

Abstract
Experimental autoimmune encephalomyelitis (EAE) is the most commonly used experimental model for the human inflammatory demyelinating disease, multiple sclerosis (MS). EAE is a complex condition in which the interaction between a variety of immunopathological and neuropathological mechanisms leads to an approximation of the key pathological features of MS: inflammation, demyelination, axonal loss and gliosis. The counter-regulatory mechanisms of resolution of inflammation and remyelination also occur in EAE, which, therefore can also serve as a model for these processes. Moreover, EAE is often used as a model of cell-mediated organ-specific autoimmune conditions in general. EAE has a complex neuropharmacology, and many of the drugs that are in current or imminent use in MS have been developed, tested or validated on the basis of EAE studies. There is great heterogeneity in the susceptibility to the induction, the method of induction and the response to various immunological or neuropharmacological interventions, many of which are reviewed here. This makes EAE a very versatile system to use in translational neuro- and immunopharmacology, but the model needs to be tailored to the scientific question being asked. While creating difficulties and underscoring the inherent weaknesses of this model of MS in straightforward translation from EAE to the human disease, this variability also creates an opportunity to explore multiple facets of the immune and neural mechanisms of immune-mediated neuroinflammation and demyelination as well as intrinsic protective mechanisms. This allows the eventual development and preclinical testing of a wide range of potential therapeutic interventions.
LINKED ARTICLES
This article is part of a themed issue on Translational Neuropharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-4
Keywords: animal models, autoimmunity, demyelination, experimental autoimmune encephalomyelitis, immunomodulation, multiple sclerosis, neurodegeneration, neuroinflammation, neuroprotection, therapy
Introduction
Multiple sclerosis
Multiple sclerosis (MS) is the prototypical inflammatory demyelinating disease of the central nervous system (CNS). It is estimated to affect up to two million people worldwide and some 100 000 people in the United Kingdom (Compston and Coles, 2008). Its clinical manifestations begin typically in the third and fourth decade of life, and it affects women preferentially, with a female : male ratio approaching 3:1. Thus, MS represents a prime cause of neurological disability in young adults and has wide health, psychological, economical and social consequences.
Clinically, MS manifests itself as neurological deficits that frequently exhibit a relapsing and remitting pattern and can resolve completely or leave residual deficits. The deficits can involve any part of the CNS alone or in combination. Somatosensory, pyramidal-motor and visual manifestations, the latter due either to inflammatory demyelination in the afferent visual pathways (optic neuritis) or in the efferent visual pathways (ocular motility disorders such as internuclear ophthalmoplegia) are among the most common manifestations. Eventually, many people with relapse-onset MS have fewer clinically recognizable relapses and develop a gradual neurological progression.
In terms of the clinical course, there are several MS subtypes: relapsing-remitting MS (RRMS), with relapses (flare-ups) of disease separated by periods without clinical progression; secondary progressive, SPMS, which represents the phase of the disease where a gradual neurological deterioration (progression) follows a period of RR disease; primary progressive, PPMS, affecting approximately 15% of people with MS where the neurological deterioration is present from the onset, most frequently without superimposed relapses. The rare variant where a few acute exacerbations are superimposed on the gradual PPMS-like course is called progressive-relapsing MS (PRMS) (Lublin and Reingold, 1996).
Individuals who have experienced a single typical episode of inflammatory demyelination suggestive of being the first attack of MS but have not had a second event are said to have clinically isolated syndrome (CIS).
There are four key pathological features of MS: (a) inflammation, of complex pathogenesis, which is generally believed to be the main trigger of the events leading to CNS tissue damage in the majority of cases, although recent evidence suggests that initial damage to neuroglial elements can trigger secondary inflammation in some cases (Barnett and Prineas, 2004); (b) demyelination, the hallmark of MS, where the myelin sheath or the oligodendrocyte cell body is destroyed by the inflammatory process; (c) axonal loss or damage; and (d) gliosis (astrocytic reaction to CNS damage). There is a certain degree of remyelination, which offers hope for therapies aimed at enhancing endogenous repair mechanisms in various experimental models (see below) but is partial and its efficiency is limited.
In addition to the clinical heterogeneity there is pathological heterogeneity, in terms of the relative proportion of the above key pathological features and the components of cellular and humoral immune response elements that mediate the inflammation.
The pathological correlate of relapses is inflammation and disruption of the blood–brain barrier (BBB), clinical relapses being thought to correspond to fresh waves of inflammatory cell infiltration in the CNS. The pathological correlate of long-term disability and progression is irreversible axonal loss. The acute MS lesion is characterized by inflammatory infiltrates with various immune cells and active demyelination (macrophages with myelin debris in their cytoplasm); when this lesion becomes chronic, there is significant loss of myelin with few if any inflammatory infiltrates and gliosis, which gives lesions their ‘plaque’ appearance (Charcot, 1868).
Axonal loss is most severe in the chronic plaques, but it is also present in what is known as the normal-appearing white matter (NAWM), or normal-appearing brain tissue (NABT), to take into account pathological changes in the normal appearing gray matter as well (Trapp et al., 1998; Peterson et al., 2001).
Diagnosis
As its name implies, the diagnosis of relapse-onset MS (also known as disseminated sclerosis) requires evidence of dissemination in time and space of the inflammatory lesions. Clinically, this has traditionally meant two or more demyelinating attacks and clinical evidence of two or more parts of the CNS being involved (Poser et al., 1983). The advent of magnetic resonance imaging (MRI), however, has greatly facilitated the diagnosis (and as a consequence the early treatment) of MS. The introduction of the International Panel (MacDonald) diagnostic criteria, which are strongly based on MRI, allows early diagnosis by substituting the appearance of new lesions for the requirement for a second demyelinating event (McDonald et al., 2001; Polman et al., 2005).
The diagnostic hallmark of MS is the presence of hyperintense lesions on T2-weighted images; the typical location is periventricular but posterior fossa, juxtacortical and spinal lesions often coexist. The T2 lesions lack pathological specificity but are very useful for diagnosis. Acute lesions show enhancement after administration of gadolinium, a paramagnetic agent, on T1-weighted images. The pathological substrate of Gadolinium enhancing T1 lesions is inflammatory infiltration with recent breakdown of the BBB (Filippi et al., 2002). Extensive evidence also shows that the brain and spinal cord undergo atrophy in MS, the pathological substrate for which is loss of axons (and myelin) (Lin et al., 2004; Edwards et al., 2007).
The above conventional MRI measures are widely used in clinical trials in MS as reliable outcome measures. In addition, a number of quantitative MRI methods have contributed to further understanding of the pathogenesis of MS. As in pathology, there is ample evidence that the NABT is also abnormal by sensitive MRI metrics. This partially explains the lack of tight correlation between clinical and MRI activity in MS.
MS is undoubtedly an immune-mediated disease with many features consistent with an autoimmune pathogenesis. In addition to its many similarities to experimental autoimmune encephalomyelitis (EAE) (discussed below), its response to immunosuppressive and immunomodulatory treatments (some of which are discussed below) and its association with other autoimmune diseases (Edwards and Constantinescu, 2004; Constantinescu and Gran, 2010), strong evidence in support of its immune mediation comes from genetics. While its association with major histocompatibility complex (MHC) genes has been well known for a long time, recent advances in genome-wide association study methodology has allowed identification of approximately another 16 genes, virtually all of which are immune response genes (International Multiple Sclerosis Genetics Consortium, 2008). Not surprisingly, an enormous amount of work has been invested so far in finding pathogenic immune pathways and immune modulation strategies, both in EAE and in MS, relative to the amount of work aimed directly at neuroprotection, repair or remyelination.
As a result, some immune response–modifying therapies have entered clinical practice (many undergoing successful translation from EAE studies) and have thus revolutionized MS treatment, care and quality of life in the last two decades. Although they are practically entirely aimed at the relapsing stages of the disease where inflammation is a predominant pathogenic mechanism, they have made a major impact (Lim and Constantinescu, 2010b). In the progressive stages of disease, axonal/ neuronal loss partially dissociated from inflammation is more prominent, although low-grade inflammation persists. Immunomodulatory/ immunosuppressive drugs may have a marginal effect against such low-grade inflammation, but overall they have not shown success in reducing progression. Neuroprotective and reparative strategies need to be found for this stages (as well as for PPMS), and, as discussed below, a few studies are promising in EAE, but so far none of these has been translated into MS treatment.
The disease-modifying treatments (DMTs) for MS have been in large part based on the concepts of MS immunopathogenesis. These concepts and consequently the therapeutic targets have evolved with time, and we will soon witness the emergence of a third generation of MS DMT.
A detailed discussion of these established and emerging drugs is beyond the scope of this review. The readers are directed to recent reviews (Lim and Constantinescu, 2010b; Rejdak et al., 2010; Yiu and Banwell, 2010).
The first line of treatment was represented by type 1 interferons (IFN) and glatiramer acetate (GA). Although initial studies showed success both with IFN-alpha and IFN-eta, the established DMT currently is IFN-beta (in several preparations, including IFNβ1a, Rebif and Avonex; and IFNβ1b, Betaferon, Betaseron, Extavia). Type I IFNs are natural antiviral molecules produced with immunoregulatory properties. GA (Copaxone), which was discovered due to studies in EAE (Teitelbaum et al., 1971), is a copolymer of four amino acids present in myelin basic protein, namely glutamic acid, lysine, alanine and tyrosine. All first-generation DMT, while varying in route and frequency of administration and side effect profile, roughly reduce the relapse rate by 30% (or more if given in CIS). They have marginal or no effects in SPMS, PPMS or PRMS.
The currently approved DMT of a second generation is natalizumab (Polman et al., 2006). This is a monoclonal antibody against VLA-4 integrin, which was shown in preclinical studies in EAE to be required for T-cell entry into the CNS (Yednock et al., 1992). Due to success in phase II and III clinical trials, natalizumab was approved and is currently the most potent licenced drug for MS, reducing relapse rate by 70% and new MRI disease activity by 90% (Kappos et al., 2007). Natalizumab has been associated with a severe complication, which prompted its transient removal from the market: progressive multifocal leukoencephalitis (PML), an opportunistic CNS infection with high mortality and morbidity, caused by the JC virus, a human specific polyoma virus (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005).
Mitoxantrone is another drug licenced for MS. It is an anthracene dione used as a cancer chemotherapeutic agent and is also very effective in more aggressive MS, reducing relapses and showing a potential effect against progressive disease. Mitoxantrone has cumulative cardiotoxicity reducing its long-term use and is associated with a risk of promyelocytic leukaemia.
Azathioprine, a less potent immunosuppressive agent, has the advantage of oral administration and is effective in reducing relapse frequency and possibly disease progression (Casetta et al., 2007). It also reduces the number of new brain inflammatory lesions (Massacesi et al., 2005). It is well tolerated and is considered appropriate maintenance treatment for patients with frequent relapses requiring steroids (Casetta et al., 2007).
Drugs that have successfully completed or in phase III studies and are promising DMT in the not so distant future, as well as some drugs successful in phase II studies and undergoing phase III studies are listed in Table 1.
Table 1.
Drugs in current or expected near-future use in MS
| Route of administration in humans | Primary mechanism of action (elucidated via animal studies?) | Secondary/alternative mechanism of action (elucidated via animal studies?) | Animal studies in EAE | Reference | |
|---|---|---|---|---|---|
| Cladribine | Oral | Immune cell depletion, apoptosis, cell cycle arrest; selectivity for lymphocytes | Cytokine suppression; adenosine receptor effects (No) | None; studies in murine leukaemia and cancer xenografts; | (Giovannoni et al., 2010) |
| (yes, in part) | |||||
| Fingolimod | Oral | Sphingosine 1 phosphate partial agonist; blocks leukocyte migration from lymph nodes | Potential remyelination effect (yes) | yes | (Brinkmann et al., 2002); (Kappos et al., 2010); (Cohen et al., 2010) |
| (yes) | (Foster et al., 2007; 2009) | ||||
| Fumarate | Oral | Potential antioxidant; inhibits inflammatory products of immune cells (yes) | Neuroprotective; potential antioxidant effect on other cells including astrocytes and oligodendrocytes (yes) | yes | (Rammohan and Shoemaker, 2010) |
| Alemtuzumab | i.v. | Anti-CD52 monoclonal antibody: depletion of immune cells bearing CD52 surface marker (No) | Immune system reset? (no) | None; transgenic mouse expressing CD52 generated recently | (Coles et al., 2008; Jones et al., 2009); (Hu et al., 2009) |
| Rituximab | i.v. | Anti-CD20 monoclonal antibody: blockade/depletion of CD20+ B cells; (yes, partially) | Antibody depletion (including potentially anti-myelin antibodies); interference with B cell antigen presentation; depletion/blockade of EBV infected cells; preferential sparing of regulatory B cells | Yes (murine equivalent) | (Hauser et al., 2008); (Kap et al., 2010b) |
| (no) | |||||
| Teriflunomide | Oral | Pyrimidine analog (yes*) | Cytostatic effect, preferentially on proliferating lymphocytes | Yes (*parent compound leflunomide) | (Korn et al., 2004); (O'Connor et al., 2006); (Tallantyre et al., 2008) |
| Laquinimod | Oral | (yes) | Cytokine modulation, inhibition of T cell migration, suppression of Th17 cells, induction of Th3 cells (yes) | Yes | (Wegner et al., 2010); (Brunmark et al., 2002); (Yang et al., 2004); (Comi et al., 2008); (Tselis, 2010) |
Slightly further at the horizon are the future cellular therapies. The only such treatment that has entered clinical practice, albeit not in large controlled studies, is haematopoietic stem cell transplantation, thought to represent a drastic form of immunosuppression, which may reset an autoimmune-prone immune system, and has at least theoretical potential for neurorepair (Muraro and Uccelli, 2010). More than 400 patients with MS, in large part SPMS, have received this treatment within or outside of trials. This approach and its relationship with knowledge derived from EAE are discussed later in this review.
The interaction between multiple components of the immune system and all elements of the CNS determine the pathogenesis of MS. The most widely accepted current concepts are schematically represented in Figure 1.
Figure 1.
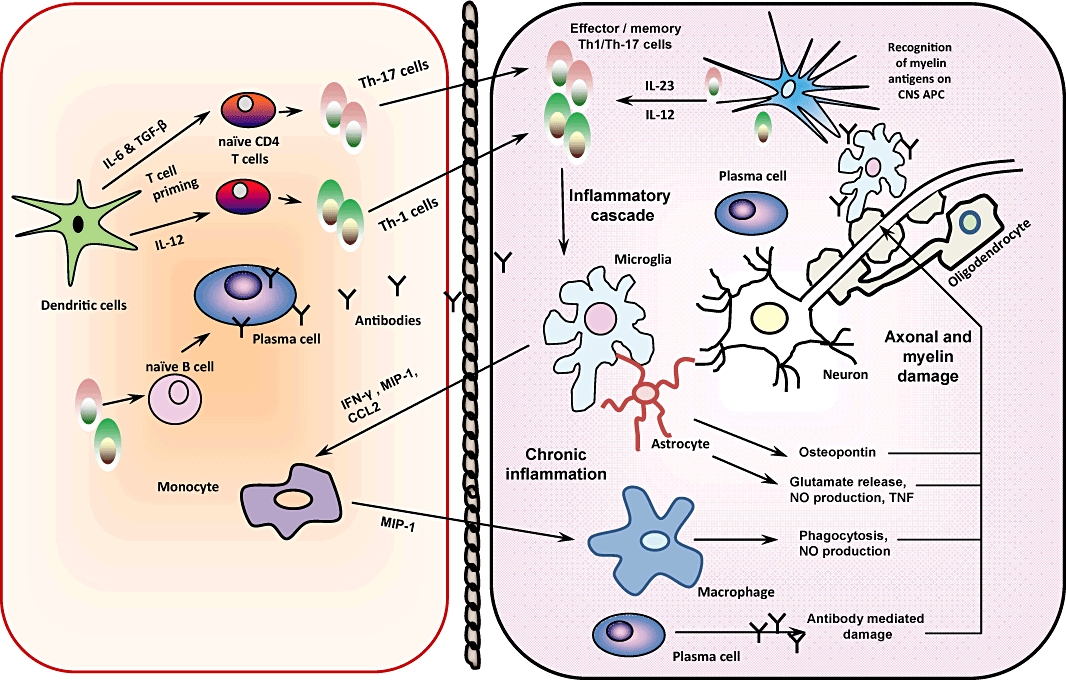
Schematic diagram of some of the key pathological features of EAE pathogenesis. Activated Th1 cells and Th17 cells are thought to be the main culprit in EAE and MS. Th1 are IFN-γ producing, and Th17 are IL-17 producing T lymphocytes. They are primed outside the CNS by dendritic cells, then cross the blood–brain barrier and encounter CNS antigen-presenting cells. They produce inflammatory products and cytokines that damage the myelin and axons. They also activate the resident microglia and produce factors that attract further inflammatory cells to the CNS and perpetuate the inflammatory cascade. Antibodies and B cells can also enter the CNS, and plasma cells produce antibodies within the CNS. Antibody mediated damage contributes to the inflammatory demyelination and neurodegeneration.
Briefly, T cells in the periphery become activated by a viral or another infectious antigen or a superantigen. These show molecular similarity (mimicry) with some CNS antigen (Sospedra and Martin, 2005). These T cells are capable of producing inflammatory cytokines and may be differentiated or have the potential to differentiate on activation into Th1 (producing IFN-gamma) or Th17 cells (IL-17, IL-22, IL-21) or cells producing both (McFarland and Martin, 2007). Activated T cells up-regulate integrins such as VLA-4 and are capable of crossing the BBB. Through the permeabilized BBB, attracted by chemokine release, other immune cells including B cells and monocytes/macrophages migrate into the CNS. There, they encounter the cognate antigen, probably derived from myelin antigen, presented by CNS resident or immigrant antigen-presenting cells (APC). These can be macrophages/microglia and in certain instances dendritic cells or astrocytes. On encountering the antigen, such autoreactive T cells are reactivated and differentiate, producing their signature cytokines, which activate the neighbouring immune or neural cells and attract further inflammatory cells into the CNS. Of these, it is especially activated macrophages that are thought to indirectly and directly damage the CNS. Myelin is phagocytosed by macrophages (Barnett et al., 2006). Elements of the humoral immune response and soluble mediators also contribute to the pathology, via complement activation, direct cytokine cytotoxicity, nitric oxide, reactive oxygen and nitrogen species (Hemmer et al., 2006). Plasma cells produce antibodies, which can bind and activate complement or induce antibody-dependent cytotoxicity. Th2 cells (producing IL-4) may enhance antibody production. CD8 (cytotoxic) T cells may enhance the damage through further cytokine production as well as granzyme and perforin production and can directly transect axons (Fletcher et al., 2010). The resolution of inflammation, which can be partial and subject to recrudescence, occurs when anti-inflammatory cytokines (e.g. IL-10) and other immunoregulatory mechanisms such as regulatory T cells (Treg) or NK cells come into play. The consequence is that the myelin is destroyed and typically lacks full regeneration potential, especially after repeated injury, and the axons degenerate, in part because they are devoid of myelin and more exposed and deprived of trophic support, in part through wallerian degeneration and metabolic injury (Piaton et al., 2009).
EAE
Many elements of this cascade of events have been identified, tested or confirmed in EAE. From the pathogenesis point of view, therefore, EAE is a good model for studying MS mechanisms, even more so than for testing or developing drugs (Farooqi et al., 2010).
A major difference between MS and EAE is that the latter requires an external immunization step to develop, whereas in humans, the sensitization to autoantigens is obviously not artificially induced (Gran et al., 2007). Sensitization to myelin antigens in EAE typically occurs through the use of adjuvant, usually containing bacterial components highly capable of activating the innate immune system via pattern recognition receptors (Libbey and Fujinami, 2010). In EAE, the inducing antigens are known, whereas in MS, there is no unique identified antigen. Thus, important differences between these conditions may be due to how autoreactive T cells are primed and activated.
More recently, however, a refined model of pathogenesis has been put forward by t Hart and colleagues, in which three rather than two compartments are considered critical to EAE and MS pathogenesis (t Hart et al., 2009). The hypothesis was derived from work in a non-human primate model of MS (Kap et al., 2010a). In this model, autoreactive T cells are actively induced by peripheral immunization (occurring in lymph nodes and spleen, ‘afferent compartment’) with antigen emulsified in an adjuvant (in EAE) or by infection with an as yet unidentified pathogen (in MS). Such T cells collect in the spleen (Flugel et al., 2001) before migrating to the ‘target compartment’ (the CNS), where they recognize their cognate antigen on local APCs, are activated and start an inflammatory cascade leading to tissue injury. Tissue debris that are cleared from the CNS are then found in APCs within a third, ‘CNS draining compartment’, comprising the cervical and lumbar lymph nodes and the spleen (t Hart et al., 2009). Draining is thought to occur by means of interstitial fluids and/or the cerebrospinal fluid. T-cell responses are then triggered in the lymph nodes of the third compartment leading to the generation of new autoreactive T-cell specificities. Such cells are then released into the afferent compartment, where they can either mitigate or exacerbate the ongoing autoimmune reaction (t Hart et al., 2008; 2009). Similar observations were reported in a transgenic mouse model of EAE (Furtado et al., 2008). Data show that APCs in the cervical lymph nodes that contain myelin breakdown products have an anti-inflammatory phenotype, whereas APCs containing neuronal antigen such as the light chain of neurofilament appear to have a pro-inflammatory phenotype. The anti-inflammatory nature of myelin-containing APCs is consistent with data showing a generally anti-inflammatory activity of myelin-fed macrophages. Both in EAE and in MS, increased numbers of professional APC-containing neuroantigens are seen in the CNS-draining lymphoid organs.
The three-compartment model of EAE pathogenesis can be extrapolated to MS immunopathogenesis. However, a specific peripheral trigger of autoreactive T cells in MS has been elusive. Thus, in accordance with the primary lesion hypothesis (Wilkin's hypothesis) (Wilkin, 1990), the attractive mechanism has been proposed whereby initial activation of encephalitogenic T cells in MS does not occur in the afferent compartment, such as in the EAE model, but in the draining compartment.
A further refinement of the primate model is the recent development of a marmoset model in which EAE can be induced by incomplete, rather than complete Freund's adjuvant (i.e. without Mycobacterium tuberculosis, a powerful inducer of pro-inflammatory cytokines) (Kap et al., 2008). Although the use of incomplete Freund's adjuvant still involves external immunization, it more accurately reflects the human immune response. We discuss EAE in primates and its role in MS treatment development below.
EAE induction
EAE is primarily used as an animal model of autoimmune inflammatory diseases of the CNS, and it resembles MS, the prototypical such disease, in many respects (Gold et al., 2006; Steinman and Zamvil, 2005; 2006; Farooqi et al., 2010). Some models are more similar to other, less common inflammatory CNS disorders, such as the monophasic acute disseminated encephalomyelitis (ADEM) or neuromyelitis optica (NMO, Devic's disease) (Furlan et al., 2009). Increasingly, the use of EAE has expanded considerably beyond the laboratory study of MS and the development of MS therapeutics. EAE has also become a very well characterized model for organ-specific autoimmune disease in general. Indeed, several recent first reports of key novel functions of immunologically important molecules, or of a novel knockout mouse were published with EAE data as the in vivo validation model. Examples include the discovery of ROR-γ (RORC) as a master transcription factor for Th17 cell development (Ivanov et al., 2006), the identification of the aryl hydrocarbon receptor (AHR) as an essential component in the development of both Treg and Th17 responses (Veldhoen et al., 2008) and the differential role of the related molecules IL-12 and IL-23 in the susceptibility to autoimmune demyelination (Becher et al., 2002; Gran et al., 2002; Cua et al., 2003).
EAE was first described over 75 years ago (Rivers et al., 1933; Rivers and Schwentker, 1935) and is still a popular and widely used model. A PubMed search [‘(experimental) autoimmune encephalomyelitis/encephalitis OR EAE OR experimental allergic encephalomyelitis/encephalitis’] identifies over 9000 citations, of which almost 6000 since 1990. Like all animal models, EAE has limitations when applied to human disease (Sriram and Steiner, 2005;Gold et al., 2006; Steinman and Zamvil, 2006; Furlan et al., 2009; Farooqi et al., 2010); it is very heterogeneous in terms of induction methods, clinical and pathological features, and amenability to treatment, all of which add to its complexity. Therefore, its usefulness is critically dependent on using appropriate models to answer the specific scientific or clinical questions that are being addressed. If, for example, the pathogenesis of spontaneous recurrence of inflammation is studied, a relapsing rather than a monophasic EAE model should be used (Baker et al., 1991; Miller et al., 2007b).
Table 2 shows a list of potential uses of EAE to explore, develop and test general neuroscience and immunology concepts, developing general therapeutic strategies, and the potential uses of EAE to answer question specifically related to inflammatory demyelination and its consequences.
Table 2.
Uses of EAE
| General | Neuroprotective strategies |
| Immunosuppressive drugs | |
| Neurotransmitters in inflammation | |
| Channel function during inflammation, demyelination and remyelination | |
| Immune responses in immunologically privileged sites | |
| Effects of cytokines in the CNS | |
| Blood–brain barrier function and dysfunction | |
| Immunological tolerance | |
| T-cell receptor restriction | |
| Epitope spreading | |
| Regulatory T cells | |
| Specific to CNS inflammation, demyelination | Development, testing and validation of MS drugs: bioavailability, pharmacokinetics, preclinical efficacy, safety |
| Development and testing of drugs with possible dual action on the CNS and immune system, for development of drugs with both immunomodulatory and neuroprotective properties | |
| Gene expression during demyelination a remyelination | |
| Gene expression profiling for discovery and validation of new targets for MS treatment | |
| Expression of genes associated with sparing of CNS elements or resolution of inflammation | |
| Mechanisms of axonal damage and loss | |
| Study of symptoms and symptomatic treatment for MS: e.g. spasticity and anti-spastic drugs, bladder dysfunction, pain |
In terms of providing clues to the MS pathogenesis and allowing development of treatments, a most exciting and rewarding approach was that of the bidirectional translational studies pioneered by the group of L Steinman (Lock et al., 2002; Robinson et al., 2003; Steinman and Zamvil, 2003; Kanter et al., 2006; Han et al., 2008). This involved gene expression profiling in MS brain, identification of a number of plausible novel targets and then testing and validating these targets in EAE. Several such targets have been identified in this fashion, some supported by small previous studies in EAE, and these targets have a potential for being translated into MS treatment soon. Such targets include osteopontin, platelet-activating factor receptor, histamine receptors and alpha-B crystallin (Lock et al., 2002; Han et al., 2008; Steinman, 2009).
Induction of EAE in different strains of rodents and monkeys
EAE can be induced in a multitude of species and strains. Interestingly, humans were the first species where sensitization with nervous tissue led to an inflammatory demyelinating CNS disease. This occurred as a rare complication of rabies vaccination with virus grown on rabbit spinal cord (Sabin and Wright, 1934). It was subsequently shown that the resultant encephalomyelitis was not due to rabies, but to an autoimmune response triggered by the spinal cord contaminant of the vaccine. Rivers in 1933 developed EAE in an attempt to understand better the pathogenesis of this post-vaccinal encephalomyelitis (Zinsser and Tang, 1926; Rivers and Stewart, 1928; no authors listed, 1931; Rivers et al., 1933; Rivers and Schwentker, 1935).
Since then, EAE has been induced in a variety of rodents and monkeys, providing models of acute monophasic, relapsing–remitting and chronic progressive CNS inflammation. Optic neuritis, often a first sign of autoimmune demyelination, has also been modelled (Rao et al., 1977; Raine et al., 1980; Mendel et al., 1995; Kornek et al., 2000; Pomeroy et al., 2005; Gold et al., 2006)
‘Active’ EAE is induced by immunization with CNS tissue or myelin peptides, such as myelin basic protein (MBP) and proteolipid protein (PLP) in CFA, with high incidence of disease induced in susceptible animal strains (Stromnes and Goverman, 2006a). Disease onset typically occurs after 9–12 days and is followed by variable clinical and pathological outcomes as mentioned above. For example, acute self-limiting or chronic relapsing–remitting disease/ progressive disease can be induced in guinea pigs by immunization with MBP or with CNS tissue homogenate respectively (Raine et al., 1977; Alvord et al., 1985). By contrast, ‘passive’ or adoptive–transfer EAE (AT-EAE) can be induced in recipient animals by transferring pathogenic, myelin-specific CD4 T cells generated in donor animals by active immunisation (Stromnes and Goverman, 2006b). The latter type of EAE was instrumental in establishing the key role of myelin-reactive T cells in disease pathogenesis (Pettinelli and McFarlin, 1981). AT-EAE has enabled researchers to focus on variables associated with the ‘effector phase’ of disease and to ‘bypass’ its induction phase. Encephalitogenic T cells can also be manipulated in vitro to characterize the role of specific cytokines and other biological agents before adoptive transfer into recipients. These cells can be conveniently labelled to follow their localization, survival and interactions with other cell types in the recipient host. In addition, adoptive transfer of cells has made it possible to address the role of a variety of inflammatory molecules in different aspects of disease development and regulation through the use of gene-targeted donor or recipient animal strains (most frequently, C57BL/6 mice).
The pathology of lesions varies in different animal strains (Gran et al., 2007; Lassmann, 2007). For example, in the C57BL/6 mouse, immunization with MOG35–55 in CFA can induce monophasic or a chronic, sustained form of EAE. The former is characterized by multifocal, confluent areas of mononuclear inflammatory infiltration and demyelination in the peripheral white matter of the spinal cord (Day, 2005). Macrophages and CD4+ T cells are the main cell types in the inflammatory infiltrate. In the brain, there is meningitis and perivascular inflammatory cuffing in the cerebellum and hindbrain white matter. The latter type of EAE, often induced with a ‘booster’ injection of the same myelin peptide 7 days after the initial immunisation (or with higher doses of peptide at the first immunization), shows similar pathology with reduced tendency to resolution of inflammation and demyelination after the peak of disease. These characteristics make this disease type a good model of chronic inflammatory demyelination, which approximates SPMS more closely (Bannerman et al., 2005).
Another frequently used EAE model is induced in SJL/J mice by immunization with PLP139–151, leading to relapsing–remitting disease in which T-cell reactivity spreads to new myelin peptide determinants with each relapse (epitope spreading) (McRae et al., 1995; Vanderlugt et al., 2000). Typical lesions appear in the optic nerve, brainstem, spinal cord, cerebellum and cerebral cortex, initially with perivascular and meningeal lymphocyte and neutrophil infiltration, followed by resolution of the inflammatory infiltrate and at the same time progression of white matter damage and gliosis, demyelinated axons and myelin debris-containing macrophages. In the Lewis rat, active and passive EAE induced by MBP or transfer of MBP-specific T cells typically produces severe CNS inflammation, with little or no demyelination (Meeson et al., 1994). Thus, the model is useful for the study of acute CNS inflammation. Interestingly, injection with antibodies to surface myelin antigens such as MOG leads to the appearance of demyelination and increased perivascular inflammation and clinical symptoms, usually followed by remyelination. In the Dark Agouti (DA) rat, syngeneic spinal cord tissue or recombinant rat MOG can be used to induce EAE characterized by demyelination and spinal cord lesions with perivascular and subpial inflammatory infiltration (Tanuma et al., 2000). Demyelination tends to appear in the dorsal column of the spinal cord only during the second disease relapse. Whereas both TCRαβ-positive T cells and ED-1-positive macrophages are observed in the acute phase, in the chronic/relapsing phases, there is a predominance of macrophages over T cells (Tanuma et al., 2000).
Therapies in EAE
Immunologically (Table 3) or neurobiologically (Table 4) based interventions in EAE have allowed the exploration of pathogenesis pathways and the development of validation of certain targets for MS therapies.
Table 3.
Immunologically based therapies in EAE
| General approach class | Treatment type | Examples (EAE) | Used/tried in MS (comments) | Reference |
|---|---|---|---|---|
| Pharmacological (traditional pharmaceutical agents) | General immunosuppressive | Mitoxantrone | Yes (licenced) | (Ridge et al., 1985; Levine and Saltzman, 1986; Lublin et al., 1987; Tischner and Reichardt, 2007; Mangano et al., 2010) |
| Cyclophosphamide | Yes | |||
| Glucocorticoids | Yes | |||
| Immunomodulatory drugs | IFN-beta | Yes (licensed, widely used) | (Abreu, 1982; Jacobs et al., 1982; Hertz and Deghenghi, 1985; Abreu et al., 1986; Paty and Li, 1993; Brod and Burns, 1994; Ruuls et al., 1996; Yu et al., 1996; Croxford et al., 1998b; van der Meide et al., 1998; Luca et al., 1999; Yasuda et al., 1999; Wender et al., 2001; Schaefer et al., 2006; Jaini et al., 2006; Martin-Saavedra et al., 2007) | |
| Other drugs | Captopril, losartan, pentoxyphyline, prazosin, antihistamines, and others | No (not as DMT) | (Babington and Wedeking, 1971; Brosnan et al., 1985; Claudio and Brosnan, 1992; Nataf et al., 1993; Constantinescu et al., 1995; Dimitriadou et al., 2000; Stegbauer et al., 2009; Jadidi-Niaragh and Mirshafiey, 2010) | |
| Immune deviation | Cytokines inducing Th1-Th2 shift | IL-4 | No | (Racke et al., 1994; Young et al., 2000) |
| Immune decoy/antigen mimicry; interference with antigen presentation | Glatiramer acetate | Yes (licensed, widely used | (Teitelbaum et al., 1971; 1973; 1974; 1996; 1999; 2004; Keith et al., 1979; Lisak et al., 1983; Aharoni et al., 1993; 1997; 1998; Johnson et al., 1995; 1998; Gran et al., 2000; Gilgun-Sherki et al., 2003; Illes et al., 2004; Stern et al., 2004; Giuliani et al., 2005a,b; Jee et al., 2007; Begum-Haque et al., 2008; Stern et al., 2008; Kala et al., 2010) | |
| (Brocke et al., 1996; Bielekova et al., 2000; Kappos et al., 2000) | Altered peptide ligands | MBP | No (trial unsuccessful; some APL induced an immune response with exacerbation of the disease and/or anaphylactoid reaction) | |
| Ongoing trial using ower doses | ||||
| Induction of immunological tolerance | i.v., p.o. administration of antigen | Oral tolerance to MBP, i.v. tolerance to MBP | No (trial of oral myelin showed interesting immune effects but largely unsuccessful) | (Weiner et al., 1993; Chen et al., 1994; Whitacre et al., 1996) |
| Blockade of second signal | CTLA4-Ig; non-depleting antiCD3 | No (trials unsuccessful or inconclusive) | (Croxford et al., 1998a; Tran et al., 2001) | |
| DNA therapy | cDNA ‘vaccination’ e.g. MBP, PLP | Yes (promising) | (Waisman et al., 1996; Lobell et al., 1998; Selmaj et al., 2000) | |
| Pathogenic (e.g. Th1, Th17) cytokine or cytokine developmental pathway blockade | Anti-cytokine monoclonal antibodies | Anti-IL-12/IL-23p40, IL-1RA, lenercept | No (worsening with lenercept, no effect with the others | (Constantinescu et al., 1998; Chen et al., 2006; Furlan et al., 2007; Martin and Near, 1995) |
| Cyokine receptor antagonists | ||||
| Targeting/depleting immune cells other than T cells | Macrophage/microglia depletion | Chlodronate | No | (Huitinga et al., 1990; Jung et al., 1993; Tran et al., 1998) |
| B cell depletion/blockade | AntiCD20/CD19 antibodies | Yes (promising) | (Hauser et al., 2008; Matsushita et al., 2008; Kap et al., 2010b) | |
| Mast cell | Luteolin | No | (Theoharides, 2009) | |
| Enhancing endogenous immune regulatory mechanisms | Up-regulation of Treg cells | Retinoic acid | Yes (small study plus IFN) | (Massacesi et al., 1991; Vergelli et al., 1997; Qu et al., 1998) |
| Gut parasites | Phase I trials ongoing; Larger trials pending | (Sewell et al., 2002; Sewell et al., 2003; Correale and Farez, 2009) | ||
| Enhancement of endogenous type 1 IFN | TLR ligands | Yes (polyI:C) | (Bever et al., 1991) | |
| (Touil et al., 2006; O'Brien et al., 2010) | ||||
| Cell-based therapies | Transfer of Treg | Transfer of CD4+CD25+ Treg cells | No | (Stephens et al., 2009) |
| Haematopoietic stem cell treatment | Immunosuppression and immune system renewal | Yes | (Karussis et al., 1992; 1993; 1999; Burt et al., 1998; van Bekkum, 2000; Burt et al., 2009; Muraro and Uccelli, 2010; Pasquini et al., 2010) | |
| Cell trafficking/based approaches | Targeting adhesion molecules required for crossing the BBB | VLA4 antibody | Yes (licenced) | (Yednock et al., 1992; Kent et al., 1995; Leger et al., 1997; Soilu-Hanninen et al., 1997; Brocke et al., 1999; Sheremata et al., 1999; Theien et al., 2001; 2003; Piraino et al., 2002; van der Laan et al., 2002; Cannella et al., 2003; Leone et al., 2003; Miller et al., 2003; O'Connor et al., 2004; 2005; Myers et al., 2005; Polman et al., 2006; Miller et al., 2007a; Havrdova et al., 2009) |
| Targeting lymphocyte egress from the lymphoid organs | Fingolimod | Yes (licence awaited soon) | (Fujino et al., 2003; Rausch et al., 2004; Webb et al., 2004; Kataoka et al., 2005; Balatoni et al., 2007; Foster et al., 2007; Foster et al., 2009; Chiba et al., 2011; Cohen et al., 2010; Kappos et al., 2010; Papadopoulos et al., 2010) | |
| Transgenic mice | Cytokine, chemokines, cell surface molecule knockouts; transgenic mice overexpressing immune molecules in CNS | Numerous | N/A but some have led to targeting immune molecules based on the results | (Glabinski et al., 1999; Hilliard et al., 1999; Izikson et al., 2000; Furlan et al., 2001; Huang et al., 2001; Cua et al., 2003; Park et al., 2004; Elhofy et al., 2005; Axtell et al., 2006; Laouar et al., 2008; Yang et al., 2009) |
Table 4.
Neurobiologically/neuropharmacologically based therapies in EAE
Of note, some of the neuroimmune molecules listed above may have dual action, immunomodulatory and neuroprotective, and may be attractive candidates as therapeutic targets in MS. A few of the above have made their way into early clinical trials. These include cannabinoids, neuropeptides and ion channels, which are present and subject to modulation also in the immune system, and some neurotransmitters such as dopamine (Rog et al., 2005; Nessler et al., 2006; Vollmar et al., 2009). This dual expression in the immune and nervous system not only certainly provides the opportunity for multiple therapeutic mechanisms but also invites caution: the favourable effects in one system should not be counteracted by detrimental effects in the other.
Correlation between EAE and MS studies
As can be already observed from the above tables, a large number of EAE studies are corroborated by results in MS. Since the immune system and the immune mechanisms in EAE are very complex, some treatments that have been successful in EAE are yet to be assessed in MS.
In EAE, and to some extent in MS, unsuccessful studies may not be published, leading to publication bias. While this can be assessed when there are a substantial number of studies with the same or very similar compound, by plotting the standard error /deviation against the effect size (funnel plot), this is not feasible for a few small exploratory studies as are often done in EAE. However, for some of the studies discussed below, where such analysis is possible, there is no evidence of major (publication) bias (Farooqi et al., 2010).
EAE studies may differ widely in terms of the experimental conditions. This includes the species, strain, and sex of the animals used; the age; specific induction method (including the neuroantigen, the type of adjuvant used, the active induction vs. AT EAE model); and the timing, frequency and dose of the therapeutic agent under study (Gold et al., 2006). Most rodent EAE experiments are done in genetically identical groups of animals. This at least eliminates an important source of variation (although this variation does exist in humans). However, genetically identical animals may differ in their susceptibility to EAE depending on environmental factors, which may not easily be controlled. For example, the degree of colonization of the gut and the type of commensal flora can determine to a great degree the susceptibility to EAE (Yokote et al., 2008).
Many of the above-discussed caveats may explain discrepancies between EAE studies.
Notwithstanding sources of variation, there are many examples of successful therapies in EAE that have also proven successful in MS.
Based on the congruence between the evidence in EAE and MS, studies of candidate DMT can be divided into several categories:
EAE and MS treatment success
The most convincing correlations between EAE and MS therapeutic success are, reassuringly, those of the currently licensed and used DMT: IFN-beta (Abreu, 1982; Paty and Li, 1993), GA (Teitelbaum et al., 1971; Johnson et al., 1995) and the anti-VLA-4 antibody (Yednock et al., 1992; Polman et al., 2006). Their use in MS is discussed above. We have recently conducted a systematic review of all EAE studies looking at type 1 IFN, GA and anti-VLA-4 (natalizumab) (Farooqi et al., 2010). The reader is referred to that review for more detail. The evidence and the role of EAE in the drug development and testing is discussed below.
IFN-beta
This is a good example of bidirectional dialogue between MS and EAE. The IFN evidence has developed almost in parallel, findings from the experimental model feeding into MS study and vice versa. In 1982, in EAE, Abreu found that IFN type 1 suppressed disease (Abreu, 1982), while Jacobs found that intrathecal IFN-beta was effective in 5 of 10 MS patients (Jacobs et al., 1982).
A total of 25 therapeutic trials in EAE with IFN type 1 (both alpha and beta, as both have been used successfully in MS) have been identified during our systematic review Meta-analysis showed that the overall effect was beneficial, though in some studies, the results were equivocal and in a small number of studies there was an actual worsening (Farooqi et al., 2010). Of note, many of the more recent studies were done with the purpose of exploring further the immunomodulatory effects of IFN-beta and elucidating mechanisms of action rather than confirming its clinical effects.
GA
The story of GA is tightly linked to EAE. As discussed above, GA a random copolymer of tyrosine, glutamate, alanine and lysine in ratios resembling myelin basic protein. It was developed in 1971 by Teitelbaum and colleagues in the laboratory of M. Sela, who was conducting extensive studies of the immunogenicity of proteins. GA was developed initially as a putative encephalitogen; however, it showed the opposite action in that it effectively blocked EAE (Teitelbaum et al., 1971; 1973; 1974).
Extensive subsequent studies were aimed mostly at elucidating mechanisms of actions of this versatile drug, but also at optimizing its pharmacology. Both pre- and post-licensing studies with GA in EAE have been successful. Our recent systematic review did not appear to detect a suggestion of publication bias (Farooqi et al., 2010).
GA is a versatile compound. Although its mechanism of action is incompletely elucidated, it is likely to be multifactorial. The understanding of GA has paralleled the advances in our understanding of fundamental immunological principles and has thus evolved. From an immune decoy mechanism, through interference with antigen presentation, cytokine shift, induction of immunological tolerance, induction of immunoregulatory mechanisms (in the form the T suppressor cells to the Treg cells of today), antioxidant effects, to the more recent attention to its suppressive effects on Th17 development and effector function, and even to even more speculative attribution of neuroprotective and remyelinative properties, there is some evidence for a potential role of GA in all of these therapeutic mechanisms (Arnon and Aharoni, 2009). Most of these have come from studies in EAE and/or related demyelinating disease models.
Altered peptide ligands
Altered peptide ligands (APL) of MBP were initially developed to treat EAE based on the concept that substitution of one or more amino acids to change MHC or T-cell receptor (TCR) binding characteristics would induce tolerance to the native peptide through mechanisms including T-cell antagonism and partial agonism as well as cytokine deviation (Evavold and Allen, 1991; Racioppi et al., 1993) and antagonism at the T-cell activation level (De Magistris et al., 1992). Brocke et al. induced EAE with a MBP87-99-specific T-cell clone and were able to induce tolerance in vivo by treating mice with an analogue of the native peptide (alanine substitution of the phenylalanine at residue 96). Paralysis was reversed, inflammatory infiltrates were regressed and brain T-cell infiltrates were depleted. Interestingly, it was also found that the simple administration of the native MBP peptide was equally effective, indicating that a ‘tolerizing’ injection before the ‘immunizing’ one was sufficient to prevent disease (Brocke et al., 1996). The approach was then tested in MS in a small phase II placebo-controlled trial, which had to be suspended because of hypersensitivity reactions in 9% of the patients. Secondary analysis of patients who completed the study showed a reduction of the volume and number of contrast-enhancing lesions in the treatment group. A regulatory Th2 response to the APL was also observed, which cross-reacted with the native peptide (Kappos et al., 2000). Major concerns, however, were raised by a parallel study by Bielekova et al. who clearly documented that APL could exacerbate MS. In a phase II clinical trial, they found that three patients developed exacerbations after administration of a MBP APL. In two patients, increased disease activity was shown to be linked to the APL immunologically and radiologically. Patients recovered well, and the therapeutic disappointment was at least partly compensated for by the scientific insight into the ability of modified MBP peptides to induce exacerbations in patients, thus confirming a long suspected link between autoimmunity to MBP and MS (Bielekova et al., 2000).
Adhesion molecule blockade
A solid body of evidence implicated VLA-4 (alpha4 beta 1 integrin) as an essential molecule in EAE, important for lymphocyte entry into the brain parenchyma. Steinman and Yednock showed that an antibody to VLA-4 suppressed EAE (Yednock et al., 1992). This eventually led to the development and production of the monoclonal antibody natalizumab (Tysabri®). Our recent meta-analysis of 19 EAE studies of VLA-4 antibodies or blockers that use clinical score as an outcome measure identified an overall beneficial effect (Farooqi et al., 2010). Interestingly, however, two studies (Theien et al., 2001; 2003) demonstrated a significant exacerbation of disease with anti-VLA-4 agents and advised caution in translation to MS. Of note, these two studies showed a discordant effect depending on the timing of administration of the agent (when administered either before EAE induction or at peak disease it induced an exacerbation), and one employed a small molecule VLA-4 antagonist. It is of interest that preliminary human studies with small molecule antagonists of integrins including VLA4 seem to be less effective than antibody blockade studies.
Clinical licence for Tysabri in MS was obtained in 2004 after remarkable success in phase II and III clinical trials (Miller et al., 2003; O'Connor et al., 2004). However, the drug was withdrawn soon thereafter following its rare association with progressive multifocal leukoencephalopathy (PML), an opportunistic infection due to the reactivation of latent John Cunningham virus (JCV) (Kleinschmidt-DeMasters and Tyler, 2005). The drug was cautiously re-introduced in 2006 after safety review, under a special prescription and observation programme, as the clinical benefits in people with aggressive RRMS are judged to outweigh the risks (Polman et al., 2006; Goodin et al., 2008). Indeed, natalizumab is currently considered the most potent licenced DMT.
Although natalizumab is another example of general concordance between EAE and MS studies, it does illustrate the potential risks of extrapolating all aspects of EAE results of a drug treatment to MS. As JCV only infects humans, side effects relating to it were not predicted on the basis of EAE experiments. This underscores the need for tailoring the appropriate experimental model experiments to the clinical/scientific question being addressed.
Generally, while hints at immune or neurobiological effects can often be provided by EAE experiments, alternative modelling may well be required for safety and pharmacokinetic/pharmacodynamic studies.
Other studies in EAE have investigated the inhibition of other adhesion molecules than VLA-4 and inhibitors other than monoclonal antibodies. None as yet have been translated to clinical use.
Other compounds
A substantial number of other studies have shown treatment success with concordant results in EAE and MS, using a variety of compounds. Some of these agents, like the immunosuppressants azathioprine (Hauser et al., 1983; Massacesi et al., 2005; Casetta et al., 2007; Elkhalifa and Weiner, 2010), mitoxantrone (Hartung et al., 2002; Weiner, 2004; Martinelli Boneschi et al., 2005) are licenced or well-established therapies for specific groups of patients with MS. Others, like laquinimod or fingolimod, have reached late phase clinical trials or are awaiting licencing decisions (please see Table 1 for examples). Table 5 lists some approaches with some degrees of evidence of success in MS with proof of concept/mechanisms supported by concordant success in EAE.
Table 5.
Studies of compounds used with some success in both EAE and MS
Discrepancies between EAE and MS treatment success
The results outlined above suggest a reassuring degree of concordance between EAE and MS treatment. Moreover, this concordance is seen for the majority of the established or soon-to-be established DMTs for MS. However, there are many examples of EAE treatment success, which has not translated into similar MS success. This has been one of the most consistent criticisms of the EAE model (Sriram and Steiner, 2005). There are also, of course, a large number of therapeutic interventions that have shown positive results in EAE and that, for many reasons, have not yet been tested in MS. Depending of the strength of evidence from EAE results and on the practicability of application to human use in early stage trials, some of these may someday emerge into the field of MS. Nevertheless, there are many examples in the literature of treatments that were successful in EAE but not in MS (see below). Some of these examples are discussed below, although the list is incomplete. There is a possibility that negative results in EAE are not published, and thus, the EAE literature predominantly contains positive results. In MS, the current trend is to publish both positive and negative large studies, industry sponsors having an obligation to do so. However, small pilot studies, in particular older ones, that have not yielded conclusive results or that were negative may still remain unpublished.
Induction of immunological tolerance by oral administration of antigen has provided very important clues to the fundamental mechanisms of immune regulation and offered hopes for the treatment of autoimmune diseases such as MS and rheumatoid arthritis (Weiner, 2000). Oral tolerance has been shown to suppress EAE (as well as animal models of arthritis) (Benson et al., 1999). A pioneering study in which oral myelin was given to patients with RRMS did not show efficacy on the clinical primary outcome measures, although it did show evidence of immune modulation that may, in the future, be harnessed more successfully to treat MS (Weiner et al., 1993).
Deoxyspergualin is a xenobiotic with immunosuppressive properties. In the early 1990s, it was shown to be successful in several models of EAE (Yamamura et al., 1987; Schorlemmer and Seiler, 1991). Despite this, when applied to a trial in MS with clinical MRI follow-up, it failed to show success on the primary outcome measure (Kappos et al., 1996).
There is ample evidence of alteration of the balance between the pro- and anti-inflammatory cytokines in MS, with great potential opportunity for disease modulation. Indeed, the existing DMTs act, in part, via their cytokine-modulatory effects. Cytokine therapy, whereby pro-inflammatory cytokines are blocked by antibodies or their soluble receptors, has been successful in many models of autoimmune disease including many EAE studies. It has also made its way in human disease where it has revolutionized the treatment of rheumatoid arthritis (RA), for example, where anti-tumour necrosis factor (TNF) biologicals are now in well-established use and have made a great impact on the quality of life and the prevention of late complications in patients (Feldmann, 2002). The IL-1 receptor antagonist, anakinra, is also used as a treatment for RA, although not on such large a scale as TNF blockers (Mertens and Singh, 2009). TNF and MS are discussed below. Despite evidence for a role of IL-1 in EAE (Martin and Near, 1995), anakinra was unsuccessful in an MS trial.
A similar approach is to enhance anti-inflammatory cytokines. Both IL-10 and transforming growth factor (TGF)-beta were shown to suppress EAE (Santambrogio et al., 1993; Rott et al., 1994); however, attempts at treating a small number of MS patients with TGF-beta were unsuccessful due to side effects (Calabresi et al., 1998).
Another immunological concept to which EAE studies have brought a major contribution is that of a restricted TCR use in immune responses (Hafler et al., 1996). This has significant implications both for the presumed pathogenesis and for therapeutic opportunities. As regards the pathogenesis, such a restricted receptor use could imply an inciting or causative infectious agent such as a virus; it could also explain why superantigens (which have predilection for specific TCRs) can trigger or exacerbate autoimmune disease. The finding of shared TCR usage across more than one model of autoimmune disease, for example EAE and its peripheral nerve counterpart, experimental autoimmune neuritis (EAN), led to the hypothesis that certain TCRs usage predisposes to autoimmune diseases in general. This also raised the possibility of immune intervention across the autoimmune spectrum. One approach was a trial of an antibody against the TCR most frequently used by MS patients, and that was unsuccessful (Killestein et al., 2002). Another approach is inducing tolerance to the autoimmune-prone TCR by T-cell vaccination in MS, which is thought to trigger an anti-idiotypic immunoregulatory network (Vandenbark et al., 2008). Such an approach is conceptually interesting but so far has not been translated into clinical success.
MS and EAE were long considered Th1-mediated diseases. Similar to other Th1/Th2 dichotomous experimental situations (e.g. the murine leishmaniasis model), modulation of both disease susceptibility and established disease activity was possible by manipulating the differentiation and maintenance of Th1/Th2 pathways. However, the discovery of IL-23, the IL-12-related cytokine that shares the p40 subunit with IL-23, led to the re-evaluation of all previous work done with neutralization of p40 or with p40 knockout mice and to the eventual description of the Th17 cells that are stimulated by IL-23 and produce IL-17 and other cytokines (IL-21, IL-22) (Cua et al., 2003; Harrington et al., 2005; Langrish et al., 2005). This represented a paradigm shift in immunology. The developmental pathway of murine Th17 cells was also shown to involve TGF-beta and IL-6, and the in vivo system in which this was shown was EAE. Subsequent work has shown that, with some possible variation, these pathways also seem to be working in the human immune system in similar ways, with IL-23 having a role in stimulating and maintaining, if perhaps not inducing, Th17 responses (Korn et al., 2009). It therefore became obvious that an intervention that targeted IL-12/23p40, thus down-regulating both Th1 and Th17 responses, is potentially beneficial in MS. The results of the clinical trial of ustekinumab, a human anti-p40 monoclonal antibody, in RRMS, were both surprising and disappointing in that respect (Segal et al., 2008). The lack of clinical or MRI effect was shown despite the fact that there was evidence that the antibody did have an immunomodulatory effect. This study led to another rethinking, and consideration of therapeutic options in MS that might be beyond the Th1/Th2/Th17 split. Some potential options are considered in a recent review article (Steinman, 2010) and some are discussed below.
IFN-gamma has been one of the most poignant examples of discrepancy between MS and EAE and a major argument in the criticism of the EAE model. Moreover, the experience with this cytokine in MS and EAE and the studies showing its amenability to inhibition by type 1 interferons have contributed to the development of the latter compounds as DMTs. The role of IFNs in EAE and MS is discussed in more detail in other reviews (Sanvito et al., 2010) but the evidence can be summarized as follows: treatment of EAE with IFN-gamma suppresses disease, while its blockade enhances disease in EAE. The opposite is true for MS, where intravenous IFN-gamma treatment in a clinical trial induced relapses in a substantial number of participants (Panitch et al., 1987). A non-placebo controlled trial of an anti-IFN-gamma antibody showed that it suppressed MS, in contrast to an anti-TNF antibody, which did not (Skurkovich et al., 2001).
There are more examples of discrepancies between therapeutic successes in EAE and MS, including some chemokine antagonists. The reasons for these translational failures are likely multiple and complicated but such results always offer the opportunity to rethink the pathogenesis of MS and the therapeutic approaches to it. Also, occasionally they provide new insights into fundamental immunological mechanisms.
Unpredicted EAE and MS treatment failure
There is an informative category of studies where most of the results of the EAE studies concur with those in MS and are both negative, despite high biological plausibility of the intervention used as potentially beneficial in EAE and MS. This is usually when there are results in EAE that are discrepant or can be interpreted differently as beneficial or ineffective (for example a transient positive effect in EAE or a difference between antibody neutralisation and knockout mouse results). The most poignant example in this category is the work related to the role of TNF in EAE and MS (reviewed in Lim and Constantinescu, 2010a). In some papers addressing the deficits of EAE as a model for MS, TNF is often used as an example of inconsistency between MS and EAE results. In EAE, disease is said to be suppressed by antibody neutralisation of TNF, but in fact a closer look at the literature shows that this only occurs in adoptive transfer EAE models. In MS, trials of anti-TNF biological lenercept showed an unexpected worsening of the disease and further study of anti-TNF agents has been discontinued (The Lenercept Multiple Sclerosis Study Group, 1999). Moreover, demyelination has been reported in recipients of anti-TNF biologicals for other inflammatory diseases; these are contraindicated when these inflammatory conditions coexist with MS (Mohan et al., 2001). The initial studies of anti-TNF biologicals in MS preceded the EAE studies with TNF (or TNF plus Lymphotoxin) knockout mice, and were only supported by passive transfer EAE studies, but not by active EAE studies. Even in a TNF knockout mouse model, a delay in EAE onset despite retained susceptibility led to the more emphasized conclusion that TNF is important for early disease, rather than that it is not absolutely required for EAE (Frei et al., 1997). Anti-TNF antibody transiently delayed superantigen induced relapses in EAE; however, it was subsequently shown that other cytokines (IL-12/IL-23 and possibly IL-6) were more important in suppressing superantigen-induced relapses (Lim and Constantinescu, 2010a).
In conclusion, the results of TNF in EAE and MS are not incongruous, as they agree more than they disagree; on balance, they seem to argue against a significant and unique pathogenic role of TNF in demyelinating disease, and against TNF as a therapeutic target in MS.
Discrepancies in therapeutic effects of in different EAE models depending on the experimental conditions
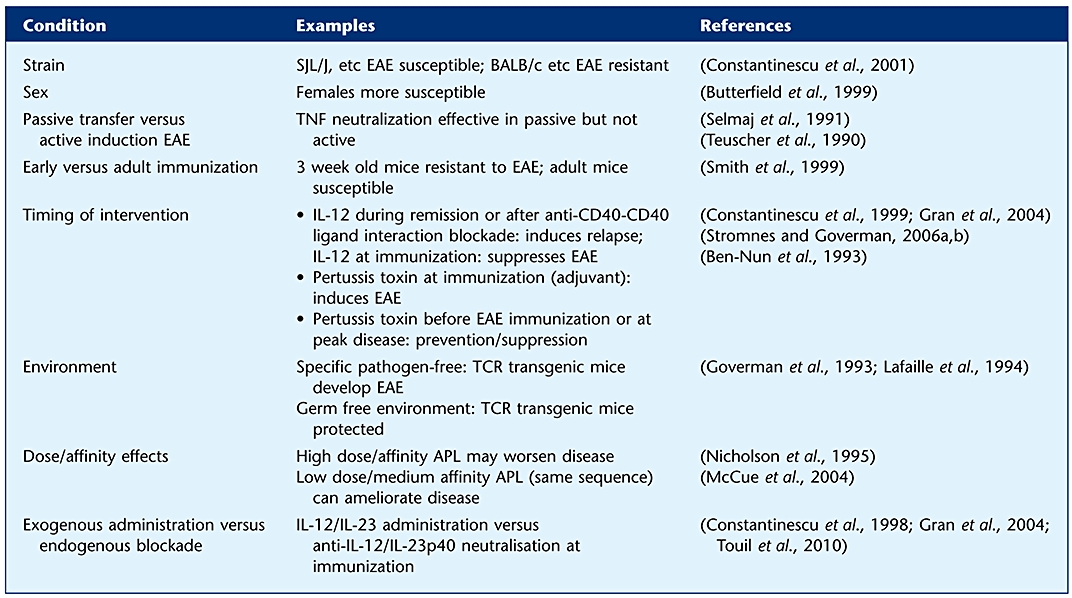
The above example with TNF neutralisation having an effect in passive but not active EAE illustrates that different results are sometimes seen in different EAE models or even in the same model depending on the experimental conditions. There are many such examples. The differential susceptibility to EAE of male versus female mice is well known. Other examples are given in Table 6.
Table 6.
Examples of conditions that determine variations in EAE outcome
 |
Besides the discrepancies that can result from the experimental conditions, other factors that must be taken into account when extrapolating from EAE to MS are the dual or multiple actions of the same target, and that some of these actions might be contradictory. Depletion of macrophages suppresses EAE but impedes nerve regeneration and remyelination (Huitinga et al., 1990; Kotter et al., 2001); a whole range of cytokines have opposing actions in EAE that need to be considered when designing MS treatments. A special issue of the Open Autoimmunity Journal is dedicated to the most representative of these cytokines (Gran and Becher, 2010).
Prostaglandin E-2 (PGE-2) enhances both Th1 and Th17 and thus augments EAE but also has BBB stabilizing effects that partially counteract the former effects (Esaki et al., 2010). Osteopontin also enhances both Th1 and Th2 responses but also has a role in remyelination and is neuroprotective (Braitch and Constantinesco, 2010). These examples and others underscore the complexity of inflammatory demyelination and the numerous factors that come into play when considering therapeutic possibilities. Moreover, some of the subtle or delayed effects may be hidden, for example interference with neural repair or remyelination may be missed in a study dealing only with immunological aspects of EAE.
Aspects of MS that cannot be tested in animal models
Some treatments that have shown success in MS and are approaching widespread clinical application have not been tested or published in EAE. Cladribine, an immunosuppressant showing remarkable results from phase II and III studies in RRMS, is not reported to have been tested in EAE (Giovannoni et al., 2010).
Alemtuzumab, a monoclonal antibody against human CD52, is an immunosuppressant that depletes immune cells achieving a highly suppressive effect on disease activity in RRMS (Coles et al., 2008). Alemtuzumab does not cross-react with mouse CD52 and its effects in EAE have not been investigated. However, there is a recently developed transgenic mouse expressing human CD52, where effects of alemtuzumab have been tested with regard to its other applications in cancer (Hu et al., 2009). Its effects in EAE would be very interesting.
Some targets cannot be tested in knockout mouse models because they are embryonic or neonatal lethal, for example TGF-beta or Retinaldehyde dehydrogenase type 3 knockout mice (Hines et al., 1994; Dubinsky et al., 2010). Also, activity dependent neuroprotective protein (ADNP) which is a neuroprotective molecule also amply expressed in the immune system and capable of immune modulation, has been reported to suppress EAE and its knockout mouse is neonatal lethal (Braitch et al., 2010).
A very important situation where animal models such as EAE cannot answer MS-related question is related to complications restricted to humans, for example infectious diseases with humans as only hosts. We mentioned the JC virus example above (Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Clifford et al., 2010).
Increasing epidemiological and biological evidence implicates the Epstein-Barr virus (EBV) in the pathogenesis of MS (Serafini et al., 2007; Ascherio and Munger, 2010). Many hypotheses have been put forward about how EBV might modify the immune system and make people more prone to MS, including its ability to immortalize B cells and enhance their antigen presenting function. EBV is also a human specific virus, and all these mechanistic studies, as well as any potential studies of EBV-targeted therapies would not be appropriate in rodent EAE (unless one were dealing with a mouse with a human immune system).
MS can affect a variety of specific human factors such as fatigue or induce subtle cognitive disturbances. It also has wide psychological, social and economic implications. None of these can be modelled properly in experimental models.
Transgenic and knockout mouse models
The development of transgenic and knockout technology has made a major impact in the understanding of the pathogenesis of EAE and has contributed to the widespread use of the mouse models of EAE (Steinman, 1997; Gold et al., 2006). Many of the emerging or future therapeutic interventions have been tested in these models, and the confirmation of a potential benefit of a (immuno)pharmacological blocking intervention through the appropriate knockout or transgenic model adds credence to the validity of the approach. However, caution is needed in interpreting the results of such studies, due to the well-known redundancy in the immune system and to some extent in the nervous system, or to some early developmental roles of some immune or neural molecules that might no longer be relevant for the adult rodent. In some instances conditional knockouts may be preferable (Korn et al., 2008).
Testing MS symptomatic treatment in EAE
EAE has served as a model for developing and validating symptomatic treatments in MS as well. A study in EAE has shown the role of cannabinoids in controlling spasticity and tremor (Baker et al., 2000). More recently, bladder symptoms of MS could be modelled in EAE and the utility of future drugs for neurogenic bladder dysfunction in MS could be tested in this model (Altuntas et al., 2008; Al-Izki et al., 2009).
Pharmacokinetics
Although rodent EAE has been an important model for understanding the pathogenesis and develop treatments for MS, relatively few published studies have included pharmacological evaluations of potential drugs, such as pharmacodynamic and pharmacokinetic studies. This is justified in part by the largely immune active rather than classical pharmaceutical repertoire of therapies used, but also by the difficulties extrapolating the results to humans. As discussed above, pharmacokinetic studies of a promising compound are sometimes done on larger animals and mechanistic studies are done in EAE. One notable study looking at retinoid modulation of EAE, however, has measured the exogenous retinoid pharmacokinetics in rats (Vergelli et al., 1997).
Requirements for translational success in MS of a putative treatment as predicted by EAE studies
EAE results, especially those taken in relative isolation, cannot easily predict the success of a given therapeutic intervention in MS. For an immunological intervention in EAE to be matched in MS, the appropriate MS population (i.e. RRMS) needs to be tested. For example, photopheresis has positive effects on the inflammatory activity in rat EAE (Cavaletti et al., 2001). In humans it was studied in progressive MS, where it was not successful (Rostami et al., 1999); but a small study shows it to be successful in RRMS, which validates the EAE results (Cavaletti et al., 2006). Many EAE treatments are tested at disease induction, therefore for a prophylactic effect, whereas, for obvious reasons, they are then tested in established MS. Of course, with the increased availability of first and second line DMTs, a major unmet need is developing effective treatments for progressive MS forms. EAE resembling secondary progressive MS can be established in some mouse models (Tsunoda et al., 2005; Hampton et al., 2008). However, the development of pure progressive EAE models to mimic primary progressive MS is problematic. Although a very interesting model of partial immunological tolerance induction shows that it cannot stop progressive neurodegeneration and thus resembles primary progressive MS (or one-attack progressive, or transitional MS) in terms of clinical phenomenology, it is not clear that is actually shares pathogenic mechanisms with primary progressive MS; however, it does create opportunities to study treatments targeted to the progressive neurodegeneration in MS (Pryce et al., 2005).
Notwithstanding the importance of the right MS population being selected for translational studies, there are some aspects of the EAE studies themselves that may help to develop a successful MS drug.
Generally, corroboration of successful results from several EAE studies is a better predictor of translational success.
Listed below are several qualities of a putative drug that we consider, with the obvious variations depending on the drug being tested, requirements for translational success in MS:
Biological plausibility
Convincing, significant differences from appropriately chosen controls in initial studies
Evidence of biological effect (e.g. down-regulation of a known pathogenic pathway for the chosen EAE model)
Validation with congruent results in further EAE models/ refinement of the same model (e.g. transfer EAE, EAE induced by another neuroantigen, EAE in another species/strain)
Clinical, immunological, histological (imaging) results are consistent
Efficacy in both males and females
Efficacy both as prophylactic and therapeutic (in established disease) treatment
Efficacy both in monophasic and relapsing EAE models
Synergistic effects of synergistic drugs/interventions
Antagonistic effects of antagonistic drugs/interventions
Where possible and appropriate, confirmation in transgenic/knockout mice
Ideally dual action, immunomodulatory and neuroprotective
Careful monitoring of CNS effects (effects on axon, myelin, brain/spinal cord volume, etc.) shows favourable effects/no adverse effects
Careful monitoring of long term effects, not only on immune/nervous system
Validation in another model of autoimmune disease if an immune-active drug; validation in another model of neurodegenerative disease if neurobiologically active drug
Where appropriate and possible, validation in primate MS model, where pharmacokinetics/ pharmacodynamics studies support use in MS; Clinical, immunological, histological (imaging) results are consistent; results favour treatment versus control despite clinical and genetic heterogeneity.
Concluding remarks
In conclusion, EAE has contributed to the development, validation, and testing of MS drugs and even more remarkably, to the understanding of the pathogenesis of MS. The multitude of models and results underscores the complexity both of MS and of this model. It also indicates that studies in the model need to be carefully tailored to the pathogenesis or therapy question, and that results showing a high degree of consistency between various models and experimental conditions are more likely to lead to translation into therapeutic success.
Acknowledgments
Work in the CSC and BG laboratory relevant to this article has been supported in part by grants from the Multiple Sclerosis Society of Great Britain and Northern Ireland (to CSC and BG), the University of Nottingham (to CSC and BG), and The European Union (Marie Curie International Reintegration Grant to BG; FP7 PRIMOCID programme to BG and CSC) and the pharmaceutical industry (see below)
Glossary
Abbreviations
- ADEM
acute disseminated encephalomyelitis
- ADNP
activity dependent neuroprotective protein
- AHR
aryl hydrocarbon receptor
- APC
antigen-presenting cells
- APL
altered peptide ligand
- AT
adoptive transfer
- C1
and CB2 receptors, cannabinoid receptors 1 and 2
- CIS
clinically isolated syndrome
- CNS
central nervous system
- DA
dark agouti
- DMT
disease-modifying treatment
- EAE
experimental autoimmune (allergic) encephalomyelitis
- EAN
experimental autoimmune (allergic) neuritis
- EBV
Epstein–Barr virus
- GA
glatiramer acetate
- IFN
interferon
- IL
interleukin
- IL-1RA
interleukin 1 receptor antagonist
- JCV
John Cunningham virus
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- MRI
magnetic resonance imaging
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NABT
normal appearing brain tissue
- NAWM
normal appearing white matter
- NMO
neuromyelitis optica
- NK1
receptor, neurokinin 1 receptor
- PGE
prostaglandin E
- PLP
proteolipid protein
- PP
primary progressive
- PR
progressive relapsing
- ROR
retinoid orphan receptor
- RR
relapsing-remitting
- SP
secondary progressive
- TCR
T-cell receptor
- TGF
transforming growth factor
- TLR
Toll-like receptor
- TNF
tumour necrosis factor
- Treg
regulatory T cell
- VLA
very late antigen
Declarations of conflict of interest
CSC has received research support from Bayer Schering, Biogen Idec, Centocor, Cephalon, GlaxoSmithKline, GW Pharma, Merck Serono, Teva, Novartis, Roche, and UCB, honoraria and consultancy fees from Almirall, Bayer Schering, Biogen Idec, Centocor, GlaxoSmithKline, GW Pharma, Merck Serono, Teva, and Novartis, and travel support from Bayer Schering, Biogen Idec, Merck Serono, and Teva. BG has received research support from Bayer Schering, Biogen Idec, and Merck Serono, and travel support to scientific meetings from Bayer Schering, Biogen Idec, Merck Serono, and TEVA. NF and KOB have nothing to disclose.
References
- Abreu SL. Suppression of experimental allergic encephalomyelitis by interferon. Immunol Commun. 1982;11:1–7. doi: 10.3109/08820138209050718. [DOI] [PubMed] [Google Scholar]
- Abreu SL, Thampoe I, Kaplan P. Interferon in experimental autoimmune encephalomyelitis: intraventricular administration. J Interferon Res. 1986;6:627–632. doi: 10.1089/jir.1986.6.627. [DOI] [PubMed] [Google Scholar]
- Achiron A, Margalit R, Hershkoviz R, Markovits D, Reshef T, Melamed E, et al. Intravenous immunoglobulin treatment of experimental T cell-mediated autoimmune disease. Upregulation of T cell proliferation and downregulation of tumor necrosis factor alpha secretion. J Clin Invest. 1994;93:600–605. doi: 10.1172/JCI117012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achiron A, Gabbay U, Gilad R, Hassin-Baer S, Barak Y, Gornish M, et al. Intravenous immunoglobulin treatment in multiple sclerosis. Effect on relapses. Neurology. 1998;50:398–402. doi: 10.1212/wnl.50.2.398. [DOI] [PubMed] [Google Scholar]
- Achiron A, Mor F, Margalit R, Cohen IR, Lider O, Miron S. Suppression of experimental autoimmune encephalomyelitis by intravenously administered polyclonal immunoglobulins. J Autoimmun. 2000;15:323–330. doi: 10.1006/jaut.2000.0433. [DOI] [PubMed] [Google Scholar]
- Aharoni R, Teitelbaum D, Arnon R. T suppressor hybridomas and interleukin-2-dependent lines induced by copolymer 1 or by spinal cord homogenate down-regulate experimental allergic encephalomyelitis. Eur J Immunol. 1993;23:17–25. doi: 10.1002/eji.1830230105. [DOI] [PubMed] [Google Scholar]
- Aharoni R, Teitelbaum D, Sela M, Arnon R. Copolymer 1 induces T cells of the T helper type 2 that crossreact with myelin basic protein and suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 1997;94:10821–10826. doi: 10.1073/pnas.94.20.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharoni R, Teitelbaum D, Sela M, Arnon R. Bystander suppression of experimental autoimmune encephalomyelitis by T cell lines and clones of the Th2 type induced by copolymer 1. J Neuroimmunol. 1998;91:135–146. doi: 10.1016/s0165-5728(98)00166-0. [DOI] [PubMed] [Google Scholar]
- Al-Izki S, Pryce G, Giovannoni G, Baker D. Evaluating potential therapies for bladder dysfunction in a mouse model of multiple sclerosis with high-resolution ultrasonography. Mult Scler. 2009;15:795–801. doi: 10.1177/1352458509104594. [DOI] [PubMed] [Google Scholar]
- Altuntas CZ, Daneshgari F, Liu G, Fabiyi A, Kavran M, Johnson JM, et al. Bladder dysfunction in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;203:58–63. doi: 10.1016/j.jneuroim.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvord EC, Jr, Driscoll BF, Kies MW. Large subpial plaques of demyelination in a new form of chronic experimental allergic encephalomyelitis in the guinea pig. Neurochem Pathol. 1985;3:195–214. doi: 10.1007/BF02834271. [DOI] [PubMed] [Google Scholar]
- Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23:2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon R, Aharoni R. Neuroprotection and neurogeneration in MS and its animal model EAE effected by glatiramer acetate. J Neural Transm. 2009;116:1443–1449. doi: 10.1007/s00702-009-0272-3. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Epstein-barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol. 2010;5:271–277. doi: 10.1007/s11481-010-9201-3. [DOI] [PubMed] [Google Scholar]
- Axtell RC, Xu L, Barnum SR, Raman C. CD5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J Immunol. 2006;177:8542–8549. doi: 10.4049/jimmunol.177.12.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babington RG, Wedeking PW. The influence of cinanserin and selected pharmacologic agents on experimental allergic encephalomyelitis (EAE) J Pharmacol Exp Ther. 1971;177:454–460. [PubMed] [Google Scholar]
- Bai L, Lennon DP, Eaton V, Maier K, Caplan AI, Miller SD, et al. Human bone marrow-derived mesenchymal stem cells induce Th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia. 2009;57:1192–1203. doi: 10.1002/glia.20841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, O'Neill JK, Turk JL. Cytokines in the central nervous system of mice during chronic relapsing experimental allergic encephalomyelitis. Cell Immunol. 1991;134:505–510. doi: 10.1016/0008-8749(91)90321-2. [DOI] [PubMed] [Google Scholar]
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, et al. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404:84–87. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- Balatoni B, Storch MK, Swoboda EM, Schonborn V, Koziel A, Lambrou GN, et al. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull. 2007;74:307–316. doi: 10.1016/j.brainresbull.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Balkowiec-Iskra E, Kurkowska-Jastrzebska I, Joniec I, Ciesielska A, Muszynska A, Przybylkowski A, et al. MPTP-induced central dopamine depletion exacerbates experimental autoimmune encephalomyelitis (EAE) in C57BL mice. Inflamm Res. 2007;56:311–317. doi: 10.1007/s00011-007-6128-0. [DOI] [PubMed] [Google Scholar]
- Bannerman PG, Hahn A, Ramirez S, Morley M, Bonnemann C, Yu S, et al. Motor neuron pathology in experimental autoimmune encephalomyelitis: studies in THY1-YFP transgenic mice. Brain. 2005;128:1877–1886. doi: 10.1093/brain/awh550. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Henderson AP, Prineas JW. The macrophage in MS: just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult Scler. 2006;12:121–132. doi: 10.1191/135248506ms1304rr. [DOI] [PubMed] [Google Scholar]
- Bar-Or A, Vollmer T, Antel J, Arnold DL, Bodner CA, Campagnolo D, et al. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol. 2007;64:1407–1415. doi: 10.1001/archneur.64.10.nct70002. [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002;110:493–497. doi: 10.1172/JCI15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtold DA, Kapoor R, Smith KJ. Axonal protection using flecainide in experimental autoimmune encephalomyelitis. Ann Neurol. 2004;55:607–616. doi: 10.1002/ana.20045. [DOI] [PubMed] [Google Scholar]
- Bechtold DA, Miller SJ, Dawson AC, Sun Y, Kapoor R, Berry D, et al. Axonal protection achieved in a model of multiple sclerosis using lamotrigine. J Neurol. 2006;253:1542–1551. doi: 10.1007/s00415-006-0204-1. [DOI] [PubMed] [Google Scholar]
- Bedoui S, Miyake S, Lin Y, Miyamoto K, Oki S, Kawamura N, et al. Neuropeptide Y (NPY) suppresses experimental autoimmune encephalomyelitis: NPY1 receptor-specific inhibition of autoreactive Th1 responses in vivo. J Immunol. 2003;171:3451–3458. doi: 10.4049/jimmunol.171.7.3451. [DOI] [PubMed] [Google Scholar]
- Beeton C, Barbaria J, Giraud P, Devaux J, Benoliel AM, Gola M, et al. Selective blocking of voltage-gated K+ channels improves experimental autoimmune encephalomyelitis and inhibits T cell activation. J Immunol. 2001;166:936–944. doi: 10.4049/jimmunol.166.2.936. [DOI] [PubMed] [Google Scholar]
- Begum-Haque S, Sharma A, Kasper IR, Foureau DM, Mielcarz DW, Haque A, et al. Downregulation of IL-17 and IL-6 in the central nervous system by glatiramer acetate in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;204:58–65. doi: 10.1016/j.jneuroim.2008.07.018. [DOI] [PubMed] [Google Scholar]
- van Bekkum DW. Stem cell transplantation in experimental models of autoimmune disease. J Clin Immunol. 2000;20:10–16. doi: 10.1023/a:1006682225181. [DOI] [PubMed] [Google Scholar]
- Ben-Nun A, Yossefi S, Lehmann D. Protection against autoimmune disease by bacterial agents. II. PPD and pertussis toxin as proteins active in protecting mice against experimental autoimmune encephalomyelitis. Eur J Immunol. 1993;23:689–696. doi: 10.1002/eji.1830230318. [DOI] [PubMed] [Google Scholar]
- Benson JM, Stuckman SS, Cox KL, Wardrop RM, Gienapp IE, Cross AH, et al. Oral administration of myelin basic protein is superior to myelin in suppressing established relapsing experimental autoimmune encephalomyelitis. J Immunol. 1999;162:6247–6254. [PubMed] [Google Scholar]
- Bever CT, Jr, Jacobson S, Mingioli ES, McFarland HF, McFarlin DE, Levy HB. Changes in leukocyte recirculation, NK cell activity, and HLA-DR expression in peripheral blood mononuclear cells of MS patients treated with Poly ICLC. Int J Immunopharmacol. 1991;13:613–618. doi: 10.1016/0192-0561(91)90084-k. [DOI] [PubMed] [Google Scholar]
- Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- Black JA, Liu S, Hains BC, Saab CY, Waxman SG. Long-term protection of central axons with phenytoin in monophasic and chronic-relapsing EAE. Brain. 2006;129:3196–3208. doi: 10.1093/brain/awl216. [DOI] [PubMed] [Google Scholar]
- Blaszczyk B, Gieldanowski J, Karakoz I. Experimental allergic encephalomyelitis in chickens. Arch Immunol Ther Exp (Warsz) 1978;26:743–746. [PubMed] [Google Scholar]
- Braitch M, Constantinescu CS. The role of osteopontin in Experimental Autoimmune Encephalomyelitis (EAE) and Multiple Sclerosis (MS) Inflamm Allergy Drug Targets. 2010;9:249–256. doi: 10.2174/187152810793358778. [DOI] [PubMed] [Google Scholar]
- Braitch M, Kawabe K, Nyirenda M, Gilles LJ, Robins RA, Gran B, et al. Expression of activity-dependent neuroprotective protein in the immune system: possible functions and relevance to multiple sclerosis. Neuroimmunomodulation. 2010;17:120–125. doi: 10.1159/000258695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand-Schieber E, Werner P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp Neurol. 2004;189:5–9. doi: 10.1016/j.expneurol.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- Brocke S, Gijbels K, Allegretta M, Ferber I, Piercy C, Blankenstein T, et al. Treatment of experimental encephalomyelitis with a peptide analogue of myelin basic protein. Nature. 1996;379:343–346. doi: 10.1038/379343a0. [DOI] [PubMed] [Google Scholar]
- Brocke S, Piercy C, Steinman L, Weissman IL, Veromaa T. Antibodies to CD44 and integrin alpha4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis by blocking secondary leukocyte recruitment. Proc Natl Acad Sci U S A. 1999;96:6896–6901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brod SA, Burns DK. Suppression of relapsing experimental autoimmune encephalomyelitis in the SJL/J mouse by oral administration of type I interferons. Neurology. 1994;44:1144–1148. doi: 10.1212/wnl.44.6.1144. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Goldmuntz EA, Cammer W, Factor SM, Bloom BR, Norton WT. Prazosin, an alpha 1-adrenergic receptor antagonist, suppresses experimental autoimmune encephalomyelitis in the Lewis rat. Proc Natl Acad Sci U S A. 1985;82:5915–5919. doi: 10.1073/pnas.82.17.5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain. 2002;125:1297–1308. doi: 10.1093/brain/awf133. [DOI] [PubMed] [Google Scholar]
- Brunmark C, Runstrom A, Ohlsson L, Sparre B, Brodin T, Astrom M, et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;130:163–172. doi: 10.1016/s0165-5728(02)00225-4. [DOI] [PubMed] [Google Scholar]
- Burt RK, Burns W, Ruvolo P, Fischer A, Shiao C, Guimaraes A, et al. Syngeneic bone marrow transplantation eliminates V beta 8.2 T lymphocytes from the spinal cord of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res. 1995;41:526–531. doi: 10.1002/jnr.490410412. [DOI] [PubMed] [Google Scholar]
- Burt RK, Padilla J, Begolka WS, Canto MC, Miller SD. Effect of disease stage on clinical outcome after syngeneic bone marrow transplantation for relapsing experimental autoimmune encephalomyelitis. Blood. 1998;91:2609–2616. [PubMed] [Google Scholar]
- Burt RK, Loh Y, Cohen B, Stefoski D, Balabanov R, Katsamakis G, et al. Autologous non-myeloablative haemopoietic stem cell transplantation in relapsing-remitting multiple sclerosis: a phase I/II study. Lancet Neurol. 2009;8:244–253. doi: 10.1016/S1474-4422(09)70017-1. [DOI] [PubMed] [Google Scholar]
- Butterfield RJ, Blankenhorn EP, Roper RJ, Zachary JF, Doerge RW, Sudweeks J, et al. Genetic analysis of disease subtypes and sexual dimorphisms in mouse experimental allergic encephalomyelitis (EAE): relapsing/remitting and monophasic remitting/nonrelapsing EAE are immunogenetically distinct. J Immunol. 1999;162:3096–3102. [PubMed] [Google Scholar]
- Calabresi PA, Fields NS, Maloni HW, Hanham A, Carlino J, Moore J, et al. Phase 1 trial of transforming growth factor beta 2 in chronic progressive MS. Neurology. 1998;51:289–292. doi: 10.1212/wnl.51.1.289. [DOI] [PubMed] [Google Scholar]
- Cannella B, Gaupp S, Tilton RG, Raine CS. Differential efficacy of a synthetic antagonist of VLA-4 during the course of chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res. 2003;71:407–416. doi: 10.1002/jnr.10487. [DOI] [PubMed] [Google Scholar]
- Casetta I, Iuliano G, Filippini G. Azathioprine for multiple sclerosis. Cochrane Database Syst Rev. 2007;(4):CD003982. doi: 10.1002/14651858.CD003982.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassiani-Ingoni R, Muraro PA, Magnus T, Reichert-Scrivner S, Schmidt J, Huh J, et al. Disease progression after bone marrow transplantation in a model of multiple sclerosis is associated with chronic microglial and glial progenitor response. J Neuropathol Exp Neurol. 2007;66:637–649. doi: 10.1097/nen.0b013e318093f3ef. [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Perseghin P, Buscemi F, Dassi M, Oggioni N, Sala F, et al. Immunomodulating effects of extracorporeal photochemotherapy in rat experimental allergic encephalomyelitis. Int J Tissue React. 2001;23:21–31. [PubMed] [Google Scholar]
- Cavaletti G, Perseghin P, Dassi M, Cavarretta R, Frigo M, Caputo D, et al. Extracorporeal photochemotherapy: a safety and tolerability pilot study with preliminary efficacy results in refractory relapsing-remitting multiple sclerosis. Neurol Sci. 2006;27:24–32. doi: 10.1007/s10072-006-0561-7. [DOI] [PubMed] [Google Scholar]
- Centonze D, Bari M, Rossi S, Prosperetti C, Furlan R, Fezza F, et al. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain. 2007;130:2543–2553. doi: 10.1093/brain/awm160. [DOI] [PubMed] [Google Scholar]
- Charcot J. Histologie de la sclerose en plaques. Gaz Hop. 1868;41:554–555. [Google Scholar]
- Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- Chen Y, Langrish CL, McKenzie B, Joyce-Shaikh B, Stumhofer JS, McClanahan T, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest. 2006;116:1317–1326. doi: 10.1172/JCI25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Kataoka H, Seki N, Shimano K, Koyama M, Fukunari A, et al. Fingolimod (FTY720), sphingosine 1-phosphate receptor modulator, shows superior efficacy as compared with interferon-beta in mouse experimental autoimmune encephalomyelitis. Int Immunopharmacol. 2011;11:366–372. doi: 10.1016/j.intimp.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Claudio L, Brosnan CF. Effects of prazosin on the blood-brain barrier during experimental autoimmune encephalomyelitis. Brain Res. 1992;594:233–243. doi: 10.1016/0006-8993(92)91130-7. [DOI] [PubMed] [Google Scholar]
- Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438–446. doi: 10.1016/S1474-4422(10)70028-4. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, Margolin DH, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786–1801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- Collin C, Davies P, Mutiboko IK, Ratcliffe S. Randomized controlled trial of cannabis-based medicine in spasticity caused by multiple sclerosis. Eur J Neurol. 2007;14:290–296. doi: 10.1111/j.1468-1331.2006.01639.x. [DOI] [PubMed] [Google Scholar]
- Comi G, Pulizzi A, Rovaris M, Abramsky O, Arbizu T, Boiko A, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;371:2085–2092. doi: 10.1016/S0140-6736(08)60918-6. [DOI] [PubMed] [Google Scholar]
- Comi G, Abramsky O, Arbizu T, Boyko A, Gold R, Havrdova E, et al. Oral laquinimod in patients with relapsing-remitting multiple sclerosis: 36-week double-blind active extension of the multi-centre, randomized, double-blind, parallel-group placebo-controlled study. Mult Scler. 2010;16:1360–1366. doi: 10.1177/1352458510378127. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Constantinescu C, Gran B. Autoimmune associations in multiple sclerosis. Nat Rev Neurol. 2010;6:1–2. doi: 10.1038/nrneurol.2010.147. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Ventura E, Hilliard B, Rostami A. Effects of the angiotensin converting enzyme inhibitor captopril on experimental autoimmune encephalomyelitis. Immunopharmacol Immunotoxicol. 1995;17:471–491. doi: 10.3109/08923979509016382. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Wysocka M, Hilliard B, Ventura ES, Lavi E, Trinchieri G, et al. Antibodies against IL-12 prevent superantigen-induced and spontaneous relapses of experimental autoimmune encephaloomyelitis. J Immunol. 1998;161:5097–5104. [PubMed] [Google Scholar]
- Constantinescu CS, Hilliard B, Wysocka M, Ventura ES, Bhopale MK, Trinchieri G, et al. IL-12 reverses the suppressive effect of the CD40 ligand blockade on experimental autoimmune encephalomyelitis (EAE) J Neurol Sci. 1999;171:60–64. doi: 10.1016/s0022-510x(99)00249-x. [DOI] [PubMed] [Google Scholar]
- Constantinescu CS, Hilliard B, Ventura E, Wysocka M, Showe L, Lavi E, et al. Modulation of susceptibility and resistance to an autoimmune model of multiple sclerosis in prototypically susceptible and resistant strains by neutralization of interleukin-12 and interleukin-4, respectively. Clin Immunol. 2001;98:23–30. doi: 10.1006/clim.2000.4944. [DOI] [PubMed] [Google Scholar]
- Correale J, Farez M. Helminth antigens modulate immune responses in cells from multiple sclerosis patients through TLR2-dependent mechanisms. J Immunol. 2009;183:5999–6012. doi: 10.4049/jimmunol.0900897. [DOI] [PubMed] [Google Scholar]
- Croxford JL, O'Neill JK, Ali RR, Browne K, Byrnes AP, Dallman MJ, et al. Local gene therapy with CTLA4-immunoglobulin fusion protein in experimental allergic encephalomyelitis. Eur J Immunol. 1998a;28:3904–3916. doi: 10.1002/(SICI)1521-4141(199812)28:12<3904::AID-IMMU3904>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Croxford JL, Triantaphyllopoulos K, Podhajcer OL, Feldmann M, Baker D, Chernajovsky Y. Cytokine gene therapy in experimental allergic encephalomyelitis by injection of plasmid DNA-cationic liposome complex into the central nervous system. J Immunol. 1998b;160:5181–5187. [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Day M. Histopathology of EAE. Experimental Models of Multiple Sclerosis. L. E and C. C. New York: Springer; 2005. pp. 25–43. [Google Scholar]
- De Magistris MT, Alexander J, Coggeshall M, Altman A, Gaeta FC, Grey HM, et al. Antigen analog-major histocompatibility complexes act as antagonists of the T cell receptor. Cell. 1992;68:625–634. doi: 10.1016/0092-8674(92)90139-4. [DOI] [PubMed] [Google Scholar]
- Dijkstra CD, van der Voort ER, De Groot CJ, Huitinga I, Uitdehaag BM, Polman CH, et al. Therapeutic effect of the D2-dopamine agonist bromocriptine on acute and relapsing experimental allergic encephalomyelitis. Psychoneuroendocrinology. 1994;19:135–142. doi: 10.1016/0306-4530(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Dimitriadou V, Pang X, Theoharides TC. Hydroxyzine inhibits experimental allergic encephalomyelitis (EAE) and associated brain mast cell activation. Int J Immunopharmacol. 2000;22:673–684. doi: 10.1016/s0192-0561(00)00029-1. [DOI] [PubMed] [Google Scholar]
- Dubinsky AN, Burt RK, Martin R, Muraro PA. T-cell clones persisting in the circulation after autologous hematopoietic SCT are undetectable in the peripheral CD34+ selected graft. Bone Marrow Transplant. 2010;45:325–331. doi: 10.1038/bmt.2009.139. [DOI] [PubMed] [Google Scholar]
- Edwards LJ, Constantinescu CS. A prospective study of conditions associated with multiple sclerosis in a cohort of 658 consecutive outpatients attending a multiple sclerosis clinic. Mult Scler. 2004;10:575–581. doi: 10.1191/1352458504ms1087oa. [DOI] [PubMed] [Google Scholar]
- Edwards LJ, Tench CR, Gilmore CP, Evangelou N, Constantinescu CS. Multiple sclerosis findings in the spinal cord. Expert Rev Neurother. 2007;7:1203–1211. doi: 10.1586/14737175.7.9.1203. [DOI] [PubMed] [Google Scholar]
- Einstein O, Karussis D, Grigoriadis N, Mizrachi-Kol R, Reinhartz E, Abramsky O, et al. Intraventricular transplantation of neural precursor cell spheres attenuates acute experimental allergic encephalomyelitis. Mol Cell Neurosci. 2003;24:1074–1082. doi: 10.1016/j.mcn.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Einstein O, Grigoriadis N, Mizrachi-Kol R, Reinhartz E, Polyzoidou E, Lavon I, et al. Transplanted neural precursor cells reduce brain inflammation to attenuate chronic experimental autoimmune encephalomyelitis. Exp Neurol. 2006;198:275–284. doi: 10.1016/j.expneurol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Elhofy A, Wang J, Tani M, Fife BT, Kennedy KJ, Bennett J, et al. Transgenic expression of CCL2 in the central nervous system prevents experimental autoimmune encephalomyelitis. J Leukoc Biol. 2005;77:229–237. doi: 10.1189/jlb.0804465. [DOI] [PubMed] [Google Scholar]
- Elkhalifa A, Weiner H. Cyclophosphamide treatment of MS: current therapeutic approaches and treatment regimens. Int MS J. 2010;17:12–18. [PubMed] [Google Scholar]
- Ephrem A, Chamat S, Miquel C, Fisson S, Mouthon L, Caligiuri G, et al. Expansion of CD4+CD25+ regulatory T cells by intravenous immunoglobulin: a critical factor in controlling experimental autoimmune encephalomyelitis. Blood. 2008;111:715–722. doi: 10.1182/blood-2007-03-079947. [DOI] [PubMed] [Google Scholar]
- Esaki Y, Li Y, Sakata D, Yao C, Segi-Nishida E, Matsuoka T, et al. Dual roles of PGE2-EP4 signaling in mouse experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:12233–12238. doi: 10.1073/pnas.0915112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science. 1991;252:1308–1310. doi: 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- Farooqi N, Gran B, Constantinescu CS. Are current disease-modifying therapeutics in multiple sclerosis justified on the basis of studies in experimental autoimmune encephalomyelitis? J Neurochem. 2010;115:829–844. doi: 10.1111/j.1471-4159.2010.06982.x. [DOI] [PubMed] [Google Scholar]
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- Filippi M, Dousset V, McFarland HF, Miller DH, Grossman RI. Role of magnetic resonance imaging in the diagnosis and monitoring of multiple sclerosis: consensus report of the White Matter Study Group. J Magn Reson Imaging. 2002;15:499–504. doi: 10.1002/jmri.10097. [DOI] [PubMed] [Google Scholar]
- Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flugel A, Berkowicz T, Ritter T, Labeur M, Jenne DE, Li Z, et al. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity. 2001;14:547–560. doi: 10.1016/s1074-7613(01)00143-1. [DOI] [PubMed] [Google Scholar]
- Foster CA, Howard LM, Schweitzer A, Persohn E, Hiestand PC, Balatoni B, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–475. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- Foster CA, Mechtcheriakova D, Storch MK, Balatoni B, Howard LM, Bornancin F, et al. FTY720 rescue therapy in the dark agouti rat model of experimental autoimmune encephalomyelitis: expression of central nervous system genes and reversal of blood-brain-barrier damage. Brain Pathol. 2009;19:254–266. doi: 10.1111/j.1750-3639.2008.00182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman RM, Adekanmi O, Waterfield MR, Waterfield AE, Wright D, Zajicek J. The effect of cannabis on urge incontinence in patients with multiple sclerosis: a multicentre, randomised placebo-controlled trial (CAMS-LUTS) Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:636–641. doi: 10.1007/s00192-006-0086-x. [DOI] [PubMed] [Google Scholar]
- Freedman MS, Bar-Or A, Atkins HL, Karussis D, Frassoni F, Lazarus H, et al. The therapeutic potential of mesenchymal stem cell transplantation as a treatment for multiple sclerosis: consensus report of the International MSCT Study Group. Mult Scler. 2010;16:503–510. doi: 10.1177/1352458509359727. [DOI] [PubMed] [Google Scholar]
- Frei K, Eugster HP, Bopst M, Constantinescu CS, Lavi E, Fontana A. Tumor necrosis factor alpha and lymphotoxin alpha are not required for induction of acute experimental autoimmune encephalomyelitis. J Exp Med. 1997;185:2177–2182. doi: 10.1084/jem.185.12.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- Fujino M, Funeshima N, Kitazawa Y, Kimura H, Amemiya H, Suzuki S, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–77. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- Furlan R, Poliani PL, Marconi PC, Bergami A, Ruffini F, Adorini L, et al. Central nervous system gene therapy with interleukin-4 inhibits progression of ongoing relapsing-remitting autoimmune encephalomyelitis in Biozzi AB/H mice. Gene Ther. 2001;8:13–19. doi: 10.1038/sj.gt.3301357. [DOI] [PubMed] [Google Scholar]
- Furlan R, Bergami A, Brambilla E, Butti E, De Simoni MG, Campagnoli M, et al. HSV-1-mediated IL-1 receptor antagonist gene therapy ameliorates MOG(35–55)-induced experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther. 2007;14:93–98. doi: 10.1038/sj.gt.3302805. [DOI] [PubMed] [Google Scholar]
- Furlan R, Cuomo C, Martino G. Animal models of multiple sclerosis. Methods Mol Biol. 2009;549:157–173. doi: 10.1007/978-1-60327-931-4_11. [DOI] [PubMed] [Google Scholar]
- Furtado GC, Marcondes MC, Latkowski JA, Tsai J, Wensky A, Lafaille JJ. Swift entry of myelin-specific T lymphocytes into the central nervous system in spontaneous autoimmune encephalomyelitis. J Immunol. 2008;181:4648–4655. doi: 10.4049/jimmunol.181.7.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garren H, Robinson WH, Krasulova E, Havrdova E, Nadj C, Selmaj K, et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann Neurol. 2008;63:611–620. doi: 10.1002/ana.21370. [DOI] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Panet H, Holdengreber V, Mosberg-Galili R, Offen D. Axonal damage is reduced following glatiramer acetate treatment in C57/bl mice with chronic-induced experimental autoimmune encephalomyelitis. Neurosci Res. 2003;47:201–207. doi: 10.1016/s0168-0102(03)00217-7. [DOI] [PubMed] [Google Scholar]
- Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- Giuliani F, Fu SA, Metz LM, Yong VW. Effective combination of minocycline and interferon-beta in a model of multiple sclerosis. J Neuroimmunol. 2005a;165:83–91. doi: 10.1016/j.jneuroim.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J Leukoc Biol. 2005b;78:135–143. doi: 10.1189/jlb.0804477. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Krakowski M, Han Y, Owens T, Ransohoff RM. Chemokine expression in GKO mice (lacking interferon-gamma) with experimental autoimmune encephalomyelitis. J Neurovirol. 1999;5:95–101. doi: 10.3109/13550289909029750. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Goodin DS, Cohen BA, O'Connor P, Kappos L, Stevens JC. Assessment: the use of natalizumab (Tysabri) for the treatment of multiple sclerosis (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;71:766–773. doi: 10.1212/01.wnl.0000320512.21919.d2. [DOI] [PubMed] [Google Scholar]
- Gordon D, Pavlovska G, Uney JB, Wraith DC, Scolding NJ. Human mesenchymal stem cells infiltrate the spinal cord, reduce demyelination, and localize to white matter lesions in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2010;69:1087–1095. doi: 10.1097/NEN.0b013e3181f97392. [DOI] [PubMed] [Google Scholar]
- Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- Gran B, Becher B. Dual and opposite actions of cytokines in autoimmune inflammatory demyelination. Open Autoimmun J. 2010;2:139–140. [Google Scholar]
- Gran B, Tranquill LR, Chen M, Bielekova B, Zhou W, Dhib-Jalbut S, et al. Mechanisms of immunomodulation by glatiramer acetate. Neurology. 2000;55:1704–1714. doi: 10.1212/wnl.55.11.1704. [DOI] [PubMed] [Google Scholar]
- Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- Gran B, Chu N, Zhang GX, Yu S, Li Y, Chen XH, et al. Early administration of IL-12 suppresses EAE through induction of interferon-gamma. J Neuroimmunol. 2004;156:123–131. doi: 10.1016/j.jneuroim.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Gran B, O'Brien K, Fitzgerald D, Rostami A. Experimental Autoimmune Encephalomyelitis. Handbook of Neurochemistry and Molecular Neurobiology A. Lajtha. Heidelberg: Springer; 2007. p. 19. [Google Scholar]
- Haas J. High dose IVIG in the post partum period for prevention of exacerbations in MS. Mult Scler. 2000;6(Suppl 2):S18–S20. discussion S33. [PubMed] [Google Scholar]
- Hafler DA, Orav J, Gertz R, Stazzone L, Weiner HL. Immunologic effects of cyclophosphamide/ACTH in patients with chronic progressive multiple sclerosis. J Neuroimmunol. 1991;32:149–158. doi: 10.1016/0165-5728(91)90007-t. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Saadeh MG, Kuchroo VK, Milford E, Steinman L. TCR usage in human and experimental demyelinating disease. Immunol Today. 1996;17:152–159. doi: 10.1016/0167-5699(96)80611-6. [DOI] [PubMed] [Google Scholar]
- Hampton DW, Anderson J, Pryce G, Irvine KA, Giovannoni G, Fawcett JW, et al. An experimental model of secondary progressive multiple sclerosis that shows regional variation in gliosis, remyelination, axonal and neuronal loss. J Neuroimmunol. 2008;201–202:200–211. doi: 10.1016/j.jneuroim.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- t Hart BA, Hintzen RQ, Laman JD. Preclinical assessment of therapeutic antibodies against human CD40 and human interleukin-12/23p40 in a nonhuman primate model of multiple sclerosis. Neurodegener Dis. 2008;5:38–52. doi: 10.1159/000109937. [DOI] [PubMed] [Google Scholar]
- t Hart BA, Hintzen RQ, Laman JD. Multiple sclerosis – a response-to-damage model. Trends Mol Med. 2009;15:235–244. doi: 10.1016/j.molmed.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Hartung HP, Gonsette R, Konig N, Kwiecinski H, Guseo A, Morrissey SP, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Dawson DM, Lehrich JR, Beal MF, Kevy SV, Propper RD, et al. Intensive immunosuppression in progressive multiple sclerosis. A randomized, three-arm study of high-dose intravenous cyclophosphamide, plasma exchange, and ACTH. N Engl J Med. 1983;308:173–180. doi: 10.1056/NEJM198301273080401. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- Havrdova E, Galetta S, Hutchinson M, Stefoski D, Bates D, Polman CH, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study. Lancet Neurol. 2009;8:254–260. doi: 10.1016/S1474-4422(09)70021-3. [DOI] [PubMed] [Google Scholar]
- Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung HP. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol. 2006;2:201–211. doi: 10.1038/ncpneuro0154. [DOI] [PubMed] [Google Scholar]
- Hertz F, Deghenghi R. Effect of rat and beta-human interferons on hyperacute experimental allergic encephalomyelitis in rats. Agents Actions. 1985;16:397–403. doi: 10.1007/BF01982879. [DOI] [PubMed] [Google Scholar]
- Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y. Experimental autoimmune encephalomyelitis in NF-kappa B-deficient mice:roles of NF-kappa B in the activation and differentiation of autoreactive T cells. J Immunol. 1999;163:2937–2943. [PubMed] [Google Scholar]
- Hines KL, Kulkarni AB, McCarthy JB, Tian H, Ward JM, Christ M, et al. Synthetic fibronectin peptides interrupt inflammatory cell infiltration in transforming growth factor beta 1 knockout mice. Proc Natl Acad Sci U S A. 1994;91:5187–5191. doi: 10.1073/pnas.91.11.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology. 2009;128:260–270. doi: 10.1111/j.1365-2567.2009.03115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 1990;172:1025–1033. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illes Z, Stern JN, Reddy J, Waldner H, Mycko MP, Brosnan CF, et al. Modified amino acid copolymers suppress myelin basic protein 85-99-induced encephalomyelitis in humanized mice through different effects on T cells. Proc Natl Acad Sci U S A. 2004;101:11749–11754. doi: 10.1073/pnas.0403833101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Refining genetic associations in multiple sclerosis. Lancet Neurol. 2008;7:567–569. doi: 10.1016/S1474-4422(08)70122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192:1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs L, O'Malley J, Freeman A, Murawski J, Ekes R. Intrathecal interferon in multiple sclerosis. Arch Neurol. 1982;39:609–615. doi: 10.1001/archneur.1982.00510220007002. [DOI] [PubMed] [Google Scholar]
- Jadidi-Niaragh F, Mirshafiey A. Histamine and histamine receptors in pathogenesis and treatment of multiple sclerosis. Neuropharmacology. 2010;59:180–189. doi: 10.1016/j.neuropharm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Jaini R, Hannaman D, Johnson JM, Bernard RM, Altuntas CZ, Delasalas MM, et al. Gene-based intramuscular interferon-beta therapy for experimental autoimmune encephalomyelitis. Mol Ther. 2006;14:416–422. doi: 10.1016/j.ymthe.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Jee Y, Piao WH, Liu R, Bai XF, Rhodes S, Rodebaugh R, et al. CD4(+)CD25(+) regulatory T cells contribute to the therapeutic effects of glatiramer acetate in experimental autoimmune encephalomyelitis. Clin Immunol. 2007;125:34–42. doi: 10.1016/j.clim.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1998;50:701–708. doi: 10.1212/wnl.50.3.701. [DOI] [PubMed] [Google Scholar]
- Jones JL, Phuah CL, Cox AL, Thompson SA, Ban M, Shawcross J, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H) J Clin Invest. 2009;119:2052–2061. doi: 10.1172/JCI37878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge SI, Yeh JZ, Mannie MD, Pope Seifert L, Paterson PY. Potassium Channel Blockers Inhibit Adoptive Transfer of Experimental Allergic Encephalomyelitis by Myelin-Basic-Protein-Stimulated Rat T Lymphocytes. J Biomed Sci. 1997;4:169–178. doi: 10.1007/BF02255646. [DOI] [PubMed] [Google Scholar]
- Jung S, Huitinga I, Schmidt B, Zielasek J, Dijkstra CD, Toyka KV, et al. Selective elimination of macrophages by dichlormethylene diphosphonate-containing liposomes suppresses experimental autoimmune neuritis. J Neurol Sci. 1993;119:195–202. doi: 10.1016/0022-510x(93)90134-k. [DOI] [PubMed] [Google Scholar]
- Kala M, Rhodes SN, Piao WH, Shi FD, Campagnolo DI, Vollmer TL. B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp Neurol. 2010;221:136–145. doi: 10.1016/j.expneurol.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, et al. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- Kap YS, Smith P, Jagessar SA, Remarque E, Blezer E, Strijkers GJ, et al. Fast progression of recombinant human myelin/oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis in marmosets is associated with the activation of MOG34-56-specific cytotoxic T cells. J Immunol. 2008;180:1326–1337. doi: 10.4049/jimmunol.180.3.1326. [DOI] [PubMed] [Google Scholar]
- Kap YS, Laman JD, t Hart BA. Experimental autoimmune encephalomyelitis in the common marmoset, a bridge between rodent EAE and multiple sclerosis for immunotherapy development. J Neuroimmune Pharmacol. 2010a;5:220–230. doi: 10.1007/s11481-009-9178-y. [DOI] [PubMed] [Google Scholar]
- Kap YS, van Driel N, Blezer E, Parren PW, Bleeker WK, Laman JD, et al. Late B cell depletion with a human anti-human CD20 IgG1kappa monoclonal antibody halts the development of experimental autoimmune encephalomyelitis in marmosets. J Immunol. 2010b;185:3990–4003. doi: 10.4049/jimmunol.1001393. [DOI] [PubMed] [Google Scholar]
- Kappos L, Radue E, Dellas S. 15 6 Deoxyspergualin (DSG) in the treatment of active multiple sclerosis: final analysis of the European multicenter study. Neurology. 1996;46A:410–411. [Google Scholar]
- Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6:1176–1182. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- Kappos L, Bates D, Hartung HP, Havrdova E, Miller D, Polman CH, et al. Natalizumab treatment for multiple sclerosis: recommendations for patient selection and monitoring. Lancet Neurol. 2007;6:431–441. doi: 10.1016/S1474-4422(07)70078-9. [DOI] [PubMed] [Google Scholar]
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- Karussis DM, Slavin S, Ben-Nun A, Ovadia H, Vourka-Karussis U, Lehmann D, et al. Chronic-relapsing experimental autoimmune encephalomyelitis (CR-EAE): treatment and induction of tolerance, with high dose cyclophosphamide followed by syngeneic bone marrow transplantation. J Neuroimmunol. 1992;39:201–210. doi: 10.1016/0165-5728(92)90254-i. [DOI] [PubMed] [Google Scholar]
- Karussis DM, Lehmann D, Slavin S, Vourka-Karussis U, Mizrachi-Koll R, Ovadia H, et al. Treatment of chronic-relapsing experimental autoimmune encephalomyelitis with the synthetic immunomodulator linomide (quinoline-3-carboxamide) Proc Natl Acad Sci U S A. 1993a;90:6400–6404. doi: 10.1073/pnas.90.14.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karussis DM, Vourka-Karussis U, Lehmann D, Ovadia H, Mizrachi-Koll R, Ben-Nun A, et al. Prevention and reversal of adoptively transferred, chronic relapsing experimental autoimmune encephalomyelitis with a single high dose cytoreductive treatment followed by syngeneic bone marrow transplantation. J Clin Invest. 1993b;92:765–772. doi: 10.1172/JCI116648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karussis D, Vourka-Karussis U, Mizrachi-Koll R, Abramsky O. Acute/relapsing experimental autoimmune encephalomyelitis: induction of long lasting, antigen-specific tolerance by syngeneic bone marrow transplantation. Mult Scler. 1999;5:17–21. doi: 10.1177/135245859900500104. [DOI] [PubMed] [Google Scholar]
- Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:1187–1194. doi: 10.1001/archneurol.2010.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassis I, Grigoriadis N, Gowda-Kurkalli B, Mizrachi-Kol R, Ben-Hur T, Slavin S, et al. Neuroprotection and immunomodulation with mesenchymal stem cells in chronic experimental autoimmune encephalomyelitis. Arch Neurol. 2008;65:753–761. doi: 10.1001/archneur.65.6.753. [DOI] [PubMed] [Google Scholar]
- Kataoka H, Sugahara K, Shimano K, Teshima K, Koyama M, Fukunari A, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2:439–448. [PubMed] [Google Scholar]
- Katz U, Kishner I, Magalashvili D, Shoenfeld Y, Achiron A. Long term safety of IVIg therapy in multiple sclerosis: 10 years experience. Autoimmunity. 2006;39:513–517. doi: 10.1080/08916930600825867. [DOI] [PubMed] [Google Scholar]
- Keith AB, Arnon R, Teitelbaum D, Caspary EA, Wisniewski HM. The effect of Cop 1, a synthetic polypeptide, on chronic relapsing experimental allergic encephalomyelitis in guinea pigs. J Neurol Sci. 1979;42:267–274. doi: 10.1016/0022-510x(79)90058-3. [DOI] [PubMed] [Google Scholar]
- Kent SJ, Karlik SJ, Cannon C, Hines DK, Yednock TA, Fritz LC, et al. A monoclonal antibody to alpha 4 integrin suppresses and reverses active experimental allergic encephalomyelitis. J Neuroimmunol. 1995;58:1–10. doi: 10.1016/0165-5728(94)00165-k. [DOI] [PubMed] [Google Scholar]
- Killestein J, Olsson T, Wallstrom E, Svenningsson A, Khademi M, Blumhardt LD, et al. Antibody-mediated suppression of Vbeta5.2/5.3(+) T cells in multiple sclerosis: results from an MRI-monitored phase II clinical trial. Ann Neurol. 2002;51:467–474. doi: 10.1002/ana.10146. [DOI] [PubMed] [Google Scholar]
- Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology. 1999;52:1230–1238. doi: 10.1212/wnl.52.6.1230. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- Korn T, Magnus T, Toyka K, Jung S. Modulation of effector cell functions in experimental autoimmune encephalomyelitis by leflunomide – mechanisms independent of pyrimidine depletion. J Leukoc Biol. 2004;76:950–960. doi: 10.1189/jlb.0504308. [DOI] [PubMed] [Google Scholar]
- Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:18460–18465. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. 2001;35:204–212. doi: 10.1002/glia.1085. [DOI] [PubMed] [Google Scholar]
- Krapf H, Morrissey SP, Zenker O, Zwingers T, Gonsette R, Hartung HP. Effect of mitoxantrone on MRI in progressive MS: results of the MIMS trial. Neurology. 2005;65:690–695. doi: 10.1212/01.wnl.0000174439.70369.7a. [DOI] [PubMed] [Google Scholar]
- van der Laan LJ, van der Goes A, Wauben MH, Ruuls SR, Dopp EA, De Groot CJ, et al. Beneficial effect of modified peptide inhibitor of alpha4 integrins on experimental allergic encephalomyelitis in Lewis rats. J Neurosci Res. 2002;67:191–199. doi: 10.1002/jnr.10095. [DOI] [PubMed] [Google Scholar]
- Lafaille JJ, Nagashima K, Katsuki M, Tonegawa S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 1994;78:399–408. doi: 10.1016/0092-8674(94)90419-7. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza C, Morando S, Voci A, Canesi L, Principato MC, Serpero LD, et al. Neuroprotective mesenchymal stem cells are endowed with a potent antioxidant effect in vivo. J Neurochem. 2009;110:1674–1684. doi: 10.1111/j.1471-4159.2009.06268.x. [DOI] [PubMed] [Google Scholar]
- Laouar Y, Town T, Jeng D, Tran E, Wan Y, Kuchroo VK, et al. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H. Experimental models of multiple sclerosis. Rev Neurol. 2007;163:651–655. doi: 10.1016/s0035-3787(07)90474-9. [DOI] [PubMed] [Google Scholar]
- Leger OJ, Yednock TA, Tanner L, Horner HC, Hines DK, Keen S, et al. Humanization of a mouse antibody against human alpha-4 integrin: a potential therapeutic for the treatment of multiple sclerosis. Hum Antibodies. 1997;8:3–16. [PubMed] [Google Scholar]
- Leone DR, Giza K, Gill A, Dolinski BM, Yang W, Perper S, et al. An assessment of the mechanistic differences between two integrin alpha 4 beta 1 inhibitors, the monoclonal antibody TA-2 and the small molecule BIO5192, in rat experimental autoimmune encephalomyelitis. J Pharmacol Exp Ther. 2003;305:1150–1162. doi: 10.1124/jpet.102.047332. [DOI] [PubMed] [Google Scholar]
- Levine S, Saltzman A. Regional suppression, therapy after onset and prevention of relapses in experimental allergic encephalomyelitis by mitoxantrone. J Neuroimmunol. 1986;13:175–181. doi: 10.1016/0165-5728(86)90063-9. [DOI] [PubMed] [Google Scholar]
- Libbey JE, Fujinami RS. Experimental autoimmune encephalomyelitis as a testing paradigm for adjuvants and vaccines. Vaccine. 2010 doi: 10.1016/j.vaccine.2010.08.103. DOI: 10.1016/j.vaccine.2010.08.103 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S-Y, Constantinescu CS. TNF-alpha: a paradigm of paradox and complexity in multiple sclerosis and its animal models. Open Autoimmun J. 2010a;2:160–170. [Google Scholar]
- Lim SY, Constantinescu CS. Current and future disease-modifying therapies in multiple sclerosis. Int J Clin Pract. 2010b;64:637–650. doi: 10.1111/j.1742-1241.2009.02261.x. [DOI] [PubMed] [Google Scholar]
- Lin X, Tench CR, Evangelou N, Jaspan T, Constantinescu CS. Measurement of spinal cord atrophy in multiple sclerosis. J Neuroimaging. 2004;14(Suppl):20S–26S. doi: 10.1177/1051228404266265. [DOI] [PubMed] [Google Scholar]
- Lisak RP, Zweiman B, Blanchard N, Rorke LB. Effect of treatment with Copolymer 1 (Cop-1) on the in vivo and in vitro manifestations of experimental allergic encephalomyelitis (EAE) J Neurol Sci. 1983;62:281–293. doi: 10.1016/0022-510x(83)90205-8. [DOI] [PubMed] [Google Scholar]
- Lobell A, Weissert R, Storch MK, Svanholm C, de Graaf KL, Lassmann H, et al. Vaccination with DNA encoding an immunodominant myelin basic protein peptide targeted to Fc of immunoglobulin G suppresses experimental autoimmune encephalomyelitis. J Exp Med. 1998;187:1543–1548. doi: 10.1084/jem.187.9.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Lu Z, Hu X, Zhu C, Wang D, Zheng X, Liu Q. Overexpression of CNTF in Mesenchymal Stem Cells reduces demyelination and induces clinical recovery in experimental autoimmune encephalomyelitis mice. J Neuroimmunol. 2009;206:58–69. doi: 10.1016/j.jneuroim.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Lavasa M, Viti C, Knobler RL. Suppression of acute and relapsing experimental allergic encephalomyelitis with mitoxantrone. Clin Immunol Immunopathol. 1987;45:122–128. doi: 10.1016/0090-1229(87)90118-8. [DOI] [PubMed] [Google Scholar]
- Luca ME, Visser L, Lucas CJ, Nagelkerken L. IFN-beta modulates specific T cell responses in vitro but does not affect Experimental Autoimmune Encephalomyelitis in the SJL mouse. J Neuroimmunol. 1999;100:190–196. doi: 10.1016/s0165-5728(99)00198-8. [DOI] [PubMed] [Google Scholar]
- Luhder F, Lee DH, Gold R, Stegbauer J, Linker RA. Small but powerful: short peptide hormones and their role in autoimmune inflammation. J Neuroimmunol. 2009;217:1–7. doi: 10.1016/j.jneuroim.2009.08.008. [DOI] [PubMed] [Google Scholar]
- McCue D, Ryan KR, Wraith DC, Anderton SM. Activation thresholds determine susceptibility to peptide-induced tolerance in a heterogeneous myelin-reactive T cell repertoire. J Neuroimmunol. 2004;156:96–106. doi: 10.1016/j.jneuroim.2004.07.010. [DOI] [PubMed] [Google Scholar]
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen LS, Christophersen P, Olesen SP. Blockade of Ca2+-activated K+ channels in T cells: an option for the treatment of multiple sclerosis? Eur J Immunol. 2005;35:1023–1026. doi: 10.1002/eji.200526078. [DOI] [PubMed] [Google Scholar]
- Makar TK, Trisler D, Bever CT, Goolsby JE, Sura KT, Balasubramanian S, et al. Stem cell based delivery of IFN-beta reduces relapses in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;196:67–81. doi: 10.1016/j.jneuroim.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Mangano K, Nicoletti A, Patti F, Donia M, Malaguarnera L, Signorelli S, et al. Variable effects of cyclophosphamide in rodent models of experimental allergic encephalomyelitis. Clin Exp Immunol. 2010;159:159–168. doi: 10.1111/j.1365-2249.2009.04050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- Martin D, Near SL. Protective effect of the interleukin-1 receptor antagonist (IL-1ra) on experimental allergic encephalomyelitis in rats. J Neuroimmunol. 1995;61:241–245. doi: 10.1016/0165-5728(95)00108-e. [DOI] [PubMed] [Google Scholar]
- Martinelli Boneschi F, Rovaris M, Capra R, Comi G. Mitoxantrone for multiple sclerosis. Cochrane Database Syst Rev. 2005;(4):CD002127. doi: 10.1002/14651858.CD002127.pub2. [DOI] [PubMed] [Google Scholar]
- Martin-Saavedra FM, Flores N, Dorado B, Eguiluz C, Bravo B, Garcia-Merino A, et al. Beta-interferon unbalances the peripheral T cell proinflammatory response in experimental autoimmune encephalomyelitis. Mol Immunol. 2007;44:3597–3607. doi: 10.1016/j.molimm.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Massacesi L, Castigli E, Vergelli M, Olivotto J, Abbamondi AL, Sarlo F, et al. Immunosuppressive activity of 13-cis-retinoic acid and prevention of experimental autoimmune encephalomyelitis in rats. J Clin Invest. 1991;88:1331–1337. doi: 10.1172/JCI115438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massacesi L, Parigi A, Barilaro A, Repice AM, Pellicano G, Konze A, et al. Efficacy of azathioprine on multiple sclerosis new brain lesions evaluated using magnetic resonance imaging. Arch Neurol. 2005;62:1843–1847. doi: 10.1001/archneur.62.12.1843. [DOI] [PubMed] [Google Scholar]
- Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeson AP, Piddlesden S, Morgan BP, Reynolds R. The distribution of inflammatory demyelinated lesions in the central nervous system of rats with antibody-augmented demyelinating experimental allergic encephalomyelitis. Exp Neurol. 1994;129:299–310. doi: 10.1006/exnr.1994.1172. [DOI] [PubMed] [Google Scholar]
- van der Meide PH, de Labie MC, Ruuls SR, Groenestein RJ, Botman CA, Olsson T, et al. Discontinuation of treatment with IFN-beta leads to exacerbation of experimental autoimmune encephalomyelitis in Lewis rats. Rapid reversal of the antiproliferative activity of IFN-beta and excessive expansion of autoreactive T cells as disease promoting mechanisms. J Neuroimmunol. 1998;84:14–23. doi: 10.1016/s0165-5728(97)00207-5. [DOI] [PubMed] [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol. 2009;36:1118–1125. doi: 10.3899/jrheum.090074. [DOI] [PubMed] [Google Scholar]
- Metz LM, Zhang Y, Yeung M, Patry DG, Bell RB, Stoian CA, et al. Minocycline reduces gadolinium-enhancing magnetic resonance imaging lesions in multiple sclerosis. Ann Neurol. 2004;55:756. doi: 10.1002/ana.20111. [DOI] [PubMed] [Google Scholar]
- Metz LM, Li D, Traboulsee A, Myles ML, Duquette P, Godin J, et al. Glatiramer acetate in combination with minocycline in patients with relapsing – remitting multiple sclerosis: results of a Canadian, multicenter, double-blind, placebo-controlled trial. Mult Scler. 2009;15:1183–1194. doi: 10.1177/1352458509106779. [DOI] [PubMed] [Google Scholar]
- Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- Miller DH, Soon D, Fernando KT, MacManus DG, Barker GJ, Yousry TA, et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology. 2007a;68:1390–1401. doi: 10.1212/01.wnl.0000260064.77700.fd. [DOI] [PubMed] [Google Scholar]
- Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 2007b;1103:179–191. doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- Mohan N, Edwards ET, Cupps TR, Oliverio PJ, Sandberg G, Crayton H, et al. Demyelination occurring during anti-tumor necrosis factor alpha therapy for inflammatory arthritides. Arthritis Rheum. 2001;44:2862–2869. doi: 10.1002/1529-0131(200112)44:12<2862::aid-art474>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Muraro PA, Uccelli A. Immuno-therapeutic potential of haematopoietic and mesenchymal stem cell transplantation in MS. Results Probl Cell Differ. 2010;51:237–257. doi: 10.1007/400_2008_14. [DOI] [PubMed] [Google Scholar]
- Muraro PA, Cassiani Ingoni R, Martin R. Hematopoietic stem cell transplantation for multiple sclerosis: current status and future challenges. Curr Opin Neurol. 2003;16:299–305. doi: 10.1097/01.wco.0000073930.19076.1b. [DOI] [PubMed] [Google Scholar]
- Muraro PA, Douek DC, Packer A, Chung K, Guenaga FJ, Cassiani-Ingoni R, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005;201:805–816. doi: 10.1084/jem.20041679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KJ, Witchell DR, Graham MJ, Koo S, Butler M, Condon TP. Antisense oligonucleotide blockade of alpha 4 integrin prevents and reverses clinical symptoms in murine experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;160:12–24. doi: 10.1016/j.jneuroim.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Nataf S, Louboutin JP, Chabannes D, Feve JR, Muller JY. Pentoxifylline inhibits experimental allergic encephalomyelitis. Acta Neurol Scand. 1993;88:97–99. doi: 10.1111/j.1600-0404.1993.tb04198.x. [DOI] [PubMed] [Google Scholar]
- Nessler S, Stadelmann C, Bittner A, Schlegel K, Gronen F, Brueck W, et al. Suppression of autoimmune encephalomyelitis by a neurokinin-1 receptor antagonist – a putative role for substance P in CNS inflammation. J Neuroimmunol. 2006;179:1–8. doi: 10.1016/j.jneuroim.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Ni X, Geller EB, Eppihimer MJ, Eisenstein TK, Adler MW, Tuma RF. Win 55212-2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult Scler. 2004;10:158–164. doi: 10.1191/1352458504ms1009oa. [DOI] [PubMed] [Google Scholar]
- Nicholson LB, Greer JM, Sobel RA, Lees MB, Kuchroo VK. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- No authors listed. Postvaccinal Encephalitis. Am J Public Health Nations Health. 1931;21:778–780. doi: 10.2105/ajph.21.7.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseworthy JH, Hopkins MB, Vandervoort MK, Karlik SJ, Lee DH, Penman M, et al. An open-trial evaluation of mitoxantrone in the treatment of progressive MS. Neurology. 1993;43:1401–1406. doi: 10.1212/wnl.43.7.1401. [DOI] [PubMed] [Google Scholar]
- O'Brien K, Fitzgerald D, Rostami A, Gran B. The TLR7 agonist, imiquimod, increases IFN-beta production and reduces the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;221:107–111. doi: 10.1016/j.jneuroim.2010.01.006. [DOI] [PubMed] [Google Scholar]
- O'Connor PW, Goodman A, Willmer-Hulme AJ, Libonati MA, Metz L, Murray RS, et al. Randomized multicenter trial of natalizumab in acute MS relapses: clinical and MRI effects. Neurology. 2004;62:2038–2043. doi: 10.1212/01.wnl.0000128136.79044.d6. [DOI] [PubMed] [Google Scholar]
- O'Connor P, Miller D, Riester K, Yang M, Panzara M, Dalton C, et al. Relapse rates and enhancing lesions in a phase II trial of natalizumab in multiple sclerosis. Mult Scler. 2005;11:568–572. doi: 10.1191/1352458505ms1205oa. [DOI] [PubMed] [Google Scholar]
- O'Connor PW, Li D, Freedman MS, Bar-Or A, Rice GP, Confavreux C, et al. A Phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. 2006;66:894–900. doi: 10.1212/01.wnl.0000203121.04509.31. [DOI] [PubMed] [Google Scholar]
- O'Malley HA, Shreiner AB, Chen GH, Huffnagle GB, Isom LL. Loss of Na+ channel beta2 subunits is neuroprotective in a mouse model of multiple sclerosis. Mol Cell Neurosci. 2009;40:143–155. doi: 10.1016/j.mcn.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaszynski KM, Liu H, Loo KK, Voskuhl RR. Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neuroimmunol. 2004;149:84–89. doi: 10.1016/j.jneuroim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Davoust N, Julien B, Hatterer E, Aguado T, Mechoulam R, et al. The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis. J Biol Chem. 2008;283:13320–13329. doi: 10.1074/jbc.M707960200. [DOI] [PubMed] [Google Scholar]
- Panitch HS, Hirsch RL, Haley AS, Johnson KP. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;1:893–895. doi: 10.1016/s0140-6736(87)92863-7. [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Rundle J, Patel R, Marshall I, Stretton J, Eaton R, et al. FTY720 ameliorates MOG-induced experimental autoimmune encephalomyelitis by suppressing both cellular and humoral immune responses. J Neurosci Res. 2010;88:346–359. doi: 10.1002/jnr.22196. [DOI] [PubMed] [Google Scholar]
- Park WS, Bae Y, Chung DH, Choi YL, Kim BK, Sung YC, et al. T cell expression of CIITA represses Th1 immunity. Int Immunol. 2004;16:1355–1364. doi: 10.1093/intimm/dxh132. [DOI] [PubMed] [Google Scholar]
- Pasquini MC, Griffith LM, Arnold DL, Atkins HL, Bowen JD, Chen JT, et al. Hematopoietic stem cell transplantation for multiple sclerosis: collaboration of the CIBMTR and EBMT to facilitate international clinical studies. Biol Blood Marrow Transplant. 2010;16:1076–1083. doi: 10.1016/j.bbmt.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- Paul C, Bolton C. Modulation of blood-brain barrier dysfunction and neurological deficits during acute experimental allergic encephalomyelitis by the N-methyl-D-aspartate receptor antagonist memantine. J Pharmacol Exp Ther. 2002;302:50–57. doi: 10.1124/jpet.302.1.50. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2− T lymphocytes. J Immunol. 1981;127:1420–1423. [PubMed] [Google Scholar]
- Piaton G, Williams A, Seilhean D, Lubetzki C. Remyelination in multiple sclerosis. Prog Brain Res. 2009;175:453–464. doi: 10.1016/S0079-6123(09)17530-1. [DOI] [PubMed] [Google Scholar]
- Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, et al. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci U S A. 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piraino PS, Yednock TA, Freedman SB, Messersmith EK, Pleiss MA, Vandevert C, et al. Prolonged reversal of chronic experimental allergic encephalomyelitis using a small molecule inhibitor of alpha4 integrin. J Neuroimmunol. 2002;131:147–159. doi: 10.1016/s0165-5728(02)00273-4. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–694. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, Salani G, et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436:266–271. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Gritti A, Blezer E, Amadio S, Brambilla E, Borsellino G, et al. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann Neurol. 2009;66:343–354. doi: 10.1002/ana.21745. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria’. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- Polman CH, O'Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- Pomeroy IM, Matthews PM, Frank JA, Jordan EK, Esiri MM. Demyelinated neocortical lesions in marmoset autoimmune encephalomyelitis mimic those in multiple sclerosis. Brain. 2005;128:2713–2721. doi: 10.1093/brain/awh626. [DOI] [PubMed] [Google Scholar]
- Poser C, Paty D, Scheinberg L, McDonald W, Davis F, Ebers G, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–231. doi: 10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock JM, Ledent C, Petzold A, Thompson AJ, Giovannoni G, Cuzner ML, Baker D. Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain. 2003;126:2191–2202. doi: 10.1093/brain/awg224. [DOI] [PubMed] [Google Scholar]
- Pryce G, O'Neill JK, Croxford JL, Amor S, Hankey DJ, East E, et al. Autoimmune tolerance eliminates relapses but fails to halt progression in a model of multiple sclerosis. J Neuroimmunol. 2005;165:41–52. doi: 10.1016/j.jneuroim.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Qu ZX, Dayal A, Jensen MA, Arnason BG. All-trans retinoic acid potentiates the ability of interferon beta-1b to augment suppressor cell function in multiple sclerosis. Arch Neurol. 1998;55:315–321. doi: 10.1001/archneur.55.3.315. [DOI] [PubMed] [Google Scholar]
- Racioppi L, Ronchese F, Matis LA, Germain RN. Peptide-major histocompatibility complex class II complexes with mixed agonist/antagonist properties provide evidence for ligand-related differences in T cell receptor-dependent intracellular signaling. J Exp Med. 1993;177:1047–1060. doi: 10.1084/jem.177.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racke MK, Bonomo A, Scott DE, Cannella B, Levine A, Raine CS, et al. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. J Exp Med. 1994;180:1961–1966. doi: 10.1084/jem.180.5.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafei M, Campeau PM, Aguilar-Mahecha A, Buchanan M, Williams P, Birman E, et al. Mesenchymal stromal cells ameliorate experimental autoimmune encephalomyelitis by inhibiting CD4 Th17 T cells in a CC chemokine ligand 2-dependent manner. J Immunol. 2009;182:5994–6002. doi: 10.4049/jimmunol.0803962. [DOI] [PubMed] [Google Scholar]
- Raine CS, Wisniewski HM, Iqbal K, Grundkeiqbal I, Norton WT. Studies on the encephalitogenic effects of purified preparations of human and bovine oligodendrocytes. Brain Res. 1977;120:269–286. doi: 10.1016/0006-8993(77)90906-4. [DOI] [PubMed] [Google Scholar]
- Raine CS, Traugott U, Nussenblatt RB, Stone SH. Optic neuritis and chronic relapsing experimental allergic encephalomyelitis: relationship to clinical course and comparison with multiple sclerosis. Lab Invest. 1980;42:327–335. [PubMed] [Google Scholar]
- Rammohan KW, Shoemaker J. Emerging multiple sclerosis oral therapies. Neurology. 2010;74(Suppl 1):S47–S53. doi: 10.1212/WNL.0b013e3181c97f89. [DOI] [PubMed] [Google Scholar]
- Rao NA, Tso MO, Zimmerman EL. Experimental allergic optic neuritis in guinea pigs: preliminary report. Invest Ophthalmol Vis Sci. 1977;16:338–342. [PubMed] [Google Scholar]
- Rausch M, Hiestand P, Foster CA, Baumann DR, Cannet C, Rudin M. Predictability of FTY720 efficacy in experimental autoimmune encephalomyelitis by in vivo macrophage tracking: clinical implications for ultrasmall superparamagnetic iron oxide-enhanced magnetic resonance imaging. J Magn Reson Imaging. 2004;20:16–24. doi: 10.1002/jmri.20057. [DOI] [PubMed] [Google Scholar]
- Reich EP, Cui L, Yang L, Pugliese-Sivo C, Golovko A, Petro M, et al. Blocking ion channel KCNN4 alleviates the symptoms of experimental autoimmune encephalomyelitis in mice. Eur J Immunol. 2005;35:1027–1036. doi: 10.1002/eji.200425954. [DOI] [PubMed] [Google Scholar]
- Reinke EK, Johnson MJ, Ling C, Karman J, Lee J, Weinstock JV, et al. Substance P receptor mediated maintenance of chronic inflammation in EAE. J Neuroimmunol. 2006;180:117–125. doi: 10.1016/j.jneuroim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Rejdak K, Jackson S, Giovannoni G. Multiple sclerosis: a practical overview for clinicians. Br Med Bull. 2010;5:79–104. doi: 10.1093/bmb/ldq017. [DOI] [PubMed] [Google Scholar]
- Ridge SC, Sloboda AE, McReynolds RA, Levine S, Oronsky AL, Kerwar SS. Suppression of experimental allergic encephalomyelitis by mitoxantrone. Clin Immunol Immunopathol. 1985;35:35–42. doi: 10.1016/0090-1229(85)90075-3. [DOI] [PubMed] [Google Scholar]
- Rivers TM, Schwentker FF. Encephalomyelitis Accompanied by Myelin Destruction Experimentally Produced in Monkeys. J Exp Med. 1935;61:689–702. doi: 10.1084/jem.61.5.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers TM, Stewart FW. Virus Iii Encephalitis. J Exp Med. 1928;48:603–613. doi: 10.1084/jem.48.5.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med. 1933;58:39–53. doi: 10.1084/jem.58.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson WH, Fontoura P, Lee BJ, de Vegvar HE, Tom J, Pedotti R, et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nat Biotechnol. 2003;21:1033–1039. doi: 10.1038/nbt859. [DOI] [PubMed] [Google Scholar]
- Rog DJ, Nurmikko TJ, Friede T, Young CA. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology. 2005;65:812–819. doi: 10.1212/01.wnl.0000176753.45410.8b. [DOI] [PubMed] [Google Scholar]
- Rostami AM, Sater RA, Bird SJ, Galetta S, Farber RE, Kamoun M, et al. A double-blind, placebo-controlled trial of extracoporeal photopheresis in chronic progressive multiople sclerosis. Mult Scler. 1999;5:198–203. doi: 10.1177/135245859900500310. [DOI] [PubMed] [Google Scholar]
- Rott O, Fleischer B, Cash E. Interleukin-10 prevents experimental allergic encephalomyelitis in rats. Eur J Immunol. 1994;24:1434–1440. doi: 10.1002/eji.1830240629. [DOI] [PubMed] [Google Scholar]
- Ruuls SR, de Labie MC, Weber KS, Botman CA, Groenestein RJ, Dijkstra CD, et al. The length of treatment determines whether IFN-beta prevents or aggravates experimental autoimmune encephalomyelitis in Lewis rats. J Immunol. 1996;157:5721–5731. [PubMed] [Google Scholar]
- Sabin AB, Wright AM. Acute ascending myelitis following a monkey bite, with the isolation of a virus capable of reproducing the disease. J Exp Med. 1934;59:115–136. doi: 10.1084/jem.59.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santambrogio L, Hochwald GM, Saxena B, Leu CH, Martz JE, Carlino JA, et al. Studies on the mechanisms by which transforming growth factor-beta (TGF-beta) protects against allergic encephalomyelitis. Antagonism between TGF-beta and tumor necrosis factor. J Immunol. 1993;151:1116–1127. [PubMed] [Google Scholar]
- Sanvito L, Constantinescu CS, Gran B, Hart BA. The multifaceted role of interferon-gamma in central nervous system autoimmune demyelination. Open Autoimmun J. 2010;2:151–159. [Google Scholar]
- Schaefer C, Hidalgo TR, Cashion L, Petry H, Brooks A, Szymanski P, et al. Gene-based delivery of IFN-beta is efficacious in a murine model of experimental allergic encephalomyelitis. J Interferon Cytokine Res. 2006;26:449–454. doi: 10.1089/jir.2006.26.449. [DOI] [PubMed] [Google Scholar]
- Schafer R, Ayturan M, Bantleon R, Kehlbach R, Siegel G, Pintaske J, et al. The use of clinically approved small particles of iron oxide (SPIO) for labeling of mesenchymal stem cells aggravates clinical symptoms in experimental autoimmune encephalomyelitis and influences their in vivo distribution. Cell Transplant. 2008;17:923–941. doi: 10.3727/096368908786576480. [DOI] [PubMed] [Google Scholar]
- Schorlemmer HU, Seiler FR. Therapeutic effects of 15-deoxyspergualin in acute and chronic relapsing experimental allergic encephalomyelitis (EAE) as models for multiple sclerosis (MS) Drugs Exp Clin Res. 1991;17:461–469. [PubMed] [Google Scholar]
- Scott CF, Jr, Cashman N, Spitler LE. Experimental allergic encephalitis; treatment with drugs which alter CNS serotonin levels. J Immunopharmacol. 1982;4:153–162. doi: 10.3109/08923978209026431. [DOI] [PubMed] [Google Scholar]
- Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. [DOI] [PubMed] [Google Scholar]
- Selmaj K, Raine CS, Cross AH. Anti-tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991;30:694–700. doi: 10.1002/ana.410300510. [DOI] [PubMed] [Google Scholar]
- Selmaj K, Kowal C, Walczak A, Nowicka J, Raine CS. Naked DNA vaccination differentially modulates autoimmune responses in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2000;111:34–44. doi: 10.1016/s0165-5728(00)00329-5. [DOI] [PubMed] [Google Scholar]
- Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204:2899–2912. doi: 10.1084/jem.20071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell DL, Reinke EK, Hogan LH, Sandor M, Fabry Z. Immunoregulation of CNS autoimmunity by helminth and mycobacterial infections. Immunol Lett. 2002;82:101–110. doi: 10.1016/s0165-2478(02)00025-1. [DOI] [PubMed] [Google Scholar]
- Sewell D, Qing Z, Reinke E, Elliot D, Weinstock J, Sandor M, et al. Immunomodulation of experimental autoimmune encephalomyelitis by helminth ova immunization. Int Immunol. 2003;15:59–69. doi: 10.1093/intimm/dxg012. [DOI] [PubMed] [Google Scholar]
- Sheremata WA, Vollmer TL, Stone LA, Willmer-Hulme AJ, Koller M. A safety and pharmacokinetic study of intravenous natalizumab in patients with MS. Neurology. 1999;52:1072–1074. doi: 10.1212/wnl.52.5.1072. [DOI] [PubMed] [Google Scholar]
- Sicotte NL, Giesser BS, Tandon V, Klutch R, Steiner B, Drain AE, et al. Testosterone treatment in multiple sclerosis: a pilot study. Arch Neurol. 2007;64:683–688. doi: 10.1001/archneur.64.5.683. [DOI] [PubMed] [Google Scholar]
- Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL. Increasing CNS noradrenaline reduces EAE severity. J Neuroimmune Pharmacol. 2010;5:252–259. doi: 10.1007/s11481-009-9182-2. [DOI] [PubMed] [Google Scholar]
- Skurkovich S, Boiko A, Beliaeva I, Buglak A, Alekseeva T, Smirnova N, et al. Randomized study of antibodies to IFN-gamma and TNF-alpha in secondary progressive multiple sclerosis. Mult Scler. 2001;7:277–284. doi: 10.1177/135245850100700502. [DOI] [PubMed] [Google Scholar]
- Smith DR, Weinstock-Guttman B, Cohen JA, Wei X, Gutmann C, Bakshi R, et al. A randomized blinded trial of combination therapy with cyclophosphamide in patients-with active multiple sclerosis on interferon beta. Mult Scler. 2005;11:573–582. doi: 10.1191/1352458505ms1210oa. [DOI] [PubMed] [Google Scholar]
- Smith ME, Eller NL, McFarland HF, Racke MK, Raine CS. Age dependence of clinical and pathological manifestations of autoimmune demyelination. Implications for multiple sclerosis. Am J Pathol. 1999;155:1147–1161. doi: 10.1016/S0002-9440(10)65218-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soilu-Hanninen M, Roytta M, Salmi A, Salonen R. Therapy with antibody against leukocyte integrin VLA-4 (CD49d) is effective and safe in virus-facilitated experimental allergic encephalomyelitis. J Neuroimmunol. 1997;72:95–105. doi: 10.1016/s0165-5728(96)00158-0. [DOI] [PubMed] [Google Scholar]
- Soldan SS, Alvarez Retuerto AI, Sicotte NL, Voskuhl RR. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J Immunol. 2003;171:6267–6274. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Sriram S, Steiner I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Ann Neurol. 2005;58:939–945. doi: 10.1002/ana.20743. [DOI] [PubMed] [Google Scholar]
- Stegbauer J, Lee DH, Seubert S, Ellrichmann G, Manzel A, Kvakan H, et al. Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc Natl Acad Sci U S A. 2009;106:14942–14947. doi: 10.1073/pnas.0903602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. Some misconceptions about understanding autoimmunity through experiments with knockouts. J Exp Med. 1997;185:2039–2041. doi: 10.1084/jem.185.12.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9:440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- Steinman L. Mixed results with modulation of TH-17 cells in human autoimmune diseases. Nat Immunol. 2010;11:41–44. doi: 10.1038/ni.1803. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil S. Transcriptional analysis of targets in multiple sclerosis. Nat Rev Immunol. 2003;3:483–492. doi: 10.1038/nri1108. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005;26:565–571. doi: 10.1016/j.it.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol. 2009;39:1108–1117. doi: 10.1002/eji.200839073. [DOI] [PubMed] [Google Scholar]
- Stern JN, Illes Z, Reddy J, Keskin DB, Sheu E, Fridkis-Hareli M, et al. Amelioration of proteolipid protein 139-151-induced encephalomyelitis in SJL mice by modified amino acid copolymers and their mechanisms. Proc Natl Acad Sci U S A. 2004;101:11743–11748. doi: 10.1073/pnas.0403832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JN, Keskin DB, Zhang H, Lv H, Kato Z, Strominger JL. Amino acid copolymer-specific IL-10-secreting regulatory T cells that ameliorate autoimmune diseases in mice. Proc Natl Acad Sci U S A. 2008;105:5172–5176. doi: 10.1073/pnas.0712131105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser-Fuchs S, Fazekas F, Deisenhammer F, Nahler G, Mamoli B. The Austrian Immunoglobulin in MS (AIMS) study: final analysis. Mult Scler. 2000;6(Suppl 2):S9–13. [PubMed] [Google Scholar]
- Strauss U, Wissel K, Jung S, Wulff H, Hansel W, Zhu J, et al. K(+) channel-blocking alkoxypsoralens inhibit the immune response of encephalitogenic T line cells and lymphocytes from Lewis rats challenged for experimental autoimmune encephalomyelitis. Immunopharmacology. 2000;48:51–63. doi: 10.1016/s0162-3109(00)00177-6. [DOI] [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc. 2006a;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006b;1:1952–1960. doi: 10.1038/nprot.2006.284. [DOI] [PubMed] [Google Scholar]
- Tallantyre E, Evangelou N, Constantinescu CS. Spotlight on teriflunomide. Int MS J. 2008;15:62–68. [PubMed] [Google Scholar]
- Tanuma N, Shin T, Matsumoto Y. Characterization of acute versus chronic relapsing autoimmune encephalomyelitis in DA rats. J Neuroimmunol. 2000;108:171–180. doi: 10.1016/s0165-5728(00)00309-x. [DOI] [PubMed] [Google Scholar]
- Teitelbaum D, Meshorer A, Hirshfeld T, Arnon R, Sela M. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur J Immunol. 1971;1:242–248. doi: 10.1002/eji.1830010406. [DOI] [PubMed] [Google Scholar]
- Teitelbaum D, Webb C, Meshorer A, Arnon R, Sela M. Suppression by several synthetic polypeptides of experimental allergic encephalomyelitis induced in guinea pigs and rabbits with bovine and human basic encephalitogen. Eur J Immunol. 1973;3:273–279. doi: 10.1002/eji.1830030505. [DOI] [PubMed] [Google Scholar]
- Teitelbaum D, Webb C, Bree M, Meshorer A, Arnon R, Sela M. Suppression of experimental allergic encephalomyelitis in Rhesus monkeys by a synthetic basic copolymer. Clin Immunol Immunopathol. 1974;3:256–262. doi: 10.1016/0090-1229(74)90012-9. [DOI] [PubMed] [Google Scholar]
- Teitelbaum D, Fridkis-Hareli M, Arnon R, Sela M. Copolymer 1 inhibits chronic relapsing experimental allergic encephalomyelitis induced by proteolipid protein (PLP) peptides in mice and interferes with PLP-specific T cell responses. J Neuroimmunol. 1996;64:209–217. doi: 10.1016/0165-5728(95)00180-8. [DOI] [PubMed] [Google Scholar]
- Teitelbaum D, Arnon R, Sela M. Immunomodulation of experimental autoimmune encephalomyelitis by oral administration of copolymer 1. Proc Natl Acad Sci U S A. 1999;96:3842–3847. doi: 10.1073/pnas.96.7.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelbaum D, Aharoni R, Klinger E, Kreitman R, Raymond E, Malley A, et al. Oral glatiramer acetate in experimental autoimmune encephalomyelitis: clinical and immunological studies. Ann N Y Acad Sci. 2004;1029:239–249. doi: 10.1196/annals.1309.055. [DOI] [PubMed] [Google Scholar]
- Teuscher C, Hickey WF, Korngold R. An analysis of the role of tumor necrosis factor in the phenotypic expression of actively induced experimental allergic orchitis and experimental allergic encephalomyelitis. Clin Immunol Immunopathol. 1990;54:442–453. doi: 10.1016/0090-1229(90)90057-w. [DOI] [PubMed] [Google Scholar]
- The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53:457–465. [PubMed] [Google Scholar]
- Theien BE, Vanderlugt CL, Eagar TN, Nickerson-Nutter C, Nazareno R, Kuchroo VK, et al. Discordant effects of anti-VLA-4 treatment before and after onset of relapsing experimental autoimmune encephalomyelitis. J Clin Invest. 2001;107:995–1006. doi: 10.1172/JCI11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theien BE, Vanderlugt CL, Nickerson-Nutter C, Cornebise M, Scott DM, Perper SJ, et al. Differential effects of treatment with a small-molecule VLA-4 antagonist before and after onset of relapsing EAE. Blood. 2003;102:4464–4471. doi: 10.1182/blood-2003-03-0974. [DOI] [PubMed] [Google Scholar]
- Theoharides TC. Luteolin as a therapeutic option for multiple sclerosis. J Neuroinflammation. 2009;6:29. doi: 10.1186/1742-2094-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischner D, Reichardt HM. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol. 2007;275:62–70. doi: 10.1016/j.mce.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Tokuhara N, Namiki K, Uesugi M, Miyamoto C, Ohgoh M, Ido K, Yoshinaga T, Yamauchi T, Kuromitsu J, Kimura S, Miyamoto N, Kasuya Y. N-type calcium channel in the pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem. 2010;285:33294–33306. doi: 10.1074/jbc.M109.089805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol. 2006;177:7505–7509. doi: 10.4049/jimmunol.177.11.7505. [DOI] [PubMed] [Google Scholar]
- Touil T, Chu N, Bohlmann U, Bargsten P, Li Y, Fitzgerald D, et al. A Paradoxical Effect of Systemic IL-23 in EAE – Limitation of Autoimmune Inflammatory Demyelination. Open Autoimmun J. 2010;2:141–150. [Google Scholar]
- Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- Tran GT, Carter N, He XY, Spicer TS, Plain KM, Nicolls M, et al. Reversal of experimental allergic encephalomyelitis with non-mitogenic, non-depleting anti-CD3 mAb therapy with a preferential effect on T(h)1 cells that is augmented by IL-4. Int Immunol. 2001;13:1109–1120. doi: 10.1093/intimm/13.9.1109. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Tselis A. Laquinimod, a new oral autoimmune modulator for the treatment of relapsing-remitting multiple sclerosis. Curr Opin Investig Drugs. 2010;11:577–585. [PubMed] [Google Scholar]
- Tsunoda I, Libbey JE, Kuang LQ, Terry EJ, Fujinami RS. Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. Am J Pathol. 2005;167:1631–1646. doi: 10.1016/S0002-9440(10)61247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark AA, Culbertson NE, Bartholomew RM, Huan J, Agotsch M, LaTocha D, et al. Therapeutic vaccination with a trivalent T-cell receptor (TCR) peptide vaccine restores deficient FoxP3 expression and TCR recognition in subjects with multiple sclerosis. Immunology. 2008;123:66–78. doi: 10.1111/j.1365-2567.2007.02703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlugt CL, Neville KL, Nikcevich KM, Eagar TN, Bluestone JA, Miller SD. Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol. 2000;164:670–678. doi: 10.4049/jimmunol.164.2.670. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Vergelli M, Olivotto J, Castigli E, Gran B, Raimondi L, Pirisino R, et al. Immunosuppressive activity of 13-cis-retinoic acid in rats: aspects of pharmacokinetics and pharmacodynamics. Immunopharmacology. 1997;37:191–197. doi: 10.1016/s0162-3109(97)00048-9. [DOI] [PubMed] [Google Scholar]
- Vollmar P, Nessler S, Kalluri SR, Hartung HP, Hemmer B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int J Neuropsychopharmacol. 2009;12:525–536. doi: 10.1017/S1461145708009425. [DOI] [PubMed] [Google Scholar]
- Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–1608. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- Waisman A, Ruiz PJ, Hirschberg DL, Gelman A, Oksenberg JR, Brocke S, et al. Suppressive vaccination with DNA encoding a variable region gene of the T-cell receptor prevents autoimmune encephalomyelitis and activates Th2 immunity. Nat Med. 1996;2:899–905. doi: 10.1038/nm0896-899. [DOI] [PubMed] [Google Scholar]
- Wallstrom E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci. 1996;137:89–96. doi: 10.1016/0022-510x(95)00339-4. [DOI] [PubMed] [Google Scholar]
- Webb M, Tham CS, Lin FF, Lariosa-Willingham K, Yu N, Hale J, et al. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol. 2004;153:108–121. doi: 10.1016/j.jneuroim.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Wegner C, Stadelmann C, Pfortner R, Raymond E, Feigelson S, Alon R, et al. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;227:133–143. doi: 10.1016/j.jneuroim.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Weiner HL. Oral tolerance, an active immunologic process mediated by multiple mechanisms. J Clin Invest. 2000;106:935–937. doi: 10.1172/JCI11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL. Immunosuppressive treatment in multiple sclerosis. J Neurol Sci. 2004;223:1–11. doi: 10.1016/j.jns.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, et al. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. 1993;259:1321–1324. doi: 10.1126/science.7680493. [DOI] [PubMed] [Google Scholar]
- Weistock M, Shoham-Moshonov S, Teitelbaum D, Arnon R. Inactivation of neurogenic 5-hydroxytryptamine receptors in guinea pigs with experimental allergic encephalomyelitis (EAE) induced paralysis. Brain Res. 1977;125:192–195. doi: 10.1016/0006-8993(77)90374-2. [DOI] [PubMed] [Google Scholar]
- Wender M, Michalak S, Wygladalska-Jernas H. The effect of short-term treatment with interferon beta 1a on acute experimental allergic encephalomyelitis. Folia Neuropathol. 2001;39:91–93. [PubMed] [Google Scholar]
- Whitacre CC, Gienapp IE, Meyer A, Cox KL, Javed N. Oral tolerance in experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 1996;778:217–227. doi: 10.1111/j.1749-6632.1996.tb21130.x. [DOI] [PubMed] [Google Scholar]
- Wilkin TJ. The primary lesion theory of autoimmunity: a speculative hypothesis. Autoimmunity. 1990;7:225–235. doi: 10.3109/08916939009087582. [DOI] [PubMed] [Google Scholar]
- Wissel J, Haydn T, Muller J, Brenneis C, Berger T, Poewe W, et al. Low dose treatment with the synthetic cannabinoid Nabilone significantly reduces spasticity-related pain: a double-blind placebo-controlled cross-over trial. J Neurol. 2006;253:1337–1341. doi: 10.1007/s00415-006-0218-8. [DOI] [PubMed] [Google Scholar]
- van de Wyngaert FA, Beguin C, D'Hooghe MB, Dooms G, Lissoir F, Carton H, et al. A double-blind clinical trial of mitoxantrone versus methylprednisolone in relapsing, secondary progressive multiple sclerosis. Acta Neurol Belg. 2001;101:210–216. [PubMed] [Google Scholar]
- Yamamura T, Da-Lin Y, Satoh J, Tabira T. Suppression of experimental allergic encephalomyelitis by 15-deoxyspergualin. J Neurol Sci. 1987;82:101–110. doi: 10.1016/0022-510x(87)90010-4. [DOI] [PubMed] [Google Scholar]
- Yamout B, Hourani R, Salti H, Barada W, El-Hajj T, Al-Kutoubi A, et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: a pilot study. J Neuroimmunol. 2010;227:185–189. doi: 10.1016/j.jneuroim.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Yang J, Jiang Z, Fitzgerald DC, Ma C, Yu S, Li H, et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J Clin Invest. 2009;119:3678–3691. doi: 10.1172/JCI37914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang JS, Xu LY, Xiao BG, Hedlund G, Link H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J Neuroimmunol. 2004;156:3–9. doi: 10.1016/j.jneuroim.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Yasuda CL, Al-Sabbagh A, Oliveira EC, Diaz-Bardales BM, Garcia AA, Santos LM. Interferon beta modulates experimental autoimmune encephalomyelitis by altering the pattern of cytokine secretion. Immunol Invest. 1999;28:115–126. doi: 10.3109/08820139909061141. [DOI] [PubMed] [Google Scholar]
- Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- Yiu EM, Banwell B. Update on emerging therapies for multiple sclerosis. Expert Rev Neurother. 2010;10:1259–1262. doi: 10.1586/ern.10.98. [DOI] [PubMed] [Google Scholar]
- Yokote H, Miyake S, Croxford JL, Oki S, Mizusawa H, Yamamura T. NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am J Pathol. 2008;173:1714–1723. doi: 10.2353/ajpath.2008.080622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DA, Lowe LD, Booth SS, Whitters MJ, Nicholson L, Kuchroo VK, et al. IL-4, IL-10, IL-13, and TGF-beta from an altered peptide ligand-specific Th2 cell clone down-regulate adoptive transfer of experimental autoimmune encephalomyelitis. J Immunol. 2000;164:3563–3572. doi: 10.4049/jimmunol.164.7.3563. [DOI] [PubMed] [Google Scholar]
- Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- Yu M, Nishiyama A, Trapp BD, Tuohy VK. Interferon-beta inhibits progression of relapsing-remitting experimental autoimmune encephalomyelitis. J Neuroimmunol. 1996;64:91–100. doi: 10.1016/0165-5728(95)00160-3. [DOI] [PubMed] [Google Scholar]
- Zabad RK, Metz LM, Todoruk TR, Zhang Y, Mitchell JR, Yeung M, et al. The clinical response to minocycline in multiple sclerosis is accompanied by beneficial immune changes: a pilot study. Mult Scler. 2007;13:517–526. doi: 10.1177/1352458506070319. [DOI] [PubMed] [Google Scholar]
- Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, et al. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet. 2003;362:1517–1526. doi: 10.1016/S0140-6736(03)14738-1. [DOI] [PubMed] [Google Scholar]
- Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106:1755–1761. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Metz LM, Yong VW, Bell RB, Yeung M, Patry DG, et al. Pilot study of minocycline in relapsing-remitting multiple sclerosis. Can J Neurol Sci. 2008;35:185–191. doi: 10.1017/s0317167100008611. [DOI] [PubMed] [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RJ, Kong W, Ganea D, et al. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J Neuroimmune Pharmacol. 2009;4:249–259. doi: 10.1007/s11481-009-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsser H, Tang FF. Immunological studies with herpes virus with a consideration of the herpes-encephalitis problem. J Exp Med. 1926;44:21–34. doi: 10.1084/jem.44.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]