischaemia. Acta Neurochir Suppl (Wien) 1987;41:97–103. doi: 10.1007/978-3-7091-8945-0_13. [DOI] [PubMed] [Google Scholar]
- Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1:53–60. doi: 10.1038/jcbfm.1981.6. [DOI] [PubMed] [Google Scholar]
- The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J. 2003;44:85–95. doi: 10.1093/ilar.44.2.85. [DOI] [PubMed] [Google Scholar]
- Traystman RJ. Effect of anesthesia in stroke models. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 121–138. Springer Protocols Neuromethods 47. [Google Scholar]
- Vesterinen HV, Egan K, Deister A, Schlattmann P, Macleod MR, Dirnagl U. Systematic survey of the design, statistical analysis, and reporting of studies published in the 2008 volume of the Journal of Cerebral Blood Flow and Metabolism. J Cereb Blood Flow Metab. 2010;31:1064–1072. doi: 10.1038/jcbfm.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virley D, Hadingham SJ, Roberts JC, Farnfield B, Elliott H, Whelan G, et al. A new primate model of focal stroke: endothelin-1-induced middle cerebral artery occlusion and reperfusion in the common marmoset. J Cereb Blood Flow Metab. 2004;24:24–41. doi: 10.1097/01.WCB.0000095801.98378.4A. [DOI] [PubMed] [Google Scholar]
- Wang R-Y, Wang PS-G, Yang Y-R. Effect of age in rats following middle cerebral artery occlusion. Gerontology. 2003;49:27–32. doi: 10.1159/000066505. [DOI] [PubMed] [Google Scholar]
- Ward NM, Sharkey J, Marston HM, Brown VJ. Simple and choice reaction-time performance following occlusion of the anterior cerebral arteries in the rat. Exp Brain Res. 1998;123:269–281. doi: 10.1007/s002210050569. [DOI] [PubMed] [Google Scholar]
- Watanabe O, Bremer AM, West CR. Experimental regional cerebral ischaemia in the middle cerebral artery territory in primates. Part 1: angio-anatomy and description of an experimental model with selective embolization of the internal carotid artery bifurcation. Stroke. 1977;8:61–70. doi: 10.1161/01.str.8.1.61. [DOI] [PubMed] [Google Scholar]
- Watson BD, Dietrich WD, Busto R, Wachtelms M, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17:497–504. doi: 10.1002/ana.410170513. [DOI] [PubMed] [Google Scholar]
- Wester P, Watson BD, Prado R, Dietrich WD. A photothrombotic ‘ring’ model of rat stroke-in-evolution displaying putative penumbral inversion. Stroke. 1995;26:444–450. doi: 10.1161/01.str.26.3.444. [DOI] [PubMed] [Google Scholar]
- Witte OW. Photochemical and endothelin models of focal brain ischaemia. In: Dirnagl U, editor. Rodent Models of Stroke. New York: Humana Press; 2010. pp. 71–83. Springer Protocols Neuromethods 47. [Google Scholar]
- Yamaguchi S, Yamakawa T, Niimi H. Microcirculatory responses to repeated embolism-reperfusion in cerebral microvessels of cat: a fluorescence videomicroscopic study. Clin Hemorheolo Microcirc. 2000;23:313–319. [PubMed] [Google Scholar]
- Yepes M, Roussel BD, Ali C, Vivien D. Tissue-type plasminogen activator in the ischemic brain: more than a thrombolytic. Trends Neurosci. 2009;32:48–55. doi: 10.1016/j.tins.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Yonas H, Wolfson SK, Dujovny M, Boehnke M, Cook E. Selective lenticulostriate occlusion in the primate. A highly focal cerebral ischemia model. Stroke. 1981;12:567–572. doi: 10.1161/01.str.12.5.567. [DOI] [PubMed] [Google Scholar]
- Yonemori F, Yamaguchi T, Yamada H, Tamura A. Spatial cognitive performance after chronic focal cerebral ischaemia in rats. J Cereb Blood Flow Metab. 1999;19:483–494. doi: 10.1097/00004647-199905000-00002. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chopp M, Zhang RI, Goussev A. A mouse model of embolic focal cerebral ischaemia. J Cereb Blood Flow Metab. 1997;17:1081–1088. doi: 10.1097/00004647-199710000-00010. [DOI] [PubMed] [Google Scholar]
- Zivin JA, DeGirolami U, Kochhar A, Lyden PD, Mazzarella V, Hemenway CC, et al. A model for quantitative evaluation of embolic stroke therapy. Brain Res. 1987;435:305–309. doi: 10.1016/0006-8993(87)91613-1. [DOI] [PubMed] [Google Scholar]