

Abstract
Developing reliable, predictive animal models for complex psychiatric disorders, such as schizophrenia, is essential to increase our understanding of the neurobiological basis of the disorder and for the development of novel drugs with improved therapeutic efficacy. All available animal models of schizophrenia fit into four different induction categories: developmental, drug-induced, lesion or genetic manipulation, and the best characterized examples of each type are reviewed herein. Most rodent models have behavioural phenotype changes that resemble ‘positive-like’ symptoms of schizophrenia, probably reflecting altered mesolimbic dopamine function, but fewer models also show altered social interaction, and learning and memory impairment, analogous to negative and cognitive symptoms of schizophrenia respectively. The negative and cognitive impairments in schizophrenia are resistant to treatment with current antipsychotics, even after remission of the psychosis, which limits their therapeutic efficacy. The MATRICS initiative developed a consensus on the core cognitive deficits of schizophrenic patients, and recommended a standardized test battery to evaluate them. More recently, work has begun to identify specific rodent behavioural tasks with translational relevance to specific cognitive domains affected in schizophrenia, and where available this review focuses on reporting the effect of current and potential antipsychotics on these tasks. The review also highlights the need to develop more comprehensive animal models that more adequately replicate deficits in negative and cognitive symptoms. Increasing information on the neurochemical and structural CNS changes accompanying each model will also help assess treatments that prevent the development of schizophrenia rather than treating the symptoms, another pivotal change required to enable new more effective therapeutic strategies to be developed.
LINKED ARTICLES
This article is part of a themed issue on Translational Neuropharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-4
Keywords: schizophrenia, animal model, antipsychotics, isolation rearing, ventral hippocampal lesion, reelin, DISC-1, neuregulin, dysbindin, phencyclidine
Introduction
Schizophrenia is a chronic debilitating neuropsychiatric disorder affecting approximately 1% of the population worldwide. Symptoms cluster into three categories: positive (including auditory and visual hallucinations, delusions, conceptual disorganization and thought disorder), negative (emotional blunting, social withdrawal, anhedonia, avolition, poverty of thought and content of speech) and cognitive dysfunction (including impaired executive function, working memory and attention) (Andreasen, 1995). Patients present with extremely heterogeneous symptom combinations, making diagnosis and treatment problematic. Many patients undergo prolonged periods of remission interspersed with relapses of psychotic episodes. Disease onset is typically post adolescence (16–25 years), with a higher incidence of psychotic symptoms in males and a bimodal later onset (40–60 years) in females. Although the aetiology of schizophrenia remains contentious, it is a multifactorial neurodevelopmental disorder influenced by both genetic and environmental factors (Lewis and Lieberman, 2000; van Os et al., 2010), such that monozygotic siblings of affected individuals show a 50–80% risk of developing the disorder.
The first drugs, found by serendipity rather than design in the 1950s, to treat the psychotic symptoms of schizophrenia (haloperidol and chlorpromazine, called classical neuroleptics) are also known as the first-generation antipsychotics. The second-generation or atypical antipsychotics, so called because of their different clinical profile (including clozapine, olanzepine, risperidone and aripiprazole) developed from the 1970s have less tendency to produce unwanted extrapyramidal side effects and hyperprolactinaemia (Remington, 2003). While first-generation antipsychotics are classified according to chemical structure, the second-generation antipsychotics are characterized according to their pharmacology. These drugs were developed to treat the positive (psychotic) symptoms and not the negative or cognitive impairments. However, multi-site, double-blind studies comparing several second-generation antipsychotics with a typical antipsychotic, perphenazine, failed to substantiate any major therapeutic advantage of the former (Lieberman et al., 2005). The cognitive symptoms of schizophrenia often precede the occurrence of psychosis, and their treatment is considered a better predictor of therapeutic outcome (Mintz and Kopelowicz, 2007). However, while positive symptoms are currently treated to a varying degree by typical and atypical antipsychotics, the negative, and in particular, the cognitive impairments, remain resistant to treatment with current antipsychotics even after remission of the psychosis (Nuechterlein et al., 2004; Keefe et al., 2007; Mintz and Kopelowicz, 2007). Consequently, there is an urgent need to develop novel compounds that demonstrate increased efficacy against cognitive dysfunction and negative symptoms most likely by the use of adjunct therapy in combination with existing antipsychotics. In recognition of this problem, the US National Institute of Mental Health, in partnership with the US Food and Drug Administration and academic partners developed the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) and Treatment Units for Research on Neurocognition and Schizophrenia (TURNS) initiatives to attempt to establish a reliable, valid and consensus-derived method of assessing cognition, and improve the likelihood of successful development of new compounds that could be used alongside existing drugs to more effectively treat the cognitive and negative symptoms of schizophrenia (see http://www.MATRICS.ucla.edu and http://www.turns.ucla.edu). The MATRICS initiative identified seven core domains of cognition: working memory, attention/vigilance, reasoning and problem solving, processing speed, visual learning and memory, verbal learning and memory, and social cognition, that are deficient in schizophrenia which have to be treated to meet therapeutic needs, and recommended a specific neuropsychological test battery to characterize these domains. A development of this initiative is the evaluation of the clinical relevance and predictive value of existing preclinical cognitive tasks and agreement for the need to develop a preclinical cognitive test battery to aid drug development (Hagan and Jones, 2005; Nuechterlein et al., 2005). Floresco et al. (2005) suggested using two approaches in experimental animals: lesions or drugs to manipulate specific systems altered in schizophrenia and developing models with cognitive deficits that resemble those seen in the disorder, to improve translational reliability of data obtained. Young et al. (2009) extensively reviewed existing animal cognitive paradigms and critically appraised their translational relevance to the seven human cognitive domains identified as being affected in schizophrenia. However, such cognitive paradigms need to be examined, not just in normal healthy animals, but in credible validated models of the disorder which will be reviewed in this paper.
Animal models of complex heterogeneous psychiatric disorders are clearly very valuable preclinical tools with which to investigate the neurobiological basis of the disorder. They offer a more rapid platform to monitor disease progression than in humans, and the opportunity to perform invasive monitoring of structural and molecular changes that underlie the cause of the disease and test novel therapeutics not possible in patients. However, a perplexing problem is how to assess some of the core symptoms of psychiatric disorders (like thoughts, and verbal learning and memory), which are uniquely human traits (Powell and Miyakawa, 2006). In general, most behaviours can only be indexed rather than directly quantified, and we are left to monitor performance in tasks designed to have translational relevance to core symptoms and make inference about the psychiatric state. A further problem with models of schizophrenia is that there is no current ‘gold standard’ medication available to treat all the symptoms that can be used as a definitive positive control in preclinical studies, although drugs like haloperidol and clozapine should reverse behavioural correlates of positive symptoms. Furthermore, many of the current antipsychotics may have a small therapeutic window of effect before sedation and other non-specific motor suppressant actions confound interpretation in tasks designed to assess negative and cognitive function (against which in any case these drugs have limited therapeutic effect).
All useful animal models should have the appropriate triad of face (symptom homology), construct (replicate the theoretical neurobiological rationale and pathology) and predictive (show the expected pharmacological response, or lack of it, to treatment by known antipsychotics and potential new adjunct therapies yet to be developed) validity to the clinical disorder being modelled. For schizophrenia, a suitable constellation of behavioural and neurochemical abnormalities would include postpubertal onset, loss of hippocampal and cortical connectivity and function, limbic dopamine dysregulation, cortical glutamatergic hypofunction, vulnerability to stress, abnormal response to reward, social withdrawal and cognitive impairment (Figure 1). Several recent articles (Floresco et al., 2005; Hagan and Jones, 2005; Fone and Porkess, 2008; Millan and Brocco, 2008; Bellon et al., 2009; Neill et al., 2010) have reviewed individual animal models of schizophrenia or compared the potential application of some of the common models with emphasis on their predictive validity to evaluate novel compounds that could improve the cognitive and negative symptoms seen in schizophrenia.
Figure 1.
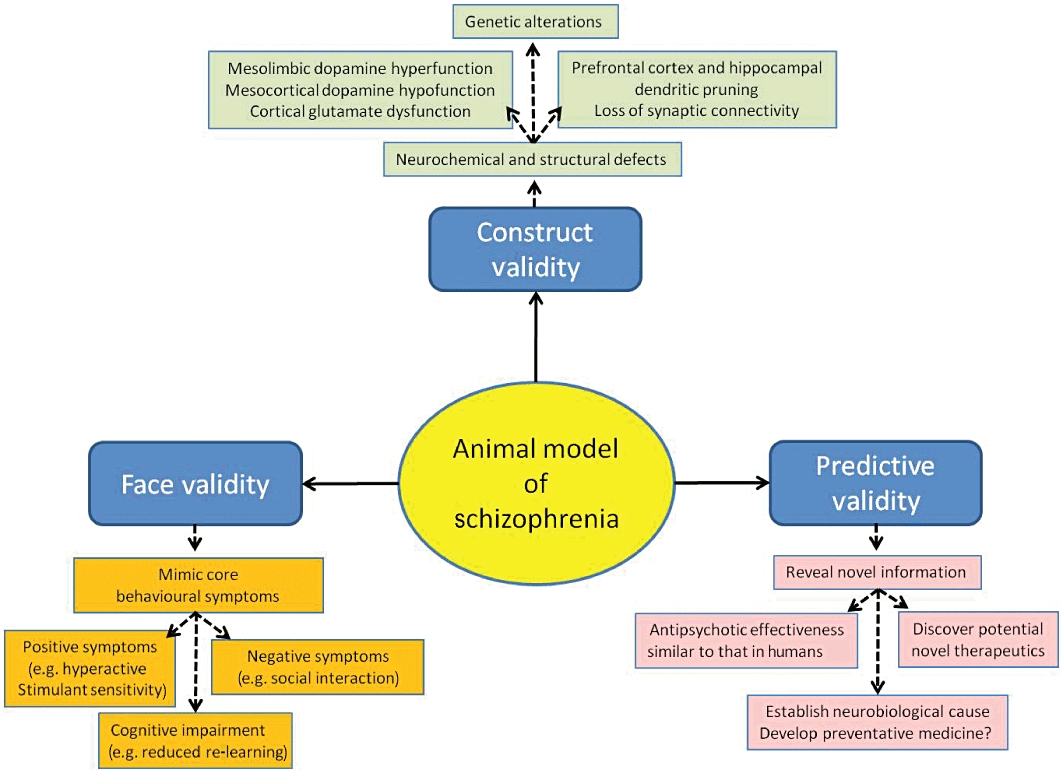
Schematic diagram of the key behavioural, neurochemical and structural changes expected be present and to have translational relevance to the three core symptom domains of schizophrenia in an animal model of the disorder.
Recently, it has been estimated that over 20 different animal models of schizophrenia have been developed (Carpenter and Koenig, 2008), although several have considerable overlap in the methodology/principle used, and all fit into four different induction categories: developmental, drug-induced, lesion or genetic manipulation, as will be discussed in this review. Initial animal models were developed on the basis of the tenet theory that dopamine dysfunction was central to the pathophysiology of schizophrenia, but with increased understanding of the genetic basis and potential involvement of glutamate animal models have also been developed to explore their involvement in the disorder. Most rodent models of schizophrenia tend to replicate aspects of the positive symptoms of schizophrenia (Table 1), such as hyperactivity probably reflecting enhanced mesolimbic dopamine function, but some, including methylazoxymethanol (MAM), neonatal hippocampal lesion, isolation rearing from weaning and chronic phencyclidine (PCP) administration, show cortical dopaminergic dysfunction and sensori-motor gating deficits that may be the consequence of altered development of frontal cortical–limbic circuits. Treatment of the negative and cognitive symptoms of schizophrenia is a vital and unmet clinical need that could have a major impact on patient recovery and re-integration into society. Therefore, the development of more comprehensive models that more adequately replicate deficits in these symptoms and help to understand causal factors is ongoing, but many of the models remain to be tested, as reviewed herein.
Table 1.
Comparative overview of the changes in basal and psychostimulant-induced locomotor activity in an open-field arena; sensorimotor gating; learning and memory; social interaction with a conspecific, structural and neurochemical changes in cortical and hippocampal areas and the reversal of these changes with antipsychotic and related drugs for selected animal models of schizophrenia
| Animal model | Basal- and drug- induced locomotor activity | Sensorimotor gating | Cognition | Social interaction | Structure and neurochemistry | Antipsychotic reversal |
|---|---|---|---|---|---|---|
| Gestational MAM (GD17) (Moore et al., 2006; Lodge et al., 2009) | Spontaneous hyperactivity in novel arena emerging at puberty. Enhanced amphetamine- and NMDA antagonist-induced locomotion. | Deficit in PPI appears at puberty. | Normal acquisition, but impaired re-learning in the Morris water maze; impaired extra-dimensional shift in attentional set-shifting task | Reduced total social interaction appears prior to puberty. | Reduced PFc and hippocampal size, enlarged ventricals, reduced hippocampal soma size and neuropil; enhanced nAcc DA release; spontaneously hyperactive VTA DA neurones; decreased PFc parvalbumin GABA interneurones | No pharmacological reversal of behaviour attempted. CLZ does not reverse change in BDNF. |
| Post-weaning social isolation (Lapiz et al., 2003; Fone and Porkess, 2008) | Hyperactivity in a novel arena appearing 2–3 weeks after commencing isolation; hyper-responsivity to amphetamine and cocaine together with increased nAcc DA release | Persistent, but strain-dependent reduction in PPI to acoustic startle appearing about 6 weeks after isolation | Deficit in novel object recognition; no effect on acquisition of spatial learning by impaired reversal learning in water maze, extra-dimensional shift in the attentional set-shifting task and fear-motivated conditioned emotional response | Increased aggression and increase in total social interaction | Reduced PFc volume; reduced dendritic spine density, cytoskeletal alteration and loss of parvalbumin-containing interneurones and reelin in the hippocampus; reduced PFc D1 binding, no change in striatal D2 density, but increased proportion of striatal D2High; increased spontaneously active VTA DA neurones | PPI reversed by atypical antipsychotics, D2 antagonists, α7-nicotinic agonists; novel object discrimination impairment reversed by 5-HT6 antagonists and mGluR2/3 agonist |
| Amphetamine models (Featherstone et al., 2007a; Featherstone et al., 2008; Sarter et al., 2009) | Sensitization of locomotor response to amphetamine | Persistent deficit in PPI dependent on dosage regimen | Deficits in attention and the attentional set-shifting task; hippocampal-dependent memory unimpaired | No reduction in social interaction | Enhanced mesolimbic DA response; altered ACh function in PFc | Locomotor sensitization blocked by CLZ and HLP; moderate attenuation of attention impairment by CLZ and HLP |
| PCP models (Jentsch and Roth, 1999; Phillips et al., 2001; Mouri et al., 2007; Neill et al., 2010) | Sensitization of locomotor response to PCP; hyper-responsive locomotor response to amphetamine and mild stress | No sustained deficit in PPI | Deficits in novel object recognition, attentional set shifting and T-maze delayed alternation | Reduced frequency and duration of primate social behaviour | Reduced basal and stress-induced PFc DA and glutamate release; decreased synaptic spines on Fc neurones and cortical and hippocampal parvalbumin-positive neurones | Deficits in reversal learning reversed by atypical antipsychotics but not HLP; locomotor sensitization attenuated by CLZ and HLP |
| Neonatal ventral hippocampal lesion (Lipska, 2004; Tseng et al., 2009) | Locomotor hyper-responsivity to stress, amphetamine and NMDA receptor antagonists; enhanced apomorphine-induced stereotypy | Adult onset deficit in PPI | Impaired acquisition of T-maze delayed alternation and water maze; impaired radial arm maze choice accuracy; selective deficit in extra-dimensional shift and reversal in the attentional set-shifting task | Deficits in social interaction with increased aggression at all developmental ages | Unaltered basal nAcc DA release, but enhanced response to stress or amphetamine; reduced mPFc NAA levels and GAD67 mRNA expression | Amphetamine-induced hyperactivity reversed by acute or chronic antipsychotic injection; social interaction deficit not reversed by CLZ |
| DISC-1 knock-out (Jaaro-Peled, 2009) | Hyperactivity seen in L100P, CaMK-ΔC mutants, but not in others; no data available regarding psychostimulant-induced locomotor activity to date | Deficits in PPI seen in some (e.g. constitutive CaMK-ΔC, L100P, Q31L), but not all mutants (e.g. inducible CaMK-ΔC, Δ25 bp); PPI not tested in CaMK cc or BAC ΔC mutants | Impaired T-maze performance seen in most strains; impaired spatial working memory only seen in female CaMK-ΔC inducible mutants | Reductions in social activity seen in some strains (e.g. Q31L) and some CaMK- ΔC transgenics | Reduced brain volume in most strains; enlarged lateral ventricles, reduced hippocampal and PFc dendritic density, structure and complexity in some strains; reduced hippocampal parvalbumin immunoreactivity in some, but not all mutants | PPI deficits in L100P mice reversed by HLP and CLZ |
| Neuregulin1 and ErbB4 knock-out (Harrison and Law, 2006a; Mei and Xiong, 2008) | Most, but not all, neuregulin and ErbB4 mutants show spontaneous locomotor hyperactivity, but inconsistent responses to psychostimulants | PPI deficits seen in most neuregulin mutants reviewed; ErbB4 mutants show normal PPI | Impaired contextual fear and mismatched negativity performance in some mutants | Some deficits in social interaction, increased aggression and reduced responses to social novelty | Increased lateral ventricles and reduced hippocampal spine density; reduction in functional forebrain NMDA receptors | Spontaneous and psychostimulant-induced locomotor hyperactivity reversed by CLZ in Nrg1(ΔTM)+/− and Nrg1(BACE)−/− mutants |
| Dysbindin knock-out (Karlsgodt et al., 2011; Papaleo et al., 2010) | Spontaneous locomotor hyperactivity and hyper-responsivity to amphetamine challenge | Increased PPI and startle response shown to be reversed by quinpirole, but not eticlopride | Increased acquisition of T-maze task; impaired spatial reference memory and novel object recognition performance | Reduced social contact during social interaction task | Hyperexcitability of PFc pyramidal neurones; altered synaptic structure and formation; elevated HVA/DA ratio in cortico-limbic regions | No data on antipsychotic reversal |
| Reelin knock-out (Krueger et al., 2006; Tueting et al., 2006) | Reduced locomotion in an open field; enhanced response to methamphetamine | Variable PPI responses, highly dependent on strain, environment and testing protocol | Few memory deficits reported; normal reversal learning and inhibitory control, normal MWM performance; some learning deficits in acquisition of operant tasks | Some modulation of social activity in novelty and/or interaction tasks | Increased neuronal packing and decreased dendritic spine density in PFc and hippocampal neurones | Normalization of reduced spontaneous activity by OLZ |
Note that some of the tasks as discussed in the review are thought to have translational relevance to particular symptom domains affected in schizophrenia. However, other experimental observations, such as drug-induced changes, do not directly relate to core symptoms, but are used as an index to test dysfunction of the neuronal pathways that are thought to contribute to schizophrenia.
5-HT6, 5-hydroxytryptamine6 receptor; BDNF, brain-derived neurotrophic factor; CLZ, clozapine; D1, dopamine D1 receptor; D2, dopamine D2 receptor; DA, dopamine; DISC-1, disrupted-in-schizophrenia 1; GAD67, glutamic acid decarboxylase enzyme 67 kDa isoform; HLP, haloperidol; MWM, Morris water maze; PFc, prefrontal cortex; PPI, prepulse inhibition of acoustic startle; NAA, N-acetylaspartic acid; nAcc, nucleus accumbens; NMDA, N-methyl-d-aspartic acid; OLZ, olanzapine; PCP, phencyclidine; VTA, ventral tegmental area.
Neurodevelopmental models
Human epidemiology provides compelling evidence that exposure of the neonate, either during gestation or the perinatal period, to adverse environmental insults increases the risk of developing schizophrenia. Thus, maternal stress, malnutrition, infection or immune activation, or obstetric complications (such as hypoxia) during birth are just some of the diverse perturbations that increase the risk of developing schizophrenia, consistent with it having a neurodevelopmental origin (Lewis and Levitt, 2002). A favoured current working hypothesis is that exposure of individuals with a genetic predisposition to an early-life adverse event could trigger an altered pattern of neuronal development and connectivity that subsequently results in the expression of a schizophrenic phenotype. While the precise nature of the early-life adverse event may not be critical, the time that this occurs is. Developmental animal models of schizophrenia utilize manipulations of environment, or drug administration during the sensitive perinatal period, to produce irreversible changes in CNS development. Disruption of neurogenesis during a critical gestational period, neonatal ventral hippocampal lesions, post-weaning social isolation of rodents and perinatal or maternal immune activation have all been proposed as neurodevelopmental models that replicate several of the core symptoms of schizophrenia. Consistent with their face validity, the long-term behavioural changes typically appear post-puberty, replicating the chronology of symptomology seen in schizophrenia.
Gestational MAM
MAM, naturally occurring in the seeds of cycad plants, is an anti-mitotic (and anti-proliferative) agent that methylates DNA (Matsumot and Higa, 1966) and specifically targets neuroblast proliferation in the CNS without affecting glial cells or causing teratogenic effects in peripheral organs (Cattabeni and DiLuca, 1997). Treatment of pregnant rat dams with MAM does not affect litter size or pup body weight (Balduini et al., 1991b; Flagstad et al., 2004), but selectively affects brain development. Indeed, administration of MAM to pregnant rat dams affects those brain structures undergoing the most rapid development in the fetus, producing long-lasting anatomical and behavioural deficits in the offspring (Moore et al., 2006; Lodge and Grace, 2009), which are dependent on the precise gestational day (GD) of administration (Talamini et al., 1998; 2000; Fiore et al., 1999). Cortical neurogenesis is at its peak during GD15 in the rat, and disruption of this process causes a marked decrease in cell number (Bayer and Altman, 1995). Thus, when administered on GD15, MAM decreases whole brain cerebellar and hippocampal volume as measured by high-field MRI (Johnson et al., 2006), and causes gross morphological changes, including microcephaly and profound cortical dysplasias, decreasing cortical mass by up to 70% (Cattabeni et al., 1989), which is much more profound than the features typically observed of schizophrenia (Shenton et al., 2001). In contrast, MAM administration on GD17, when cortical cell proliferation is much reduced, results in a more restricted preferential size reduction in neocortical and limbic structures, including the medial prefrontal (PFc), entorhinal and occipital cortices and the hippocampus, and increased neuronal density in the perirhinal cortex (Moore et al., 2006; Matricon et al., 2010). A careful immunohistochemical analysis has shown a specific reduction in neuronal number in the CA2 subfield of the hippocampus, but reduced soma size and neuropil, without cell loss, in other subfields (Matricon et al., 2010). However, ventricular enlargement (one of the more consistent findings in schizophrenia) is less consistent in GD17 MAM rat pups and does not reach significance (Matricon et al., 2010).
Likewise, the behavioural alterations seen in MAM offspring vary according to the GD of MAM administration in a sequential manner: GD14 increases exploration, GD15 causes nocturnal hyperactivity, while GD16 and 17 decrease activity, which could reflect a switch from destruction of striatal cholinergic to dopaminergic neurones from GD14 to 17 treatment (Balduini et al., 1991a), the latter being more similar to changes seen in schizophrenia. Furthermore, the extent of motor impairment in the rotorod test progressively reduces with increase in GD age of MAM treatment (Balduini et al., 1991b), and only >GD17 administration reduced active avoidance in a shuttle box (Balduini et al., 1991a), suggesting impaired acquisition of learning occurs with later administration times (Balduini et al., 1991a; Fiore et al., 1999). The observation of disorganization, sporadic density and heterotopias within the pyramidal CA3 region of the hippocampus with GD17, but not GD15 administration, which has also be observed in schizophrenia patients, lends weight to the use of GD17 MAM exposure as a preclinical model for schizophrenia (Moore et al., 2006). Interestingly, GD17 MAM offspring show reduced nerve growth factor (NGF) and brain-derived nerve growth factor (BDNF) in the parietal cortex at adulthood (Fiore et al., 2004), but whether this is a cause or consequence of the neurodevelopmental changes observed is unclear. Similarly, both an increase and a decrease in BDNF levels have been reported in schizophrenic patients, so the relevance of this observation to the disorder is unclear (Takahashi et al., 2000; Shoval and Weizman, 2005).
Thus, not only does GD17 MAM produce a pattern of histopathology similar to that observed in schizophrenia, but also behavioural abnormalities with more specific translational homology to dysfunction of the frontal cortex and limbic dopaminergic inputs (Jongen-Relo et al., 2004; Moore et al., 2006) accompanied by increased neural density rather than neuronal loss. Specifically, GD17 MAM reduces the thickness of the hippocampus, thalamus and several cortical regions, as well as decreasing total brain weight by approximately 11%, without producing striatal cell loss (Flagstad et al., 2004). GD17 MAM also enhances the locomotor response to amphetamine (Moore et al., 2006), and increases microdialysate dopamine release in the nucleus accumbens (nAcc), but not the frontal cortex (Flagstad et al., 2004), consistent with the production of a hyperactive subcortical dopamine system thought to contribute to psychosis in humans. Elegant electrophysiological studies by the Grace research group has shown that the spontaneous firing rate of ventral tegmental area (VTA) dopamine neurones is enhanced in GD17 MAM rats, which like the augmented amphetamine-induced locomotion is reversed by inactivation of the ventral hippocampus (Lodge and Grace, 2007). Indeed, these workers suggest that the hyperactivity of VTA dopamine neurones may result from hyperactivity of neurones in the ventral subiculum of the hippocampus (Lodge and Grace, 2008; Lodge et al., 2009), which in turn may be the consequence of MAM causing a loss of parvalbumin-containing GABAergic interneurones in this area (Penschuck et al., 2006), another neurochemical feature seen in schizophrenia (Beasley et al., 2002; Guidotti et al., 2005). GD17 MAM-treated rats also exhibit a spontaneous hyperactivity when placed in a novel arena that only emerges at puberty, and have enhanced sensitivity to the N-methyl-d-aspartic acid (NMDA) receptor antagonist, dizolcipine (MK-801) which thus causes a greater hyperactivity than seen in controls (Le Pen et al., 2006). PCP-induced orofacial dyskinesias (thought to be an index of frontocortical lesions) were also enhanced in GD17 MAM offspring (Moore et al., 2006). Although not unique to schizophrenia, impaired prepulse inhibition of the acoustic startle (PPI) response occurs in this disorder (Braff et al., 2001), and the high cross-species neurobiological homology of the reflex has resulted in this becoming a common test to validate rodent models (Swerdlow et al., 2000; Geyer et al., 2001). PPI is thought to reflect the sensori-motor gating process that occurs in the first few hundred milliseconds prior to conscious attention, filtering out weak and unimportant stimuli during a sensory task, and is therefore, thought to reflect pre-attention processing (Young et al., 2009). Most groups report that PPI is also impaired in GD17, but not GD15 MAM rat pups (Moore et al., 2006; Hazane et al., 2009) (although others only find PPI impairment with MAM injection on GD10 or 11 Talamini et al., 2000), and like the enhanced locomotor activity to a novel arena, the PPI deficit shows age-dependent development (Le Pen et al., 2006; Hazane et al., 2009).
Several studies have attempted to characterize the cognitive changes that occur in MAM offspring, but many of these have been conducted in GD15 rather than GD17 MAM rats and when this is taken into account results are less consistent. Several studies have failed to show alteration in cognitive paradigms when MAM is administered up to GD15; such as lack of change in fear-conditioned freezing or two-way active avoidance behaviour (Jongen-Relo et al., 2004), a form of associative learning which is impaired in schizophrenia (Rushe et al., 1999). In a food-motivated alternating Y-maze paradigm (thought to represent spatial working memory which is impaired in schizophrenia, Park and Holzman, 1992) GD17 MAM rats learned the initial rule more quickly than the controls, but took more trials to attain rule reversal (Moore et al., 2006). While in a spatial recognition Y-maze task, where the third arm is only available on the second trial, GD17 MAM offspring failed to show the normal control preference for the novel arm at post-puberty (>PND60) and not when pre-pubertal (Le Pen et al., 2006; Hazane et al., 2009). In an eight-arm radial arm maze while GD15 MAM offspring could not learn the rule, GD17 offspring were able to learn the task, but were impaired in this hippocampal-dependent spatial working memory task with the introduction of a 30 min delay between baiting the first and second four arms (Gourevitch et al., 2004), consistent with impairment of prefrontal cortical–hippocampal connectivity. In contrast, in the Morris water maze (thought to map to the visual learning and memory domain in humans), GD15 MAM offspring (of both genders) were able to acquire the location of a hidden platform in a fixed position as well as controls, but took longer to re-learn a new fixed platform location, suggesting that it may have produced behavioural rigidity. In another study, GD17 MAM offspring showed impairment in acquisition to find a fixed platform position in the Morris water maze (Hazane et al., 2009), but reversal learning was not examined. GD17 MAM Sprague-Dawley rats took significantly more trials to reach criterion on the extra-dimensional shift and reversal trials (Featherstone et al., 2007b) in the attentional set-shifting task (thought to be a rodent analogue of the Wincosin card sorting task that maps to the reasoning and problem-solving cognitive domain and was identified by the TURNS initiative as useful to determine problem-solving deficits) (Birrell and Brown, 2000), consistent with the known effect of MAM on the parietal cortex and/or PFc, which are involved in this response. In contrast, in the five-choice serial reaction test of attention processing (thought to map to the attention and speed of processing cognitive domain in humans), rats receiving the same treatment regimen failed to show a difference in any parameter (e.g. accuracy, premature responding, omissions. . . .) from controls (Featherstone et al., 2007b), suggesting that MAM may not affect sustained attention even though this is also dependent on PFc function. Very few studies have examined neurotransmitter release in the MAM model. Early studies showed that both basal and potassium-induced glutamate release from hippocampal synaptosomes was elevated (without concomitant alteration in GABA) in GD15 MAM-treated rat offspring (DiLuca et al., 1997), which could contribute to the change in long-term potentiation and cognition observed in these rats. Similar elevated levels of glutamate have been found in the PFc and hippocampus of patients with schizophrenia (van Elst et al., 2005). Although decreased reelin hypermethylation (discussed further in the genetics section later) and expression have been associated with schizophrenia, these effects do not appear to be replicated in the whole hippocampus of GD17 MAM rats (Matricon et al., 2010).
The general consensus is that MAM administration at or before GD15 produces too widespread a disruption of brain morphology and behaviour to provide a useful model of changes seen in schizophrenia and that GD17 MAM is the optimal strategy. However, the effects may be very dependent on rat strain. As the effects of MAM are critically dependent on the GD of treatment, the usual practice of monitoring a vaginal plug to determine conception and calculate the MAM treatment day is inaccurate and may result in considerable variation in the resultant neurodevelopmental changes produced. The MAM model appears to have reasonable face validity for positive and cognitive symptoms, and has construct validity in terms of structural and dopaminergic changes observed. Surprisingly, few behavioural studies have been performed to carefully evaluate the GD17 model, and no studies have used pharmacological agents to attempt to reverse any of these behaviours, so the predictive validity of this paradigm to detect existing antipsychotic drugs or novel pro-cognitive compounds that might be useful to treat schizophrenia is unknown. In the only study to attempt to modify MAM-induced effects (GD12 MAM 20 mg·kg−1) with antipsychotics, neither clozapine (20 mg·kg−1) nor haloperidol (2 mg·kg−1 once per day i.p. for 8 days from PND28) reversed the changes in hippocampal, striatal or entorhinal cortex BDNF or NGF levels measured by Western blots, although some complex interactions were observed (Fiore et al., 2008). Similar neurochemical observation can only be made in post-mortem tissue usually following long-term antipsychotic medication, so it is difficult to evaluate the clinical relevance of such findings.
Post-weaning social isolation
Within a colony, rats display a defined social structure and develop a hierarchy that plays a critical impact on their development. Thus, social deprivation of rat pups from the age of weaning (by placing them in separate cages from littermates) alters brain development and causes behavioural deficits at adulthood (Lapiz et al., 2003; Fone and Porkess, 2008), which are unaltered by social re-integration in later life (Pascual et al., 2006). For instance, post-weaning social isolation of rodents induces spontaneous locomotor hyperactivity, enhanced responses to novelty (neophobia), sensorimotor gating deficits, cognitive impairments, and heightened anxiety states and aggression (Valzelli, 1973; Einon and Morgan, 1977; Heidbreder et al., 2000; Weiss et al., 2004; Fone and Porkess, 2008; Marsden et al., 2011). Collectively, these behavioural changes have been termed the ‘isolation syndrome’, and several of these features resemble some of the core symptoms of schizophrenia. Where available, this review reports behaviours where reversal with current antipsychotic drugs or potential pro-cognitive adjunct therapeutic agents has been utilized, so that the predictive validity of the paradigm can be evaluated.
Isolation-reared rats are consistently more active than group-housed littermates when placed in a mildly aversive novel arena (Fone et al., 1996; Dalley et al., 2002; Silva-Gomez et al., 2003; Del Arco et al., 2004). This hyperactivity is typically expressed as increased horizontal activity and rears particularly evident after the first 15 min in the arena, suggesting an inability to habituate, which probably reflects mesolimbic dopamine hyperactivity (as discussed later) and may serve as an index for the positive symptoms in schizophrenia. Although hyperactivity appears within 2–3 weeks of commencing isolation (Bakshi and Geyer, 1999) is easily measured, relatively robust and well sustained with repeated testing (Hall et al., 1998a; Fabricius et al., 2010a), few groups have examined the sensitivity of this behaviour to drugs used to treat schizophrenia. A recent study has shown that the isolation-induced hyperactivity is reduced by the preferential dopamine D3 receptor antagonists, S33084 and S33138, and less markedly by the D2 receptor antagonist, L741 626, none of which attenuate activity in group-housed controls (Watson et al., 2011). In addition, the known antipsychotics haloperidol, olanzapine, risperidone and the putative anti-schizophrenia agent and mGluR2/3 agonist, LY404039, and its analogue, LY379268, also reverse isolation-induced hyperactivity (Fabricius et al., 2010a; Jones et al., 2011) as does addition of the NMDA receptor modulator, l-serine [which is reduced in cerebrospinal fluid in schizophrenia (Bendikov et al., 2007)], to the drinking water (Shigemi et al., 2010). Given the clinical propensity of existing antipsychotic drugs to reverse positive symptoms (Patil et al., 2007) and that isolation-induced locomotor hyperactivity results from mesolimbic dopamine hyperactivity, this behavioural test may be a useful model with high predictive validity to test drug reversal of the positive symptoms of schizophrenia.
Isolation-reared rats consistently show impaired PPI of acoustic startle compared to group-housed controls, thought to reflect sensorimotor gating deficits (Varty et al., 1999; Cilia et al., 2001; 2005b; Schubert et al., 2009). Although this phenomenon is strain dependent (Varty and Geyer, 1998), it gradually appears with development (Bakshi and Geyer, 1999; Cassidy et al., 2010a), and it was this observation that led to the proposal that this could be a developmental model of schizophrenia (Geyer et al., 1993). Once established, the isolation-induced PPI deficit persists for weeks over multiple tests (Cilia et al., 2001; Weiss and Feldon, 2001) and is restored, or at least partially reversed, by acute injection of the atypical antipsychotics, quetiapine, olanzapine, clozapine and risperidone (Wilkinson et al., 1994; Varty and Higgins, 1995; Bakshi et al., 1998; Cilia et al., 2001); the dopamine D2 receptor antagonist, raclopride (Geyer et al., 1993); α7nicotinic receptor agonists (Cilia et al., 2005a); and the 5-HT2A receptor antagonist, M100907 (volinanserin) (Geyer et al., 1999). Furthermore, bilateral injection of the neurotoxin, 6-hydroxydopamine to deplete dopamine into the nAcc also attenuates the PPI deficit seen in isolates (Powell et al., 2003), consistent with this involving hyperactivity in the mesolimbic dopamine system. On a cautionary note, several of these compounds, such as the 5-HT2A antagonist, volinanserin, only showed modest efficacy in acute clinical trials, which was not as good as haloperidol (Gray and Roth, 2007); the atypical antipsychotic, iloperidone, failed to reverse isolation-induced PPI deficits (Barr et al., 2006); and the mGluR2/3 agonist, LY379268, further impaired PPI deficits in isolates (Jones et al., 2011). However, the ability of antipsychotics to reverse PPI deficits in patients with schizophrenia is also inconsistent (see reviews by Braff et al., 2001; Hagan and Jones, 2005). Several clinical reports have found both typical and atypical antipsychotics improve PPI deficits in patients responsive to treatment. Yet, in drug-free first-episode schizophrenic patients, neither the typical antipsychotic, zuclopenthixolor, nor the atypical antipsychotic, risperidone, had any effect (Mackeprang et al., 2002), and compared to unmedicated controls, neither olanzapine nor haloperidol treatment produce any significant improvement in PPI (Duncan et al., 2003). So, the predictive reliability of using this test alone is clearly questionable.
Humans and rodents alike have an innate curiosity to preferentially explore novel over familiar objects, and assessment of this differential exploration forms the basis of the novel object recognition task, which is thought to assess visual episodic memory (Dere et al., 2007; Winters et al., 2008) and to map in a translational manner to the visual learning and memory domain affected in schizophrenia (Young et al., 2009). In the classic two-trial novel object recognition task, several groups have shown that both male and female isolation-reared rats show premature time delay-induced forgetting, so that they are unable to discriminate between novel and familiar objects in the second-choice trial typically after a 2 h inter-trial interval (Bianchi et al., 2006; King et al., 2009; McLean et al., 2010a; Marsden et al., 2011). The impairment in object recognition is likely to reflect deficits in recognition memory, rather than cognitive inflexibility reducing attention to the new stimulus as has been suggested by some, given that no impairment in novel object discrimination occurs in isolation-reared rats using short inter-trial intervals such as 1–15 min (Lapiz et al., 2000; McLean et al., 2010a). Recently, some studies have shown the ability of 5-HT6 receptor antagonists (King et al., 2007), dopamine D3 receptor antagonists (Watson et al., 2011) and mGluR2/3 agonists (Jones et al., 2011) to reverse isolation-induced deficits in object recognition, so this could be a very promising behavioural task to help predictive evaluation of potential novel pro-cognitive drugs for use as adjuncts to antipsychotics (Marsden et al., 2011), but whether this will translate to their ability to reverse cognitive deficits in schizophrenia is unknown.
Most studies find that isolation rearing does not affect the rate of acquisition in visuo-spatial learning tasks such as the rotating T-maze (Li et al., 2007a) or Morris water maze in rats (Schrijver et al., 2002; Quan et al., 2010) or mice (Ibi et al., 2008), thought to map to visual learning and memory cognitive domain impaired in schizophrenia. Although one group found increased retention (Lapiz et al., 2001) in learning a fixed location platform position in the water maze, most studies find impaired retention or persistence of spatial memory (Quan et al., 2010). Furthermore, isolation rearing appears to impair cognitive flexibility in reversal learning tasks (Schrijver et al., 2004; Li et al., 2007a), and this deficit is accompanied by alteration in long-term potentiation both in the hippocampus (Ibi et al., 2008) and in the PFc (Quan et al., 2010). Isolates may also be preferentially impaired in tasks thought to be relevant to executive function that require shifting from spatial to non-spatial cues (Schrijver and Wurbel, 2001), and in the extra-dimensional shift in the attentional set-shifting task (McLean et al., 2010a) thought to have translational relevance to the reasoning and problem-solving cognitive domain affected in schizophrenic patients. Indeed, in an analogous manner, schizophrenic patients exhibit a selective deficit in the extra-dimensional shift of the Wisconsin card sorting task (Tyson et al., 2004; Jazbec et al., 2007). While spatial learning and acquisition are highly dependent on hippocampal–neocortical pathways, performing an attentional shift to learn a new rule depends primarily on PFc–striatal pathways, which would appear to be preferentially affected by social isolation from weaning (Quan et al., 2010). Unfortunately, few studies have examined whether antipsychotics can reverse any of these cognitive impairments, although chronic clozapine administration restored the reversal learning deficit in the T-maze task (Li et al., 2007a), a finding that questions the predictive validity of the model for cognitive symptoms given the notorious poor efficacy of current antipsychotics to perform this in the clinic. Alterations in catecholaminergic neurotransmission in mesolimbic and other brain regions are thought to underlie many of the behavioural changes induced by isolation rearing (Fulford and Marsden, 1998a,b; 2007; Hall et al., 1998b; Lapiz et al., 2003; Fone and Porkess, 2008). Similar to changes thought to occur in schizophrenia, both in vivo and ex vivo studies on isolates show increased dopamine turnover in the amygdala and nAcc, and decreased turnover in the infralimbic PFc (Jones et al., 1992; Hall et al., 1998b; Fone and Porkess, 2008). Isolates also show enhanced PFc dopamine release in response to olanzapine and clozapine, but not haloperidol (Heidbreder et al., 2001) and an increased number of spontaneously active neurones with a more irregular bursting firing pattern in the VTA (Fabricius et al., 2010b). While some changes in glutamate and amino acid release and turnover have been reported (Melendez et al., 2004), these still remain relatively unexplored and further work on the construct validity of the findings is required.
Several of the neurobiological changes in the brain of isolation-reared rats resemble those seen in the schizophrenic patient, hence the model has good construct validity. For example, as in schizophrenic patients (Hirayasu et al., 2001; Harrison, 2004), a selective reduction in PFc volume occurs in isolation-reared rats (Day-Wilson et al., 2006; Schubert et al., 2009) accompanied by decreased dendritic spine density and morphology (Silva-Gomez et al., 2003; Pascual and Zamora-Leon, 2007), cytoskeletal alterations (Bianchi et al., 2006) and reduced parvalbumin and calbindin-containing GABAergic chandelier cartridges (Harte et al., 2007; Bloomfield et al., 2008) of hippocampal and PFc interneurones. The age of commencing isolation rearing and gender both interact to affect the extent of change in dendritic spine morphology and complexity that develops in PFc, anterior cingulate and orbitofrontal cortices (Ferdman et al., 2007). A micro-array gene expression study in the PFc supports the idea that altered synaptic connectivity occurs in isolation-reared rats, as several genes involved in glutamatergic signalling, apoptosis, cell differentiation and some immediate early genes, including c-Fos, Arc, NGF1-B, Erg4 and Erg2, are all down-regulated (Levine et al., 2007).
Reelin is an extracellular matrix protein secreted by cortical GABAergic neurones in both rodents and humans, thought to be involved in synaptic formation, stability and plasticity, and has been associated with cognitive impairment in patients (Guidotti et al., 2005). Furthermore, both reelin mRNA and GAD67 mRNA expression have been found to be significantly decreased in GABAergic interneurones in the superficial layers of the PFc in patients with schizophrenia (Guidotti et al., 2000). Unfortunately, changes in post-mortem tissue from schizophrenic patients tell us little about any potential developmental role the protein may have in the disorder. Interestingly, reelin is also reduced in the ventral dentate gyrus of the hippocampus in isolation-reared rats to an extent that correlates with impairment in conditioned avoidance learning (Cassidy et al., 2010b). Furthermore, increased expression of reelin-immunoreactive cells in layer 1 of the PFc occurs at the same time ∼PND60 as the emergence of PPI deficits in isolates, which may be linked with synaptic remodelling in this area at this age (Cassidy et al., 2010a).
In addition to the structural changes, isolation rearing induces several receptor changes relevant to observations in schizophrenia. Isolates have reduced PFc dopamine D1 receptor density (Toua et al., 2010), a change which has been reported to correlate with cognitive deficits in schizophrenia (Goldman-Rakic et al., 2004; Scott and Aperia, 2009). However, changes in mesolimbic dopamine D2 receptor expression are inconsistent; down-regulation in striatum (Hall et al., 1998b), but no change in mesolimbic (Del Arco et al., 2004), hippocampal, PFc or amygdala areas (Malone et al., 2008) have been found. Interestingly, as reported in several other rodent models of the psychotic ‘positive-like’ symptoms of schizophrenia (see Seeman et al., 2006), an increased proportion of striatal D2High receptors has been documented in isolation-reared rats (King et al., 2009), which may contribute to dopamine supersensitivity.
Consistent with considerable evidence of hyperactive mesolimbic dopamine activity, a recent in vivo electrophysiology study has confirmed an increased number of spontaneously active neurones with a more irregular bursting firing pattern in the VTA of isolation-reared compared to group-housed rats (Fabricius et al., 2010b). Isolation rearing also induces a hyper-responsiveness in dopamine release in the PFc in response to systemic administration of the atypical antipsychotics clozapine and olanzapine, but not haloperidol (Heidbreder et al., 2001). Isolation-reared rats show increased d-amphetamine- and cocaine-induced dopamine release in the nAcc and striatum compared to group-housed controls measured by in vivo microdialysis (Jones et al., 1992; Hall et al., 1998b; Howes et al., 2000; Lapiz et al., 2003), and enhanced basal and stimulated dopamine and 5-HT release in the nAcc when exposed to conditioned and contextual cues in a conditioned emotional response paradigm (Fulford and Marsden, 1998a; 2007). Collectively, these findings show that the extensive neurochemical imbalances occur in a number of key cortico-limbic brain regions in isolation-reared rats consistent with the validity and usefulness of this paradigm to investigate the aberrant neurobiology underlying schizophrenia.
Much less work has focused on change in glutamatergic receptor density in isolates, and findings are inconsistent. Both down- and up-regulation of PFc NMDA receptor NR2A mRNA expression and no change in PFc, striatal or hippocampal NR1A, NR2B, NR2C, NR2D, NR3A or NR3B subunit expression have all been reported in isolates (Hall et al., 2002; Turnock-Jones et al., 2009; Toua et al., 2010). Although Turnock-Jones reported no change in NR2B subunit protein, down-regulation of the NR2B gene has been reported in the PFc, coupled with increases in NR2A, NR2B, PSD-95 and SAP-102 genes in the hippocampus of isolation-reared rats (Zhao et al., 2009). Increased PFc metabotropic mGlurR6 and ionotropic AMPA3 receptor subunit gene expression (Levine et al., 2007), and reduced mGluR1 and mGlur5 expression (Melendez et al., 2004) are consistent with the proposal that dysregulation of glutamatergic activity may contribute to the behavioural/cognitive deficits associated with social isolation. Indeed, hippocampal synaptophysin (a putative marker of presynaptic glutamatergic activity) is also decreased in isolates (Varty et al., 1999). Although alteration in ionotropic and metabotropic glutamate receptor expression has been reported in schizophrenic patients (Meador-Woodruff and Healy, 2000), changes are not all consistent with the rodent findings. Changes in glutamate release and turnover in isolation-reared rats are relatively unexplored. While attenuated PFc glutamate release following injection of mGluR1 and mGluR2 agonists has been reported (Melendez et al., 2004), Heidbreder et al. (2001) found basal levels of many amino acids (glutamate, glutamine, glycine, GABA, aspartate, alanine, arginine, tyrosine, threonine, taurine and histidine) in the medial PFc to be unaltered by isolation rearing. Furthermore, olanzapine and haloperidol had no effect on any amino acid levels, except glycine, arginine and threonine, while clozapine selectively increased glutamate, alanine and histidine in isolates (Heidbreder et al., 2001). Collectively, these findings show that isolation rearing of rat pups from weaning produces subtle, selective and translationally relevant neurobiological alterations in both gene and protein targets, in regions centrally implicated in schizophrenia.
Given the large array of neurochemical and structural changes that have been characterized in the isolation-reared rat, incredibly few studies have attempted to reverse any of these changes with long-term antipsychotic medication. In theory, neurodevelopmental models of schizophrenia offer the ability to perform behavioural, electrophysiological and neurochemical investigations without confounding drug effects, and have potential to detect reversal by agents operating on diverse pharmacological mechanisms. A major weakness of isolation rearing is the relative fragility of behavioural effects that can be reversed by repeated handling or exposure to too many other tests during the developmental period (Weiss et al., 1999) and which do not universally occur in every cohort (Cilia et al., 2005b; Fone and Porkess, 2008), and the long duration and associated cost of the experiments. However, unlike many other models, post-weaning social isolation is a pure environmental model that requires no physical intervention to either mother or pup, and is relatively simple to execute. This model can also be easily combined with other interventions that could potentially expand the robustness and utility of the paradigm.
Other developmental models
While MAM and isolation rearing have been extensively used as animal models, several other early-life interventions cause neurodevelopmental alterations. For instance, maternal exposure to either bacterial or viral infection during pregnancy elevates circulatory pro-inflammatory cytokines and other mediators of inflammation, affects brain development in the offspring and is associated with an increased risk of schizophrenia (Brown and Derkits, 2010; Brown, 2011). Based on this observation, many groups have attempted to replicate this process in the rodent, via maternal immune activation during a critical mid-gestation window (GD15-19; which approximates to human late third trimester), by the systemic administration of bacterial or viral (-like) agents, and then monitoring the offspring for schizophrenia-like pathologies (for reviews, see Meyer et al., 2009; Meyer and Feldon, 2010). The Boksa group compared the effect of prenatal challenge with a variety of immune-activating agents on the development of altered PPI in the offspring, and showed that maternal infection during GD15-19 produced effects dependent on the agent used (Fortier et al., 2007; Boksa, 2010; see also Meyer et al., 2006a). For instance, an early study showed that exposure of pregnant rats to the bacterial endotoxin, lipopolysaccharide (LPS), increased tyrosine hydroxylase in the nAcc, enhanced amphetamine-induced locomotion (Fortier et al., 2004) and impaired PPI, and the authors suggested this might model aspects of schizophrenia (Borrell et al., 2002), especially as the deficits in PPI and altered cytokine serum levels were reversed by haloperidol (Romero et al., 2007). Furthermore, rats exposed post-natally to LPS (500 µg·kg−1 i.p. PND 7 and 9) were less active and had impaired object recognition on PND 70 (but not PND 35), and reduced hippocampal (but not PFc) parvalbumin-immunoreactive neurones in the CA1-CA3 (Jenkins et al., 2010). Interestingly, other studies found 100 µg·kg−1 i.p. LPS on GD15–16 reduced the number of ultrasonic vocalizations at PND 3 and 5, impaired nest-seeking behaviour and odour-stroke associative learning at PND 8 and 9, suggesting it reduced social/communicative behaviour in offspring that may relate to childhood and pre-morbid abnormalities reported in schizophrenic subjects (Baharnoori et al., 2010). Such early changes may offer the possibility of using neurodevelopmental models of schizophrenia to develop prophylactic rather than curative drug strategies.
Other studies found prenatal immune activation by systemic administration of a by-product of viral replication, polyriboinosinic–polyribocytidilic acid (poly I : C, 4 mg·kg−1 on GD15) to pregnant dams causes acute cytokine elevation and, in resultant offspring, the developmental appearance of sensitization to the locomotor effects of both amphetamine (Zuckerman et al., 2003) and the NMDA receptor antagonist, MK-801, and increased time to reach the platform specifically in the reversal learning component of both a T-maze and water maze paradigm (Zuckerman and Weiner, 2005). Similar studies with poly I : C administration to pregnant mice showed that this produced deficits in PPI (Shi et al., 2003), and enhanced methamphetamine-induced locomotion and reduced novel object discrimination, the latter being reversed by 14 days pretreatment with clozapine, but not haloperidol (Ozawa et al., 2006), leading Meyer's group to propose this as a model of schizophrenia (Meyer et al., 2005). Many negative symptoms of schizophrenia, such as alogia, affective flattening and apathy, are virtually impossible to model in laboratory animals (Ellenbroek and Cools, 2000). Offspring from maternal immune-activated mouse dams also show reduced social interaction and anhedonic behaviour (apparent reduced ability to experience pleasure) in a sucrose preference test (thought to resemble the human negative ‘symptom-like’ domain), together with reduced PFc and hippocampal DA and glutamate levels (Bitanihirwe et al., 2010b). Interestingly, maternal poly I : C administration appears to selectively alter non-spatial information processing, such as novel object recognition, but not novel location learning in the Morris water maze, in resultant offspring (Ito et al., 2010). This poly I : C treatment also disrupts latent inhibition (Zuckerman and Weiner, 2003; Meyer et al., 2006b; Bitanihirwe et al., 2010a) (a behaviour with translational relevance to the pre-attention-processing cognitive domain in humans), which can be restored by acute pretreatment with haloperidol (0.1 mg·kg−1) or clozapine (5 mg·kg−1) (Zuckerman et al., 2003). In animals, latent inhibition describes the phenomenon where after pre-exposure to a cue having no relevance, they are less able to learn an association of this cue with an aversive unconditioned stimulus (such as a footshock). Patients with acute schizophrenia (and individuals that score high on psychometrically defined schizotypy) have disrupted latent inhibition, which is restored by antipsychotics (see reviews Dunn et al., 1993; Moser et al., 2000), consistent with the animal observations, suggesting strong potential predictive validity of this test. As mentioned previously, an advantage of neurodevelopmental models is the ability to combine this approach with pharmacological and/or genetic manipulations. This is highlighted in a recent study combining maternal immune activation with poly I : C to DISC1 mutant mice (Abazyan et al., 2010), which showed an apparently enhanced ‘schizophrenia-related’ phenotype compared to either intervention alone, reduced amygdala and periaquaductal grey volume, dendritic spine density and reduced social interaction (Abazyan et al., 2010). Although requiring further characterization before concrete conclusions can be drawn, the increased use of such approaches to neuropsychopharmacological research will only enhance knowledge and reliability of preclinical schizophrenia research.
Finally, simple exposure of pregnant dams to unpredictable stress during the third week of gestation also enhances the locomotor response to amphetamine, impairs PPI and reduces social behaviour (Koenig et al., 2005; Lee et al., 2007) akin to that which would be predicted for a model of schizophrenia. Many similar interventions have been examined, but these have not been extensively characterized, and so are not covered in detail in this review (see Meyer and Feldon, 2010).
Pharmacological models
Amphetamine model of schizophrenia
As dopamine dysregulation with hyperfunction of the mesolimbic dopamine system was the original tenet theory underlying the basis of schizophrenia (Murray et al., 2008), the first animal models were developed on the basis of pharmacological manipulation to attempt to mimic this feature. Amphetamine-induced psychosis was first described in the 1950s with a clinical picture of auditory hallucinations and persecutory delusions resembling positive symptoms of schizophrenia. In rodents, chronic amphetamine administration induces a persistent sensitization, exaggerating the hyperactivity caused by acute amphetamine challenge (Robinson and Becker, 1986; Featherstone et al., 2008), which is thought to more robustly model symptoms than a single injection (Featherstone et al., 2007a). Pre-administration of a low dose of either haloperidol or clozapine prevents the induction of sensitization (Meng et al., 1998). However, Sams-Dodd (1995; 1998) reported that chronic amphetamine did not induce deficits in social interaction in rats (a task thought to map to the social cognition domain, but also used to model negative symptoms of schizophrenia). This failure to induce negative symptoms in animals is in accordance with data in humans (Javitt and Zukin, 1991; although see Srisurapanont et al., 2003). Long-lasting PPI deficits also occur in rats (Tenn et al., 2005; Peleg-Raibstein et al., 2006), although the dosage regimen may influence the deficit (Featherstone et al., 2007a). Amphetamine sensitization may be accompanied by deficits in PFc-dependent cognitive tasks, including deficits in the extra-dimensional shift and reversal learning in the attentional set-shifting task (Fletcher et al., 2005; Featherstone et al., 2008), an increase in omissions in the five-choice serial reaction time task and reduced accuracy with shorter stimulus duration (Fletcher et al., 2007). Furthermore, clozapine, and to a lesser extent haloperidol, attenuates an amphetamine-induced impairment in attention (Martinez and Sarter, 2008). However, repeated amphetamine administration has no effect on either delayed alternation (Stefani and Moghaddam, 2002) or delayed non-match to position (Featherstone et al., 2008) tasks. Hippocampal-dependent cognition also appears to be spared, as repeated amphetamine has no effect on acquisition or retention of spatial visual learning and memory in the Morris water maze (Russig et al., 2003; Featherstone et al., 2008). Thus, cognitive impairment following chronic amphetamine appears to be restricted to some PFc-dependent tasks while hippocampal function is unaltered.
Repeated amphetamine administration causes a number of neurochemical and structural changes that may account for some of the behavioural changes seen. Locomotor sensitization to a challenge of amphetamine is accompanied by an increase in dopamine efflux from the nAcc and dorsal striatum (Robinson and Becker, 1986; Featherstone et al., 2007a). Repeated amphetamine increases both the number of dendritic branches and spine density in the nAcc shell and PFc (Robinson and Kolb, 1999). Autoradiography shows that expression of the AMPA receptor subunits, GluR1 and GluR2, is decreased in the nAcc, and GluR1 was transiently increased in the PFc (Lu and Wolf, 1999). Furthermore, increased PFc acetylcholine associated with performance of a sustained attentional task is absent in chronic amphetamine-treated rats following a challenge dose (Sarter et al., 2009).
Overall, chronic amphetamine induces psychotic-like changes, but does not replicate the negative or cognitive symptoms seen in schizophrenia. As the model is based on manipulation of the dopaminergic system, it may primarily respond to drugs that affect this neurotransmitter. However, it should be noted that the hyperlocomotion following amphetamine is sensitive to other classes of drugs, including mGluR2/3 agonists (Kim and Vezina, 2002).
PCP models of schizophrenia
In recent years, increasing evidence supports the idea that dysfunction of the glutamatergic system is a primary pathophysiological change seen in schizophrenia (see Olney and Farber, 1995; Tsai and Coyle, 2002; Coyle et al., 2003; Konradi and Heckers, 2003). Pharmacological evidence for the role of glutamate in schizophrenia centres on findings that blockade of the NMDA receptor by non-competitive antagonists, such as ketamine or PCP, induces delusions and hallucinations in otherwise healthy subjects, symptoms commonly seen in schizophrenia (Cohen et al., 1962; Krystal et al., 1994). Furthermore, in both stabilized chronic and acute schizophrenic patients, PCP rekindles and exacerbates positive symptoms (Javitt and Zukin, 1991), and even at low doses, it produces psychotic symptoms in normal volunteers accompanied by progressive withdrawal and poverty of speech, akin to the negative symptoms of schizophrenia (Luby et al., 1959). Additionally, both acute low-dose and chronic recreational use of PCP impair cognitive performance, which is reversed with cessation of drug administration (Cosgrove and Newell, 1991; Javitt and Zukin, 1991).
As PCP induces several symptoms in humans akin to those seen in schizophrenia, it has been used to attempt to produce a pharmacological rodent model of schizophrenia. Acute PCP administration causes hyperlocomotion (Kalinichev et al., 2007), social withdrawal (Sams-Dodd, 1995), and impairment of both PPI (Mansbach and Geyer, 1989) and cognition (Egerton et al., 2005) in rodents. While it is not possible to perform controlled chronic PCP studies in humans, it has been reported that recreational abuse of PCP produces symptoms that persist beyond the end of treatment (Rainey and Crowder, 1975). Additionally, early PET scans suggested that PCP abuse was accompanied by deficits in the temporal and frontal lobes, which parallels changes seen in schizophrenic patients (Hertzmann et al., 1990). Thus, it has been suggested that chronic PCP use may be used to more accurately mimic the symptoms of schizophrenia (Jentsch and Roth, 1999). This has been the basis for evaluating of the effects of chronic PCP administration in rodents; most commonly using twice-daily administration for 7 days followed by a 7 day washout period before the start of experimentation herein described as subchronic. However, different research groups have developed their own variant of the subchronic PCP treatment, detailed analysis of which is beyond the scope of this review, but some distinguishing features are discussed in Table 2. Notably, variations in the period of administration, dose, gender and strain all affect the peak concentration of PCP in the brain, which could account for many differences reported with these various protocols (Table 2).
Table 2.
Comparison of the effects of various subchronic PCP administration protocols on cognitive paradigms in rats and mice as indicated
| Dose | Species/Strain/Sex | Time of test | Behavioural test | Antipsychotic drug effect | Reference |
|---|---|---|---|---|---|
| Subchronic (2 mg·kg−1) | Rat/Lister hooded/♀ | >7 days | Deficit in reversal learning | Reversed by acute ASN, CLZ, OLZ, SRT, ZPD and repeated ASN, RSP and OLZ; no effect of acute HLP or CPM | (Abdul-Monim et al., 2006; Idris et al., 2010; McLean et al., 2010b) |
| 7 days | Deficit in novel object recognition | Reversed by acute CLZ, MLP, OLZ, RSP, SRT, but not HLP | (Grayson et al., 2007; Idris et al., 2010; Snigdha et al., 2010) | ||
| Subchronic (2 mg·kg−1) | Rat/Sprague-Dawley/♂ | 7 days | Deficit in reversal learning | (Jentsch and Taylor, 2001) | |
| Deficit in novel object recognition | No effect of concurrent RSP | (McKibben et al., 2010) | |||
| Subchronic (4.5 mg·kg−1) | Rat/Sprague-Dawley/♂ | 7 days | Deficit in performance in double Y-maze | (Beninger et al., 2010) | |
| Subchronic (5 mg·kg−1) | Rat/Lister hooded/♂ | 7 days | Deficit in episodic memory | No effect of CLZ | (Le Cozannet et al., 2010) |
| Deficit in attentional set shifting | Reversed by acute SRT, but not RSP or HLP | (Rodefer et al., 2005; Broberg et al., 2009; Goetghebeur and Dias, 2009) | |||
| Rat/Wistar/♂ | 72 h | Deficit in delayed alternation task | (Seillier and Giuffrida, 2009) | ||
| Mice/C57BL/6J/♂ | 7 days | No deficit in operant behaviour or reversal learning | (Brigman et al., 2009) | ||
| Chronic intermittent (2.6 mg·kg−1, 28 days) | Rat/Long-Evans/♂ | 72 h | Impaired attentional set shifting | (Egerton et al., 2008) | |
| 24 h | Deficit in novel object recognition | (Spano et al., 2010) | |||
| 3 days per week for 5 weeks (3 mg·kg−1) | Rat/Sprague-Dawley/♂ | 4 weeks | No effect on attentional set shifting | (Fletcher et al., 2005) | |
| Osmotic minipump (15 mg·kg−1·day−1, 14 days) | Rat/Lister hooded/♂ | 7 days | Impaired attentional set shifting | (Pedersen et al., 2009) | |
| 12 days (0.5–4 mg·kg−1) | Mice/C57Bl/6J/♂ | 15 min | Impaired spatial learning at low dose | Reversed by repeated CLZ, but not HLP | (Beraki et al., 2008) |
| 14 days (10 mg·kg−1) | Rat/Sprague-Dawley and Long-Evans/♂ | 48 h | Deficit in spatial delay alternation task (at longer delays) | (Jentsch et al., 1997b) (Marquis et al., 2007) | |
| Mice/ICR/♂ | 5 days | Deficit in novel object recognition | Reversed by acute and repeated ARP, but not HLP | (Nagai et al., 2009) | |
| Mice/C57BL/6J and Rat/Sprague-Dawley/♂ | 7 days | No deficit in spatial performance | (Li et al., 2003) | ||
| 10 days (with 2 day break) (10 mg·kg−1) | Mice/ICR/♂ | 14 days | Deficit in novel object recognition | Reversed by repeated QTP | (Tanibuchi et al., 2009) |
| 6 days (1.3 mg·kg−1) | Rat/Wistar/♂ | 30 min | Deficit in spatial learning and memory | Reversed by acute CLZ, SRT and RSP; no effect of HLP | (Didriksen et al., 2007) |
| 5 days (b.i.d. 5 mg·kg−1) | Rat/Sprague-Dawley/♂ | 9 days | No deficit in spatial delay alternation task (short delays) | (Stefani and Moghaddam, 2002) | |
| 5 days (2 mg·kg−1) | Rat/Wistar/♂ | 30 min | Deficit in attention, cognitive flexibility and speed of processing | Partially attenuated by chronic CLZ, but not QTP | (Amitai et al., 2007; Amitai and Markou, 2009b) |
Time of test indicates duration after the last PCP injection that the task was evaluated. Subchronic: PCP twice daily for 7 days, chronic intermittent: PCP once daily for 5 days followed by three times a week for 3 weeks.
Abbreviations: ARP, aripirazole; ASN, asenapine; CLZ, clozapine; CPM, chlorpromazine; HLP, haloperidol; MLP, melparone; OLZ, olanzapine; QTP, quetiapine; RSP, risperidone; SRT, sertindole; ZPD, ziprasidone. Note: As no current antipsychotic drugs reverse cognitive deficits seen in schizophrenia, the translational relevance of these observations in rodents is currently difficult to evaluate, only in future if cognitive enhancers are developed will the true predictive value of these tasks become apparent.
As with the neurodevelopmental models, hyperlocomotion is frequently used as an index thought to have translational relevance to positive symptoms. Chronic PCP regimes (including 4–10 days, either repeated or intermittent) do not cause spontaneous hyperactivity, but result in locomotor sensitization to a subsequent challenge dose of PCP (Scalzo and Holson, 1992; Xu and Domino, 1994; Johnson et al., 1998; Hanania et al., 1999; Abekawa et al., 2002; Clark et al., 2002; Fletcher et al., 2005; Tenn et al., 2005; McLean et al., 2009). This mirrors the clinic where ‘positive symptoms’ in normal patients are seen while PCP is on-board (Luby et al., 1959). Sensitization to PCP is attenuated by both typical and atypical antipsychotics, such as haloperidol and clozapine, respectively (Phillips et al., 2001), providing predictive validity to the modelling of positive symptoms. Unlike amphetamine, PCP induces changes reminiscent of not only positive, but also negative symptoms seen in patients with schizophrenia (Jentsch and Roth, 1999). Similarly in the rat, chronic PCP (3–21 days) reduces social interaction (thought to reflect social withdrawal; a negative symptom) (Sams-Dodd, 1996), which is also reversed by both acute haloperidol and clozapine injection (Sams-Dodd, 1998). Additionally, 14 day chronic PCP reduces social behaviour in both rats (Lee et al., 2005) and mice (Qiao et al., 2001), the deficit in mice being reversed by clozapine, but not haloperidol. In contrast, Jenkins et al. (2008) reported no overall decrease in social interaction, but an increase in non-contact behaviour in rats following subchronic PCP. These discrepancies may be due to variations in the dosing regimen or the time after administration that social interaction was recorded. Another negative symptom exhibited by schizophrenic patients is dysfunctional reward processing or anhedonia. Interestingly, a patient with schizophrenia typically shows a normal response to an immediate pleasurable stimuli, but they cannot maintain hedonic value, which results in loss of anticipatory pleasure, sometimes referred to as the ‘anhedonia paradox’ (Pizzagalli, 2010). In rodents, subchronic PCP fails to cause any significant difference in sucrose intake, commonly used to evaluate change in reward (Jenkins et al., 2010), and thus thought to relate to anhedonia seen in schizophrenia.
Although acute NMDA antagonist injection impairs PPI in rodents (Mansbach and Geyer, 1989), the deficit induced by chronic PCP is not sustained, such that the PPI impairment diminishes within days of PCP cessation (Ehrhardt et al., 1999; Martinez et al., 1999; Schwabe et al., 2005; Tenn et al., 2005; Egerton et al., 2008; Tunstall et al., 2009). This recovery may explain some of the discrepancies in cognition tasks observed following PCP administration in rodents. Chronic PCP usually produces cognitive impairment in both rats and mice, irrespective of strain (Table 2, but note Li et al., 2003; Fletcher et al., 2005; Brigman et al., 2009). Fletcher et al. (2005) failed to see an impairment in attentional set-shifting with a 5 week (3 mg·kg−1) intermittent dosing regimen that induced locomotor sensitization (Tenn et al., 2005). In contrast, Egerton et al. (2008) observed impaired performance in the extra-dimensional shift of the attentional set-shifting task following a similar chronic intermittent PCP regimen. However, Egerton et al. began with 5 consecutive days of PCP followed by 3 weeks of intermittent dosing, and commenced behavioural testing 3 rather than 7 days after the last dose. This is consistent with the suggestion that cognitive impairment may not be permanent, following an intermittent treatment protocol. It is also possible that the initial 5 consecutive days of PCP injection is crucial to establishing a cognitive deficit, because Egerton et al. showed that 5 days of consecutive dosing alone was sufficient to cause cognitive impairment. It is important for a model to induce changes that are stable over time both because this has face validity to the disease and enables predictive evaluation of drug reversal. In addition, with pharmacological models, a suitable ‘drug-free washout’ ensures results are the consequence of the chronic regime and not due to the presence of the pharmacological effects. However, it appears that some cognitive tasks (such as the five-choice serial reaction time task) may only be impaired shortly after the end of the dosing regimen with the drug on-board, but in these cases, chronic administration may produce a more robust deficit than a single dose of PCP (Amitai et al., 2007). Although schizophrenia affects both males and females, there are notable differences in age of onset and response to antipsychotics, which should be replicated in animal models with good face validity. In rats, gender affects both the pharmacokinetics of PCP (Gartlon et al., 2006) and cognitive ability (Sutcliffe et al., 2007), but the same subchronic PCP dosage regimen impairs reversal learning and novel object recognition equally in male (Jentsch and Taylor, 2001; McKibben et al., 2010) and female (Abdul-Monim et al., 2007; Grayson et al., 2007) rats. As discussed previously, animal tests of cognition have been evaluated to mirror most of the seven cognitive domains thought to be affected in schizophrenia (Hagan and Jones, 2005) and PCP appears to cause deficits in at least five of these (see Table 2 and Neill et al., 2010). Chronic PCP impairs working memory (delayed alternation task) (Jentsch et al., 1997b; Marquis et al., 2007; Seillier and Giuffrida, 2009), attention/vigilance and speed of processing (five-choice serial reaction time task) (Amitai et al., 2007; Amitai and Markou, 2009a), visual learning and memory (object recognition) (Grayson et al., 2007; McKibben et al., 2010; Spano et al., 2010) and reasoning and problem solving (attentional set-shifting, operant reversal learning and maze tasks) (Rodefer et al., 2005; Abdul-Monim et al., 2006; Didriksen et al., 2007; Beraki et al., 2008; Egerton et al., 2008; Pedersen et al., 2009; Idris et al., 2010), but to date we are unaware of any studies evaluating social recognition following chronic PCP. Although a few of these tasks are performed shortly after the last dose of PCP, such that the results are the combination of acute PCP after a chronic regimen, the cognitive deficits in others are seen after delays of 7 days and persist for weeks afterwards (Neill et al., 2010). The persistence of cognitive deficits may have face validity with the disease, but would seem to differ from humans where cognitive deficits appear to reduce after cessation of long-term recreational PCP use (Fauman and Fauman, 1978; Cosgrove and Newell, 1991).
Acute administration of the typical antipsychotic, haloperidol, is unable to reverse deficits in novel object recognition, reversal learning, attentional set shifting and spatial learning induced by chronic PCP (see Table 2 for references). In contrast, many of the PCP-induced cognitive deficits appear to be reversed by several atypical antipsychotics. However, acute clozapine failed to reverse a subchronic PCP-induced impairment in episodic memory (Le Cozannet et al., 2010), and only sertindole, but not risperidone, restored performance in the extra-dimensional shift of the attentional set-shifting task (Goetghebeur and Dias, 2009). Few studies have evaluated the effect of repeated or chronic antipsychotic drug treatment on cognitive impairment in PCP models. Repeated risperidone, commenced after subchronic PCP, reversed the impairment in reversal learning (McLean et al., 2010b), but when given concurrently, it failed to attenuate a deficit in novel object recognition (McKibben et al., 2010). Chronic quetiapine did not improve performance in the five-choice serial reaction time task (Amitai and Markou, 2009b), whereas chronic clozapine partially attenuated the impairment (Amitai et al., 2007). In mice, both repeated quetiapine and aripiprazole restored performance in the novel object recognition task (Nagai et al., 2009; Tanibuchi et al., 2009).
The reversal of PCP-induced cognitive impairments produced by atypical antipsychotics is in marked contrast with clinical evidence, suggesting that these drugs have a relatively small, if any, beneficial cognitive effect and little difference in effectiveness compared with typical antipsychotic drugs (Keefe et al., 2007). This raises questions about the predictive validity of the PCP models and the ability to screen out false positives. A recent study has shown that co-administration of the selective 5-HT2A receptor inverse agonists, primavanserin and volinanserin, with ineffective doses of atypical antipsychotics, reversed the subchronic PCP-induced deficit in novel object recognition (Snigdha et al., 2010). While the move to testing adjunct therapy is encouraging, use of atypical antipsychotics may confound these experiments, providing false positives. In this case, their effect alongside typical antipsychotics, such as haloperidol, may have more relevance to the clinical treatment of cognitive deficits.
Chronic PCP induces several neurochemical changes that correlate well with those thought to occur in schizophrenia. For instance, the mesolimbic dopamine system in the rat is hyper-responsive to amphetamine and mild stress following chronic PCP (Jentsch et al., 1998). Microdialysis data show that both basal and stress-induced PFc dopamine levels are reduced in rats chronically treated with PCP (Jentsch et al., 1997b; 1998), consistent with the suggestion of decreased PFc dopamine in schizophrenia patients (Akil et al., 1999). Similarly, chronic PCP (10 mg·kg−1·day−1 for 14 days) reduces basal PFc glutamate release in freely moving rats (Fattorini et al., 2008) and mice, and increases PFc glutamate–aspartate transporter (GLAST) levels in the latter, consistent with cortical glutamatergic hypofunction (Murai et al., 2007). Decreased synaptic spines on frontal cortex neurones (Flores et al., 2007; although see Hajszan et al., 2006) and a reduced number of cortical and hippocampal parvalbumin-immunoreactive neurones are observed following subchronic PCP (Reynolds et al., 2004; Abdul-Monim et al., 2007; Jenkins et al., 2008; 2010; McKibben et al., 2010), mirroring deficits seen in schizophrenia. Interestingly, rats treated chronically with MK-801 show a similar reduction in the number of parvalbumin-containing neurones in the dentate gyrus and CA1 region of the hippocampus, but no change in the PFc (Braun et al., 2007), which supports the preferential use of PCP in pharmacological models. In the chronic intermittent PCP model, reduced basal glucose utilization indicative of hypometabolism occurs in the PFc, reticular nucleus of the thalamus and auditory cortex (Cochran et al., 2003). Furthermore, there is decreased thalamic and PFc parvalbumin mRNA, which is reversed by chronic clozapine, while haloperidol only reversed the effect in the thalamus (Cochran et al., 2003). Interestingly, clozapine reversed the hypometabolism observed in the auditory cortex, but neither drug reversed it in the PFc. The inability of either clozapine or haloperidol to reverse the PCP-induced prefrontal hypometabolism may reflect the inability to restore cognitive deficits in patients (Cochran et al., 2003). Chronic intermittent PCP reduces N-acetylaspartate (NAA) and N-acetylaspartylglutamate (NAAG) levels in the temporal cortex, and increases NAAG in the hippocampus, which is thought to reflect neuronal dysfunction and closely resembles post-mortem changes seen in schizophrenia (Reynolds et al., 2005). Reductions in mRNA of the GABA-synthesizing enzymes GAD67 and GAD65, and the pre-synaptic transporter GAT-1 also occur along with increases in GABAA subunits in the cerebellum, following chronic intermittent PCP, findings also akin to those seen in schizophrenia patients (Bullock et al., 2009). Multiple changes in receptor expression have been reported following chronic PCP including: decreased dopamine D1 expression in the medial and lateral striatum (without changes in D2 or D4 receptors), increased 5-HT1A expression in medial–prefrontal and dorsolateral frontal cortices, and altered GABA expression in the Fc, hippocampus and striatum (Choi et al., 2009; Beninger et al., 2010). Long-term decreases in NMDA receptor binding occur in many areas, including the hippocampus, nAcc, caudate putamen, thalamus and cortex, although this pattern of binding was considerably different from that seen immediately after the cessation of dosing, emphasizing the importance of the washout period (Newell et al., 2007).
One of the advantages of chronic PCP models over others is the ability to translate findings to primates. Again, chronic PCP is thought to be a better model than acute PCP in part due to the absence of impaired motor function and motivation (Jentsch and Roth, 1999). PCP twice a day for 14 days in monkeys induced a deficit in a PFc-dependent object retrieval task, which was reversed by acute clozapine (Jentsch et al., 1997a). Additionally, a reduction in PFc parvalbumin-containing neurones also occurs in primates (Morrow et al., 2007). Over a 7 month period, PCP significantly reduced the frequency and duration of primate social behaviour, mirroring the negative symptoms seen in schizophrenia patients (Mao et al., 2008). Interestingly, the ‘negative symptoms’ produced following 56 days of osmotic minipump infusion of PCP seen by Linn et al. (2007) were attenuated by concurrent glycine administration, a class of treatment that has had some benefits as an adjunctive therapy on cognitive and negative symptoms in clinical trials. Thus, chronic PCP models do appear to have some translational relevance across rodents, non-human primates and humans.
One criticism of chronic PCP models is that the intervention is given to adult rats, which does not have construct validity to the proposed neurodevelopmental origin of schizophrenia. The neonatal PCP model of schizophrenia attempts to address this issue. Typically, rat pups receive 10 mg·kg−1 PCP on PND 7, 9 and 11. There is mounting evidence that neonatal PCP administration produces enduring behavioural changes in adulthood (Mouri et al., 2007). Corresponding to chronic PCP, locomotor sensitization to PCP challenge has been reported following neonatal PCP, which is reversed by acute administration of the atypical antipsychotics, olanzapine and risperidone (Wang et al., 2001; Anastasio and Johnson, 2008a; Boctor and Ferguson, 2010). Deficits in PPI have been reported by some groups to be attenuated by acute olanzapine and risperidone (Wang et al., 2001; Takahashi et al., 2006; Anastasio and Johnson, 2008a), but not seen following neonatal PCP in other studies (Rasmussen et al., 2007; Boctor and Ferguson, 2009). It should be noted that when PCP was given only once on PND 7, neither deficits in PPI nor locomotor sensitization were reported, emphasizing the importance of the repeated dosing regimen (Anastasio and Johnson, 2008a).
Enduring cognitive deficits have also been reported in the social recognition task (Depoortere et al., 2005; Harich et al., 2007), performance in the Morris water maze (Sircar, 2003; Andersen and Pouzet, 2004), acquisition of a delayed spatial alternation task (Wiley et al., 2003), disrupted performance in a continuous spatial alternation task (Boctor and Ferguson, 2010) and impaired flexibility in a set-shifting task (Stefani and Moghaddam, 2005) with the latter being reversed by acute sertindole (Broberg et al., 2009). That performance in the Morris water maze was improved by chronic d-serine treatment suggests a hypoglutamatergic state occurs in the neonatal PCP model (Andersen and Pouzet, 2004). In contrast, other groups giving repeated neonatal PCP have found no enduring effect on cognition nor any sensitization to the lomotor activity response. Several independent research groups have also shown long-lasting behavioural changes, including increased spontaneous (Harris et al., 2003) (but see Stefani and Moghaddam, 2005; Kawabe et al., 2007) and methamphetamine-induced (Uehara et al., 2010) locomotion, attentuated PPI (provided sufficient treatment length is used) (Uehara et al., 2009; 2010) and deficits in cognition, such as non-matching to position (Kawabe and Miyamoto, 2008) and radial arm maze (spatial working memory) learning and attentional set shifting (Stefani and Moghaddam, 2005) following neonatal administration of MK-801 (typically 0.13–0.4 mg·kg−1 s.c. or i.p., PND 7–10 or 20), which are not seen when the drug is given to adult rats (Kawabe et al., 2007). However, neonatal MK-801 (Kawabe et al., 2007; Uehara et al., 2009; 2010) and PCP (Boctor and Ferguson, 2010) cause a significant decrease in body weight across development, a feature not seen in schizophrenia.
The finding that pro-apoptotic genes are up-regulated and anti-apoptotic genes are down-regulated on PND 12 (Wang et al., 2001; Liu et al., 2010) supports the suggestion that the changes following neonatal NMDA receptor antagonists are the result of neurotoxicity preferentially in the frontal cortex (Wang and Johnson, 2005). This neurotoxicity can be prevented by enhancing NMDA receptor function (Lei et al., 2009). Alterations in glutamate function have been reported following neonatal PCP with increased levels of NMDA NR1, NR2A and NR2B subunits in the Fc (Wang et al., 2001; Anastasio and Johnson, 2008a,b), and increased NMDA receptors in the Fc and hippocampus (Sircar, 2000). Morphological changes in PCP rats including decreased hippocampal volume and neuronal number, and decreased synaptophysin mRNA all support the suggestion of synaptic dysfunction (Wiseman Harris et al., 2003). Furthermore, neonatal PCP also produces a sustained elevation in hippocampal and entorhinal BDNF in 8-week-old rats (Takahashi et al., 2006) similar to clinical observations in the corticolimbic system of patients with chronic schizophrenia (Takahashi et al., 2000). While most studies have occurred in rats, one notable study in mice observed hyperlocomotion, a deficit in spatial working memory and decreased social interaction, the latter being reversed by clozapine (Nakatani-Pawlak et al., 2009). Additionally, the behaviour was associated with decreases in parvalbumin-immunoreactive neurones in the Fc and hippocampus, similar to those seen in chronic PCP-treated rats and schizophrenic patients.
Lesion models
Neonatal ventral hippocampal lesion
Neonatal lesion (PND 7) of the ventral hippocampus (vHip) of the rat (corresponding to the anterior hippocampus in humans) by local injection of the excitotoxin, ibotenic acid (typically 3–5 µg in 0.3 µL (Lipska et al., 1993; Becker et al., 1999) under anaesthesia with hypothermia), causes behavioural abnormalities that emerge after puberty and compromises the architectural integrity of the developing medial PFc and nAcc, which both receive a dense innervation from the former structure (Tseng et al., 2009). The model was developed by the Lipska–Weinberger group in the early 1990s to attempt to mirror emerging brain imaging evidence for the presence of ventricular enlargement and hippocampal changes seen in first-episode schizophrenic patients (Lipska et al., 1993; 1995; Lipska and Weinberger, 1994). As with other developmental models, the time of performing the lesion is critical, and lesions on or after PND 14 produce less pronounced, but immediate onset changes failing, for instance, to enhance apomorphine-induced stereotypy (Wood et al., 1997). Furthermore, the lesion also needs to be bilateral to produce the full spectrum of changes, such as enhanced self-administration of cocaine (Chambers and Self, 2002). The behavioural changes produced by vHip lesions appear progressively with development; deficits in social interaction (thought to have relevance to negative symptoms in schizophrenia) and increased aggression occur by PND 35 and remain (Sams-Dodd et al., 1997), impairment in spatial learning and working memory also appears around PND 25 (Chambers et al., 1996). However, the full behavioural constellation, including locomotor hyper-responsivity to stress (Lipska et al., 1993); enhanced sensitivity to dopamine agonists, such as apomorphine (Lipska and Weinberger, 1993) or amphetamine (Wan et al., 1996; Beninger et al., 2009), and NMDA receptor antagonists, such as MK-801 (Al-Amin et al., 2000) or PCP (Hori et al., 2000), deficits in PPI (Le Pen et al., 2000) and reward (Lipska et al., 1995); and enhanced sensitivity to drugs of abuse, such as cocaine (as reviewed elsewhere Lipska, 2004; Tseng et al., 2009) are not present until around PND 56.
Antipsychotic drug administration either at adulthood [1 mg·kg−1 i.p. haloperidol (Sams-Dodd et al., 1997)] or from adolescence [45 µg·kg−1 i.p. risperidone, PND35-56 (Richtand et al., 2006)] prevents the hyper-responsivity to amphetamine produced in vHip rats. Interestingly, the increase in aggression and reduction in total social interaction shown by neonatal vHip lesion rats are independent of sexual maturity (Becker et al., 1999) and are not reversed by clozapine (Sams-Dodd et al., 1997).
Although behavioural studies strongly suggest increased mesolimbic dopamine activity in vHip rats, in vivo microdialysis shows that nAcc dopamine levels are unaltered under basal conditions (Wan and Corbett, 1997; Brake et al., 1999), and no enhanced dopamine release occurs in response to stress or amphetamine administration from that seen in control rats (Wan et al., 1996; 1998) despite their elevated locomotor response. However, in vHip lesioned rats, raclopride and SCH23390 prevent amphetamine-induced hyperlocomotion, suggesting that this hyper-responsiveness is probably the consequence of increased postsynaptic sensitivity of the dopamine D2 receptor (Wan and Corbett, 1997) and not altered presynaptic dopamine release. vHip rats also show enhanced acquisition of sucrose and cocaine self-administration, thus demonstrating enhance reinforcement to reward (Chambers and Self, 2002). However, as schizophrenic patients would be expected to show anhedonia and reduced reward, this does not appear to replicate the human condition.
Adult vHip lesioned rats have reduced mPFc levels of NAA (a marker of neuronal integrity) and GAD67 mRNA expression (located in GABAergic neurones) (Lipska et al., 2003), and increased GABAA receptor expression (Endo et al., 2007), together with a reduction in spine density and dendritic length of pyramidal neurones in both the mPFc and nAcc medium spiny neurones (Flores et al., 2005; Marquis et al., 2008). vHip rats also show reduced potassium-induced glutamate release from ex vivo PFc slices (Beninger et al., 2009), consistent with cortical glutamatergic hypofunction. Indeed, pulsed stimulation of the VTA source of dopaminergic innervation to the PFc caused enhanced pyramidal neuronal firing in neonatal vHip lesioned rats that was not observed when the lesion was applied to adult rats (O'Donnell et al., 2002). Taken together, this is consistent with vHip causing a developmental alteration in PFc neuronal integrity, which may contribute to the behavioural alterations seen.
vHip lesions in rats cause persistent and marked impairment in several spatial working memory tasks including impaired acquisition of the T-maze delayed alternation (Lipska et al., 2002; Marquis et al., 2006) and Morris water maze (Le Pen et al., 2000; Beninger et al., 2009) tasks, radial arm maze choice accuracy (Chambers et al., 1996) and retention of passive avoidance learning (Le Pen et al., 2000), even with prolonged learning. Recently, it has been shown that vHip rats are impaired in a spatial delayed win-shift radial arm maze task, which is dependent on communication between hippocampus and PFc, but unimpaired in a non-delayed random foraging radial arm maze task requiring connections between the hippocampus and nAcc (Brady et al., 2010), suggesting that disruption of the former pathway may be the primary mediator of spatial working deficits. vHip lesioned rats also show selective impairment in the extra-dimension phase shift and the following reversal in the attentional set-shift paradigm (Marquis et al., 2008) consistent with behavioural rigidity and impaired attention, and visual processing that may reflect medial PFc dysfunction in this model. Chronic administration of clozapine (2.5 mg·kg−1·day−1 i.p.) did not reverse, but exacerbated the radial arm maze choice accuracy deficit in male vHip lesioned rats (Levin and Christopher, 2006), so further pharmacological evaluation is required to determine the predictive utility of this model.
Interestingly, combining social isolation from weaning with vHip lesions further enhances the reduction in dendritic length and spine density of both PFc and Acc neurones seen with either intervention alone (Alquicer et al., 2008; Marquis et al., 2008) and causes an increase in dopamine content of the PFc, which is not seen with vHip lesion alone (Alquicer et al., 2004). Similarly, repeated administration of PCP at adulthood (7.5 mg·kg−1 i.p. daily from PND42–55) to neonatal vHip lesioned rats elevated striatal D1 receptor levels not seen with the lesion and enhanced the hyperactivity response from that of the lesion alone (Hori et al., 2000), consistent with the idea that dual neurodevelopmental and subsequent pharmacological stress may precipitate long-term psychiatric changes akin to those seen in schizophrenia.
Few manuscripts document the success rate, but overall mortality with the vHip lesion appears to be approximately 15% (Richtand et al., 2006), and in some studies as many as 30–33% of treated animals may have unilateral hippocampal damage, which fails to reach histological criteria (Chambers and Self, 2002; Beninger et al., 2009).
In a modification of this neurodevelopmental lesion approach, it has been shown that acute injection of the GABAA antagonist, picrotoxin into (Bast et al., 2001), or stimulation of the NMDA receptor in (Peleg-Raibstein et al., 2005) the vHip of the adult rat can produce locomotor hyperactivity, attenuation of PPI and enhanced PFc dopamine release analogous to that thought to occur in schizophrenia. However, the short-lasting nature of these effects, and the need to have indwelling cannulae, make these models best suited to examine the neurobiological role of enhanced hippocampal–cortical activity in pyschosis-related behaviour rather than as a tool to evaluate drug reversal.
Genetic models
Knock-out mice
Twin studies unequivocally demonstrate that schizophrenia is predominantly a genetic disorder with heritability estimated to be around 80%; however, no single genetic alteration is causal or sufficient to explain this complex heterogeneous disorder. A large array of candidate genes have been associated with an increased risk of schizophrenia (see reviews by Harrison and Weinberger, 2005; Gogos and Gerber, 2006; Allen et al., 2008b). Meta-analyses of genome-wide studies have identified several linkage regions containing some of the putative susceptibility genes. Furthermore, most disrupted genes predominantly segregate to proteins involved in neuronal plasticity, glutamatergic or dopaminergic function and synaptogenesis (Harrison and Weinberger, 2005). Multiple susceptibility genes acting synergistically and in conjunction with epigenetic processes and early-life environmental adverse effects are thought to contribute to the risk of developing schizophrenia. However, there may be multiple very rare genetic mutations that are highly penetrant that may contribute to the illness (Walsh et al., 2008).
The majority of genetic models have been developed on the basis of replicating changes in mRNA and protein seen in schizophrenia. Furthermore, particular emphasis has been placed on developing models of individual schizophrenia endophenotypes, which have been proposed to be present stable, measurable, intermediate disease features that bridge the gap between the overt manifestations of schizophrenia and underlying risk genes (Gottesman and Gould, 2003; Braff et al., 2007; O'Tuathaigh and Waddington, 2010).
The progress made from genetic models of schizophrenia has recently been extensively reviewed by Waddington and colleagues (Arguello and Gogos, 2010; van den Buuse, 2010; Jaaro-Peled et al., 2010; O'Tuathaigh and Waddington, 2010; O'Tuathaigh et al., 2010). The vast array of genetic models produced is beyond the scope of this review, but a selection of models which have contributed significantly to the understanding of aberrant role of dopamine and glutamate function in the pathophysiology of schizophrenia, are discussed herein.
DISC1
One of the earliest genes implicated in the development of schizophrenia was disrupted-in-schizophrenia 1 (DISC1). DISC1 is a synaptic protein expressed early in development playing a crucial role in pre- and post-natal neurone development, which is particularly active during synaptogenesis, neuronal migration and synaptic plasticity (Jaaro-Peled, 2009). Although the link between a mutated DISC1 gene and increased susceptibility for schizophrenia is contentious, studies have found positive linkage (Millar et al., 2001). Development of a knock-out mouse has proven challenging; however, seven different strains of transgenic mice containing inducible and/or partial DISC1 gene mutations resulting in a (partial) loss of DISC-1 function have been created and used to investigate schizophrenia (see Jaaro-Peled, 2009 for review).
Several pathological and behavioural alterations in DISC-1 mice resemble symptoms of schizophrenia. DISC-1 transgenic mice have enlarged lateral ventricles, and reduced cortical thickness and brain volume (Jaaro-Peled et al., 2010). Some, but not all, DISC-1 mutants show reduced parvalbumin immunoreactivity in the mPFC and hippocampus – two regions centrally implicated in the cognitive dysfunction associated with psychosis (Clapcote et al., 2007; Hikida et al., 2007; Shen et al., 2008; Jaaro-Peled et al., 2010). Furthermore, reductions in hippocampal dendritic complexity, structure and density occur in some DISC-1 mutants, consistent with a genetic link to cognitive impairment seen in schizophrenia (Li et al., 2007b; Kvajo et al., 2008). Behaviourally, subtle deficits in PPI reported in some DISC-1 transgenic mice are reversed by both haloperidol and clozapine (Clapcote et al., 2007; Hikida et al., 2007), which is in keeping with clinical findings, but not in other studies (Pletnikov et al., 2008). Similar discrepancies occur in open field/spontaneous locomotor activity paradigms, where some DISC-1 transgenics show locomotor hyperactivity (Clapcote et al., 2007; Hikida et al., 2007), while others do not (Koike et al., 2006; Li et al., 2007b; Pletnikov et al., 2008). As an index of negative symptoms, there are conflicting reports of the impact on social interaction with some studies showing reduced sociability in DISC-1 mutants (Clapcote et al., 2007; Li et al., 2007b; Desbonnet et al., 2009), whereas others report no difference (Hikida et al., 2007). Some deficits in working memory and executive function have been reported in some DISC-1 mutant mice (Pletnikov et al., 2008), while performance in the Morris water maze task, novel object recognition and fear-conditioning tasks were essentially normal (Hikida et al., 2007; Kvajo et al., 2008; Arguello and Gogos, 2010). Some of these discrepancies may relate to the method of production of the transgenic animal rather than DISC-1 changes per se. No studies to date have examined whether these cognitive changes can be reversed by existing antipsychotic drugs or novel pro-cognitive agents.
Neuregulin1 and ErbB4
Another leading candidate ‘risk’ gene is neuregulin-1 (NRG1) and its receptor ERBB4 (Harrison and Law, 2006a; Mei and Xiong, 2008). The NRG1 gene gives rise to a number of different proteins (I–VI) with diverse isoforms (31 in total) of neuregulin, all having distinct expression patterns and functions (Mei and Xiong, 2008). NRG1 is a pleiotropic growth factor containing an epidermal growth factor (EGF)-like domain critically involved in the development and functioning of the nervous system. As well as being involved in both excitatory and inhibitory neurotransmission in the mature brain, NRG1 is also involved in synaptogenesis, neuronal migration, myelination, neurone–glial interactions and glial cell formation in the developing brain (Harrison and Law, 2006a; Mei and Xiong, 2008; van den Buuse et al., 2009). NRG1 interacts with tyrosine kinase receptors, of which ErbB4 is its predominant partner. Distinct patterns and levels of expression of NRG1 isoforms are present in all tissues (Mei and Xiong, 2008).
Homozygous knock-out of NRG1 is developmentally lethal in mice; however, viable heterozygous, hypomorphic/conditional knock-outs that can modulate neuregulin–ErbB4 signalling have been developed, all with distinct ‘schizophrenia-like’ alterations (Harrison and Law, 2006a; Mei and Xiong, 2008). For example, mice with heterozygous deletion of the EGF-like domain [Nrg1(ΔEGF)+/−], heterozygous deletion of the transmembrane domain of NRG1 [Nrg1(ΔTM)+/−], deletions of the immunoglobulin (Ig) domain [Nrg1(ΔIg)+/−] and homozygous deletions of NRG1-cleaving enzyme BACE [Nrg1(BACE)−/−] all show distinct, varied schizophrenia-related alterations in behaviour and pathophysiology (Mei and Xiong, 2008; Jaaro-Peled et al., 2010). The post-mortem findings associating NRG1 changes with schizophrenia are varied; however, studies have reported increased expression of both NRG1 and NRG4 in the PFc and hippocampus (Hahn et al., 2006; Harrison and Law, 2006b; Law et al., 2006). To this end, two heterozygous, hypermorphic transgenic mouse strains have been recently described, which express GFP-tagged type-1 NRG1 cDNA (Kato et al., 2010). In addition, a homozygous deletion of the primary receptor of NRG1, ErbB4, has also been developed (Erbb4−/−).
Nrg1(ΔTM)+/− mice are hyperactive in an open-field and in an alternating Y-maze task, an effect reversed by clozapine. They also show subtle impairments in PPI of acoustic startle (Stefansson et al., 2002; O'Tuathaigh et al., 2007; van den Buuse et al., 2009). However, upon administration of psychostimulants, such as amphetamine, these mice are no longer hyperactive and do not show impaired PPI compared to wild type (Stefansson et al., 2002; van den Buuse et al., 2009). These mice also show increased aggression, impaired social novelty, but normal social interaction compared to wild-type controls and normal working memory in a spontaneous alteration task (O'Tuathaigh et al., 2007; 2010). Some of these traits, including increased spontaneous locomotion and impaired PPI, are replicated in the Nrg1(ΔEGF)+/− mouse (Stefansson et al., 2002), but no difference in the mutant and wild-type mouse response to psychostimulants occurred in either task (Duffy et al., 2008). Nrg1(ΔEGF)+/− mice show robust deficits in social interaction, and are cognitively impaired in contextual fear conditioning and mismatched negativity, but not novel object recognition tasks (Ehrlichman et al., 2009; Arguello and Gogos, 2010; O'Tuathaigh et al., 2010). In contrast, Nrg1(ΔIg)+/− mice are not spontaneously hyperactive and show normal working memory in a spontaneous alteration task, but have impaired latent inhibition (Rimer et al., 2005). Proteolytic processing of NRG1 has profound effects on the behavioural phenotype. Nrg1(BACE)−/− mice show both spontaneous and psychostimulant-induced locomotor hyperactivity, impaired PPI and reduced responses to social novelty compared to wild-type mice (Savonenko et al., 2008; van den Buuse, 2010), the first two of which are reversed by clozapine (Savonenko et al., 2008). Interestingly, the hypermorphic strains over-expressing GFP-tagged type-1 NRG1 cDNA showed considerable overlap with traits seen in the hypomorphic models, including increased spontaneous locomotor activity, reduced PPI, impaired contextual fear conditioning and reduced social interaction (Kato et al., 2010). Conversely, these mice have increased parvalbumin-positive neurones in the PFc and reduced, rather than the expected elevated, hippocampal and PFc levels of dopamine and tyrosine hydroxylase activity (Kato et al., 2010). In relation to ErbB4, homozygous knock-out mice have subtle elevations in locomotor activity, but no change in PPI compared to wild type (Stefansson et al., 2002; Golub et al., 2004), but show significantly reduced social interaction (Roy et al., 2007). Although these genetic models are interesting tools to study ‘schizophrenia-like’ phenotypes, it should be noted that their construct validity, with the possible exception of the NRG1-type 1 cDNA strains, is questionable because the clinical manifestation of schizophrenia is associated with NRG1 hyper- rather than hypo-function (Hahn et al., 2006; Harrison and Law, 2006b; Law et al., 2006). In addition, the diversity and lack of consistency of effects in hypomorphic models, and the similarity in effects between hyper- and hypomorphic models, together with the lack of pharmacological studies, means that much more work is required before the functional relevance of NRG1 changes can be evaluated.
In addition, the diversity and lack of consistency of effects, together with a lack of pharmacological studies, mean that much more work is required before the functional relevance of NRG1 changes can be evaluated from a drug discovery perspective.
Dysbindin
One of the many potential molecular targets thought to underlie alterations in neurotransmitter release seen in schizophrenia is dysbindin. Dysbindin is a synaptic protein thought to regulate exocytosis, vesicle biogenesis and receptor trafficking involved in excitatory synaptic neurotransmission (Karlsgodt et al., 2011; Papaleo et al., 2010). The protein is encoded by the gene dystobrevin-binding protein 1 (DTNBP1) currently thought to be one of the most promising candidate genes for schizophrenia susceptibility, mutations of which show strong correlations with schizophrenia susceptibility and onset in patients (Williams et al., 2005; Allen et al., 2008a; van den Buuse et al., 2009). Reduced expression of dysbindin in the dorsolateral PFc and hippocampus occurs in post-mortem tissue from patients with schizophrenia, and reduced protein expression has been linked to negative symptoms (Weickert et al., 2004; 2008; Papaleo et al., 2010).
A naturally occurring mutant [known as the ‘sandy’ (sdy)] mouse has a homozygous, spontaneous deletion in exon 2 of the DTNBP1 gene, resulting in complete loss of dysbindin protein expression (Papaleo et al., 2010). A back-cross of the spontaneous dysbindin mutant onto the C57BL/6J strain causes homo- and hetero-zygous mutants (Papaleo et al., 2010), showing a number of pathological hallmarks relevant to schizophrenia. Such mutants display alterations in dendritic spines of excitatory asymmetric synapses in the hippocampal CA1 region with narrower synaptic clefts in excitatory junctions, broader postsynaptic densities and a reduced number of larger presynaptic glutamatergic vesicles (Chen et al., 2008; Feng et al., 2008; Jaaro-Peled et al., 2010). The phenotype of Dys−/− and/or Dys+/− mice has yet to be thoroughly characterized, but recent studies show them to be hyperactive in the open field (Cox et al., 2009; van den Buuse, 2010; Papaleo et al., 2010) and hyper-responsive to amphetamine-induced locomotion (Papaleo et al., 2010). Interestingly, Dys−/− mice showed increased basal startle and PPI responses inconsistent with other mutant mice (Papaleo et al., 2010). Furthermore, these effects were reversed by the dopamine D2 receptor agonist, quinpirole, but not the antagonist, eticlopride (Papaleo et al., 2010). Altered working memory, including increased acquisition of a spatial T-maze paradigm, and impaired performance during a discrete paired-trial task under more demanding conditions (further enhanced by mild stress) occur in Dys−/− mice (Papaleo et al., 2010). Reduced social contact in a dyadic social interaction test (Feng et al., 2008; O'Tuathaigh et al., 2010), impaired spatial reference memory and novel object recognition, but enhanced contextual fear conditioning, have also been reported in dysbindin mutants (Feng et al., 2008; Takao et al., 2008; Bhardwaj et al., 2009; Arguello and Gogos, 2010).
Reelin
As previously discussed, reelin is implicated in synaptic formation and plasticity within the CNS (Cassidy et al., 2010a), and reelin mRNA and protein are significantly reduced in the cerebellum, hippocampus and Fc of patients with schizophrenia. The homozygous ‘reeler’ knock-out mouse has gross behavioural abnormalities, including aberrant gait, far more extreme than behavioural changes seen in schizophrenia (Bellon et al., 2009). In contrast, a spontaneous mutation – a deletion of one allele encoding the reeler gene – results in a viable heterozygous mutant mouse (O'Tuathaigh et al., 2010). As well as showing schizophrenia-related pathology including increased neuronal packing and reduced dendritic spine density in the frontal cortex and hippocampus (Liu et al., 2001; Krueger et al., 2006), reeler mice have a behavioural phenotype with some relevance to symptoms seen in schizophrenia.
Reeler mice show increased social dominance potentially akin to negative-like symptoms, but demonstrate normal social interaction (Podhorna and Didriksen, 2004; Tueting et al., 2006). However, modulation of reelin expression in wild-type mice via promoter hypermethylation does reduce social interaction in a novel environment (Tremolizzo et al., 2005). Given that reductions in reelin expression occur in areas implicated in cognition, such as the hippocampus and PFc, it is surprising that reeler mice show relatively few prefrontal-related cognitive deficits (Krueger et al., 2006; Arguello and Gogos, 2010). Some inconsistencies in retention of conditioned fear (Qiu et al., 2006) (associative learning) have been reported, but reeler mice showed normal behavioural flexibility in a reversal learning and inhibitory control task (Krueger et al., 2006), normal acquisition and retention of spatial reference memory in the Morris water maze (Krueger et al., 2006; Arguello and Gogos, 2010), normal performance in a three-choice serial reaction time task and normal working memory in a delayed matched to position task (Krueger et al., 2006). At most, reeler mice show subtle deficits in acquisition of reversal learning and inhibitory control tasks (Krueger et al., 2006; Arguello and Gogos, 2010), so they fail to replicate the cognitive impairment seen in schizophrenia. The reports of ‘positive-like’ phenotype symptoms in the reeler mouse are also inconsistent. For example, the heterozygous reeler mouse shows an enhanced locomotor response to the psychostimulant, methamphetamine (Matsuzaki et al., 2007; van den Buuse, 2010); however, spontaneous locomotion is reduced in this mutant and interestingly only the spontaneous hypoactivity is reversed by olanzapine (Ognibene et al., 2008). Mixed effects on PPI have also been reported, altered PPI and basal startle responses occurring in homozygous, but not heterozygous mutant mice (Salinger et al., 2003), while others have reported an age-dependent impairment in heterozygous reeler mice, that is, highly sensitive to environmental stress (Tueting et al., 2006). Furthermore, disrupted PPI in reeler mice appears highly dependent on the type and duration of the testing protocol used (Barr et al., 2008; van den Buuse, 2010). Additional studies have shown the heterozygous reeler mouse to be less sensitive to early maternal separation than wild type (Ognibene et al., 2008), suggesting that this mutation may confer resistance to phenotypic changes driven by early-life adversity. Thus, data suggest that although the reelin protein may be implicated in disrupted processes centrally involved in schizophrenia, it appears that the use of reeler mice as a potential genetic model to improve our understanding of the neurobiological basis of schizophrenia requires further validation.
Apomorphine susceptibility and apomorphine unsusceptibility rats
The dopamine D1/D2 receptor agonist, apomorphine, induces several typical dopamine-mediated behaviours, such as locomotor hyperactivity; climbing behaviour; and stereotype grooming, licking and gnawing. Selective breeding of Wistar rats, which show a heightened stereotype behavioural response to systemic administration of apomorphine, was used to develop the apomorphine susceptible (APO-SUS) rat line, and behaviour compared with those showing a diminished response, the apomorphine unsusceptible (APO-UNSUS) rat (Cools et al., 1990; Ellenbroek and Cools, 2002). These two strains showed ‘mirror image’ responses in terms of behaviour, neurobiology and immune responses (Cools et al., 1990; 1993a,b; Ellenbroek and Cools, 2002). This approach highlights how valuable spontaneous genetic variation in strains and breeding lines can be manipulated to research the contribution that interactions between gene and environmental stressors can make to disease (van Loo and Martens, 2007). Similar to that expected in schizophrenic patients, APO-SUS rats show impaired PPI and latent inhibition in a conditioned taste aversion paradigm, enhanced responses to novelty in an open field and to psychostimulants, and increased aggression towards a resident intruder (Cools et al., 1990; Ellenbroek and Cools, 2002). The enhanced response to novelty in the APO-SUS rat is accompanied by potentiation in amplitude and time of nAcc dopamine release compared to the APO-UNSUS rats (van Elst et al., 2005). Such animals have a higher level of tyrosine hydroxylase in the striatum indicative of increased dopamine synthesis and increased dopamine D2 receptor levels in the nAcc (van der Elst et al., 2005) (although similar changes seen in post-mortem tissue from patients with schizophrenia may be secondary to antipsychotic medication). Interestingly, further supporting the construct validity of APO-SUS rats as a model of schizophrenia, their pups have a somewhat retarded somatic development compared with APO-UNSUS rats, which also compares to findings in pre-schizophrenic infants (Degen et al., 2005). Few, if any, studies have reported the predictive validity of the APO-SUS/APO-UNSUS rats, and thus further work is required before judgement can be made on the utility of this animal model in the drug discovery process. However, the insight provided by a model utilizing natural gene and environmental stressor interactions could be invaluable in understanding disease pathology.
From the models reviewed above, it is apparent that creating and/or characterizing rodents with genetic mutations in several molecular targets thought to be relevant to schizophrenia is a worthwhile strategy to improve understanding of the neurobiological contribution that each gene product may have to phenotypic alterations seen in schizophrenia. Given the rich pharmacology of antipsychotic therapies currently tested in these models, it is difficult to assess their true predictive validity, and as with other animal models of schizophrenia, there is some way to go before they are fully validated and can be used as tools with any predictive validity to help develop novel therapeutic compounds acting against current drug resistant features of the disorder. Given the inability of current antipsychotics to treat all symptoms of the disorder, and the as-yet limited availability of true cognitive enhancers, treatment strategies may shift so that ‘prevention rather than cure’ approaches may be adopted. In which case, the information gleaned from genetic animal models would greatly assist targeting selective susceptibility genes. Individual genetic mutations may confer susceptibility by converging to alter a common pathophysiological process, but this is only likely to be identified if multiple genetic alterations are induced simultaneously, but will go undetected in simple genetic knock-out animals. Given the complex nature of gene–gene and gene–environment interactions, using models where a single gene with a potential link to schizophrenia has been permanently altered is unlikely to represent the true complex nature of the disorder. To address these issues, more complex models, with altered genetic approaches (such as inducible and tissue-specific knock-outs) are currently in development, which will go some way to addressing some of the confounds associated with compensatory mechanisms and will help expand the potential use of genetic models in schizophrenia research. By adopting a more conditional approach, modulating selected genes in a temporal and/or tissue-specific manor, the effect of compensatory mechanisms may be reduced and the relevance of genetic manipulation to schizophrenia enhanced. As mentioned previously, another potential beneficial future development would be to focus on combining mutations and integrating such transgenic animals with pre-existing environmental or pharmacological models of the disease to attempt to produce more embracing models of the disorder.
Conclusion
In schizophrenia, the poor patient response to current antipsychotics may largely be due to the focus on reversal of the symptoms rather than addressing prevention of development of the underlying causal structural alterations that could be evaluated in neurodevelopmental models. Similarly, one of the greatest impediments to the development of new therapeutics is the lack of understanding of the mechanisms underlying disease pathology in schizophrenia, resulting in a lack of molecular targets. A major reason for the high attrition of potential new antipsychotics in the clinic is the unreliable predictive power of animal models of schizophrenia, which tend to overestimate actual clinical efficacy. Focus on the ability of acute, rather than chronic, drug administration to reverse changes seen in animal models may have contributed to their unreliability, but the development of new behavioural tests with greater translational relevance to the negative and cognitive symptoms is also essential. No single animal model will be able to mirror all the complex sequelae manifest of such a complex and heterogeneous disorder. However, as highlighted in this review, most of the proposed rodent models of schizophrenia replicate changes in mesolimbic dopamine function, which may contribute to the positive symptoms seen in schizophrenia. In contrast, far fewer, such as MAM, neonatal vHip lesion, isolation rearing from weaning and chronic PCP, also cause changes reminiscent of negative and cognitive impairments probably as a consequence of alterations in frontal cortical–limbic circuits. A major advantage of neurodevelopmental over pharmacological or lesion models of schizophrenia is the ability to perform behavioural, electrophysiological and neurochemical investigations in the absence of confounding drug or surgical interventions, and the potential to be able to detect reversal by agents operating on multiple pharmacological mechanisms that are vital to identify new classes of antipsychotic agents and pro-cognitive compounds to be used as adjunct therapeutics. In contrast, neurodevelopmental changes and compensatory mechanisms brought into play by constitutive knock-out of genes may limit the utility of genetic models of schizophrenia. Furthermore, given the heterogeneity of changes observed in schizophrenia, animal models based on manipulation of a single gene have not surprisingly struggled to replicate the full syndrome of this complex disorder. Instead, the approach is intended to delineate the relationship between individual genes and the underlying pathophysiology and specific endophenotypes of the disorder. Genetic models have also contributed to understanding some causal disease factors and may help identify potential biomarkers of therapeutic relevance. Despite this, there is no evidence that any genetic model is used in the drug discovery process or in routine use to screen potential new antipsychotic agents.
The use of animal models to improve understanding of the neurochemical and structural CNS changes that precipitate development of schizophrenia, rather than a focus on treating the symptoms, is a prerequisite to enable new more effective therapeutic strategies to be developed. The complex and unclear nature of gene–gene and gene–environment interactions in the aetiology of schizophrenia means that the challenge to develop more reliable predictive animal models of this disorder, probably through using multiple early-life intervention, is still ongoing.
Acknowledgments
The authors would like to thank the BBSRC, MRC, Eli Lilly, Epix Pharmaceuticals Inc, GlaxoSmithKline, F. Hoffmann-La Roche, Institut de Recherches Servier and Laboratorios Dr Esteve for current or previous funding of their work on animal models of schizophrenia.
Glossary
Abbreviations
- 5-HT6
5-hydroxytryptamine6 receptor
- BDNF
brain-derived neurotrophic factor
- D2
dopamine D2 receptor
- DISC-1
disrupted-in-schizophrenia 1
- DTNBP1
dystobrevin-binding protein 1
- EGF
epidermal growth factor
- GAD65
glutamic acid decarboxylase enzyme 65 kDa isoform
- GAD67
glutamic acid decarboxylase enzyme 67 kDa isoform
- GAT-1
GABA transporter 1
- GD
gestational day
- GLAST
glutamate–aspartate transporter
- Ig
immunoglobulin
- LPS
lipopolysaccharide
- MAM
methylazoxymethanol
- NAA
N-acetylaspartic acid
- nAcc
nucleus accumbens
- NAAG
N-acetylaspartylglutamate
- NGF
nerve growth factor
- NMDA
N-methyl-d-aspartic acid
- NRG1
neuregulin 1
- PCP
phencyclidine
- PFc
prefrontal cortex
- PND
postnatal day
- PPI
prepulse inhibition of acoustic startle
- vHip
ventral hippocampal
- VTA
ventral tegmental area
Conflict of interest
The only conflict to declare is the source of funding; there are no others.
References
- Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, et al. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Monim Z, Reynolds GP, Neill JC. The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav Brain Res. 2006;169:263–273. doi: 10.1016/j.bbr.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Abdul-Monim Z, Neill JC, Reynolds GP. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol. 2007;21:198–205. doi: 10.1177/0269881107067097. [DOI] [PubMed] [Google Scholar]
- Abekawa T, Honda M, Ito K, Inoue T, Koyama T. Effect of MS-153 on the development of behavioral sensitization to locomotion- and ataxia-inducing effects of phencyclidine. Psychopharmacology (Berl) 2002;160:122–131. doi: 10.1007/s00213-001-0958-1. [DOI] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, et al. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Al-Amin HA, Weinberger DR, Lipska BK. Exaggerated MK-801-induced motor hyperactivity in rats with the neonatal lesion of the ventral hippocampus. Behav Pharmacol. 2000;11:269–278. doi: 10.1097/00008877-200006000-00010. [DOI] [PubMed] [Google Scholar]
- Allen N, Bagade S, McQueen M, Ioannidis J, Kavvoura F, Khoury M, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008a;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- Allen NC, Bagade S, McQueen MB, Ioannidis JPA, Kavvoura FK, Khoury MJ, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008b;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- Alquicer G, Silva-Gomez A, Peralta F, Flores G. Neonatal ventral hippocampus lesion alters the dopamine content in the limbic regions in postpubertal rats. Int J Dev Neurosci. 2004;22:103–111. doi: 10.1016/j.ijdevneu.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Alquicer G, Morales-Medina JC, Quirion R, Flores G. Postweaning social isolation enhances morphological changes in the neonatal ventral hippocampal lesion rat model of psychosis. J Chem Neuroanat. 2008;35:179–187. doi: 10.1016/j.jchemneu.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Amitai N, Markou A. Chronic nicotine improves cognitive performance in a test of attention but does not attenuate cognitive disruption induced by repeated phencyclidine administration. Psychopharmacology (Berl) 2009a;202:275–286. doi: 10.1007/s00213-008-1246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai N, Markou A. Increased impulsivity and disrupted attention induced by repeated phencyclidine are not attenuated by chronic quetiapine treatment. Pharmacol Biochem Behav. 2009b;93:248–257. doi: 10.1016/j.pbb.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai N, Semenova S, Markou A. Cognitive–disruptive effects of the psychotomimetic phencyclidine and attenuation by atypical antipsychotic medications in rats. Psychopharmacology (Berl) 2007;193:521–537. doi: 10.1007/s00213-007-0808-x. [DOI] [PubMed] [Google Scholar]
- Anastasio NC, Johnson KM. Atypical anti-schizophrenic drugs prevent changes in cortical N-methyl-d-aspartate receptors and behavior following sub-chronic phencyclidine administration in developing rat pups. Pharmacol Biochem Behav. 2008a;90:569–577. doi: 10.1016/j.pbb.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasio NC, Johnson KM. Differential regulation of the NMDA receptor by acute and sub-chronic phencyclidine administration in the developing rat. J Neurochem. 2008b;104:1210–1218. doi: 10.1111/j.1471-4159.2007.05047.x. [DOI] [PubMed] [Google Scholar]
- Andersen JD, Pouzet B. Spatial memory deficits induced by perinatal treatment of rats with PCP and reversal effect of d-serine. Neuropsychopharmacology. 2004;29:1080–1090. doi: 10.1038/sj.npp.1300394. [DOI] [PubMed] [Google Scholar]
- Andreasen NC. Symptoms, signs, and diagnosis of schizophrenia. Lancet. 1995;346:477–481. doi: 10.1016/s0140-6736(95)91325-4. [DOI] [PubMed] [Google Scholar]
- Arguello P, Gogos J. Cognition in mouse models of schizophrenia susceptibility genes. Schizophr Bull. 2010;36:289–300. doi: 10.1093/schbul/sbp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharnoori M, Bhardwaj S, Srivastava L. Maternal lipopolysaccharide administration leads to developmental alterations in hippocampal neuronal density and expression of cortical dopaminergic receptors in ratoffspring. Schizophr Res. 2010;117:271–272. [Google Scholar]
- Bakshi VP, Geyer MA. Ontogeny of isolation rearing-induced deficits in sensorimotor gating in rats. Physiol Behav. 1999;67:385–392. doi: 10.1016/s0031-9384(99)00082-7. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, Swerdlow NR, Braff DL, Geyer MA. Reversal of isolation rearing-induced deficits in prepulse inhibition by seroquel and olanzapine. Biol Psychiatry. 1998;43:436–445. doi: 10.1016/s0006-3223(97)00246-1. [DOI] [PubMed] [Google Scholar]
- Balduini W, Elsner J, Lombardelli G, Peruzzi G, Cattabeni F. Teatment with methylazoxymethanol at different gestational days – 2-way shuttle box avoidance and residual maze activity in rat offspring. Neurotoxicology. 1991a;12:677–686. [PubMed] [Google Scholar]
- Balduini W, Lombardelli G, Peruzzi G, Cattabeni F. Treatment with methylazoxymethanol at different gestational days – physical, reflex development and spontaneous activity in the offspring. Neurotoxicology. 1991b;12:179–188. [PubMed] [Google Scholar]
- Barr AM, Powell SB, Markou A, Geyer MA. Iloperidone reduces sensorimotor gating deficits in pharmacological models, but not a developmental model, of disrupted prepulse inhibition in rats. Neuropharmacology. 2006;51:457–465. doi: 10.1016/j.neuropharm.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Barr A, Fish K, Markou A, Honer W. Heterozygous reeler mice exhibit alterations in sensorimotor gating but not presynaptic proteins. Eur J Neurosci. 2008;27:2568–2574. doi: 10.1111/j.1460-9568.2008.06233.x. [DOI] [PubMed] [Google Scholar]
- Bast T, Zhang WN, Feldon J. Hyperactivity, decreased startle reactivity, and disrupted prepulse inhibition following disinhibition of the rat ventral hippocampus by the GABA(A) receptor antagonist picrotoxin. Psychopharmacology (Berl) 2001;156:225–233. doi: 10.1007/s002130100775. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Neurogenesis and neuronal migration. In: Paxinos G, editor. The Rat Nervous System. 2nd edn. London: Academic Press Inc; 1995. pp. 1041–1078. [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry. 2002;52:708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Bernstein HG, Hollt V, Bogerts B. Social behaviour in rats lesioned with ibotenic acid in the hippocampus: quantitative and qualitative analysis. Psychopharmacology. 1999;144:333–338. doi: 10.1007/s002130051015. [DOI] [PubMed] [Google Scholar]
- Bellon A, Le Pen G, Matricon J, Jay TM, Krebs MO. Potential application as screening and drug designing tools of cytoarchitectural deficiencies present in three animal models of schizophrenia. Expert Opin Drug Discov. 2009;4:257–278. doi: 10.1517/17460440902762794. [DOI] [PubMed] [Google Scholar]
- Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, et al. A CSF and postmortem brain study of d-serine metabolic parameters in schizophrenia. Schizophr Res. 2007;90:41–51. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Beninger RJ, Tuerke KJ, Forsyth JK, Giles A, Xue LH, Boegman RJ, et al. Neonatal ventral hippocampal lesions in male and female rats: effects on water maze, locomotor activity, plus-maze and prefrontal cortical GABA and glutamate release in adulthood. Behav Brain Res. 2009;202:198–209. doi: 10.1016/j.bbr.2009.03.044. [DOI] [PubMed] [Google Scholar]
- Beninger RJ, Beuk J, Banasikowski TJ, van Adel M, Boivin GA, Reynolds JN. Subchronic phencyclidine in rats: alterations in locomotor activity, maze performance, and GABA(A) receptor binding. Behav Pharmacol. 2010;21:1–10. doi: 10.1097/FBP.0b013e3283347091. [DOI] [PubMed] [Google Scholar]
- Beraki S, Kuzmin A, Tai F, Ogren SO. Repeated low dose of phencyclidine administration impairs spatial learning in mice: blockade by clozapine but not by haloperidol. Eur Neuropsychopharmacol. 2008;18:486–497. doi: 10.1016/j.euroneuro.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Bhardwaj S, Baharnoori M, Sharif-Askari B, Kamath A, Williams S, Srivastava L. Behavioral characterization of dysbindin-1 deficient sandy mice. Behav Brain Res. 2009;197:435–441. doi: 10.1016/j.bbr.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Fone KFC, Azmi N, Heidbreder CA, Hagan JJ, Marsden CA. Isolation rearing induces recognition memory deficits accompanied by cytoskeletal alterations in rat hippocampus. Eur J Neurosci. 2006;24:2894–2902. doi: 10.1111/j.1460-9568.2006.05170.x. [DOI] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. 2010a;35:2462–2478. doi: 10.1038/npp.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitanihirwe BKY, Weber L, Feldon J, Meyer U. Cognitive impairment following prenatal immune challenge in mice correlates with prefrontal cortical AKT1 deficiency. Int J Neuropsychopharmacol. 2010b;13:981–996. doi: 10.1017/S1461145710000192. [DOI] [PubMed] [Google Scholar]
- Bloomfield C, French SJ, Jones DNC, Reavill C, Southam E, Cilia J, et al. Chandelier cartridges in the prefrontal cortex are reduced in isolation reared rats. Synapse. 2008;62:628–631. doi: 10.1002/syn.20521. [DOI] [PubMed] [Google Scholar]
- Boctor SY, Ferguson SA. Neonatal NMDA receptor antagonist treatments have no effects on prepulse inhibition of postnatal day 25 Sprague-Dawley rats. Neurotoxicology. 2009;30:151–154. doi: 10.1016/j.neuro.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Boctor SY, Ferguson SA. Altered adult locomotor activity in rats from phencyclidine treatment on postnatal days 7, 9 and 11, but not repeated ketamine treatment on postnatal day 7. Neurotoxicology. 2010;31:42–54. doi: 10.1016/j.neuro.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897. doi: 10.1016/j.bbi.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Borrell J, Vela JM, Arevalo-Martin A, Molina-Holgado E, Guaza C. Prenatal immune challenge disrupts sensorimotor gating in adult rats: implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Brady AM, Saul RD, Wiest MK. Selective deficits in spatial working memory in the neonatal ventral hippocampal lesion rat model of schizophrenia. Neuropharmacology. 2010;59:605–611. doi: 10.1016/j.neuropharm.2010.08.012. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braff D, Freedman R, Schork N, Gottesman I. Deconstructing schizophrenia: an overview of the use of endophenotypes in order to understand a complex disorder. Schizophr Bull. 2007;33:21–32. doi: 10.1093/schbul/sbl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brake WG, Sullivan RM, Flores G, Srivastava LK, Gratton A. Neonatal ventral hippocampal lesions attenuate the nucleus accumbens dopamine response to stress: an electrochemical study in the adult rat. Brain Res. 1999;831:25–32. doi: 10.1016/s0006-8993(99)01477-8. [DOI] [PubMed] [Google Scholar]
- Braun I, Genius J, Grunze H, Bender A, Moller HJ, Rujescu D. Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA. Schizophr Res. 2007;97:254–263. doi: 10.1016/j.schres.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Ihne J, Saksida LM, Bussey TJ, Holmes A. Effects of subchronic phencyclidine (PCP) treatment on social behaviors, and operant discrimination and reversal learning in C57BL/6J mice. Front Behav Neurosci. 2009;3:2. doi: 10.3389/neuro.08.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberg BV, Glenthoj BY, Dias R, Larsen DB, Olsen CK. Reversal of cognitive deficits by an ampakine (CX516) and sertindole in two animal models of schizophrenia – sub-chronic and early postnatal PCP treatment in attentional set-shifting. Psychopharmacology (Berl) 2009;206:631–640. doi: 10.1007/s00213-009-1540-5. [DOI] [PubMed] [Google Scholar]
- Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2011;93:23–58. doi: 10.1016/j.pneurobio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock WM, Bolognani F, Botta P, Valenzuela CF, Perrone-Bizzozero NI. Schizophrenia-like GABAergic gene expression deficits in cerebellar Golgi cells from rats chronically exposed to low-dose phencyclidine. Neurochem Int. 2009;55:775–782. doi: 10.1016/j.neuint.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Buuse M. Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr Bull. 2010;36:246–270. doi: 10.1093/schbul/sbp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Buuse M, Wischhof L, Lee R, Martin S, Karl T. Neuregulin 1 hypomorphic mutant mice: enhanced baseline locomotor activity but normal psychotropic drug-induced hyperlocomotion and prepulse inhibition regulation. Int J Neuropsychopharmacol. 2009;12:1383–1393. doi: 10.1017/S1461145709000388. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology. 2008;33:2061–2079. doi: 10.1038/sj.npp.1301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy AW, Mulvany SK, Pangalos MN, Murphy KJ, Regan CM. Developmental emergence of reelin deficits in the prefrontal cortex of Wistar rats reared in social isolation. Neuroscience. 2010a;166:377–385. doi: 10.1016/j.neuroscience.2009.12.045. [DOI] [PubMed] [Google Scholar]
- Cassidy AW, Mulvany SK, Pangalos MN, Murphy KJ, Regan CM. Reduced reelin protein synthesis in ventral hippocampus of isolation reared Wistar rats accompanies impaired avoidance conditioning. Behav Brain Res. 2010b;213:130–134. doi: 10.1016/j.bbr.2010.04.040. [DOI] [PubMed] [Google Scholar]
- Cattabeni F, DiLuca M. Developmental models of brain dysfunctions induced by targeted cellular ablations with methylazoxymethanol. Physiol Rev. 1997;77:199–215. doi: 10.1152/physrev.1997.77.1.199. [DOI] [PubMed] [Google Scholar]
- Cattabeni F, Abbracchio MP, Cimino M, Cocchi D, Diluca M, Mennuni L, et al. Methylazoxymethanol-induced microencephaly: persistent increase of cortical somatostatin-like immunoreactivity. Brain Res Dev Brain Res. 1989;47:156–159. doi: 10.1016/0165-3806(89)90120-x. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Self DW. Motivational responses to natural and drug rewards in rats with neonatal ventral hippocampal lesions: an animal model of dual diagnosis schizophrenia. Neuropsychopharmacology. 2002;27:889–905. doi: 10.1016/S0893-133X(02)00365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers RA, Moore J, McEvoy JP, Levin ED. Cognitive effects of neonatal hippocampal lesions in a rat model of schizophrenia. Neuropsychopharmacology. 1996;15:587–594. doi: 10.1016/S0893-133X(96)00132-7. [DOI] [PubMed] [Google Scholar]
- Chen X, Feng Y, Hao C, Guo X, He X, Zhou Z, et al. DTNBP1, a schizophrenia susceptibility gene, affects kinetics of transmitter release. J Cell Biol. 2008;181:791–801. [Google Scholar]
- Choi YK, Snigdha S, Shahid M, Neill JC, Tarazi FI. Subchronic effects of phencyclidine on dopamine and serotonin receptors: implications for schizophrenia. J Mol Neurosci. 2009;38:227–235. doi: 10.1007/s12031-009-9204-9. [DOI] [PubMed] [Google Scholar]
- Cilia J, Reavill C, Hagan JJ, Jones DNC. Long-term evaluation of isolation-rearing induced prepulse inhibition deficits in rats. Psychopharmacology. 2001;156:327–337. doi: 10.1007/s002130100786. [DOI] [PubMed] [Google Scholar]
- Cilia J, Cluderay JE, Robbins MJ, Reavill C, Southam E, Kew JN, et al. Reversal of isolation-rearing-induced PPI deficits by an alpha7 nicotinic receptor agonist. Psychopharmacology (Berl) 2005a;182:214–219. doi: 10.1007/s00213-005-0069-5. [DOI] [PubMed] [Google Scholar]
- Cilia J, Hatcher PD, Reavill C, Jones DNC. Long-term evaluation of isolation-rearing induced prepulse inhibition deficits in rats: an update. Psychopharmacology. 2005b;180:57–62. doi: 10.1007/s00213-004-2139-5. [DOI] [PubMed] [Google Scholar]
- Clapcote S, Lipina T, Millar J, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Clark M, Johnson BG, Wright RA, Monn JA, Schoepp DD. Effects of the mGlu2/3 receptor agonist LY379268 on motor activity in phencyclidine-sensitized rats. Pharmacol Biochem Behav. 2002;73:339–346. doi: 10.1016/s0091-3057(02)00848-1. [DOI] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- Cohen BD, Rosenbaum G, Luby ED, Gottlieb JS. Comparison of phencyclidine hydrochloride (sernyl) with other drugs: simulation of schizophrenic performance with phencyclidine hydrochloride (sernyl), lysergic acid diethylamide (LSD-25), and amobarbital (amytal) sodium; II. Symbolic and sequential thinking. Arch Gen Psychiatry. 1962;6:395–401. doi: 10.1001/archpsyc.1962.01710230063007. [DOI] [PubMed] [Google Scholar]
- Cools AR, Brachten R, Heeren D, Willemen A, Ellenbroek B. Search after neurobiological profile of individual-specific features of Wistar rats. Brain Res Bull. 1990;24:49–69. doi: 10.1016/0361-9230(90)90288-b. [DOI] [PubMed] [Google Scholar]
- Cools AR, Dierx J, Coenders C, Heeren D, Ried S, Jenks BG, et al. Apomorphine-susceptible and apomorphine-unsusceptible Wistar rats differ in novelty-induced changes in hippocampal dynorphin-B expression and 2-way active avoidance – a new key in the search for the role of the hippocampal accumbens axis. Behav Brain Res. 1993a;55:213–221. doi: 10.1016/0166-4328(93)90117-9. [DOI] [PubMed] [Google Scholar]
- Cools AR, Rots NY, Ellenbroek B, Dekloet ER. Bimodal shape of individual variation in behavior of Wistar rats – the overall outcome of a fundamentally different make-up and reactivity of the brain, the endocrinological and the immunological system. Neuropsychobiology. 1993b;28:100–105. doi: 10.1159/000119009. [DOI] [PubMed] [Google Scholar]
- Cosgrove J, Newell TG. Recovery of neuropsychological functions during reduction in use of phencyclidine. J Clin Psychol. 1991;47:159–169. doi: 10.1002/1097-4679(199101)47:1<159::aid-jclp2270470125>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Cox M, Tucker A, Tang J, Talbot K, Richer D, Yeh L, et al. Neurobehavioral abnormalities in the dysbindin-1 mutant, sandy, on a C57BL/6J genetic background. Genes Brain Behav. 2009;8:390–397. doi: 10.1111/j.1601-183X.2009.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci. 2003;1003:318–327. doi: 10.1196/annals.1300.020. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Pereira EAC, Li P, Robbins TW. Specific abnormalities in serotonin release in the prefrontal cortex of isolation-reared rats measured during behavioural performance of a task assessing visuospatial attention and impulsivity. Psychopharmacology. 2002;164:329–340. doi: 10.1007/s00213-002-1215-y. [DOI] [PubMed] [Google Scholar]
- Day-Wilson KM, Jones DNC, Southam E, Cilia J, Totterdell S. Medial prefrontal cortex volume loss in rats with isolation rearing-induced deficits in prepulse inhibition of acoustic startle. Neuroscience. 2006;141:1113–1121. doi: 10.1016/j.neuroscience.2006.04.048. [DOI] [PubMed] [Google Scholar]
- Degen SB, Ellenbroek BA, Wiegant VM, Cools AR. The development of various somatic markers is retarded in an animal model for schizophrenia, namely apomorphine-susceptible rats. Behav Brain Res. 2005;157:369–377. doi: 10.1016/j.bbr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Del Arco A, Zhu SW, Terasmaa A, Mohammed AH, Fuxe K. Hyperactivity to novelty induced by social isolation is not correlated with changes in D2 receptor function and binding in striatum. Psychopharmacology. 2004;171:148–155. doi: 10.1007/s00213-003-1578-8. [DOI] [PubMed] [Google Scholar]
- Depoortere R, Dargazanli G, Estenne-Bouhtou G, Coste A, Lanneau C, Desvignes C, et al. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology. 2005;30:1963–1985. doi: 10.1038/sj.npp.1300772. [DOI] [PubMed] [Google Scholar]
- Dere E, Huston JP, De Souza Silva MA. The pharmacology, neuroanatomy and neurogenetics of one-trial object recognition in rodents. Neurosci Biobehav Rev. 2007;31:673–704. doi: 10.1016/j.neubiorev.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Desbonnet L, Waddington JL, O'Tuathaigh CM. Mutant models for genes associated with schizophrenia. Biochem Soc Trans. 2009;37:308–312. doi: 10.1042/BST0370308. [DOI] [PubMed] [Google Scholar]
- Didriksen M, Skarsfeldt T, Arnt J. Reversal of PCP-induced learning and memory deficits in the Morris' water maze by sertindole and other antipsychotics. Psychopharmacology (Berl) 2007;193:225–233. doi: 10.1007/s00213-007-0774-3. [DOI] [PubMed] [Google Scholar]
- DiLuca M, Caputi A, Cattabeni F, DeGraan PNE, Gispen WH, Raiteri M, et al. Increased presynaptic protein kinase C activity and glutamate release in rats with a prenatally induced hippocampal lesion. Eur J Neurosci. 1997;9:472–479. doi: 10.1111/j.1460-9568.1997.tb01624.x. [DOI] [PubMed] [Google Scholar]
- Duffy L, Cappas E, Scimone A, Schofield P, Karl T. Behavioral profile of a heterozygous mutant mouse model for EGF-like domain neuregulin 1. Behav Neurosci. 2008;122:748–759. doi: 10.1037/0735-7044.122.4.748. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, Szilagyi S, Efferen TR, Schwartz MP, Parwani A, Chakravorty S, et al. Effect of treatment status on prepulse inhibition of acoustic startle in schizophrenia. Psychopharmacology. 2003;167:63–71. doi: 10.1007/s00213-002-1372-z. [DOI] [PubMed] [Google Scholar]
- Dunn LA, Atwater GE, Kilts CD. Effects of antipsychotic-drugs on latent inhibition – sensitivity and specificity of an animal behavioral-model of clinical drug action. Psychopharmacology. 1993;112:315–323. doi: 10.1007/BF02244927. [DOI] [PubMed] [Google Scholar]
- Egerton A, Reid L, McKerchar CE, Morris BJ, Pratt JA. Impairment in perceptual attentional set-shifting following PCP administration: a rodent model of set-shifting deficits in schizophrenia. Psychopharmacology (Berl) 2005;179:77–84. doi: 10.1007/s00213-004-2109-y. [DOI] [PubMed] [Google Scholar]
- Egerton A, Reid L, McGregor S, Cochran SM, Morris BJ, Pratt JA. Subchronic and chronic PCP treatment produces temporally distinct deficits in attentional set shifting and prepulse inhibition in rats. Psychopharmacology (Berl) 2008;198:37–49. doi: 10.1007/s00213-008-1071-5. [DOI] [PubMed] [Google Scholar]
- Ehrhardt J, Sabel BA, Schroeder U. Subchronically applied phencyclidine fails to disrupt prepulse inhibition in rats. Eur Neuropsychopharmacol. 1999;10:69–74. doi: 10.1016/s0924-977x(99)00058-9. [DOI] [PubMed] [Google Scholar]
- Ehrlichman R, Luminais S, White S, Rudnick N, Ma N, Dow H, et al. Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Res. 2009;1294:116–127. doi: 10.1016/j.brainres.2009.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einon DF, Morgan MJ. A critical period for social isolation in the rat. Dev Psychobiol. 1977;10:123–132. doi: 10.1002/dev.420100205. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Cools AR. Animal models for the negative symptoms of schizophrenia. Behav Pharmacol. 2000;11:223–233. doi: 10.1097/00008877-200006000-00006. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Cools AR. Apomorphine susceptibility and animal models for psychopathology: genes and environment. Behav Genet. 2002;32:349–361. doi: 10.1023/a:1020214322065. [DOI] [PubMed] [Google Scholar]
- van der Elst MCJ, Roubos EW, Ellenbroek BA, Veening JG, Cools AR. Apomorphine-susceptible rats and apomorphine-unsusceptible rats differ in the tyrosine hydroxylase-immunoreactive network in the nucleus accumbens core and shell. Exp Brain Res. 2005;160:418–423. doi: 10.1007/s00221-004-2025-8. [DOI] [PubMed] [Google Scholar]
- van Elst LT, Valerius G, Buchert M, Thiel T, Rusch N, Bubl E, et al. Increased prefrontal and hippocampal glutamate concentration in schizophrenia: evidence from a magnetic resonance spectroscopy study. Biol Psychiatry. 2005;58:724–730. doi: 10.1016/j.biopsych.2005.04.041. [DOI] [PubMed] [Google Scholar]
- Endo K, Hori T, Abe S, Asada T. Alterations in GABA(A) receptor expression in neonatal ventral hippocampal lesioned rats: comparison of prepubertal and postpubertal periods. Synapse. 2007;61:357–366. doi: 10.1002/syn.20393. [DOI] [PubMed] [Google Scholar]
- Fabricius K, Helboe L, Fink-Jensen A, Wortwein G, Steiniger-Brach B. Pharmacological characterization of social isolation-induced hyperactivity. Psychopharmacology (Berl) 2010a doi: 10.1007/s00213-010-2128-9. DOI: 10.1007/s00213-00010-02128-00219. [DOI] [PubMed] [Google Scholar]
- Fabricius K, Helboe L, Fink-Jensen A, Wortwein G, Steiniger-Brach B, Sotty F. Increased dopaminergic activity in socially isolated rats: an electrophysiological study. Neurosci Lett. 2010b;482:117–122. doi: 10.1016/j.neulet.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Fattorini G, Melone M, Bragina L, Candiracci C, Cozzi A, Pellegrini Giampietro DE, et al. GLT-1 expression and Glu uptake in rat cerebral cortex are increased by phencyclidine. Glia. 2008;56:1320–1327. doi: 10.1002/glia.20700. [DOI] [PubMed] [Google Scholar]
- Fauman MA, Fauman BJ. The psychiatric aspects of chronic phencyclidine use: a study of chronic PCP users. NIDA Res Monogr. 1978;21:183–200. [PubMed] [Google Scholar]
- Featherstone RE, Kapur S, Fletcher PJ. The amphetamine-induced sensitized state as a model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2007a;31:1556–1571. doi: 10.1016/j.pnpbp.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Featherstone RE, Rizos Z, Nobrega JN, Kapur S, Fletcher PJ. Gestational methylazoxymethanol acetate treatment impairs select cognitive functions: parallels to schizophrenia. Neuropsychopharmacology. 2007b;32:483–492. doi: 10.1038/sj.npp.1301223. [DOI] [PubMed] [Google Scholar]
- Featherstone RE, Rizos Z, Kapur S, Fletcher PJ. A sensitizing regimen of amphetamine that disrupts attentional set-shifting does not disrupt working or long-term memory. Behav Brain Res. 2008;189:170–179. doi: 10.1016/j.bbr.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Feng Y, Zhou Z, He X, Wang H, Guo X, Hao C, et al. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res. 2008;106:218–228. doi: 10.1016/j.schres.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Ferdman N, Murmu RP, Bock J, Braun K, Leshem M. Weaning age, social isolation, and gender, interact to determine adult explorative and social behavior, and dendritic and spine morphology in prefrontal cortex of rats. Behav Brain Res. 2007;180:174–182. doi: 10.1016/j.bbr.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Fiore M, Talamini L, Angelucci F, Koch T, Aloe L, Korf J. Prenatal methylazoxymethanol acetate alters behavior and brain NGF levels in young rats: a possible correlation with the development of schizophrenia-like deficits. Neuropharmacology. 1999;38:857–869. doi: 10.1016/s0028-3908(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Fiore M, Grace AA, Korf J, Stampachiacchiere B, Aloe L. Impaired brain development in the rat following prenatal exposure to methylazoxymethanol acetate at gestational day 17 and neurotrophin distribution. Neuroreport. 2004;15:1791–1795. doi: 10.1097/01.wnr.0000135934.03635.6a. [DOI] [PubMed] [Google Scholar]
- Fiore M, Di Fausto V, Iannitelli A, Aloe L. Clozapine or haloperidol in rats prenatally exposed to methylazoxymethanol, a compound inducing entorhinal–hippocampal deficits, alter brain and blood neurotrophins' concentrations. Ann Ist Super Sanita. 2008;44:167–177. [PubMed] [Google Scholar]
- Flagstad P, Mork A, Glenthoj BY, van Beek J, Michael-Titus AT, Didriksen M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29:2052–2064. doi: 10.1038/sj.npp.1300516. [DOI] [PubMed] [Google Scholar]
- Fletcher PJ, Tenn CC, Rizos Z, Lovic V, Kapur S. Sensitization to amphetamine, but not PCP, impairs attentional set shifting: reversal by a D1 receptor agonist injected into the medial prefrontal cortex. Psychopharmacology (Berl) 2005;183:190–200. doi: 10.1007/s00213-005-0157-6. [DOI] [PubMed] [Google Scholar]
- Fletcher PJ, Tenn CC, Sinyard J, Rizos Z, Kapur S. A sensitizing regimen of amphetamine impairs visual attention in the 5-choice serial reaction time test: reversal by a D1 receptor agonist injected into the medial prefrontal cortex. Neuropsychopharmacology. 2007;32:1122–1132. doi: 10.1038/sj.npp.1301221. [DOI] [PubMed] [Google Scholar]
- Flores G, Alquicer G, Silva-Gomez AB, Zaldivar G, Stewart J, Quirion R, et al. Alterations in dendritic morphology of prefrontal cortical and nucleus accumbens neurons in post-pubertal rats after neonatal excitotoxic lesions of the ventral hippocampus. Neuroscience. 2005;133:463–470. doi: 10.1016/j.neuroscience.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Flores C, Wen XL, Labelle-Dumais C, Kolb B. Chronic phencyclidine treatment increases dendritic spine density in prefrontal cortex and nucleus accumbens neurons. Synapse. 2007;61:978–984. doi: 10.1002/syn.20452. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Geyer MA, Gold LH, Grace AA. Developing predictive animal models and establishing a preclinical trials network for assessing treatment effects on cognition in schizophrenia. Schizophr Bull. 2005;31:888–894. doi: 10.1093/schbul/sbi041. [DOI] [PubMed] [Google Scholar]
- Fone KCF, Porkess MV. Behavioural and neurochemical effects of post-weaning social isolation in rodents – relevance to developmental neuropsychiatric disorders. Neurosci Biobehav Rev. 2008;32:1087–1102. doi: 10.1016/j.neubiorev.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Fone KC, Shalders K, Fox ZD, Arthur R, Marsden CA. Increased 5-HT2C receptor responsiveness occurs on rearing rats in social isolation. Psychopharmacology (Berl) 1996;123:346–352. doi: 10.1007/BF02246645. [DOI] [PubMed] [Google Scholar]
- Fortier ME, Joober R, Luheshi GN, Boksa P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J Psychiatr Res. 2004;38:335–345. doi: 10.1016/j.jpsychires.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Fortier ME, Luheshi GN, Boksa P. Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res. 2007;181:270–277. doi: 10.1016/j.bbr.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Fulford AJ, Marsden CA. Conditioned release of 5-hydroxytryptamine in vivo in the nucleus accumbens following isolation-rearing in the rat. Neuroscience. 1998a;83:481–487. doi: 10.1016/s0306-4522(97)00423-5. [DOI] [PubMed] [Google Scholar]
- Fulford AJ, Marsden CA. Effect of isolation-rearing on conditioned dopamine release in vivo in the nucleus accumbens of the rat. J Neurochem. 1998b;70:384–390. doi: 10.1046/j.1471-4159.1998.70010384.x. [DOI] [PubMed] [Google Scholar]
- Fulford AJ, Marsden CA. An intact dopaminergic system is required for context-conditioned release of 5-HT in the nucleus accumbens of postweaning isolation-reared rats. Neuroscience. 2007;149:392–400. doi: 10.1016/j.neuroscience.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Gartlon JE, Barnes SA, Jones DNC. Comparison of subchronic PCP dosing regimes as animal models of schizophrenia. J Psychopharmacol. 2006;20:A44–A44. [Google Scholar]
- Geyer MA, Wilkinson LS, Humby T, Robbins TW. Isolation rearing of rats produces a deficit in prepulse inhibition of acoustic startle similar to that in schizophrenia. Biol Psychiatry. 1993;34:361–372. doi: 10.1016/0006-3223(93)90180-l. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Varty GB. The effects of M100907 in pharmacological and developmental animal models of prepulse inhibition deficits in schizophrenia. Neuropsychopharmacology. 1999;21:S134–S142. [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology. 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Goetghebeur P, Dias R. Comparison of haloperidol, risperidone, sertindole, and modafinil to reverse an attentional set-shifting impairment following subchronic PCP administration in the rat – a back translational study. Psychopharmacology (Berl) 2009;202:287–293. doi: 10.1007/s00213-008-1132-9. [DOI] [PubMed] [Google Scholar]
- Gogos JA, Gerber DJ. Schizophrenia susceptibility genes: emergence of positional candidates and future directions. Trends Pharmacol Sci. 2006;27:226–233. doi: 10.1016/j.tips.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D-1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology. 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- Golub M, Germann S, Lloyd K. Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav Brain Res. 2004;153:159–170. doi: 10.1016/j.bbr.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Gottesman I, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gourevitch R, Rocher C, Le Pen G, Krebs MO, Jay TM. Working memory deficits in adult rats after prenatal disruption of neurogenesis. Behav Pharmacol. 2004;15:287–292. doi: 10.1097/01.fbp.0000135703.48799.71. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. Molecular targets for treating cognitive dysfunction in schizophrenia. Schizophr Bull. 2007;33:1100–1119. doi: 10.1093/schbul/sbm074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson B, Idris NF, Neill JC. Atypical antipsychotics attenuate a sub-chronic PCP-induced cognitive deficit in the novel object recognition task in the rat. Behav Brain Res. 2007;184:31–38. doi: 10.1016/j.bbr.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Gerevini VD, Dwivedi Y, Grayson DR, et al. Decrease in reelin and glutamic acid decarboxylase(67) (GAD(67)) expression in schizophrenia and bipolar disorder – a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Dong EB, Grayson DR, Veldic M, et al. GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacology. 2005;180:191–205. doi: 10.1007/s00213-005-2212-8. [DOI] [PubMed] [Google Scholar]
- Hagan JJ, Jones DNC. Predicting drug efficacy for cognitive deficits in schizophrenia. Schizophr Bull. 2005;31:830–853. doi: 10.1093/schbul/sbi058. [DOI] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Hajszan T, Leranth C, Roth RH. Subchronic phencyclidine treatment decreases the number of dendritic spine synapses in the rat prefrontal cortex. Biol Psychiatry. 2006;60:639–644. doi: 10.1016/j.biopsych.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Hall FS, Huang S, Fong GW, Pert A, Linnoila M. Effects of isolation rearing on locomotion, anxiety and responses to ethanol in Fawn Hooded and Wistar rats. Psychopharmacology. 1998a;139:203–209. doi: 10.1007/s002130050705. [DOI] [PubMed] [Google Scholar]
- Hall FS, Wilkinson LS, Humby T, Inglis W, Kendall DA, Marsden CA, et al. Isolation rearing in rats: pre- and postsynaptic changes in striatal dopaminergic systems. Pharmacol Biochem Behav. 1998b;59:859–872. doi: 10.1016/s0091-3057(97)00510-8. [DOI] [PubMed] [Google Scholar]
- Hall FS, Ghaed S, Pert A, Xing G. The effects of isolation rearing on glutamate receptor NMDAR1A mRNA expression determined by in situ hybridization in Fawn hooded and Wistar rats. Pharmacol Biochem Behav. 2002;73:185–191. doi: 10.1016/s0091-3057(02)00796-7. [DOI] [PubMed] [Google Scholar]
- Hanania T, Hillman GR, Johnson KM. Augmentation of locomotor activity by chronic phencyclidine is associated with an increase in striatal NMDA receptor function and an upregulation of the NR1 receptor subunit. Synapse. 1999;31:229–239. doi: 10.1002/(SICI)1098-2396(19990301)31:3<229::AID-SYN8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Harich S, Gross G, Bespalov A. Stimulation of the metabotropic glutamate 2/3 receptor attenuates social novelty discrimination deficits induced by neonatal phencyclidine treatment. Psychopharmacology (Berl) 2007;192:511–519. doi: 10.1007/s00213-007-0742-y. [DOI] [PubMed] [Google Scholar]
- Harris LW, Sharp T, Gartlon J, Jones DN, Harrison PJ. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur J Neurosci. 2003;18:1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl) 2004;174:151–162. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- Harrison P, Law A. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006a;60:132–140. doi: 10.1016/j.biopsych.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Law AJ. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006b;60:132–140. doi: 10.1016/j.biopsych.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm. 2007;114:893–898. doi: 10.1007/s00702-007-0627-6. [DOI] [PubMed] [Google Scholar]
- Hazane F, Krebs MO, Jay TM, Le Pen G. Behavioral perturbations after prenatal neurogenesis disturbance in female rat. Neurotox Res. 2009;15:311–320. doi: 10.1007/s12640-009-9035-z. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Weiss IC, Domeney AM, Pryce C, Homberg J, Hedou G, et al. Behavioral, neurochemical and endocrinological characterization of the early social isolation syndrome. Neuroscience. 2000;100:749–768. doi: 10.1016/s0306-4522(00)00336-5. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Foxton R, Cilia J, Hughes ZA, Shah AJ, Atkins A, et al. Increased responsiveness of dopamine to atypical, but not typical antipsychotics in the medial prefrontal cortex of rats reared in isolation. Psychopharmacology (Berl) 2001;156:338–351. doi: 10.1007/s002130100760. [DOI] [PubMed] [Google Scholar]
- Hertzmann M, Reba RC, Kotlyarov EV. Single photon emission computed tomography in phencyclidine and related drug abuse. Am J Psychiatry. 1990;147:255–256. doi: 10.1176/ajp.147.2.255b. [DOI] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadr iS, Oishi K, Hookway C, Kong S, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayasu Y, Tanaka S, Shenton ME, Salisbury DF, DeSantis MA, Levitt JJ, et al. Prefrontal gray matter volume reduction in first episode schizophrenia. Cereb Cortex. 2001;11:374–381. doi: 10.1093/cercor/11.4.374. [DOI] [PubMed] [Google Scholar]
- Hori T, Subramaniam S, Srivastava LK, Quirion R. Behavioral and neurochemical alterations following repeated phencyclidine administration in rats with neonatal ventral hippocampal lesions. Neuropharmacology. 2000;39:2478–2491. doi: 10.1016/s0028-3908(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Howes SR, Dalley JW, Morrison CH, Robbins TW, Everitt BJ. Leftward shift in the acquisition of cocaine self-administration in isolation-reared rats: relationship to extracellular levels of dopamine, serotonin and glutamate in the nucleus accumbens and amygdala-striatal FOS expression. Psychopharmacology (Berl) 2000;151:55–63. doi: 10.1007/s002130000451. [DOI] [PubMed] [Google Scholar]
- Ibi D, Takuma K, Koike H, Mizoguchi H, Tsuritani K, Kuwahara Y, et al. Social isolation rearing-induced impairment of the hippocampal neurogenesis is associated with deficits in spatial memory and emotion-related behaviors in juvenile mice. J Neurochem. 2008;105:921–932. doi: 10.1111/j.1471-4159.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- Idris N, Neill J, Grayson B, Bang-Andersen B, Witten LM, Brennum LT, et al. Sertindole improves sub-chronic PCP-induced reversal learning and episodic memory deficits in rodents: involvement of 5-HT(6) and 5-HT (2A) receptor mechanisms. Psychopharmacology (Berl) 2010;208:23–36. doi: 10.1007/s00213-009-1702-5. [DOI] [PubMed] [Google Scholar]
- Ito HT, Smith SEP, Hsiao E, Patterson PH. Maternal immune activation alters nonspatial information processing in the hippocampus of the adult offspring. Brain Behav Immun. 2010;24:930–941. doi: 10.1016/j.bbi.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H. Gene models of schizophrenia: DISC1 mouse models. Prog Brain Res. 2009;179:75–86. doi: 10.1016/S0079-6123(09)17909-8. [DOI] [PubMed] [Google Scholar]
- Jaaro-Peled H, Ayhan Y, Pletnikov M, Sawa A. Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophr Bull. 2010;36:301–313. doi: 10.1093/schbul/sbp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jazbec S, Pantelis C, Robbins T, Weickert T, Weinberger DR, Goldberg TE. Intra-dimensional/extra-dimensional set-shifting performance in schizophrenia: impact of distractors. Schizophr Res. 2007;89:339–349. doi: 10.1016/j.schres.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Jenkins TA, Harte MK, McKibben CE, Elliott JJ, Reynolds GP. Disturbances in social interaction occur along with pathophysiological deficits following sub-chronic phencyclidine administration in the rat. Behav Brain Res. 2008;194:230–235. doi: 10.1016/j.bbr.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Jenkins TA, Harte MK, Reynolds GP. Effect of subchronic phencyclidine administration on sucrose preference and hippocampal parvalbumin immunoreactivity in the rat. Neurosci Lett. 2010;471:144–147. doi: 10.1016/j.neulet.2010.01.028. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR. Impaired inhibition of conditioned responses produced by subchronic administration of phencyclidine to rats. Neuropsychopharmacology. 2001;24:66–74. doi: 10.1016/S0893-133X(00)00174-3. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Redmond DE, Jr, Elsworth JD, Taylor JR, Youngren KD, Roth RH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science. 1997a;277:953–955. doi: 10.1126/science.277.5328.953. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Tran A, Le D, Youngren KD, Roth RH. Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology. 1997b;17:92–99. doi: 10.1016/S0893-133X(97)00034-1. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR, Roth RH. Subchronic phencyclidine administration increases mesolimbic dopaminergic system responsivity and augments stress- and psychostimulant-induced hyperlocomotion. Neuropsychopharmacology. 1998;19:105–113. doi: 10.1016/S0893-133X(98)00004-9. [DOI] [PubMed] [Google Scholar]
- Johnson KM, Phillips M, Wang C, Kevetter GA. Chronic phencyclidine induces behavioral sensitization and apoptotic cell death in the olfactory and piriform cortex. J Neurosci Res. 1998;52:709–722. doi: 10.1002/(SICI)1097-4547(19980615)52:6<709::AID-JNR10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Johnson K, Ryan L, Davis J, Elmore A, Guenther B, Marcus J, et al. Application of magnetic resonance imaging in developmental neurotoxicity testing: a pilot study. Neurotoxicology. 2006;27:846–851. doi: 10.1016/j.neuro.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Jones C, Brown AM, Auer DP, Fone KCF. The mGluR2/3 agonist LY379268 reverses post-weaning social isolation induced recognition memory deficits in the rat. Psychopharmacology (Berl) 2011;214:269–283. doi: 10.1007/s00213-010-1931-7. [DOI] [PubMed] [Google Scholar]
- Jones GH, Hernandez TD, Kendall DA, Marsden CA, Robbins TW. Dopaminergic and serotonergic function following isolation rearing in rats: study of behavioural responses and postmortem and in vivo neurochemistry. Pharmacol Biochem Behav. 1992;43:17–35. doi: 10.1016/0091-3057(92)90635-s. [DOI] [PubMed] [Google Scholar]
- Jongen-Relo AL, Leng A, Luber M, Pothuizen HHJ, Weber L, Feldon J. The prenatal methylazoxymethanol acetate treatment: a neurodevelopmental animal model for schizophrenia? Behav Brain Res. 2004;149:159–181. doi: 10.1016/s0166-4328(03)00228-6. [DOI] [PubMed] [Google Scholar]
- Kalinichev M, Robbins MJ, Hartfield EM, Maycox PR, Moore SH, Savage KM, et al. Comparison between intraperitoneal and subcutaneous phencyclidine administration in Sprague-Dawley rats: a locomotor activity and gene induction study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;32:414–422. doi: 10.1016/j.pnpbp.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Karlsgodt K, Robleto K, Trantham-Davidson H, Jairl C, Cannon T, Lavin A, et al. Reduced dysbindin expression mediates N-methyl-d-aspartate receptor hypofunction and impaired working memory performance. Biol Psychiatry. 2011;69:28–34. doi: 10.1016/j.biopsych.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Kasai A, Mizuno M, Liang FY, Shintani N, Maeda S, et al. Phenotypic characterization of transgenic mice overexpressing neuregulin-1. PLoS ONE. 2010;5:e14185. doi: 10.1371/journal.pone.0014185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe K, Miyamoto E. Effects of neonatal repeated MK-801 treatment on delayed non matching-to-position responses in rats. Neuroreport. 2008;19:969–973. doi: 10.1097/WNR.0b013e328302ee31. [DOI] [PubMed] [Google Scholar]
- Kawabe K, Iwasaki T, Ichitani Y. Repeated treatment with N-methyl-d-aspartate antagonists in neonatal, but not adult, rats causes long-term deficits of radial-arm maze learning. Brain Res. 2007;1169:77–86. doi: 10.1016/j.brainres.2007.06.062. [DOI] [PubMed] [Google Scholar]
- Keefe RS, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- Kim JH, Vezina P. The mGlu2/3 receptor agonist LY379268 blocks the expression of locomotor sensitization by amphetamine. Pharmacol Biochem Behav. 2002;73:333–337. doi: 10.1016/s0091-3057(02)00827-4. [DOI] [PubMed] [Google Scholar]
- King MV, Fone KCF, Shacham S, Gannon KS. The novel 5-HT6 receptor antagonist, PRX-07037, enhances memory and reduces food intake in a neurodevelopmental model of schizophrenia. J Psychopharmacol. 2007;21:A57. [Google Scholar]
- King MV, Seeman P, Marsden CA, Fone KCF. Increased dopamine D-2(high) receptors in rats reared in social isolation. Synapse. 2009;63:476–483. doi: 10.1002/syn.20624. [DOI] [PubMed] [Google Scholar]
- Koenig JI, Elmer GI, Shepard PD, Lee PR, Mayo C, Joy B, et al. Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav Brain Res. 2005;156:251–261. doi: 10.1016/j.bbr.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Koike H, Arguello P, Kvajo M, Karayiorgou M, Gogos J. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci USA. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97:153–179. doi: 10.1016/s0163-7258(02)00328-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger D, Howell J, Hebert B, Olausson P, Taylor J, Nairn A. Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology (Berl) 2006;189:95–104. doi: 10.1007/s00213-006-0530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello P, Drew L, Moore H, MacDermott A, et al. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci USA. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapiz MD, Mateo Y, Parker T, Marsden C. Effects of noradrenaline depletion in the brain on response on novelty in isolation-reared rats. Psychopharmacology (Berl) 2000;152:312–320. doi: 10.1007/s002130000534. [DOI] [PubMed] [Google Scholar]
- Lapiz MD, Mateo Y, Durkin S, Parker T, Marsden CA. Effects of central noradrenaline depletion by the selective neurotoxin DSP-4 on the behaviour of the isolated rat in the elevated plus maze and water maze. Psychopharmacology (Berl) 2001;155:251–259. doi: 10.1007/s002130100702. [DOI] [PubMed] [Google Scholar]
- Lapiz MD, Fulford A, Muchimapura S, Mason R, Parker T, Marsden CA. Influence of postweaning social isolation in the rat on brain development, conditioned behavior, and neurotransmission. Neurosci Behav Physiol. 2003;33:13–29. doi: 10.1023/a:1021171129766. [DOI] [PubMed] [Google Scholar]
- Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, et al. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc Natl Acad Sci USA. 2006;103:6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Cozannet R, Fone KC, Moran PM. Phencyclidine withdrawal disrupts episodic-like memory in rats: reversal by donepezil but not clozapine. Int J Neuropsychopharmacol. 2010;13:1011–1020. doi: 10.1017/S1461145710000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pen G, Grottick AJ, Higgins GA, Martin JR, Jenck F, Moreau JL. Spatial and associative learning deficits induced by neonatal excitotoxic hippocampal damage in rats: further evaluation of an animal model of schizophrenia. Behav Pharmacol. 2000;11:257–268. doi: 10.1097/00008877-200006000-00009. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Gourevitch R, Hazane F, Hoareau C, Jay TM, Krebs MO. Peri-pubertal maturation after developmental disturbance: a model for psychosis onset in the rat. Neuroscience. 2006;143:395–405. doi: 10.1016/j.neuroscience.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI. Social interaction deficits caused by chronic phencyclidine administration are reversed by oxytocin. Neuropsychopharmacology. 2005;30:1883–1894. doi: 10.1038/sj.npp.1300722. [DOI] [PubMed] [Google Scholar]
- Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI. Prenatal stress generates deficits in rat social behavior: reversal by oxytocin. Brain Res. 2007;1156:152–167. doi: 10.1016/j.brainres.2007.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G, Anastasio NC, Fu Y, Neugebauer V, Johnson KM. Activation of dopamine D1 receptors blocks phencyclidine-induced neurotoxicity by enhancing N-methyl-d-aspartate receptor-mediated synaptic strength. J Neurochem. 2009;109:1017–1030. doi: 10.1111/j.1471-4159.2009.06030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Christopher NC. Effects of clozapine on memory function in the rat neonatal hippocampal lesion model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:223–229. doi: 10.1016/j.pnpbp.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Levine JB, Youngs RM, MacDonald ML, Chu M, Leeder AD, Berthiaume F, et al. Isolation rearing and hyperlocomotion are associated with reduced immediate early gene expression levels in the medial prefrontal cortex. Neuroscience. 2007;145:42–55. doi: 10.1016/j.neuroscience.2006.11.063. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Li NX, Wu XH, Li L. Chronic administration of clozapine alleviates reversal-learning impairment in isolation-reared rats. Behav Pharmacol. 2007a;18:135–145. doi: 10.1097/FBP.0b013e3280d3ee83. [DOI] [PubMed] [Google Scholar]
- Li W, Zhou Y, Jentsch J, Brown R, Tian X, Ehninger D, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci USA. 2007b;104:18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Kim CH, Ichikawa J, Meltzer HY. Effect of repeated administration of phencyclidine on spatial performance in an eight-arm radial maze with delay in rats and mice. Pharmacol Biochem Behav. 2003;75:335–340. doi: 10.1016/s0091-3057(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Linn GS, O'Keeffe RT, Lifshitz K, Schroeder C, Javitt DC. Behavioral effects of orally administered glycine in socially housed monkeys chronically treated with phencyclidine. Psychopharmacology (Berl) 2007;192:27–38. doi: 10.1007/s00213-007-0771-6. [DOI] [PubMed] [Google Scholar]
- Lipska BK. Using animal models to test a neurodevelopmental hypothesis of schizophrenia. Rev Psychiatry Neurosci. 2004;29:282–286. [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Delayed-effects of neonatal hippocampal damage on haloperidol-induced catalepsy and apomorphine-induced stereotypic behaviours in the rat. Brain Res Dev Brain Res. 1993;75:213–222. doi: 10.1016/0165-3806(93)90026-7. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Subchronic treatment with haloperidol and clozapine in rats with neonatal excitotoxic hippocampal damage. Neuropsychopharmacology. 1994;10:199–205. doi: 10.1038/npp.1994.22. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage – a potential animal-model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Swerdlow NR, Geyer MA, Jaskiw GE, Braff DL, Weinberger DR. Neonatal excitotoxic hippocampal damage in rats causes post pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacology. 1995;122:35–43. doi: 10.1007/BF02246439. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Aultman JM, Verma A, Weinberger DR, Moghaddam B. Neonatal damage of the ventral hippocampus impairs working memory in the rat. Neuropsychopharmacology. 2002;27:47–54. doi: 10.1016/S0893-133X(02)00282-8. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weickert CS, Weinberger DR. Gene expression in dopamine and GABA systems in an animal model of schizophrenia: effects of antipsychotic drugs. Eur J Neurosci. 2003;18:391–402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- Liu F, Zou X, Sadovova N, Zhang X, Shi L, Guo L, et al. Changes in gene expression after phencyclidine administration in developing rats: a potential animal model for schizophrenia. Int J Dev Neurosci. 2010;29:351–358. doi: 10.1016/j.ijdevneu.2010.07.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Pesold C, Rodriguez M, Carboni G, Auta J, Lacor P, et al. Down-regulation of dendritic spine and glutamic acid decarboxylase 67 expressions in the reelin haploinsufficient heterozygous reeler mouse. Proc Natl Acad Sci USA. 2001;98:3477–3482. doi: 10.1073/pnas.051614698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Hippocampal dysfunction and disruption of dopamine system regulation in an animal model of schizophrenia. Neurotox Res. 2008;14:97–104. doi: 10.1007/BF03033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res. 2009;204:306–312. doi: 10.1016/j.bbr.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29:2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo KM, Martens GJ. Identification of genetic and epigenetic variations in a rat model for neurodevelopmental disorders. Behav Genet. 2007;37:697–705. doi: 10.1007/s10519-007-9164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Wolf ME. Repeated amphetamine administration alters AMPA receptor subunit expression in rat nucleus accumbens and medial prefrontal cortex. Synapse. 1999;32:119–131. doi: 10.1002/(SICI)1098-2396(199905)32:2<119::AID-SYN5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug; sernyl. AMA Arch Neurol Psychiatry. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- Mackeprang T, Kristiansen KT, Glenthoj BY. Effects of antipsychotics on prepulse inhibition of the startle response in drug-naive schizophrenic patients. Biol Psychiatry. 2002;52:863–873. doi: 10.1016/s0006-3223(02)01409-9. [DOI] [PubMed] [Google Scholar]
- McKibben CE, Jenkins TA, Adams HN, Harte MK, Reynolds GP. Effect of pretreatment with risperidone on phencyclidine-induced disruptions in object recognition memory and prefrontal cortex parvalbumin immunoreactivity in the rat. Behav Brain Res. 2010;208:132–136. doi: 10.1016/j.bbr.2009.11.018. [DOI] [PubMed] [Google Scholar]
- McLean SL, Woolley ML, Neill JC. Effects of subchronic phencyclidine on behaviour of female rats on the elevated plus maze and open field. J Psychopharmacol. 2009;24:787–790. doi: 10.1177/0269881109103112. [DOI] [PubMed] [Google Scholar]
- McLean SL, Grayson B, Harris M, Protheroe C, Bate S, Woolley ML, et al. Isolation rearing impairs novel object recognition and attentional set shifting performance in female rats. J Psychopharmacol. 2010a;24:57–63. doi: 10.1177/0269881108093842. [DOI] [PubMed] [Google Scholar]
- McLean SL, Neill JC, Idris NF, Marston HM, Wong EH, Shahid M. Effects of asenapine, olanzapine, and risperidone on psychotomimetic-induced reversal-learning deficits in the rat. Behav Brain Res. 2010b;214:240–247. doi: 10.1016/j.bbr.2010.05.043. [DOI] [PubMed] [Google Scholar]
- Malone DT, Kearn CS, Chongue L, Mackie K, Taylor DA. Effect of social isolation on CB1 and D-2 receptor and fatty acid amide hydrolase expression in rats. Neuroscience. 2008;152:265–272. doi: 10.1016/j.neuroscience.2007.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology. 1989;2:299–308. doi: 10.1016/0893-133x(89)90035-3. [DOI] [PubMed] [Google Scholar]
- Mao CV, Hori E, Maior RS, Ono T, Nishijo H. A primate model of schizophrenia using chronic PCP treatment. Rev Neurosci. 2008;19:83–89. doi: 10.1515/revneuro.2008.19.2-3.83. [DOI] [PubMed] [Google Scholar]
- Marquis JP, Goulet S, Dore FY. Neonatal lesions of the ventral hippocampus in rats lead to prefrontal cognitive deficits at two maturational stages. Neuroscience. 2006;140:759–767. doi: 10.1016/j.neuroscience.2006.02.048. [DOI] [PubMed] [Google Scholar]
- Marquis JP, Audet MC, Dore FY, Goulet S. Delayed alternation performance following subchronic phencyclidine administration in rats depends on task parameters. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1108–1112. doi: 10.1016/j.pnpbp.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Marquis JP, Goulet S, Dore FY. Neonatal ventral hippocampus lesions disrupt extra-dimensional shift and alter dendritic spine density in the medial prefrontal cortex of juvenile rats. Neurobiol Learn Mem. 2008;90:339–346. doi: 10.1016/j.nlm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Marsden CA, King MV, Fone KCF. Influence of social isolation in the rat on serotonergic function and memory – relevance to models of schizophrenia and the role of 5-HT6 receptors. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.03.003. DOI: 10.1016/j.neuropharm.2011.1003.1003. [DOI] [PubMed] [Google Scholar]
- Martinez V, Sarter M. Detection of the moderately beneficial cognitive effects of low-dose treatment with haloperidol or clozapine in an animal model of the attentional impairments of schizophrenia. Neuropsychopharmacology. 2008;33:2635–2647. doi: 10.1038/sj.npp.1301661. [DOI] [PubMed] [Google Scholar]
- Martinez ZA, Ellison GD, Geyer MA, Swerdlow NR. Effects of sustained phencyclidine exposure on sensorimotor gating of startle in rats. Neuropsychopharmacology. 1999;21:28–39. doi: 10.1016/S0893-133X(98)00137-7. [DOI] [PubMed] [Google Scholar]
- Matricon J, Bellon A, Frieling H, Kebir O, Le Pen G, Beuvon F, et al. Neuropathological and reelin deficiencies in the hippocampal formation of rats exposed to MAM; differences and similarities with schizophrenia. PLoS ONE. 2010;5:e10291. doi: 10.1371/journal.pone.0010291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumot H, Higa HH. Studies on methylazoxymethanol aglycone of cycasin – methylation of nucleic acids in vitro. Biochem J. 1966;98:C20–C22. doi: 10.1042/bj0980020c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Minabe Y, Nakamura K, Suzuki K, Iwata Y, Sekine Y, et al. Disruption of reelin signaling attenuates methamphetamine-induced hyperlocomotion. Eur J Neurosci. 2007;25:3376–3384. doi: 10.1111/j.1460-9568.2007.05564.x. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Res Brain Res Rev. 2000;31:288–294. doi: 10.1016/s0165-0173(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong W. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez RI, Gregory ML, Bardo MT, Kalivas PW. Impoverished rearing environment alters metabotropic glutamate receptor expression and function in the prefrontal cortex. Neuropsychopharmacology. 2004;29:1980–1987. doi: 10.1038/sj.npp.1300507. [DOI] [PubMed] [Google Scholar]
- Meng ZH, Feldpaush DL, Merchant KM. Clozapine and haloperidol block the induction of behavioral sensitization to amphetamine and associated genomic responses in rats. Brain Res Mol Brain Res. 1998;61:39–50. doi: 10.1016/s0169-328x(98)00196-x. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiology. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, et al. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006a;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Schwendener S, Feldon J, Yee BK. Prenatal and postnatal maternal contributions in the infection model of schizophrenia. Exp Brain Res. 2006b;173:243–257. doi: 10.1007/s00221-006-0419-5. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Brocco M. Cognitive impairment in schizophrenia: a review of developmental and genetic models, and pro-cognitive profile of the optimised D-3 > D-2 antagonist, S33138. Therapie. 2008;63:187–229. doi: 10.2515/therapie:2008041. [DOI] [PubMed] [Google Scholar]
- Millar J, Christie S, Anderson S, Lawson D, Hsiao-Wei Loh D, Devon R, et al. Genomic structure and localisation within a linkage hotspot of disrupted in schizophrenia 1, a gene disrupted by a translocation segregating with schizophrenia. Mol Psychiatry. 2001;6:173–178. doi: 10.1038/sj.mp.4000784. [DOI] [PubMed] [Google Scholar]
- Mintz J, Kopelowicz A. CUtLASS confirms CATIE. Arch Gen Psychiatry. 2007;64:978–978. doi: 10.1001/archpsyc.64.8.978-a. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60:253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology. 2007;192:283–290. doi: 10.1007/s00213-007-0708-0. [DOI] [PubMed] [Google Scholar]
- Moser PC, Hitchcock JM, Lister S, Moran PM. The pharmacology of latent inhibition as an animal model of schizophrenia. Brain Res Brain Res Rev. 2000;33:275–307. doi: 10.1016/s0165-0173(00)00026-6. [DOI] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Enomoto T, Nabeshima T. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem Int. 2007;51:173–184. doi: 10.1016/j.neuint.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Murai R, Noda Y, Matsui K, Kamei H, Mouri A, Matsuba K, et al. Hypofunctional glutamatergic neurotransmission in the prefrontal cortex is involved in the emotional deficit induced by repeated treatment with phencyclidine in mice: implications for abnormalities of glutamate release and NMDA-CaMKII signaling. Behav Brain Res. 2007;180:152–160. doi: 10.1016/j.bbr.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Murray RM, Lappin J, Di Forti M. Schizophrenia: from developmental deviance to dopamine dysregulation. Eur Neuropsychopharmacol. 2008;18:S129–S134. doi: 10.1016/j.euroneuro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Nagai T, Murai R, Matsui K, Kamei H, Noda Y, Furukawa H, et al. Aripiprazole ameliorates phencyclidine-induced impairment of recognition memory through dopamine D(1) and serotonin 5-HT (1A) receptors. Psychopharmacology (Berl) 2009;202:315–328. doi: 10.1007/s00213-008-1240-6. [DOI] [PubMed] [Google Scholar]
- Nakatani-Pawlak A, Yamaguchi K, Tatsumi Y, Mizoguchi H, Yoneda Y. Neonatal phencyclidine treatment in mice induces behavioral, histological and neurochemical abnormalities in adulthood. Biol Pharm Bull. 2009;32:1576–1583. doi: 10.1248/bpb.32.1576. [DOI] [PubMed] [Google Scholar]
- Neill JC, Barnes S, Cook S, Grayson B, Idris NF, McLean SL, et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128:419–432. doi: 10.1016/j.pharmthera.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Newell KA, Zavitsanou K, Huang XF. Opposing short- and long-term effects on muscarinic M1/4 receptor binding following chronic phencyclidine treatment. J Neurosci Res. 2007;85:1358–1363. doi: 10.1002/jnr.21247. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Barch DM, Gold JM, Goldberg TE, Green MF, Heaton RK. Identification of separable cognitive factors in schizophrenia. Schizophr Res. 2004;72:29–39. doi: 10.1016/j.schres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Robbins TW, Einat H. Distinguishing separable domains of cognition in human and animal studies: what separations are optimal for targeting interventions? A summary of recommendations from breakout group 2 at the Measurement and Treatment Research to Improve Cognition in Schizophrenia New Approaches Conference. Schizophr Bull. 2005;31:870–874. doi: 10.1093/schbul/sbi047. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cereb Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- O'Tuathaigh C, Babovic D, O'Sullivan G, Clifford J, Tighe O, Croke D, et al. Phenotypic characterization of spatial cognition and social behavior in mice with ‘knockout’ of the schizophrenia risk gene neuregulin 1. Neuroscience. 2007;147:18–27. doi: 10.1016/j.neuroscience.2007.03.051. [DOI] [PubMed] [Google Scholar]
- O'Tuathaigh C, Waddington J. Mutant mouse models: phenotypic relationships to domains of psychopathology and pathobiology in schizophrenia. Schizophr Bull. 2010;36:243–245. doi: 10.1093/schbul/sbq004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Tuathaigh C, Kirby B, Moran P, Waddington J. Mutant mouse models: genotype–phenotype relationships to negative symptoms in schizophrenia. Schizophr Bull. 2010;36:271–288. doi: 10.1093/schbul/sbp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ognibene E, Adriani W, Caprioli A, Ghirardi O, Ali S, Aloe L, et al. The effect of early maternal separation on brain derived neurotrophic factor and monoamine levels in adult heterozygous reeler mice. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1269–1276. doi: 10.1016/j.pnpbp.2008.03.023. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- van Os J, Kenis G, Rutten BPF. The environment and schizophrenia. Nature. 2010;468:203–212. doi: 10.1038/nature09563. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley J, et al. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.106. DOI: 10.1038/mp.2010.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Holzman PS. Schizophrenics show spatial working memory deficits. Arch Gen Psychiatry. 1992;49:975–982. doi: 10.1001/archpsyc.1992.01820120063009. [DOI] [PubMed] [Google Scholar]
- Pascual R, Zamora-Leon SP. Chronic (–)-deprenyl administration attenuates dendritic developmental impairment induced by early social isolation in the rat. Dev Neurosci. 2007;29:261–267. doi: 10.1159/000096413. [DOI] [PubMed] [Google Scholar]
- Pascual R, Zamora-Leon SP, Valero-Cabre A. Effects of postweaning social isolation and re-socialization on the expression of vasoactive intestinal peptide (VIP) and dendritic development in the medial prefrontal cortex of the rat. Acta Neurobiol Exp (Wars) 2006;66:7–14. doi: 10.55782/ane-2006-1582. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Pedersen CS, Goetghebeur P, Dias R. Chronic infusion of PCP via osmotic mini-pumps: a new rodent model of cognitive deficit in schizophrenia characterized by impaired attentional set-shifting (ID/ED) performance. J Neurosci Methods. 2009;185:66–69. doi: 10.1016/j.jneumeth.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Peleg-Raibstein D, Pezze MA, Ferger B, Zhang WN, Murphy CA, Feldon J, et al. Activation of dopaminergic neurotransmission in the medial prefrontal cortex by N-methyl-d-aspartate stimulation of the ventral hippocampus in rats. Neuroscience. 2005;132:219–232. doi: 10.1016/j.neuroscience.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Peleg-Raibstein D, Sydekum E, Russig H, Feldon J. Withdrawal from repeated amphetamine administration leads to disruption of prepulse inhibition but not to disruption of latent inhibition. J Neural Transm. 2006;113:1323–1336. doi: 10.1007/s00702-005-0390-5. [DOI] [PubMed] [Google Scholar]
- Penschuck S, Flagstad P, Didriksen M, Leist M, Michael-Titus AT. Decrease in parvalbumin-expressing neurons in the hippocampus and increased phencyclidine-induced locomotor activity in the rat methylazoxymethanol (MAM) model of schizophrenia. Eur J Neurosci. 2006;23:279–284. doi: 10.1111/j.1460-9568.2005.04536.x. [DOI] [PubMed] [Google Scholar]
- Phillips M, Wang C, Johnson KM. Pharmacological characterization of locomotor sensitization induced by chronic phencyclidine administration. J Pharmacol Exp Ther. 2001;296:905–913. [PubMed] [Google Scholar]
- Pizzagalli DA. The ‘anhedonia paradox’ in schizophrenia: insights from affective neuroscience. Biol Psychiatry. 2010;67:899–901. doi: 10.1016/j.biopsych.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov M, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov M, Huang H, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- Podhorna J, Didriksen M. The heterozygous reeler mouse: behavioural phenotype. Behav Brain Res. 2004;153:43–54. doi: 10.1016/j.bbr.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Powell CM, Miyakawa T. Schizophrenia-relevant behavioral testing in rodent models: a uniquely human disorder? Biol Psychiatry. 2006;59:1198–1207. doi: 10.1016/j.biopsych.2006.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SB, Geyer MA, Preece MA, Pitcher LK, Reynolds GP, Swerdlow NR. Dopamine depletion of the nucleus accumbens reverses isolation-induced deficits in prepulse inhibition in rats. Neuroscience. 2003;119:233–240. doi: 10.1016/s0306-4522(03)00122-2. [DOI] [PubMed] [Google Scholar]
- Qiao H, Noda Y, Kamei H, Nagai T, Furukawa H, Miura H, et al. Clozapine, but not haloperidol, reverses social behavior deficit in mice during withdrawal from chronic phencyclidine treatment. Neuroreport. 2001;12:11–15. doi: 10.1097/00001756-200101220-00010. [DOI] [PubMed] [Google Scholar]
- Qiu S, Korwek K, Pratt-Davis A, Peters M, Bergman M, Weeber E. Cognitive disruption and altered hippocampus synaptic function in reelin haploinsufficient mice. Neurobiol Learn Mem. 2006;85:228–242. doi: 10.1016/j.nlm.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Quan MN, Tian YT, Xu KH, Zhang T, Yang Z. Post weaning social isolation influences spatial cognition, prefrontal cortical synaptic plasticity and hippocampal potassium ion channels in Wistar rats. Neuroscience. 2010;169:214–222. doi: 10.1016/j.neuroscience.2010.04.048. [DOI] [PubMed] [Google Scholar]
- Rainey JM, Jr, Crowder MK. Prolonged psychosis attributed to phencyclidine: report of three cases. Am J Psychiatry. 1975;132:1076–1078. doi: 10.1176/ajp.132.10.1076. [DOI] [PubMed] [Google Scholar]
- Rasmussen BA, O'Neil J, Manaye KF, Perry DC, Tizabi Y. Long-term effects of developmental PCP administration on sensorimotor gating in male and female rats. Psychopharmacology. 2007;190:43–49. doi: 10.1007/s00213-006-0584-z. [DOI] [PubMed] [Google Scholar]
- Remington G. Understanding antipsychotic ‘atypicality’: a clinical and pharmacological moving target. J Psychiatry Neurosci. 2003;28:275–284. [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP, Abdul-Monim Z, Neill JC, Zhang ZJ. Calcium binding protein markers of GABA deficits in schizophrenia – postmortem studies and animal models. Neurotox Res. 2004;6:57–61. doi: 10.1007/BF03033297. [DOI] [PubMed] [Google Scholar]
- Reynolds LM, Cochran SM, Morris BJ, Pratt JA, Reynolds GP. Chronic phencyclidine administration induces schizophrenia-like changes in N-acetylaspartate and N-acetylaspartylglutamate in rat brain. Schizophr Res. 2005;73:147–152. doi: 10.1016/j.schres.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Richtand NM, Taylor B, Welge JA, Ahlbrand R, Ostrander MM, Burr J, et al. Risperidone pretreatment prevents elevated locomotor activity following neonatal hippocampal lesions. Neuropsychopharmacology. 2006;31:77–89. doi: 10.1038/sj.npp.1300791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimer M, Barrett D, Maldonado M, Vock V, Gonzalez-Lima F. Neuregulin-1 immunoglobulin-like domain mutant mice: clozapine sensitivity and impaired latent inhibition. Neuroreport. 2005;16:271–275. doi: 10.1097/00001756-200502280-00014. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Becker JB. Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res. 1986;396:157–198. doi: 10.1016/s0006-8993(86)80193-7. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Rodefer JS, Murphy ER, Baxter MG. PDE10A inhibition reverses subchronic PCP-induced deficits in attentional set-shifting in rats. Eur J Neurosci. 2005;21:1070–1076. doi: 10.1111/j.1460-9568.2005.03937.x. [DOI] [PubMed] [Google Scholar]
- Romero E, Ali C, Molina-Holgado E, Castellano B, Guaza C, Borrell J. Neurobehavioral and immunological consequences of prenatal immune activation in rats. Influence of antipsychotics. Neuropsychopharmacology. 2007;32:1791–1804. doi: 10.1038/sj.npp.1301292. [DOI] [PubMed] [Google Scholar]
- Roy K, Murtie J, El-Khodor B, Edgar N, Sardi S, Hooks B, et al. Loss of ErbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proc Natl Acad Sci USA. 2007;104:8131–8136. doi: 10.1073/pnas.0702157104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushe TM, Woodruff PWR, Murray RM, Morris RG. Episodic memory and learning in patients with chronic schizophrenia. Schizophr Res. 1999;35:85–96. doi: 10.1016/s0920-9964(98)00117-0. [DOI] [PubMed] [Google Scholar]
- Russig H, Durrer A, Yee BK, Murphy CA, Feldon J. The acquisition, retention and reversal of spatial learning in the Morris water maze task following withdrawal from an escalating dosage schedule of amphetamine in Wistar rats. Neuroscience. 2003;119:167–179. doi: 10.1016/s0306-4522(03)00045-9. [DOI] [PubMed] [Google Scholar]
- Salinger W, Ladrow P, Wheeler C. Behavioral phenotype of the reeler mutant mouse: effects of RELN gene dosage and social isolation. Behav Neurosci. 2003;117:1257–1275. doi: 10.1037/0735-7044.117.6.1257. [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F. Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav Pharmacol. 1995;6:55–65. [PubMed] [Google Scholar]
- Sams-Dodd F. Phencyclidine-induced stereotyped behaviour and social isolation in rats: a possible animal model of schizophrenia. Behav Pharmacol. 1996;7:3–23. [PubMed] [Google Scholar]
- Sams-Dodd F. A test of the predictive validity of animal models of schizophrenia based on phencyclidine and d-amphetamine. Neuropsychopharmacology. 1998;18:293–304. doi: 10.1016/S0893-133X(97)00161-9. [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F, Lipska BK, Weinberger DR. Neonatal lesions of the rat ventral hippocampus result in hyperlocomotion and deficits in social behaviour in adulthood. Psychopharmacology. 1997;132:303–310. doi: 10.1007/s002130050349. [DOI] [PubMed] [Google Scholar]
- Sarter M, Martinez V, Kozak R. A neurocognitive animal model dissociating between acute illness and remission periods of schizophrenia. Psychopharmacology (Berl) 2009;202:237–258. doi: 10.1007/s00213-008-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savonenko A, Melnikova T, Laird F, Stewart K, Price D, Wong P. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci USA. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalzo FM, Holson RR. The ontogeny of behavioral sensitization to phencyclidine. Neurotoxicol Teratol. 1992;14:7–14. doi: 10.1016/0892-0362(92)90023-4. [DOI] [PubMed] [Google Scholar]
- Schrijver NCA, Wurbel H. Early social deprivation disrupts attentional, but not affective, shifts in rats. Behav Neurosci. 2001;115:437–442. [PubMed] [Google Scholar]
- Schrijver NC, Bahr NI, Weiss IC, Wurbel H. Dissociable effects of isolation rearing and environmental enrichment on exploration, spatial learning and HPA activity in adult rats. Pharmacol Biochem Behav. 2002;73:209–224. doi: 10.1016/s0091-3057(02)00790-6. [DOI] [PubMed] [Google Scholar]
- Schrijver NC, Pallier PN, Brown VJ, Wurbel H. Double dissociation of social and environmental stimulation on spatial learning and reversal learning in rats. Behav Brain Res. 2004;152:307–314. doi: 10.1016/j.bbr.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Schubert MI, Porkess MV, Dashdorj N, Fone KCF, Auer DP. Effects of social isolation rearing on the limbic brain: a combined behavioral and magnetic resonance imaging volumetry study in rats. Neuroscience. 2009;159:21–30. doi: 10.1016/j.neuroscience.2008.12.019. [DOI] [PubMed] [Google Scholar]
- Schwabe K, Brosda J, Wegener N, Koch M. Clozapine enhances disruption of prepulse inhibition after sub-chronic dizocilpine- or phencyclidine-treatment in Wistar rats. Pharmacol Biochem Behav. 2005;80:213–219. doi: 10.1016/j.pbb.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Scott L, Aperia A. Interaction between N-methyl-d-aspartic acid receptors and D1 dopamine receptors: an important mechanism for brain plasticity. Neuroscience. 2009;158:62–66. doi: 10.1016/j.neuroscience.2008.10.020. [DOI] [PubMed] [Google Scholar]
- Seeman P, Schwarz J, Chen JF, Szechtman H, Perreault M, McKnight GS, et al. Psychosis pathways converge via D2(high) dopamine receptors. Synapse. 2006;60:319–346. doi: 10.1002/syn.20303. [DOI] [PubMed] [Google Scholar]
- Seillier A, Giuffrida A. Evaluation of NMDA receptor models of schizophrenia: divergences in the behavioral effects of sub-chronic PCP and MK-801. Behav Brain Res. 2009;204:410–415. doi: 10.1016/j.bbr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan S, et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LM, Fatemi H, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemi K, Tsuneyoshi Y, Yamada S, Kabuki Y, Hayamizu K, Denbow DM, et al. Oral administration of l-serine reduces the locomotor activity of socially isolated rats. Neurosci Lett. 2010;468:75–79. doi: 10.1016/j.neulet.2009.10.068. [DOI] [PubMed] [Google Scholar]
- Shoval G, Weizman A. The possible role of neurotrophins in the pathogenesis and therapy of schizophrenia. Eur Neuropsychopharmacol. 2005;15:319–329. doi: 10.1016/j.euroneuro.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Silva-Gomez AB, Rojas D, Juarez I, Flores G. Decreased dendritic spine density on prefrontal cortical and hippocampal pyramidal neurons in postweaning social isolation rats. Brain Res. 2003;983:128–136. doi: 10.1016/s0006-8993(03)03042-7. [DOI] [PubMed] [Google Scholar]
- Sircar R. Developmental maturation of the N-methyl-d-aspartic acid receptor channel complex in postnatal rat brain. Int J Dev Neurosci. 2000;18:121–131. doi: 10.1016/s0736-5748(99)00069-6. [DOI] [PubMed] [Google Scholar]
- Sircar R. Postnatal phencyclidine-induced deficit in adult water maze performance is associated with N-methyl-d-aspartate receptor upregulation. Int J Dev Neurosci. 2003;21:159–167. doi: 10.1016/s0736-5748(03)00026-1. [DOI] [PubMed] [Google Scholar]
- Snigdha S, Horiguchi M, Huang M, Li Z, Shahid M, Neill JC, et al. Attenuation of phencyclidine-induced object recognition deficits by the combination of atypical antipsychotic drugs and pimavanserin (ACP 103), a 5-hydroxytryptamine(2A) receptor inverse agonist. J Pharmacol Exp Ther. 2010;332:622–631. doi: 10.1124/jpet.109.156349. [DOI] [PubMed] [Google Scholar]
- Spano MS, Fadda P, Frau R, Fattore L, Fratta W. Cannabinoid self-administration attenuates PCP-induced schizophrenia-like symptoms in adult rats. Eur Neuropsychopharmacol. 2010;20:25–36. doi: 10.1016/j.euroneuro.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Srisurapanont M, Ali R, Marsden J, Sunga A, Wada K, Monteiro M. Psychotic symptoms in methamphetamine psychotic in-patients. Int J Neuropsychopharmacol. 2003;6:347–352. doi: 10.1017/S1461145703003675. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Effects of repeated treatment with amphetamine or phencyclidine on working memory in the rat. Behav Brain Res. 2002;134:267–274. doi: 10.1016/s0166-4328(02)00040-2. [DOI] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Transient N-methyl-d-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry. 2005;57:433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Marshall KM, Neill JC. Influence of gender on working and spatial memory in the novel object recognition task in the rat. Behav Brain Res. 2007;177:117–125. doi: 10.1016/j.bbr.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. Animal models of deficient sensorimotor gating: what we know, what we think we know and what we hope to know soon. Behav Pharmacol. 2000;11:185–204. doi: 10.1097/00008877-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Shirakawa O, Toyooka K, Kitamura N, Hashimoto T, Maeda K, et al. Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol Psychiatry. 2000;5:293–300. doi: 10.1038/sj.mp.4000718. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Kakita A, Futamura T, Watanabe Y, Mizuno M, Sakimura K, et al. Sustained brain-derived neurotrophic factor up-regulation and sensorimotor gating abnormality induced by postnatal exposure to phencyclidine: comparison with adult treatment. J Neurochem. 2006;99:770–780. doi: 10.1111/j.1471-4159.2006.04106.x. [DOI] [PubMed] [Google Scholar]
- Takao K, Toyama K, Nakanishi K, Hattori S, Takamura H, Takeda M, et al. Impaired long-term memory retention and working memory in sdy mutant mice with a deletion in Dtnbp1, a susceptibility gene for schizophrenia. Mol Brain. 2008;1:11. doi: 10.1186/1756-6606-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talamini LM, Koch T, Ter Horst GJ, Korf J. Methylazoxymethanol acetate-induced abnormalities in the entorhinal cortex of the rat; parallels with morphological findings in schizophrenia. Brain Res. 1998;789:293–306. doi: 10.1016/s0006-8993(98)00020-1. [DOI] [PubMed] [Google Scholar]
- Talamini LM, Ellenbroek B, Koch T, Korf J. Impaired sensory gating and attention in rats with developmental abnormalities of the mesocortex – implications for schizophrenia. Ann N Y Acad Sci. 2000;911:486–494. doi: 10.1111/j.1749-6632.2000.tb06751.x. [DOI] [PubMed] [Google Scholar]
- Tanibuchi Y, Fujita Y, Kohno M, Ishima T, Takatsu Y, Iyo M, et al. Effects of quetiapine on phencyclidine-induced cognitive deficits in mice: a possible role of alpha1-adrenoceptors. Eur Neuropsychopharmacol. 2009;19:861–867. doi: 10.1016/j.euroneuro.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Tenn CC, Kapur S, Fletcher PJ. Sensitization to amphetamine, but not phencyclidine, disrupts prepulse inhibition and latent inhibition. Psychopharmacology (Berl) 2005;180:366–376. doi: 10.1007/s00213-005-2253-z. [DOI] [PubMed] [Google Scholar]
- Toua C, Brand L, Moller M, Emsley RA, Harvey BH. The effects of sub-chronic clozapine and haloperidol administration on isolation rearing induced changes in frontal cortical N-methyl-d-aspartate and D1 receptor binding in rats. Neuroscience. 2010;165:492–499. doi: 10.1016/j.neuroscience.2009.10.039. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Doueiri M, Dong E, Grayson D, Davis J, Pinna G, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res. 2009;204:295–305. doi: 10.1016/j.bbr.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tueting P, Doueiri M, Guidotti A, Davis J, Costa E. Reelin down-regulation in mice and psychosis endophenotypes. Neurosci Biobehav Rev. 2006;30:1065–1077. doi: 10.1016/j.neubiorev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Tunstall B, Beckett S, Mason R. Ultrasonic vocalisations explain unexpected effects on pre-pulse inhibition responses in rats chronically pre-treated with phencyclidine. Behav Brain Res. 2009;202:184–191. doi: 10.1016/j.bbr.2009.03.035. [DOI] [PubMed] [Google Scholar]
- Turnock-Jones JJ, Jennings CA, Robbins MJ, Cluderay JE, Cilia J, Reid JL, et al. Increased expression of the NR2A NMDA receptor subunit in the prefrontal cortex of rats reared in isolation. Synapse. 2009;63:836–846. doi: 10.1002/syn.20665. [DOI] [PubMed] [Google Scholar]
- Tyson PJ, Laws KR, Roberts KH, Mortimer AM. Stability of set-shifting and planning abilities in patients with schizophrenia. Psychiatry Res. 2004;129:229–239. doi: 10.1016/j.psychres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Uehara T, Sumiyoshi T, Seo T, Itoh H, Matsuoka T, Suzuki M, et al. Long-term effects of neonatal MK-801 treatment on prepulse inhibition in young adult rats. Psychopharmacology. 2009;206:623–630. doi: 10.1007/s00213-009-1527-2. [DOI] [PubMed] [Google Scholar]
- Uehara T, Sumiyoshi T, Seo T, Matsuoka T, Itoh H, Suzuki M, et al. Neonatal exposure to MK-801, an N-methyl-d-aspartate receptor antagonist, enhances methamphetamine-induced locomotion and disrupts sensorimotor gating in pre- and postpubertal rats. Brain Res. 2010;1352:223–230. doi: 10.1016/j.brainres.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Valzelli L. Isolation syndrome in mice. Psychopharmacologia. 1973;31:305–320. doi: 10.1007/BF00421275. [DOI] [PubMed] [Google Scholar]
- Varty GB, Geyer MA. Effects of isolation rearing on startle reactivity, habituation, and prepulse inhibition in male Lewis, Sprague-Dawley, and Fischer F344 rats. Behav Neurosci. 1998;112:1450–1457. doi: 10.1037//0735-7044.112.6.1450. [DOI] [PubMed] [Google Scholar]
- Varty GB, Higgins GA. Examination of drug-induced and isolation-induced disruptions of prepulse inhibition as models to screen antipychotic drugs. Psychopharmacology. 1995;112:15–26. doi: 10.1007/BF02246437. [DOI] [PubMed] [Google Scholar]
- Varty GB, Marsden CA, Higgins GA. Reduced synaptophysin immunoreactivity in the dentate gyrus of prepulse inhibition-impaired isolation-reared rats. Brain Res. 1999;824:197–203. doi: 10.1016/s0006-8993(99)01173-7. [DOI] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Wan RQ, Corbett R. Enhancement of postsynaptic sensitivity to dopaminergic agonists induced by neonatal hippocampal lesions. Neuropsychopharmacology. 1997;16:259–268. doi: 10.1016/S0893-133X(96)00217-5. [DOI] [PubMed] [Google Scholar]
- Wan RQ, Giovanni A, Kafka SH, Corbett R. Neonatal hippocampal lesions induced hyperresponsiveness to amphetamine: behavioral and in vivo microdialysis studies. Behav Brain Res. 1996;78:211–223. doi: 10.1016/0166-4328(95)00251-0. [DOI] [PubMed] [Google Scholar]
- Wan RQ, Hartman H, Corbett R. Alteration of dopamine metabolites in CSF and behavioral impairments induced by neonatal hippocampal lesions. Physiol Behav. 1998;65:429–436. doi: 10.1016/s0031-9384(98)00179-6. [DOI] [PubMed] [Google Scholar]
- Wang CZ, Johnson KM. Differential effects of acute and subchronic administration on phencyclidine-induced neuro degeneration in the perinatal rat. J Neurosci Res. 2005;81:284–292. doi: 10.1002/jnr.20559. [DOI] [PubMed] [Google Scholar]
- Wang C, McInnis J, Ross-Sanchez M, Shinnick-Gallagher P, Wiley JL, Johnson KM. Long-term behavioral and neurodegenerative effects of perinatal phencyclidine administration: implications for schizophrenia. Neuroscience. 2001;107:535–550. doi: 10.1016/s0306-4522(01)00384-0. [DOI] [PubMed] [Google Scholar]
- Watson DJG, Marsden CA, Millan MJ, Fone KCF. Blockade of dopamine D3 but not D2 receptors reverses the novel object discrimination impairment produced by post-weaning social isolation: implications for schizophrenia and its treatment. Int J Neuropsychopharmacol. 2011 doi: 10.1017/S1461145711000435. DOI: 10.1017/S1461145711000435. [DOI] [PubMed] [Google Scholar]
- Weickert C, Straub R, McClintock B, Matsumoto M, Hashimoto R, Hyde T, et al. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry. 2004;61:544–555. doi: 10.1001/archpsyc.61.6.544. [DOI] [PubMed] [Google Scholar]
- Weickert C, Rothmond D, Hyde T, Kleinman J, Straub R. Reduced DTNBP1 (dysbindin-1) mRNA in the hippocampal formation of schizophrenia patients. Schizophr Res. 2008;98:105–110. doi: 10.1016/j.schres.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss IC, Feldon J. Environmental animal models for sensorimotor gating deficiencies in schizophrenia: a review. Psychopharmacology. 2001;156:305–326. doi: 10.1007/s002130100800. [DOI] [PubMed] [Google Scholar]
- Weiss IC, Feldon J, Domeney AM. Isolation rearing-induced disruption of prepulse inhibition: further evidence for fragility of the response. Behav Pharmacol. 1999;10:139–149. doi: 10.1097/00008877-199903000-00003. [DOI] [PubMed] [Google Scholar]
- Weiss IC, Pryce CR, Jongen-Relo AL, Nanz-Bahr NI, Feldon J. Effect of social isolation on stress-related behavioural and neuroendocrine state in the rat. Behav Brain Res. 2004;152:279–295. doi: 10.1016/j.bbr.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Buhler KG, Lavecchia KL, Johnson KM. Pharmacological challenge reveals long-term effects of perinatal phencyclidine on delayed spatial alternation in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:867–873. doi: 10.1016/S0278-5846(03)00146-5. [DOI] [PubMed] [Google Scholar]
- Wilkinson LS, Killcross SS, Humby T, Hall FS, Geyer MA, Robbins TW. Social-isolation in the rat produces developmentally specific deficits in prepulse inhibition of the acoustic startle response without disrupting latent inhibition. Neuropsychopharmacology. 1994;10:61–72. doi: 10.1038/npp.1994.8. [DOI] [PubMed] [Google Scholar]
- Williams N, O'Donovan M, Owen M. Is the dysbindin gene (DTNBP1) a susceptibility gene for schizophrenia? Schizophr Bull. 2005;31:800–805. doi: 10.1093/schbul/sbi061. [DOI] [PubMed] [Google Scholar]
- Winters BD, Saksida LM, Bussey TJ. Object recognition memory: neurobiological mechanisms of encoding, consolidation and retrieval. Neurosci Biobehav Rev. 2008;32:1055–1070. doi: 10.1016/j.neubiorev.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Wiseman Harris L, Sharp T, Gartlon J, Jones DN, Harrison PJ. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur J Neurosci. 2003;18:1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- Wood GK, Lipska BK, Weinberger DR. Behavioral changes in rats with early ventral hippocampal damage vary with age at damage. Brain Res Dev Brain Res. 1997;101:17–25. doi: 10.1016/s0165-3806(97)00050-3. [DOI] [PubMed] [Google Scholar]
- Xu X, Domino EF. Phencyclidine-induced behavioral sensitization. Pharmacol Biochem Behav. 1994;47:603–608. doi: 10.1016/0091-3057(94)90165-1. [DOI] [PubMed] [Google Scholar]
- Young JW, Powell SB, Risbrough V, Marston HM, Geyer MA. Using the MATRICS to guide development of a preclinical cognitive test battery for research in schizophrenia. Pharmacol Ther. 2009;122:150–202. doi: 10.1016/j.pharmthera.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XH, Sun L, Jia HX, Meng QX, Wu S, Li NX, et al. Isolation rearing induces social and emotional function abnormalities and alters glutamate and neurodevelopment-related gene expression in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1173–1177. doi: 10.1016/j.pnpbp.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Weiner I. Post-pubertal emergence of disrupted latent inhibition following prenatal immune activation. Psychopharmacology. 2003;169:308–313. doi: 10.1007/s00213-003-1461-7. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]