

Abstract
Traumatic brain injury (TBI) is the leading cause of death and disability in young adults. Survivors of TBI frequently suffer from long-term personality changes and deficits in cognitive and motor performance, urgently calling for novel pharmacological treatment options. To date, all clinical trials evaluating neuroprotective compounds have failed in demonstrating clinical efficacy in cohorts of severely injured TBI patients. The purpose of the present review is to describe the utility of animal models of TBI for preclinical evaluation of pharmacological compounds. No single animal model can adequately mimic all aspects of human TBI owing to the heterogeneity of clinical TBI. To successfully develop compounds for clinical TBI, a thorough evaluation in several TBI models and injury severities is crucial. Additionally, brain pharmacokinetics and the time window must be carefully evaluated. Although the search for a single-compound, ‘silver bullet’ therapy is ongoing, a combination of drugs targeting various aspects of neuroprotection, neuroinflammation and regeneration may be needed. In summary, finding drugs and prove clinical efficacy in TBI is a major challenge ahead for the research community and the drug industry. For a successful translation of basic science knowledge to the clinic to occur we believe that a further refinement of animal models and functional outcome methods is important. In the clinical setting, improved patient classification, more homogenous patient cohorts in clinical trials, standardized treatment strategies, improved central nervous system drug delivery systems and monitoring of target drug levels and drug effects is warranted.
LINKED ARTICLES
This article is part of a themed issue on Translational Neuropharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-4
Keywords: Traumatic brain injury, review, neuroprotection, magnesium, cyclosporin, glutamate, reactive oxygen species, inflammation, plasticity, animal modelling, contusion
Life brings on unexpected changes
But we must carry on despite it
– Lenny Kravitz, the Future Song
Introduction
Traumatic brain injury (TBI) caused by, for example, transportation, falls, assault, sports, mainly affects young individuals and is the leading cause of mortality and disability in the population below 50 years of age (Ghajar, 2000; Corrigan et al., 2010). In Europe, TBI is the cause for 60 000 deaths annually and the incidence of fatal and hospitalized TBI combined was estimated to be 235/100 000 inhabitants. Although the global magnitude of TBI is unknown, available data suggest that the number of TBI victims globally is rising sharply (Tagliaferri et al., 2006; Maas et al., 2008; Corrigan et al., 2010). The TBI frequently causes persistent functional deficits, leading to a loss of decades of productive life with a huge cost to the patient, his or her family and the society. Common consequences of TBI include personality changes, seizures, cognitive problems, impaired motor function and a reduced quality of life calling for long-term rehabilitation and treatment (Masel and DeWitt, 2010). A range of psychiatric disorders, where depression may be the most common, also occur after TBI (Bryant et al., 2010). Currently, treatment of TBI according to established guidelines consists of supportive measures including the avoidance of prehospital hypoxia and hypotension, rapid surgery of mass lesions when needed, and neurocritical care (NCC) (Marion, 2006; Brain Trauma Foundation et al., 2007). Improved NCC has been a major factor for improving outcome of severely brain-injured patients (Elf et al., 2002). The outcome of TBI patients may also reflect the economic status of the region or country in which the patient is treated (Mauritz et al., 2008). Further improvements in TBI outcomes may be achieved by a continued refinement of NCC based on research and development in the areas listed in Table 1. However, to date there are no specific neuroprotective pharmacological treatment options with proven clinical efficacy available for TBI patients (Maas et al., 2008). Importantly, prolonged and supportive neurorehabilitation following the initial post-injury phase may also aid in improving outcome where a number of cognitive ‘stimulants’ have been evaluated to ameliorate post-injury disabilities (Napolitano et al., 2005).
Table 1.
Key scientific approaches for further improvement of neurocritical care in traumatic brain injury
| Pharmacological neuroprotection | Drugs blocking specific secondary injury mechanisms |
| Drugs stimulating reparative mechanisms | |
| Monitoring of the brain injury process | Biomarkers of specific secondary injury mechanisms |
| Refined/novel neuroimaging methods | |
| Neurorepair | Axonal regeneration |
| Neurogenesis/stem cell therapy | |
| Neurorehabilitation/plasticity |
Traumatic brain injury may be the most complicated disease of the most complex organ of the body. This statement reflects the many features and distribution patterns of TBI as well as the sophisticated structure and function of the brain. For a successful translation of basic science knowledge to the clinic to occur in this context we believe that a further refinement of animal models and functional outcome methods as well as TBI patient classification and standardized treatment strategies is warranted (also see Morales et al., 2005).
The purpose of this review is to discuss the current use of animal modelling of TBI as a tool in preclinical drug development with a focus on closed head injury (CHI) models in rodents, excluding the rapidly growing field of blast injury models.
Basic pathophysiology of TBI
The primary injury to the head causes rapid deformation of brain tissue with destruction of brain parenchyma and blood vessels causing damage to cell membranes with the immediate release of intracellular contents (McIntosh, 1994; Rink et al., 1995). This initial injury cannot be treated, only prevented. Not all neuronal and glial damage occurs at the time of primary injury which is markedly exacerbated by a complex cascade of pathophysiological and neurochemical events during the course of the initial hours and days (Figure 1). Importantly, several reports have shown a progressive encephalomalacia for many years post-injury (Greenberg et al., 2008; Ng et al., 2008). Although the duration and magnitude of this secondary injury cascade may be highly variable among subtypes of TBI and among TBI patients, the concept of secondary brain injury is central to modern TBI management and the goal to improve outcome of TBI patients using novel pharmacological treatment options. A detailed overview of all aspects of the secondary injury cascade is beyond the scope of this review but key factors are highlighted below and in Figure 1.
Figure 1.
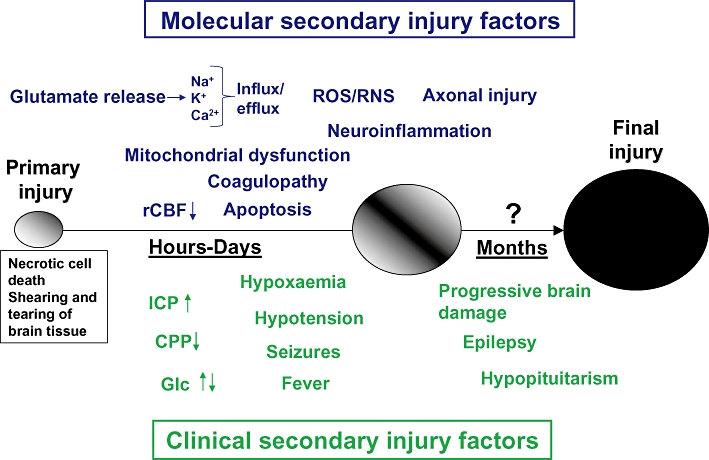
Basic concept of primary and secondary injury in traumatic brain injury. Simplistic illustration of major preclinical ‘molecular’ secondary injury factors as well as clinical secondary injury factors (named ‘avoidable factors’ in the neurocritical care setting). Early post-injury, glutamate release and ionic disturbances (Na+, Ca2+ and K+) cause an energy metabolic disturbance complicated by an early decrease in cerebral blood flow. At this time, mitochondrial disturbance is marked and a large increase in reactive oxygen/nitrogen species (ROS/RNS) is observed. Hyper- or hypocoagulation may also be present either causing microthrombosis or increased haemorrhages respectively. Neuroinflammation and axonal injury is emerging in the immediate post-injury phase. Clinically, an increased ICP and/or decreased CPP must urgently be treated and both too low and too high blood glucose levels corrected. It is also crucial that hypoxaemia and hypotension, seizures and fever is detected and treated. Chronically, marked hormone disturbance may be observed. These factors have been shown to contribute to the progression of the primary injury (indicated by the enlarging circles) and may be suitable targets for pharmacological intervention to reduce the extent of final injury. Ca2+, calcium ions; CPP, cerebral perfusion pressure; GLc, Glucose; ICP, intracranial pressure; K+, potassium; Na+, sodium; rCBF, regional cerebral blood flow.
In the immediate period following the primary injury, there is a massive disturbance of the cellular ion homeostasis initiated by excessive release of the excitatory amino acid neurotransmitters glutamate and aspartate with the subsequent activation of glutamate receptors, a process named excitotoxicity. The release of glutamate results in cellular influx of Na+ and Ca2+ and efflux of K+ (Faden et al., 1989; Katayama et al., 1990; Nilsson et al., 1990; 1993). The influx of calcium ions is regarded to be a key event early post-TBI leading to mitochondrial damage, an increase in free radical production, changes in gene expression and activation of calcium-dependent proteases including caspases, calpains and phospholipases resulting in extensive cytoskeletal damage (Marklund et al., 2001; Singleton et al., 2001; Israelsson et al., 2008). The marked mitochondrial perturbation post-TBI (Verweij et al., 2000; Lifshitz et al., 2004) leads to uncoupling of mitochondrial ATP synthesis at the time of increased energy demand due to activation of energy-consuming ion transport systems and cell repair enzymes. Conversely, an increase in glucose utilization or hyperglycolysis, has been observed across animal models and in humans early following TBI (Bergsneider et al., 1997; Giri et al., 2000). Unfortunately, the high demand for glucose and increase in local cerebral metabolic rate of glucose occurs at a time of reduced regional cerebral blood flow (rCBF) (Nilsson et al., 1996; Ginsberg et al., 1997; Marklund et al., 2002) and this uncoupling of blood flow-cerebral metabolism may be deleterious to the injured brain post-TBI (Chen et al., 2004). Increased oxidative stress is another key feature of the post-injury process because the brain is highly sensitive to free radicals or reactive oxygen/nitrogen species (ROS/RNS). Following TBI, there are several potential sources for overproduction of ROS/RNS, including the mitochondrial respiratory chain, increased free iron resulting from breakdown of extravasated haemoglobin, oxidation of catecholamines, breakdown of membrane phospholipids, NADPH (the reduced form of nicotinamide adenine dinucleotide phosphate) oxidase activation, infiltrating neutrophils and activation of nitric oxide synthetase occurring at a time when the intra- and extracellular antioxidant defence systems are challenged and may become exhausted (Shohami et al., 1997; Hall et al., 2010). Oxidative stress is currently thought to be a major contributor to the secondary injury cascade following TBI by ROS/RNS-induced damage to cellular membranes and organelles by lipid peroxidation, protein oxidation and nucleotide breakdown (Lewen et al., 2000).
The TBI induces a robust immune activation including an acute inflammatory response with breakdown of the blood–brain barrier (BBB), oedema formation, infiltration of peripheral immune cells, activation of resident microglia and astrocytes and intrathecal release of cytokines (Schmidt et al., 2005; Clausen et al., 2009; Ziebell and Morganti-Kossmann, 2010). A prominent up-regulation of several chemokine-related gene transcripts was recently observed following focal TBI in mice (Israelsson et al., 2008) and activated glial cells and leukocytes secrete a variety of neurotoxic molecules including tumour necrosis factor and the interleukin family of peptides (Adamchik et al., 2000; Luheshi et al., 2009; Lu et al., 2009a). Importantly, infiltrating leukocytes secreting myeloperoxidase may be an important source for ROS by producing hypochlorous acid (see Lewen et al., 2000). Conversely, numerous anti-inflammatory compounds have been evaluated in the experimental TBI setting and repeatedly shown to attenuate the behavioural and histological deficits post-TBI (Clausen et al., 2009; Ziebell and Morganti-Kossmann, 2010). However, some inflammatory pathways may be important for regenerative responses and repair suggesting that inflammation may be a double-edged sword following TBI (Lenzlinger et al., 2001; Morganti-Kossmann et al., 2002; Whitney et al., 2009).
Neural injury has been well documented in the first few hours after human TBI in important brain regions such as the cerebral cortex, hippocampus and thalamus and parts of the brain stem (Kotapka et al., 1994). The TBI-induced mitochondrial damage causes initiation of apoptotic cell death via an opening of the mitochondrial permeability transition pore, the release and activation of pro-apoptotic factors including soluble cytochrome c, apoptosis inducing factor and caspases (Lifshitz et al., 2004; Mazzeo et al., 2009a). It should be emphasized that TBI causes not only apoptotic but also necrotic neuronal cell death (Raghupathi, 2004). Regardless of the mechanisms for cell death, widespread neuronal damage occurs and may be observed even remote from the site of impact, where the hippocampal region may be particularly sensitive (Saatman et al., 2006).
Although neuronal cell death has received the predominating attention in the TBI field it is obvious that traumatic axonal injury, often referred to as diffuse axonal injury (DAI), is common following TBI. Importantly, DAI is a dominant contributor to the functional deficits observed in TBI patients and is observed with high frequency in those unfortunate patients remaining in a persistent vegetative state (Graham et al., 2005). Axonal injury is increasingly observed across injury severities and TBI subtypes due to the refinement of, for example, the magnetic resonance imaging technique (Inglese et al., 2005). Acute axonal disconnection at the time of impact is rarely observed and only in patients with very severe TBI dying at the scene of the accident. Instead, stretching and shearing of axons caused by the impact may occur in areas remote to the impact and has been linked to the intra-axonal cytoskeleton damage that ultimately leads to axonal failure and disconnection (Ferrand-Drake et al., 2003; Buki and Povlishock, 2006). Axonal swellings and axonal bulbs are the histological hallmarks of traumatic axonal injury, implying that axonal injury is an ongoing process that evolves over hours to days (Buki and Povlishock, 2006). When axons are disconnected, CNS axons do not spontaneously regrow following injury in contrast to axons in the peripheral nervous system. The reasons for the inability of CNS axons to regenerate are likely multifactorial although the most important factor may be the myelin-associated inhibitors of axonal growth including myelin-associated glycoprotein, oligodendrocyte-myelin glycoprotein and Nogo-A all binding to the neuronal Nogo-66 receptor (Sandvig et al., 2004; Walmsley and Mir, 2007; Gonzenbach and Schwab, 2008). It should be remembered that unmyelinated axons comprise a numerical majority of fibres in subcortical white matter and these fibres may be particularly vulnerable to TBI (Reeves et al., 2005).
Although damaged axons fail to regenerate, damaged neurons are not replaced and there is a marked glial scar counteracting any attempts for recovery, animals and patients often show a surprisingly high degree of spontaneous recovery over the initial months post-TBI. This spontaneous behavioural improvement is thought to partly reflect a remaining ability for neurophysiological and neuroanatomical changes in regions remote from a focal brain injury. Neuroanatomical plasticity, or the restructuring of neural connections in response to lesions of the CNS, is a well-documented phenomenon in the neonatal age group but highly restricted in adults. Nevertheless, an increased sprouting of uninjured corticospinal tract fibres into the injured tracts was observed 6 weeks following TBI in the rat (Lenzlinger et al., 2005; Marklund et al., 2007), suggesting that TBI may elicit yet unidentified mechanisms causing spontaneous sprouting that may be linked to the observed recovery post-injury. Additionally, increased expression of trophic factors as a part the inflammatory response, increased synapse formation and sprouting of hippocampal mossy fibres have all been observed following TBI although their role in the recovery process of TBI remains to be established (Scheff et al., 2005; Hanell et al., 2010). It should be emphasized that yet other factors, including disturbances in the neurotrophin, coagulation, endocrinological and neurotransmitter systems, may contribute to the pathology of TBI and be manipulated pharmacologically (Marklund et al., 2006). In general terms, current pharmacological approaches may be roughly categorized into neuroprotection, inflammatory modulation and enhancement of regeneration.
Clinical TBI
Classification of human TBI is traditionally based on the symptoms and level of consciousness present on admission to hospital, generally using the Glasgow Coma Scale (GCS) and patients with severe TBI, that is, unconscious patients with a GCS score of ≤8, are frequently selected for severe TBI in clinical trials. Although the basic pathophysiology mentioned in the previous section may be common to most TBI patients, we strongly emphasize that TBI is not one disease (Figure 2). Instead, patients with similar clinical signs, symptoms and level of consciousness may have markedly different radiological appearance (including skull fractures, contusions, lacerations, axonal injury, BBB disruption, neurovascular injuries and haematoma with epidural, subdural, subarachnoid, intra-ventricular and/or intracerebral location; exemplified in Figure 2). Currently, acute treatment options for clinical TBI comprise optimal prehospital management and emergency room stabilization, surgery for space-occupying mass lesions, measurement and treatment of increased intracranial pressure (ICP) and the detection and treatment of secondary injury factors, for example, fever, seizures, hypoxia, hypotension (Figure 1) in a NCC setting (Elf et al., 2002; Meyer et al., 2010). The evidence-based guidelines for the treatment of TBI patients were recently published in revised form by the Brain Trauma Foundation et al. (2007). Pharmacological options in NCC include mannitol for reduction of emergent intracranial hypertension, sodium pentobarbital or other sedative drugs for reduction of brain metabolism and lowering of ICP in selected patients and antiepileptic drugs. However, none of these available compounds can be considered to have Class I evidence in the treatment of TBI. Due to the long-term and frequently life-long disabilities experienced by survivors of TBI (Masel and DeWitt, 2010), the development of novel pharmacological treatment options is of highest priority. Animal models of TBI are crucial in the preclinical drug development phase, reviewed in the next section.
Figure 2.
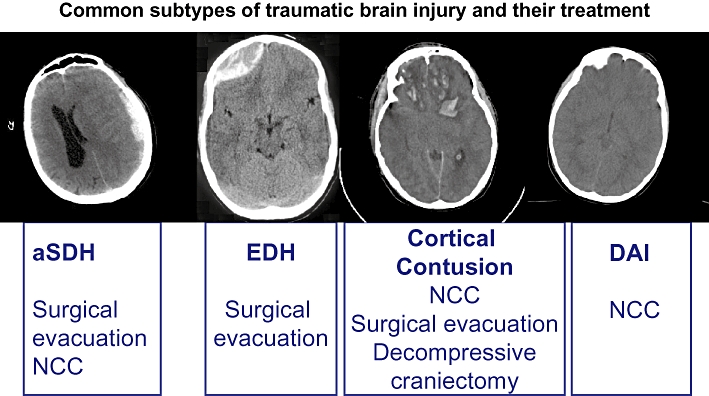
Key issue in clinical traumatic brain injury (TBI) research. TBI is not one disease as exemplified with initial computerized tomography scans of patients with severe TBI treated in our unit. These patients all had a decreased level of consciousness upon arrival in our unit. Typical primary treatment options for the individual TBI subtype are shown. aSDH, acute subdural haematoma; DAI, diffuse axonal injury; EDH, epidural haematoma; NCC, neurocritical care.
Animal models of TBI
In view of the heterogeneous clinical situation, numerous TBI models have been developed. Mimicking all aspects of TBI in a single animal model is impossible and for that reason, a variety of TBI models are being used in animals of various ages and injury severity levels. Rodent models are the most common in TBI research due to their low cost and small size (Finnie and Blumbergs, 2002). In addition to the heterogeneity of TBI, the difficulty in evaluating subtle cognitive and psychiatric impairments in small animal species is a major challenge in the preclinical evaluation of neuroprotective drug candidates. Ideally, for an animal model to be useful in preclinical development of pharmacological compounds it needs to mimic the injury characteristics and severity observed in the clinical setting. Additional features of a useful preclinical TBI model are reproducibility, low costs, applicability to both rats and mice, technically easy to perform and, perhaps most important, production of long-lasting behavioural deficits (Morales et al., 2005). Although even predominantly focal TBI shows a substantial degree of diffuse injury, we categorize the animal models in the next section into ‘focal’, ‘mixed’, ‘diffuse’, ‘complex’ and ‘other’ models of TBI for the sake of simplicity.
Focal TBI models
Controlled cortical impact
The most common brain lesion following human TBI is cortical contusion, defined as a focal destruction of brain tissue with micro haemorrhages, either with intact pia mater (contusion) or a torn pia mater (laceration). Importantly, contusions frequently enlarge markedly during the initial days post-TBI (Stein et al., 1993). Cortical contusions predominantly occur in the frontal and temporal regions and are often observed also at brain regions opposite to the initial impact (contre-coup contusion). A common TBI model used to mimic this feature of principally focal human TBI is the controlled cortical impact (CCI; Figure 3) model, producing extensive cortical tissue loss, hippocampal and thalamic damage (Saatman et al., 2006), cortical spreading depression (von Baumgarten et al., 2008) and signs of post-traumatic epilepsy (Hunt et al., 2009; Yang et al., 2010). Using silver staining methodology, widespread ipsilateral but also contralateral axonal damage in the cortex, hippocampus and thalamus was also observed (Hall et al., 2008). To induce CCI, a craniectomy over one hemisphere is performed to avoid skull fractures and brain injury is produced by a pneumatically driven rigid impactor striking the intact dura mater where the deformation of brain tissue including the time of compression, impactor velocity and depth of impact can be easily controlled using a computer-based monitoring device. Different designs of the impactor tip can also be used to vary the mechanical impact features. The CCI has been used in the ferret, sheep and swine, but rat (Dixon et al., 1991) and mouse (Smith et al., 1995) are currently the predominant species investigated with this model. Although CCI is rather fast and simple to use, reproducible and the injury severity can easily be adjusted, this TBI model also has some disadvantages. First, the contusion injury used in many reports is huge, frequently destroying the majority of the ipsilateral cortex and clearly not comparable with the extent of brain injury observed in survivors of human TBI. Second, a large craniectomy is often produced and if the bone flap is not replaced after injury, the effect of secondary brain swelling may be attenuated thus mimicking a decompressive craniectomy used for alleviation of raised ICP in humans (Zweckberger et al., 2006; De Bonis et al., 2010). Finally, the lack of brain stem damage in CCI will not produce long-lasting unconsciousness and carries a very low mortality, limiting the clinical relevance of this model. However, the rather low variability of the model, the reproducibility among centres and the long-lasting behavioural impairments still make the CCI model suitable for evaluation of pharmacological interventions.
Figure 3.
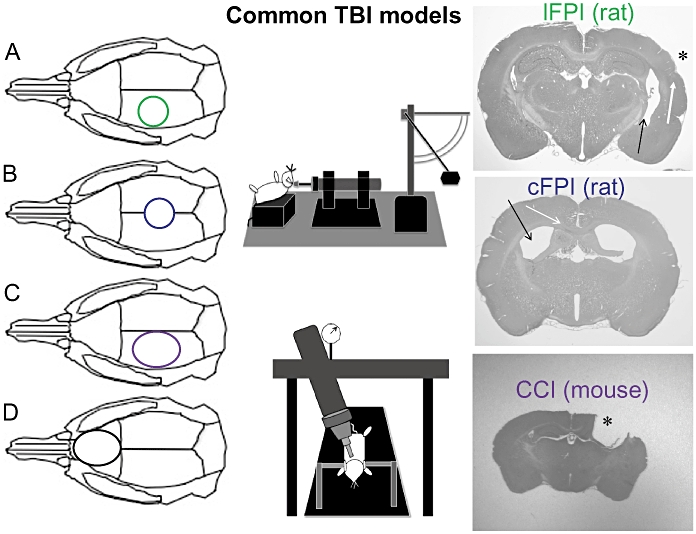
Examples of common preclinical traumatic brain injury (TBI) models mentioned in this review. Left panel: A = craniotomy position for lateral fluid percussion (lFPI) injury and B = craniotomy for the central/midline fluid percussion injury (cFPI). The fluid percussion device (top, middle panel) is used to produce the injury in both the lFPI and cFPI models (see text for details). In C and D, the controlled cortical impact (CCI) device is used (bottom, middle panel). In C, the craniotomy is placed over the parietal cortex and gives a chiefly focal contusion injury. In D, the craniotomy position is placed anterior to the bregma to produce a bifrontal contusion injury. Right panel; examples of the histological appearance from the models (A–C) from our institution. Upper right, examples from a mild lFPI injury in a rat (contusion site marked with * and the rather small cortical contusion with a white arrow). Note the ipsilateral ventricular dilatation (dark arrow). Right panel, middle image; example of a cFPI injury (rat). Note the bilateral ventricular dilatation (black arrow) and haemorrhages in the corpus callosum (white arrow). Right panel, lower image; example from a CCI brain injury in the mouse (impact site marked with *). Note the extensive cortical and hippocampal damage ipsilateral to the injury.
Weight drop injury
In the weight drop injury (WDI) model, an anaesthetized animal is subjected to TBI by a free-falling weight dropped from a predetermined height onto the exposed dura mater (open skull WDI) or the exposed skull (closed skull WDI). In the open skull WDI model the weight is dropped onto a piston resting on the exposed dura. Injury severity can be graded by varying the mass of the weight, the height or, in the open skull WDI model, by varying the depth of compression by different designs of the piston. To minimize variability the head is fixed in a restraint to prevent movement. Frequently, the device has the measures to prevent bouncing of the weight which may be difficult to control. The WDI model is used in the rat using an open skull technique and in both the mouse and rat using a closed skull technique (Chen et al., 1996; Henninger et al., 2005; Morales et al., 2005; Flierl et al., 2009) where a skull fracture may be needed to produce a cortical contusion (Flierl et al., 2009).
The open skull WDI model shows several similarities to the CCI model, producing a chiefly focal brain injury with a cortical contusion and ipsilateral hippocampal and thalamic damage, although some bilateral components to this injury exist (Lewen et al., 1996; Marklund et al., 2001; Clausen et al., 2005). Important information on the neurochemistry of TBI and the comparison between clinical and rodent TBI has been achieved using cerebral microdialysis in this model (Katayama et al., 1990; Nilsson et al., 1990), including post-traumatic seizure activity (Nilsson et al., 1994). Because the vast majority of clinical TBI is CHI, concerns were raised to the clinical validity of the open WDI (Flierl et al., 2009). On the other hand, when using the closed WDI model the incidence of skull fractures increases with rising injury severity and may also cause a substantial degree of diffuse injury (vide infra). As an example, a cerebral concussion, clearly a diffuse type of TBI was simulated in a rat closed WDI model using head fixation (Henninger et al., 2005).
In all, the WDI models are fast and reliable and produce a significant degree of brain damage, neuroinflammation and behavioural deficits including cognitive impairment and provide important information on focal TBI in both rats and mice.
Bifrontal contusion
Contusions commonly occur in the frontal region and are an important contributor to the observed personality changes following human TBI (Fork et al., 2005). Due to the clinical importance of this injury type, a bifrontal contusion rat model was developed by Dr Stein's group that showed histological damage in the medial prefrontal cortex and impairment in cognitive and motor function (Hoffman et al., 1994). This model may be considered a modification of the CCI model and following the central craniectomy the impactor is placed over the midline centred anterior to bregma. This model represent a preclinical approach to an important clinical problem and has been repeatedly used in numerous pharmacological studies (Goss et al., 2003; Djebaili et al., 2005), particularly on endocrinological treatment options. The ongoing progesterone clinical trial (vide infra) is to a large extent based on preclinical data obtained in this model.
Acute subdural haematoma
Acute subdural haematoma (aSDH) is a major cause for the acute mortality observed following clinical TBI. Improved outcome was clearly demonstrated if surgery was initiated within 4 h after the injury suggesting that time is crucial in the management of aSDH (Seelig et al., 1981). Following surgery, many patients still need NCC owing to persisting depression of the level of consciousness, focal deficits and cerebral swelling. There are rodent models of aSDH developed for both the rat and mouse in which autologous blood is injected into the subdural space. Common findings include uncoupling of metabolism/blood flow, development of ischaemia and, for example, dextromethorphan, sodium channel antagonists and glutamate receptor antagonists have been evaluated using these models (Miller et al., 1990; Tsuchida and Bullock, 1995; Tsuchida et al., 1996; Wang et al., 2010). A model of aSDH in the mouse has also been developed (Sasaki and Dunn, 2001). To date, only a minority of TBI studies in general and pharmacological treatments in particular are conducted in these models despite the huge clinical importance of aSDH.
Epidural haematoma
Epidural haematoma (EDH) is commonly associated with skull fractures and is caused by either arterial or venous haemorrhage or a combination of both. In the clinical setting, by far the most important treatment is rapid surgery which is indicated for the evacuation of space-occupying haematomas. The brain compression caused by the EDH may cause secondary oedema that may require NCC and ICP monitoring. To date, the mortality of EDH is still high. Only rarely used animal models of EDH employed in rats, dogs and pigs exist where EDH is simulated by either an inflated balloon or injection of autologous blood into the epidural space (Ganz et al., 1990; Ebmeyer et al., 1998; Balikci et al., 2008). Pharmacological treatment has currently no role in the management of EDH in the clinical setting although vitamin E-supplemented rats showed increased blood flow in the compressed cortex following EDH evacuation (Busto et al., 1984).
Mixed TBI models
The lateral fluid percussion injury (lFPI) model is an important TBI model used worldwide following its introduction by McIntosh et al. (1989), reviewed by Thompson et al. (2005; see Figure 3). In this model, the skull is exposed and a craniotomy is performed, typically over the parietal cortex, and a plastic cap is secured over the craniotomy using dental cement. Injury is produced by the release of a pendulum, hitting the end of a saline-filled reservoir producing a pressure wave creating a fluid bolus which strikes the intact dural surface to extend into the epidural space causing brain deformation (Sullivan et al., 1976). In humans, the duration of the force creating brain dislocation after falls is estimated to be approximately 25 ms (Lindgren, 1966) which is similar to the pressure wave produced by lFPI. Although mostly used in rats, the lFPI model has also been adapted to the mouse (Carbonell et al., 1998). Importantly, the severity of the injury produced by the lFPI model may easily be varied to evaluate also aspects of mild TBI in both mice (Spain et al., 2010) and rats (Li et al., 2006). At the time of injury, a short-lasting apnoea and concomitant seizures are frequently observed and seizures are common at long-term post-injury (D'Ambrosio et al., 2004; Kharatishvili et al., 2006; Kharatishvili and Pitkanen, 2010). The lFPI model produces consistent behavioural (cognitive, motor, complex) deficits and necrotic and apoptotic cell death in the cortex, hippocampus and thalamus (Thompson et al., 2005). In addition to a focal cortical contusion ipsilateral to the impact, there is also a significant degree of axonal injury in the capsula interna and externa and the corpus callosum making this injury model clinically relevant (Graham et al., 2000). Limitations of the lFPI model include some variability and it may be, at least initially, technically challenging with marked differences in outcome among technicians and centres at similar injury severity levels. Even minor changes in craniotomy position may result in large differences in histological and behavioural outcome (Floyd et al., 2002). One important shortcoming of this model, similar to the focal TBI models, is the lack of long-lasting coma and brain stem damage. However, the lFPI model remains very important in the preclinical evaluation of drug candidates.
Diffuse TBI models
A large proportion of patients suffer from diffuse TBI, evidenced by traumatic subarachnoid haemorrhage, diffuse oedema, small intraparenchymal haemorrhages and DAI (Adams, 1982). In fact, the majority of TBI patients have at least a degree of diffuse injury (Maas et al., 2007). Predominant locations for DAI are the parasagittal white matter, corpus callosum, internal capsule, the thalami, and parts of the brainstem and ventricular dilatation is commonly observed (Ai et al., 2007). Mechanistically, angular acceleration/rotation and, to an uncertain extent, linear acceleration is the primary cause of axonal injury (Smith et al., 2003; Fijalkowski et al., 2009). Some diffuse TBI models produce a degree of angular acceleration without an associated impact and inertial acceleration injury models have been developed using the non-human primate, pig, sheep, cats and rabbits (see Morales et al., 2005). These models produce clinically relevant damage to deep white matter tracts and grey-white matter junction sliding contusions but their use in pharmacological studies remain to be established. Because rotational acceleration forces are required to induce axonal damage, rotational models have also been developed for the rat (Xiao-Sheng et al., 2000; Ellingson et al., 2005; Fijalkowski et al., 2007; Kilbourne et al., 2009; Li et al., 2010). Although these modifications of diffuse TBI models have many theoretical advantages, they still need to be defined in terms of long-term behavioural deficits and more extensive pathophysiological evaluation in order to be used as tools for pharmacological evaluation. To date, the impact/acceleration (I/A), midline (central) fluid percussion injury (cFPI) and what we refer to as the ‘CCI-based’ models are the most commonly used diffuse TBI models as discussed in the following section.
The I/A (‘Marmarou’) model
Since its introduction, the I/A weight drop model (Foda and Marmarou, 1994; Marmarou et al., 1994), commonly called the ‘Marmarou model’, has become a widely used diffuse head injury model in rat. The trauma device used in this model consists of a weight falling from a designated height through a tube onto a stainless steel disc glued to the skull of the rat. The head of the anaesthetized rat rests on a platform covered by a foam with a carefully defined ‘spring constant’ to allow a controlled movement of the head after impact. The model may be considered a high-level weight drop model where the use of the steel disc distributes the energy diffusely over the brain, and the foam enables dorsal/ventral acceleration of the unrestrained head. With this relatively easy to perform model, widespread axonal injury is observed and, importantly, this model is one of few TBI models that can result in prolonged unconsciousness. Behavioural deficits have been reported after I/A TBI in the rat up to 1 month post-injury (Beaumont et al., 1999; Cernak et al., 2001; Rancan et al., 2001; O'Connor et al., 2003). One limitation of the Marmarou model is that it is unavailable for mice, although a similar technique of a weight dropped on a disc was used in mice (Kupina et al., 2001). The Marmarou model has provided much useful information on the pathobiology of DAI and results in an injury similar to what is observed in humans. Although pharmacological studies have been conducted in this model, mainly from the lab of Drs Povlishock and Vink (see Table 2; Buki et al., 2003; Thornton et al., 2006; O'Connor et al., 2007; Harford-Wright et al., 2010), its use in such experiments is still rather scarce particularly in terms of behavioural outcome. Thus, this model could and should be used more frequently for long-term pharmacological evaluation.
Table 2.
Magnesium and cyclosporin A evaluation in rodent TBI models
| Drug | TBI model | Route of administration | Time window | Repeated administration | Outcome measure | Time for outcome |
|---|---|---|---|---|---|---|
| Magnesium | lFPI1–6,13–14,16,22 | i.v.1–7,9–11,13–16,18,19,22 | Pre-injury3,22 | Yes8,18 | Cognition1,2,11,14,16,18,19,22 | ≤7 days1,6,8–10,12,13,17,18,20 |
| I/A7–11,15,17,19–20 | i.p.17,20,21 | ≤1 h1,2,4–7,9–21 | No1–7,9–17,19–22 | Motor/ | 7–28 days2–5,11,14,19,21,22 | |
| CHI12 | s.c.12 | 1–6 h8 | Sensorymotor2–4,6–12,14,18,19,21 | >28 days15,16 | ||
| Bifrontal contusion18 | i.m.8,10 | >6 h8 | Oedema2,17,20 | |||
| CCI23,24,27,28 | Lesion vol/cell death5,6,13,16,19,22 | |||||
| Rat1–22 | Complex11,16 | |||||
| Mouse | ||||||
| Cyclosporin | lFPI25,26,28 | i.v.25,27 | Pre-injury23,25 | Yes23–26,28* | Cognition25,26 | ≤7 days23,24,27,28 |
| CCI23,24,27,28 | i.p.23,26,28 | ≤1 h23,24,26,27 | No27 | Motor/Sensorymotor25,26 | 7–28 days25,26 | |
| Rat25–28 | s.c.24 | 1–6 h28 | >28 days | |||
| Mouse23,24 | i.m. | >6 h28 | Oedema27 | |||
| Lesion vol/cell death23,24,28 | ||||||
| Complex |
References to Table 2:
Overview of preclinical studies evaluating Mg2+ or cyclosporin A in rodent models of TBI.
Prolonged, >1 h infusion, included in the Repeated administration column.
CCI, controlled cortical contusion injury; CHI, closed head injury; i.m., intramuscular; i.p., intraperitoneal; i.v., intravenous; I/A, impact-acceleration; lFPI, lateral fluid percussion injury; s.c., subcutaneous; TBI, traumatic brain injury.
Midline (central) fluid percussion injury
Originally used in the cat and rabbit and later adapted to pigs, dogs and sheep, the popularity of the cFPI model increased when it was modified for use in the rat (Dixon et al., 1987; McIntosh et al., 1987). The model uses a similar set-up as the lateral fluid percussion (vide supra) with the exception that the craniotomy and cap placement is centred at the midline between the bregma and lambda sutures and over the sagittal sinus (Figure 3). In addition to the production of widespread axonal injury and hippocampal damage with accompanying brain stem damage, persistent motor and cognitive impairment has been demonstrated (Morales et al., 2005). The cFPI model produces a clinically relevant diffuse brain injury with TBI-induced hypertension, increased ICP and a reduced cerebral blood flow (Kabadi et al., 2010). An additional strength of this model is the recent murine application (the lab of Dr J. Povlishock at the Medical College of Virginia, pers. comm.; as well as our own lab, N. Marklund and L. Hillered, unpubl. results). Shortcomings of the cFPI model include a rather challenging and time consuming surgical preparation and that only a limited injury severity can be achieved because of acute mortality owing to brain stem damage. Although there are numerous pharmacological studies using the cFPI model, particularly on cognitive function, more long-term evaluation of the behavioural consequences and treatment efficacy is warranted.
Diffuse TBI models using a pneumatically driven device, ‘CCI-based’ models
The CCI device used to produce a focal TBI and described in a previous section of this review, may also be modified to create a diffuse injury by placing the impact over the midline (Lighthall, 1988) or using bilateral craniotomies (Meaney et al., 1994). However, other similar models have been introduced and achieved a higher popularity. Common for these models is that the pneumatically driven impactor strikes a steel disc fixed on the skull of the animal that rests on a gel-filled base allowing for some movement of the head at impact. These models have been used in both rats and mice (Cernak et al., 2004; Laskowitz et al., 2007; Maruichi et al., 2009). Advantages of this setup include the ease and speed of the surgical preparation and the addition of the steel disc enables the impact energy to be distributed diffusely over the brain tissue resulting in hippocampal, brain stem and axonal damage with motor and cognitive deficits. These models have some impact variability and the relevance to the human situation needs to be better defined. However, these models have been successfully used in preclinical pharmacological research and represent an interesting addition to the ‘classical’ TBI models.
In conclusion, diffuse injury models produce a significant degree of axonal injury, of highest importance due to the vast clinical problem of DAI, and should in our opinion be available in most labs evaluating the efficacy of pharmacological compounds for TBI.
Complex TBI models
It has been demonstrated beyond doubt that additional, concomitant systemic injuries markedly exacerbate the outcome following TBI. Hypoxia (PaO2 < 60 mmHg) and, in particular, hypotension (systolic blood pressure <90 mmHg) are the most common and occur in about one third of patients with severe TBI (Chesnut et al., 1993). A common notion has been that animals are far more resistant to secondary insults than humans (Lammie et al., 1999). However, other studies have demonstrated an impaired motor and histological outcome in animals subjected to TBI and hypoxia (Ishige et al., 1987; Clark et al., 1997) and the time period between the injury and the secondary insults may be highly important (Geeraerts et al., 2008). Thus, the CCI and I/A models have been used in combination with hypoxia (Hellewell et al., 2010), hypoxia and hypotension (Robertson et al., 2000; Stiefel et al., 2005; Taya et al., 2010) and the lFPI model with hypoxia (Bramlett et al., 1999; Bauman et al., 2000) and/or hypotension (Matsushita et al., 2001; Aoyama et al., 2008). The secondary insults were shown to exacerbate histological and behavioural outcome in these studies and were also implemented in a model of aSDH (Sawauchi et al., 2004). In all, these models represent an excellent example of a true translational approach where an important clinical problem is addressed in the experimental setting. One elegant additional model is the combination of the lFPI model with a tibial fracture (Maegele et al., 2005; Maegele et al., 2007). The effects of hypothermia have been evaluated in these models (e.g. Robertson et al., 2000; Gao et al., 2010) although pharmacological intervention studies are scarce. We suggest that pharmacological treatment options be evaluated also in the combined TBI models prior to launching full-scale clinical trials enrolling patients with multiple injuries.
Other TBI models
The use of larger animals such as the pig or piglet in TBI research is increasing (Smith et al., 1999; Missios et al., 2009) and examples of such TBI models being used in pharmacological evaluation is emerging (e.g. Zhang et al., 2008; Armstead et al., 2010). These TBI models may gain wider use than those using non-human primates and may hopefully be more commonly employed for pharmacological evaluation in the future. Although outside the primary scope of this review, additional TBI models only rarely evaluated in pharmacological studies are briefly mentioned in the following section.
Repetitive models
Epidemiological data suggest an increased risk for onset of neurodegenerative diseases, such as Alzheimer's disease, in people who has suffered repetitive head injury. In addition, repeated mild TBI commonly observed in athletes (soccer, boxing, ice hockey, etc.) may have cumulative adverse effects on cognitive function. Several models aim at reproducing the clinical consequences of repetitive mild TBI (see Morales et al., 2005). Repetitive concussive brain injury models have also been developed for use in the mouse using the weight drop model (DeFord et al., 2002; Creeley et al., 2004) and in the rat using repeated, up to four, mild concussive insults using the lFPI model (DeRoss et al., 2002). For instance, using the CCI device modified using a rounded silicone impactor on the intact skull in the mouse, the animals were subjected to a mild injury and then to a second injury at various intervals post-injury. Results from these studies showed that a second injury impaired motor and histological outcome with increased axonal pathology if the second injury occurs within a 3- to 5-day period of the first (Laurer et al., 2001; Longhi et al., 2005). Models of repetitive TBI have also been used in mice over-expressing human Amyloid precursor protein (Tg2576 mice), in which vitamin E treatment resulted in less amyloidosis and improved cognitive function (Conte et al., 2004). Although these models provide insight into the field of repeated head injury of value for, for example, athletes at risk for repeated concussions, pharmacological evaluation has only rarely been performed.
Blast injury models
Rapidly growing field of interest is based on the recent experience from combat field activities in Iraq and Afghanistan where an increasing number of soldiers exposed to blast waves from detonations suffer functional sequelae requiring long periods of treatment. Rodent models of blast TBI have recently been established (Cheng et al., 2010; Risling et al., 2011; Saljo et al., 2010; Svetlov et al., 2010). However, to our knowledge, no post-injury pharmacological intervention studies have been conducted to date and long-term behavioural and morphological consequences need to be established. These models of blast injury are still at their infancy although future studies will likely provide novel information on the mechanisms of, for example, axonal injury and its treatment.
Penetrating injury models
A model of penetrating brain injury has been characterized in the rat (Williams et al., 2005; Williams et al., 2006a,b) and shown to produce cognitive impairment (Davis et al., 2010). In this model, a metal rod covered with an elastic tube that rapidly (10 ms) inflates and deflates is used to produce a shock wave along the injury tract to mimic a penetrating ballistic injury. Marked grey and white tissue damage, brain swelling, seizures, cortical spreading depression and neuroinflammation with a resulting neurological impairment was demonstrated. This model has been recently used in pharmacological evaluation (Shear et al., 2009; Lu et al., 2009b) and future studies are needed to define the role of this model in the development of neuroprotective compounds.
Although these models have not, or rarely, been used in pharmacological studies, it is important to emphasize that treatment efficacy of a compound in one TBI model does not in any way guarantee efficacy in other TBI models. Thus, more studies of efficacy of treatment compounds across a range of TBI models are needed.
Outcome measures
Following human TBI, long-term changes in personality, cognitive performance and motor function is common and leads to a marked reduction in the quality of life. Spontaneous recovery is most pronounced within the first 6 months after the TBI but may vary with different types of TBI (Consensus conference, 1999). Usually, outcome is assessed in the clinical situation at 3 or, more commonly, 6 months post-injury using increasingly complex batteries of neuropsychological tests (Bagiella et al., 2010). However, the Glasgow Outcome Scale (or its extended version GOSe) remains a basic outcome measure included in the majority of clinical trials (Maas et al., 2008). In the experimental setting, there are numerous behavioural tests in use where the evaluation periods are relatively short and only rarely extending beyond 1 month post-injury (Marklund et al., 2006). To date, cognitive and motor function tests are routinely included in outcome evaluations (Fujimoto et al., 2004). Widely used tests for cognition include the Morris water maze, object recognition test, memory task, freezing response and others. Common motor function tests include the Rotarod, cylinder test, skilled forelimb reach, grip strength tests and staircase tests. In view of the complex personality and psychological disturbances experienced by many TBI patients, more complex behavioural assessment test have also been used in experimental TBI research, albeit less frequently. These tests include the open field, elevated plus maze, spontaneous motor activity, exploratory activity and emotional activity tests. In addition, using lFPI in rats, a pervasive hyperanxious phenotype was observed (Jones et al., 2008). Depression is also a major clinical problem post-TBI that has not been thoroughly studied in animal models although there are reports using the Porsolt test of forced swimming (Tweedie et al., 2007). There is concern that the outcome measures in clinical trials and the behavioural tests used in preclinical research are not well matched (Fujimoto et al., 2004) calling for a continual refinement of experimental outcome methods. As an example, a novel behavioural model for testing numerous spontaneous complex tasks was recently established for TBI in mice (Ekmark-Lewen et al., 2010) using a test situation with a free choice of different optional environmental settings that may be useful for behavioural evaluation in pharmacological research. Although it is obvious that evaluation of rodents cannot directly be compared to that of humans, scientists evaluating pharmacological compounds are encouraged to incorporate more complex behavioural assessment tools in addition to standard motor and cognitive tests into the study protocols.
Neuroprotective pharmacology in TBI – why no success thus far?
As outlined in the short overview in the previous sections, there is a plethora of TBI models and outcome measures used in many institutions worldwide, frequently employed for evaluation of neuropharmacological interventions. In our opinion, the key to successful preclinical development of pharmacological compounds for the future lies not within the development of yet more TBI models, but instead that existing animal models be modified to better mimic the complex clinical situation. It is also evident that features of TBI observed in the clinical setting should be added to existing TBI models to enhance clinical applicability. Although some previous clinical trials in TBI have been conducted based on preclinical efficacy only in stroke models it should be emphasized that TBI and ischemic stroke, although sharing some common mechanisms, are truly two very different diseases of the CNS (Bramlett and Dietrich, 2004). It appears logical that clinical and experimental TBI research work together in a translational fashion in order to evaluate experimental findings clinically and vice versa, and that clinical experience and knowledge be incorporated into preclinical TBI research. In the treatment of TBI there are numerous pharmacological aspects to consider; for example, treatment dose and time window, target brain concentration, route of administration and efficacy in modifying certain aspects of the pathophysiology of TBI. Most drug companies and scientist focus on the development of a single, ‘silver-bullet’ therapy although a more realistic approach may be a combination of compounds using different targets (Margulies and Hicks, 2009; Loane and Faden, 2010). In fact, several compounds such as minocycline, erythropoietin and 2-sulfophenyl-N-tert-butyl nitrone (S-PBN), have been shown to act as multifunctional therapeutics influencing multiple targets in the secondary injury cascade. In the next section, we outline some common shortcomings and problems of existing TBI models and provide suggestions on how to further improve these models.
Time window, route of administration and the blood–brain barrier
In the vast majority of preclinical protocols, the treatment compounds are administered early and, frequently, even prior to the TBI. The administration of a compound early by prehospital care personnel may be problematic because of the difficulty in obtaining informed consent (Menon, 2009). For that reason, the time allowed for drug administration in clinical trials has often been extended way beyond the time window suggested by preclinical trials. In fact, time window issues may be a major cause of the failure of the randomized clinical trials evaluating neuroprotective compounds. More research on extended time windows in the experimental setting is needed. Specifically, the temporal appearance of the secondary injury mechanism at target must be studied in detail. Furthermore, numerous experimental studies have employed oral, intraperitoneal or intracerebroventricular administration routes that may have restricted clinical utility. Additionally, single-dose treatment using high drug doses has commonly been employed experimentally, in contrast to the clinical setting where continuous infusion of a lower treatment dose, in order to avoid adverse side effects, is standard. In the NCC setting, numerous drugs, such as pentobarbital and phenytoin, are used which may cause drug interaction problems and influence clearance and distribution volume of the study drugs (Empey et al., 2006). In addition, altered cerebral metabolism, changed gene expression and protein synthesis may influence the clearance, brain penetration and distribution of a compound (see Boucher and Hanes, 1998; Lo et al., 2001; Kalsotra et al., 2003). Thus, a correct translation of dosage of a compound from the experimental to the clinical setting requires careful pharmacokinetic evaluation in humans.
In terms of pharmacology of TBI, the BBB must be considered. In fact, knowledge about in which patient and at what time the BBB is open may be key factor in future clinical trials (Blyth et al., 2009). The BBB is a physical barrier, composed of capillary endothelial cells connected by tight junctions together with astrocytes, that actively controls the penetration of molecules and microscopic objects from the blood into the brain. Small hydrophobic molecules (e.g. O2, CO2, anaesthetics, barbiturates, ethanol and hormones) and water readily pass through the BBB whereas diffusion of microscopic objects (e.g. bacteria), large molecules (e.g. plasma proteins) or hydrophilic molecules (e.g. electrolyte ions) is restricted. Metabolic products such as glucose and lactate, pyruvate are actively transported across the BBB by specific proteins (Glucose transporter 1 and monocarboxylate transporters respectively). In numerous studies, TBI has been shown to alter the status of the BBB allowing for the passage of substances normally restricted to the blood stream. The duration of this BBB opening in TBI models is variable but typically exists for a minimum of 3–7 days and the BBB disturbance may appear in certain brain regions only and with a different temporal profile for different molecular weight substances (Cortez et al., 1989; Habgood et al., 2007; Kelley et al., 2007). Due to an altered BBB, pharmacological compounds hindered from entering the normal brain may thus reach the injured brain. To take advantage of this situation in the NCC setting novel tools for monitoring BBB function in TBI patients is urgently needed. The possibility that TBI-induced oedema and reduced blood flow may influence drug transport into the injured brain should also be considered. Ongoing research using chemically modified pharmacological compounds (Banks, 2008) with improved brain penetration may be an additional method for the treatment of TBI.
Thus, knowledge of brain penetration and pharmacokinetics is highly relevant although only rarely evaluated in preclinical research. In fact, most neuroprotective trials have been conducted without such preclinical documentation. We would like to encourage scientist to measure the penetration of the study compound into target brain regions. In this context, monitoring of a suitable surrogate end point biomarker by cerebral microdialysis may enable evaluation of the appropriate therapeutic window, dosage and brain penetration of the study compound also in clinical TBI research. Another key issue is to establish whether a drug has the desired effect on a specified mechanism in vivo. Again, such questions may be addressed using microdialysis to monitor a suitable surrogate end point biomarker in TBI patients. In a previous report, the free interstitial concentration of the glutamate release inhibitor Topiramate as well as glutamate was measured and a significant lowering of interstitial glutamate was achieved only after the Topiramate dose was elevated to a dose much higher than anticipated at the planning stage (Alves et al., 2003). These data strongly support the importance of determining adequate target drug concentration and proof-of-principle testing before moving to large scale clinical trials (Alves et al., 2003; Helmy et al., 2007). The proof-of-principle approach also helps to optimize the timing of drug administration in relation to the time course of the injury mechanism at target.
Finally, it should be noted that it may not be mandatory for all drugs to penetrate the BBB to exert a neuroprotective effect. Important information was obtained when the brain penetration of two ROS scavengers, the nitrone spin traps S-PBN and α-Phenyl-N-tert -butyl nitrone (PBN), were compared. Although S-PBN did not reach the injured brain in detectable amounts in contrast to PBN, equal ROS scavenging properties and behavioural outcome improvement were obtained suggesting that brain penetration was not an absolute requirement for efficacy (Marklund et al., 2001). This notion is also supported by the preclinical information obtained using tirilazad and polyethylene glycol-conjugated superoxide dismutase both without significant brain penetration but with neuroprotective properties (see Marklund et al., 2006). These results suggest that the cerebral microvascular endothelium may be an important target for pharmacological intervention following TBI.
Animal models of TBI – are they good enough?
One crucial question is – do current experimental TBI models adequately mimic clinical head injury? Rodents are vastly different from humans in terms of brain size and geometry, white-to-grey matter ratio and they lack the cortical gyri observed in higher species (Gennarelli, 1994). Major clinical problems such as emotional and language difficulties are obviously not possible to mimic using rodent models. There is also a lack of TBI models producing long-lasting periods with a decreased level of consciousness or coma, commonly observed in severely injured TBI patients. Most laboratories also typically employ TBI models that, in reality, represent injuries on the mild-moderate scale, in sharp contrast to the severely brain-injured patients evaluated in clinical trials (see Thompson et al., 2005). Another treatment strategy for severe clinical TBI that is increasingly recognized is decompressive craniotomy (Piek, 2002), which may prove difficult to evaluate in rodent models due to differences in brain conformation and anatomy, degree of cerebral swelling and skull anatomy. In contrast, animal models appear to adequately mimic certain aspects of blood flow-energy metabolic disturbances post-injury. In patients dying from severe TBI, 90% were found to have ischemic lesions upon autopsy (Graham et al., 1978; Kotapka et al., 1994). However, observations in TBI patients in the NCC setting suggest that ischaemia may be a less prominent feature in TBI than previously thought and, instead, other types of energy metabolic perturbations unrelated to hypoxia/ischaemia appear to be common (Hlatky et al., 2004; Vespa et al., 2005; Dusick et al., 2007; Hillered and Enblad, 2008). The reduction of CBF in TBI models is usually modest, not reaching ischemic levels (Muir et al., 1992; Nilsson et al., 1996), in agreement with the clinical observations.
Despite several shortcomings of rodent models, only a minority of all published TBI studies involves higher species and mice and rats will likely continue to be the dominant animal species in the future. In the next section, we discuss some additional factors influencing animal modelling in TBI.
Gender
In practically all cohort studies on TBI patients, a clear majority of TBI victims are males. In the clinical TBI literature, initial reports suggested that women did better following TBI than males although this has been questioned (Groswasser et al., 1998; Farace and Alves, 2000; Bramlett and Dietrich, 2001; Ponsford et al., 2008). Apparently, gender may play an important role because progesterone repeatedly improved outcome in experimental TBI studies and progesterone is currently undergoing clinical trial evaluation. Although most rodent studies have been performed in male rats, female animals are increasingly used to incorporate gender differences into experimental TBI models (Roof and Hall, 2000; Bramlett and Dietrich, 2001). Gender may also influence numerous aspects of the pathophysiology of TBI including the response to enriched environment following TBI in the rat and the degree of neurodegeneration (Kupina et al., 2003; Wagner et al., 2004; 2007; Bonatti et al., 2008). Increased attention to gender aspects in TBI is clearly warranted.
Age
The mortality following TBI in the elderly patients is more than twice that of young patients (Mosenthal et al., 2002) and age per se is one of the most important predictors of outcome after human TBI (Mosenthal et al., 2002; Hukkelhoven et al., 2003). In fact, the incidence of elderly patients with TBI appears to be increasing (Maas et al., 2008). The vast majority of rodent TBI reports have been performed in adolescent or young adult animals with a rather narrow age range despite the fact that the past decade has seen a 21% increase in individuals over the age of 65 (Adekoya et al., 2002). In the experimental setting, aged rats are more impaired in motor and cognitive performance compared to younger animals following lFPI and bifrontal contusions (Hamm et al., 1992; Hoane et al., 2004). Age-dependent injury-induced differences in gene expression in the hippocampus may contribute to the increased vulnerability of the aged rat brain to lFPI and mitochondria from aged animals are more perturbed compared with those of young animals (Shimamura et al., 2004; Gilmer et al., 2010). Pharmacological studies on aged animals are rare following TBI (Cutler et al., 2007) and age-related preclinical TBI research still remains a severely underexplored area.
Species/strain differences and epigenetic changes
Strain differences in the response to TBI (lFPI) were recently reported between 3-month-old male Sprague–Dawley and Fisher rats, the latter showing a higher ICP, more seizure activity, longer ictal durations and more pronounced motor deficits, but surprisingly a better cognitive performance in a Morris Water Maze task (Reid et al., 2010). Another study showed differences in cerebral immune cell infiltration and extent of brain damage following open skull WDI in two rat strains (Bellander et al., 2010) and Sprague–Dawley rats had a more rapid behavioural recovery compared to Long–Evans rats following lFPI (Tan et al., 2009; Reid et al., 2010). Importantly, in a study on cyclosporine treatment following CCI, two different mice strains were evaluated for efficacy (Scheff and Sullivan, 1999). Accumulating evidence also suggest that various genetic factors are markedly important contributors to the pathophysiology and outcome of TBI patients (see Dardiotis et al., 2010).
Epigenetic changes induced by TBI are also increasingly recognized (Gao et al., 2006; Dash et al., 2009) as a potentially important part of the secondary brain injury process. Thus, both epigenetic studies and the influence of strain on the outcome following experimental (and clinical) TBI is of very high interest in the TBI field and should be considered from a pharmacological view.
Neurocritical care and secondary insults
During the initial NCC period following human TBI, numerous secondary insults threaten the injured and vulnerable brain. These secondary injury factors can be divided into systemic (hypoxia, hypotension, anaemia, acid-base, electrolyte or glucose disturbances) or intracranial (raised ICP, seizures, impaired rCBF, hyperthermia) factors. These insults have only rarely been incorporated into existing TBI models. Additionally, good physiological monitoring (pO2, pCO2, pH, blood pressure, blood glucose, etc.) before and after TBI is often not used in the preclinical TBI models. Another commonly used clinical monitoring device is brain tissue oxygen monitoring, that is, Licox®, Paratrend® (Sarrafzadeh et al., 2003) which has not, to date, been evaluated in experimental TBI. In contrast, in vivo cerebral microdialysis is used worldwide in the clinical setting and also in experimental TBI providing a possibility for translational research on, for example, energy metabolic perturbations following TBI (see Hillered et al., 2005). A novel application of microdialysis is sampling of protein biomarkers directly in the injured brain following TBI using high molecular cut-off membrane catheters and is receiving increasing attention in the NCC setting (Hillman et al., 2005; Brody et al., 2008; Helmy et al., 2009; Marklund et al., 2009; Dahlin et al., 2010). Biomarker sampling directly in the injured brain and more closely to the pathoneurochemical process may improve the spatial and temporal resolution of the biomarker signals compared to traditional sampling in ventricular CSF or blood (Hillered et al., 2005). Thus, microdialysis biomarker sampling may avoid many problems associated with long-distance transport, dilution and degradation causing a delay and reduction of the biomarker signals in CSF and blood. In support of this working hypothesis, our recent data in TBI patients indicate that the levels of the F2-isoprostane 8-iso-PGF2α, a widely used biomarker of oxidative stress, were markedly higher in microdialysate compared with ventricular CSF and blood samples (F. Clausen et al, submitted).
Clinical NCC also uses numerous strategies to lower ICP, including administration of mannitol and hyperventilation, targeting physiological parameters. Sedatives and anaesthetics are commonly used in intensive care management of TBI patients. One elegant study using CCI in the rat showed that ‘sub-optimal’ cerebral perfusion pressure (CPP < 70 mmHg) resulted in an increased lesion volume (Kroppenstedt et al., 1999) showing a good example of how scientists can take clinical observations for testing in the TBI laboratory. In the clinical setting, there is an ongoing search for prognostic and pathophysiological biomarkers (Dash et al., 2010) that also should be evaluated in the experimental setting.
We emphasize that not only preclinical scientists are responsible for translating preclinical knowledge into clinical treatments. In numerous aspects of the management of TBI and, in particular, NCC there are discrepancies regarding, for example, sedation, fluid management and strategies to correct changes in ICP and CPP levels. These differences among neurosurgical and NCC centres indicate a lack of consensus and evidence-based guidelines making the animal modelling even more difficult. Clinicians obviously need to create international platforms working towards a more unified treatment strategy in the overall management of TBI. Encouragingly, international consortia and workshops have been created with the aim of creating, for example, management guidelines and uniform classification for TBI (e.g. Consensus conference, 1999; Saatman et al., 2008; Compagnone et al., 2005; Brain Trauma Foundation et al., 2007; Margulies and Hicks, 2009), efforts that will likely be a key step in the improvement in TBI care worldwide.
Neurorehabilitation
Without doubt, neurorehabilitation is a very important part of the treatment of TBI victims. However, neurorehabilitation varies tremendously among countries and also regionally within many countries. Despite the differences in clinical rehabilitation, efforts for mimicking this aspect of the clinical management have been made. In the laboratory, numerous studies have evaluated enriched environment as a treatment option and consistently shown to enhance functional and histological recovery after both FPI and CCI in the adult rat (Hamm et al., 1996; Passineau et al., 2001; Hoffman et al., 2008; Sozda et al., 2010). Additionally, late effects of enriched environment plus multimodal early onset stimulation after TBI in rats resulted in an improved motor function without reduction of lesion volume (Lippert-Gruner et al., 2007). The efficacy of drugs in TBI animals also subjected to enriched environment (Kline et al., 2007) would be an interesting approach in order to further improve the clinical similarity of preclinical research.
Animal modelling in some previous and ongoing clinical trials
In the past, numerous extensive and expensive clinical trials have evaluated pharmacological compounds aiming to improve the recovery of TBI patients. Invariably, they all failed to demonstrate clinical efficacy. The reasons for these failures are multifactorial and have been addressed in the previous sections of this review (see also Maas et al., 2008). Common shortcomings have included incomplete preclinical evaluation, incorrect timing and dosing of the selected compounds and the inclusion of too mildly or severely injured patients and inclusion of all the various subtypes of TBI. Still, numerous clinical trials in TBI are ongoing, examples being the ProTECT trial (progesterone; Wright et al., 2007), aiming to enrol 1140 patients), the citicoline brain injury treatment trial (Zafonte et al., 2009), aiming to enrol 1292 patients) and erythropoietin Phase II and III trials based on solid preclinical studies (NCT00375869 and NCT00313716; see Nichol et al., 2009). Thus, it is important to learn lessons from past trials and also continuously evaluate trials that are ongoing or being initiated. Here we selected two examples, the magnesium and cyclosporine A trials with the aim of reviewing the published preclinical documentation. The reasons for selecting these two trials are that they both have unusually solid preclinical documentation prior to embarking on the clinical trials and the compounds are both in clinical use for other medical conditions. One of the trials is completed and the other is ongoing.
Magnesium
Magnesium (Mg2+) is the second most abundant cation in the body, involved in more than 300 enzyme reactions (McKee et al., 2005). Following experimental TBI, brain intracellular free Mg2+ concentration significantly declines in number of animal models evaluating mild-severe TBI in addition to focal and diffuse TBI and this decline was shown to correlate with the functional outcome (Heath and Vink, 1999a). In addition, in both humans and animals, TBI is associated with decreased serum Mg2+ levels. Mg2+ was suggested to enhance neuronal survival by, for example, blocking of the NMDA receptor ion channels. In a number of preclinical TBI studies, supplemental Mg2+ treatment consistently improved outcome in several different TBI models using a battery of functional outcome tests (Table 2). Because continuous Mg2+ infusion was safely used for other medical conditions including eclampsia and considered cheap and readily available, these encouraging animal data suggested that Mg2+ should be evaluated in a clinical setting. In a randomized, double-blind placebo-controlled monocentre clinical trial, 499 patients with moderate-severe TBI (GCS score of 3–12) were enrolled within 8 h post-injury (Temkin et al., 2007). Mg2+ was administered by an initial i.v. dose followed by an i.v. infusion for 5 days and similar to the experimental findings, initial serum Mg2+ levels were low in 60% of patients. Due to initially high mortality and low blood pressure at the target serum Mg2+ level of 1.25–2.5 mmol·L−1 (normal levels are 0.75–1.0 mmol·L−1), target serum levels were decreased to 1.0–1.85 mmol·L−1. The included patients had various subtypes of TBI or a combination thereof; including DAI (36% of Mg2+-treated patients), subdural haematoma (56%), EDH (21%), intracerebral haematoma (9%), depressed skull fracture (18%) and cortical contusions (60%) and a few patients had a penetrating TBI. Primary outcome was a composite of mortality, seizures, functional measures and neuropsychological tests. No improvement of Mg2+ treatment was observed, instead a clear negative trend in the outcome of Mg2+-treated patients was observed. Numerous possible explanations for these disappointing and unexpected results were suggested, including a possible interaction with phenytoin and a possible negative effect of prolonged treatment (contrary to the preclinical setting) resulting in excessive NMDA receptor blockade. In our opinion, other possible explanations of the study results included the inclusion of all TBI subtypes, a too wide range of TBI severity and a prolonged time window without prior experimental supportive data. Importantly, rather high serum Mg2+ levels are well tolerated but produce a very modest increase in Mg2+ levels in the CSF (McKee et al., 2005). The CSF Mg2+ was suggested to be increased in TBI per se (Kafadar et al., 2007), suggesting a complex brain pharmacodynamic situation with regard to Mg2+ in humans. These aspects need to be considered in future TBI clinical trials.
Cyclosporin A
Cyclosporin A (CsA), known to inhibit T-cell lymphocytes by binding to cyclophilin A, has long been used in the clinical setting as an immunosuppressant to, for example, inhibit graft rejections following transplantation procedures. The CsA was suggested to influence TBI pathophysiology by binding to calcineurin, a known causative factor in the damage to the axonal cytoskeleton following TBI and positively influenced several aspects of cytoskeletal damage following TBI (Buki et al., 1999; Okonkwo and Povlishock, 1999). The CsA was also suggested to inhibit the opening of the mitochondrial permeability transition pore although this mechanism of action has been questioned (Marmarou and Povlishock, 2006). The role of CsA as a neuroprotectant has been evaluated in several animal models of TBI (summarized in Table 2). The CsA does not reach the brain in high concentrations in non-TBI patients, since it is highly bound in the serum and is a substrate for multidrug resistance efflux pumps, eliminating CsA from the CNS compartment (Cook et al., 2009). In TBI patients, CsA is detectable in the CSF for up to 6 days, suggesting that the increased permeability of the BBB after TBI may result in increased access for CsA to injured brain regions (Hatton et al., 2008). Recently, the safety, tolerability and pharmacokinetics of CsA in TBI patients were evaluated. In 30 patients with severe TBI included within 8 h post-injury, CsA was injected twice daily using escalating doses up to 2.5 mg·kg−1 dose−1 (Empey et al., 2006). Compared with CsA pharmacokinetics in other disease states, a more rapid clearance and a larger distribution volume of CsA was demonstrated. Additionally, no significant differences between the placebo- and CsA-treated patients with regard to immunological parameters such as total lymphocyte count, the incidence of infections and T-cell subtypes were observed (Mazzeo et al., 2006). In an additional study (Mazzeo et al., 2009b), 50 patients with severe TBI received 5 mg·kg−1 infused over 24 h using continuous i.v. infusion initiated within 12 h of the injury. Overall, small and likely not clinically significant differences in renal function and white blood cell counts were observed in CsA-treated patients and the safety profile of CsA was good. However, no improvements in neurological outcome were observed at 3 or 6 months post-injury. Still, brain pharmacokinetics need to be carefully determined and it is yet unclear if all subtypes of TBI will be included in the next trial. A large Phase III efficacy trial of CsA in severe TBI is currently under peer review by the National Institute of Health-National Institute of Neurological Disorders and Stroke Clinical Trials study section, and if approved, will be performed in about 50 centres throughout the USA. The results of this study will be eagerly awaited by the TBI research community.
Conclusions
The lack of drugs with proven clinical efficacy in TBI is a major challenge ahead for the research community and the drug industry. So, where do we go from here? As outlined in this review, a successful translation of basic science knowledge to the clinic requires numerous refinements of the existing preclinical TBI models that may be achieved without extensive efforts or costs. We believe that the current experimental TBI models adequately reflect many aspects of human TBI. However, to more adequately mimic the clinical situation, modification of injury severity, refined functional outcome tests, addition of secondary insults and multimodality monitoring may be needed. In addition, more research into the effect of age, gender and species/strain on the outcome of TBI is warranted. On the clinical side the ongoing international effort to come up with a novel classification system for TBI patients is widely appreciated (see Saatman et al., 2008) and may enable selection of more homogenous patient cohorts in future clinical trials. We also suggest that clinicians work toward an international consensus with a more homogenous treatment strategy for TBI patients in the NCC setting to facilitate multicentre comparisons. In addition, improved CNS drug delivery systems and monitoring of target drug levels and drug effects is warranted. Numerous promising treatment options have emerged in recent years, including neuroprotective, neurorestorative and anti-inflammatory compounds that should be subjected to a rigorous preclinical dose–response analysis of their efficacy on the target mechanism and the ability to reduce post-traumatic neurodegeneration and to improve behavioural and neurological recovery. Based on the complexity of injury mechanisms involved in TBI pharmacological combination treatment strategies may be an important option to consider.
Acknowledgments
The Swedish Research Council, Uppsala University, Uppsala University Hospital, the Selander Foundation and the Åhlén Foundation are acknowledged for financial support, and Anders Hånell and Johanna Hedin for helpful comments and aid with figures.
Glossary
Abbreviations
- aSDH
acute subdural haematoma
- BBB
blood–brain barrier
- CCI
controlled cortical impact
- cFPI
midline (central) fluid percussion injury
- CHI
closed head injury
- CNS
central nervous system
- CPP
cerebral perfusion pressure
- CsA
cyclosporin A
- CSF
cerebrospinal fluid
- EDH
epidural haematoma
- GCS
Glasgow Coma Scale
- I/A
impact-acceleration
- ICP
intracranial pressure
- lFPI
lateral fluid percussion injury
- NCC
neurocritical care
- NMDA
N-methyl-D-aspartic acid
- PBN
α-Phenyl-N-tert -butyl nitrone
- rCBF
regional cerebral blood flow
- S-PBN
2-sulfophenyl-N-tert-butyl nitrone
- TBI
traumatic brain injury
- WDI
weight drop injury
Conflict of interest
None.
References
- Adamchik Y, Frantseva MV, Weisspapir M, Carlen PL, Perez Velazquez JL. Methods to induce primary and secondary traumatic damage in organotypic hippocampal slice cultures. Brain Res Brain Res Protoc. 2000;5:153–158. doi: 10.1016/s1385-299x(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Adams JH. Diffuse axonal injury in non-missile head injury. Injury. 1982;13:444–445. doi: 10.1016/0020-1383(82)90105-x. [DOI] [PubMed] [Google Scholar]
- Adekoya N, Thurman DJ, White DD, Webb KW. Surveillance for traumatic brain injury deaths – United States, 1989-1998. MMWR Surveill Summ. 2002;51:1–14. [PubMed] [Google Scholar]
- Ai J, Liu E, Wang J, Chen Y, Yu J, Baker AJ. Calpain inhibitor MDL-28170 reduces the functional and structural deterioration of corpus callosum following fluid percussion injury. J Neurotrauma. 2007;24:960–978. doi: 10.1089/neu.2006.0224. [DOI] [PubMed] [Google Scholar]
- Alessandri B, Rice AC, Levasseur J, DeFord M, Hamm RJ, Bullock MR. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. doi: 10.1089/08977150260190429. [DOI] [PubMed] [Google Scholar]
- Alves OL, Doyle AJ, Clausen T, Gilman C, Bullock R. Evaluation of topiramate neuroprotective effect in severe TBI using microdialysis. Ann N Y Acad Sci. 2003;993:25–34. doi: 10.1111/j.1749-6632.2003.tb07508.x. discussion 48–53. [DOI] [PubMed] [Google Scholar]
- Aoyama N, Lee SM, Moro N, Hovda DA, Sutton RL. Duration of ATP reduction affects extent of CA1 cell death in rat models of fluid percussion injury combined with secondary ischemia. Brain Res. 2008;1230:310–319. doi: 10.1016/j.brainres.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead WM, Kiessling JW, Kofke WA, Vavilala MS. SNP improves cerebral hemodynamics during normotension but fails to prevent sex dependent impaired cerebral autoregulation during hypotension after brain injury. Brain Res. 2010;12:142–150. doi: 10.1016/j.brainres.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagiella E, Novack TA, Ansel B, Diaz-Arrastia R, Dikmen S, Hart T, et al. Measuring outcome in traumatic brain injury treatment trials: recommendations from the traumatic brain injury clinical trials network. J Head Trauma Rehabil. 2010;25:375–382. doi: 10.1097/HTR.0b013e3181d27fe3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balikci M, Koc K, Anik I, Anik Y, Cekmen MB, Yazir Y, et al. Biochemical effects of experimental epidural hematoma on brain parenchyma of rats. Neurol Res. 2008;30:450–456. doi: 10.1179/016164107X251637. [DOI] [PubMed] [Google Scholar]
- Banks WA. Delivery of peptides to the brain: emphasis on therapeutic development. Biopolymers. 2008;90:589–594. doi: 10.1002/bip.20980. [DOI] [PubMed] [Google Scholar]
- Barbre AB, Hoane MR. Magnesium and riboflavin combination therapy following cortical contusion injury in the rat. Brain Res Bull. 2006;69:639–646. doi: 10.1016/j.brainresbull.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Saatman KE, Helfaer MA, Sinson G, Weisser JD, Brown AL, et al. Alterations in ionized and total blood magnesium after experimental traumatic brain injury: relationship to neurobehavioral outcome and neuroprotective efficacy of magnesium chloride. J Neurochem. 1999;73:271–280. doi: 10.1046/j.1471-4159.1999.0730271.x. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Saatman KE, Raghupathi R, McIntosh TK. Postinjury treatment with magnesium chloride attenuates cortical damage after traumatic brain injury in rats. J Neurotrauma. 2000;17:1029–1039. doi: 10.1089/neu.2000.17.1029. [DOI] [PubMed] [Google Scholar]
- Bauman RA, Widholm JJ, Petras JM, McBride K, Long JB. Secondary hypoxemia exacerbates the reduction of visual discrimination accuracy and neuronal cell density in the dorsal lateral geniculate nucleus resulting from fluid percussion injury. J Neurotrauma. 2000;17:679–693. doi: 10.1089/089771500415427. [DOI] [PubMed] [Google Scholar]
- von Baumgarten L, Trabold R, Thal S, Back T, Plesnila N. Role of cortical spreading depressions for secondary brain damage after traumatic brain injury in mice. J Cereb Blood Flow Metab. 2008;28:1353–1360. doi: 10.1038/jcbfm.2008.30. [DOI] [PubMed] [Google Scholar]
- Beaumont A, Marmarou A, Czigner A, Yamamoto M, Demetriadou K, Shirotani T, et al. The impact-acceleration model of head injury: injury severity predicts motor and cognitive performance after trauma. Neurol Res. 1999;21:742–754. doi: 10.1080/01616412.1999.11741008. [DOI] [PubMed] [Google Scholar]
- Bellander BM, Lidman O, Ohlsson M, Meijer B, Piehl F, Svensson M. Genetic regulation of microglia activation, complement expression, and neurodegeneration in a rat model of traumatic brain injury. Exp Brain Res. 2010;205:103–114. doi: 10.1007/s00221-010-2342-z. [DOI] [PubMed] [Google Scholar]
- Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997;86:241–251. doi: 10.3171/jns.1997.86.2.0241. [DOI] [PubMed] [Google Scholar]
- Blyth BJ, Farhavar A, Gee C, Hawthorn B, He H, Nayak A, et al. Validation of serum markers for blood-brain barrier disruption in traumatic brain injury. J Neurotrauma. 2009;26:1497–1507. doi: 10.1089/neu.2008.0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonatti E, Zamarian L, Wagner M, Benke T, Hollosi P, Strubreither W, et al. Making decisions and advising decisions in traumatic brain injury. Cogn Behav Neurol. 2008;21:164–175. doi: 10.1097/WNN.0b013e318184e688. [DOI] [PubMed] [Google Scholar]
- Boucher BA, Hanes SD. Pharmacokinetic alterations after severe head injury. Clinical relevance. Clin Pharmacokinet. 1998;35:209–221. doi: 10.2165/00003088-199835030-00004. [DOI] [PubMed] [Google Scholar]
- Brain Trauma Foundation; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24(Suppl 1):S1–S106. doi: 10.1089/neu.2007.9999. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Neuropathological protection after traumatic brain injury in intact female rats versus males or ovariectomized females. J Neurotrauma. 2001;18:891–900. doi: 10.1089/089771501750451811. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133–150. doi: 10.1097/01.WCB.0000111614.19196.04. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD, Green EJ. Secondary hypoxia following moderate fluid percussion brain injury in rats exacerbates sensorimotor and cognitive deficits. J Neurotrauma. 1999;16:1035–1047. doi: 10.1089/neu.1999.16.1035. [DOI] [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esrarza TJ, Stocchetti N, et al. Amyloid-β dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne KD, Leoni MJ, Iwata A, Chen XH, Smith DH. Acute treatment with MgSO4 attenuates long-term hippocampal tissue loss after brain trauma in the rat. J Neurosci Res. 2004;77:878–883. doi: 10.1002/jnr.20215. [DOI] [PubMed] [Google Scholar]
- Bryant RA, O'Donnell ML, Creamer M, McFarlane AC, Clark CR, Silove D. The psychiatric sequelae of traumatic injury. Am J Psychiatry. 2010;167:312–320. doi: 10.1176/appi.ajp.2009.09050617. [DOI] [PubMed] [Google Scholar]
- Buki A, Povlishock JT. All roads lead to disconnection? – Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–193. doi: 10.1007/s00701-005-0674-4. discussion 193–194. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511–521. doi: 10.1089/neu.1999.16.511. [DOI] [PubMed] [Google Scholar]
- Buki A, Farkas O, Doczi T, Povlishock JT. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma. 2003;20:261–268. doi: 10.1089/089771503321532842. [DOI] [PubMed] [Google Scholar]
- Busto R, Yoshida S, Ginsberg MD, Alonso O, Smith DW, Goldberg WJ. Regional blood flow in compression-induced brain edema in rats: effect of dietary vitamin E. Ann Neurol. 1984;15:441–448. doi: 10.1002/ana.410150507. [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Maris DO, McCall T, Grady MS. Adaptation of the fluid percussion injury model to the mouse. J Neurotrauma. 1998;15:217–229. doi: 10.1089/neu.1998.15.217. [DOI] [PubMed] [Google Scholar]
- Cernak I, O'Connor C, Vink R. Activation of cyclo-oxygenase-2 contributes to motor and cognitive dysfunction following diffuse traumatic brain injury in rats. Clin Exp Pharmacol Physiol. 2001;28:922–925. doi: 10.1046/j.1440-1681.2001.03549.x. [DOI] [PubMed] [Google Scholar]
- Cernak I, Vink R, Zapple DN, Cruz MI, Ahmed F, Chang T, et al. The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol Dis. 2004;17:29–43. doi: 10.1016/j.nbd.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J Neurotrauma. 1996;13:557–568. doi: 10.1089/neu.1996.13.557. [DOI] [PubMed] [Google Scholar]
- Chen SF, Richards HK, Smielewski P, Johnstrom P, Salvador R, Pickard JD, et al. Relationship between flow-metabolism uncoupling and evolving axonal injury after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2004;24:1025–1036. doi: 10.1097/01.WCB.0000129415.34520.47. [DOI] [PubMed] [Google Scholar]
- Cheng J, Gu J, Ma Y, Yang T, Kuang Y, Li B, et al. Development of a rat model for studying blast-induced traumatic brain injury. J Neurol Sci. 2010;294:23–28. doi: 10.1016/j.jns.2010.04.010. [DOI] [PubMed] [Google Scholar]
- Chesnut RM, Marshall SB, Piek J, Blunt BA, Klauber MR, Marshall LF. Early and late systemic hypotension as a frequent and fundamental source of cerebral ischemia following severe brain injury in the Traumatic Coma Data Bank. Acta Neurochir Suppl (Wien) 1993;59:121–125. doi: 10.1007/978-3-7091-9302-0_21. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Dixon CE, Chen M, Marion DW, Heineman S, et al. Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J Neurotrauma. 1997;14:179–189. doi: 10.1089/neu.1997.14.179. [DOI] [PubMed] [Google Scholar]
- Clausen F, Lewen A, Marklund N, Olsson Y, McArthur DL, Hillered L. Correlation of hippocampal morphological changes and morris water maze performance after cortical contusion injury in rats. Neurosurgery. 2005;57:154–163. doi: 10.1227/01.neu.0000163412.07546.57. [DOI] [PubMed] [Google Scholar]
- Clausen F, Hanell A, Bjork M, Hillered L, Mir AK, Gram H, et al. Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci. 2009;30:385–396. doi: 10.1111/j.1460-9568.2009.06820.x. [DOI] [PubMed] [Google Scholar]
- Compagnone C, Murray GD, Teasdale GM, Maas AI, Esposito D, Princi P, et al. The management of patients with intradural post-traumatic mass lesions: a multicenter survey of current approaches to surgical management in 729 patients coordinated by the European Brain Injury Consortium. Neurosurgery. 2005;57:1183–1192. doi: 10.1227/01.neu.0000279218.53504.fe. [DOI] [PubMed] [Google Scholar]
- Consensus Conference. Rehabilitation of persons with traumatic brain injury. NIH Consensus Development Panel on Rehabilitation of Persons With Traumatic Brain Injury. JAMA. 1999;282:974–983. [PubMed] [Google Scholar]
- Conte V, Uryu K, Fujimoto S, Yao Y, Rokach J, Longhi L, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- Cook AM, Whitlow J, Hatton J, Young B. Cyclosporine A for neuroprotection: establishing dosing guidelines for safe and effective use. Expert Opin Drug Saf. 2009;8:411–419. doi: 10.1517/14740330903066742. [DOI] [PubMed] [Google Scholar]
- Corrigan JD, Selassie AW, Orman JA. The epidemiology of traumatic brain injury. J Head Trauma Rehabil. 2010;25:72–80. doi: 10.1097/HTR.0b013e3181ccc8b4. [DOI] [PubMed] [Google Scholar]
- Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- Creeley CE, Wozniak DF, Bayly PV, Olney JW, Lewis LM. Multiple episodes of mild traumatic brain injury result in impaired cognitive performance in mice. Acad Emerg Med. 2004;11:809–819. doi: 10.1111/j.1553-2712.2004.tb00761.x. [DOI] [PubMed] [Google Scholar]
- Cutler SM, Cekic M, Miller DM, Wali B, VanLandingham JW, Stein DG. Progesterone improves acute recovery after traumatic brain injury in the aged rat. J Neurotrauma. 2007;24:1475–1486. doi: 10.1089/neu.2007.0294. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, Miller JW. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127:304–314. doi: 10.1093/brain/awh038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin AP, Wetterhall M, Caldwell KD, Larsson A, Bergquist J, Hillered L, et al. Methodological aspects on microdialysis protein sampling and quantification in biological fluids: an in vitro study on human ventricular CSF. Anal Chem. 2010;82:4376–4385. doi: 10.1021/ac1007706. [DOI] [PubMed] [Google Scholar]
- Dardiotis E, Fountas KN, Dardioti M, Xiromerisiou G, Kapsalaki E, Tasiou A, et al. Genetic association studies in patients with traumatic brain injury. Neurosurg Focus. 2010;28:E9. doi: 10.3171/2009.10.FOCUS09215. [DOI] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience. 2009;163:1–8. doi: 10.1016/j.neuroscience.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Zhao J, Hergenroeder G, Moore AN. Biomarkers for the diagnosis, prognosis, and evaluation of treatment efficacy for traumatic brain injury. Neurotherapeutics. 2010;7:100–114. doi: 10.1016/j.nurt.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AR, Shear DA, Chen Z, Lu XC, Tortella FC. A comparison of two cognitive test paradigms in a penetrating brain injury model. J Neurosci Methods. 2010;189:84–87. doi: 10.1016/j.jneumeth.2010.03.012. [DOI] [PubMed] [Google Scholar]
- De Bonis P, Pompucci A, Mangiola A, D'Alessandris QG, Rigante L, Anile C. Decompressive craniectomy for the treatment of traumatic brain injury: does an age limit exist. J Neurosurg. 2010;112:1150–1153. doi: 10.3171/2009.7.JNS09505. [DOI] [PubMed] [Google Scholar]
- DeFord SM, Wilson MS, Rice AC, Clausen T, Rice LK, Barabnova A, et al. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma. 2002;19:427–438. doi: 10.1089/08977150252932389. [DOI] [PubMed] [Google Scholar]
- DeRoss AL, Adams JE, Vane DW, Russell SJ, Terella AM, Wald SL. Multiple head injuries in rats: effects on behavior. J Trauma. 2002;52:708–714. doi: 10.1097/00005373-200204000-00017. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, et al. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- Djebaili M, Guo Q, Pettus EH, Hoffman SW, Stein DG. The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J Neurotrauma. 2005;22:106–118. doi: 10.1089/neu.2005.22.106. [DOI] [PubMed] [Google Scholar]
- Dusick JR, Glenn TC, Lee WN, Vespa PM, Kelly DF, Lee SM, et al. Increased pentose phosphate pathway flux after clinical traumatic brain injury: a [1,2-13C2]glucose labeling study in humans. J Cereb Blood Flow Metab. 2007;27:1593–1602. doi: 10.1038/sj.jcbfm.9600458. [DOI] [PubMed] [Google Scholar]
- Ebmeyer U, Safar P, Radovsky A, Obrist W, Alexander H, Pomeranz S. Moderate hypothermia for 48 hours after temporary epidural brain compression injury in a canine outcome model. J Neurotrauma. 1998;15:323–336. doi: 10.1089/neu.1998.15.323. [DOI] [PubMed] [Google Scholar]
- Ekmark-Lewen S, Lewen A, Meyerson BJ, Hillered L. The Multivariate Concentric Square Field Test reveals behavioral profiles regarding risk taking, risk assessment and exploration in mice subjected to traumatic brain injury. J Neurotrauma. 2010;27:1643–1655. doi: 10.1089/neu.2009.0953. [DOI] [PubMed] [Google Scholar]
- Elf K, Nilsson P, Enblad P. Outcome after traumatic brain injury improved by an organized secondary insult program and standardized neurointensive care. Crit Care Med. 2002;30:2129–2134. doi: 10.1097/00003246-200209000-00029. [DOI] [PubMed] [Google Scholar]
- Ellingson BM, Fijalkowski RJ, Pintar FA, Yoganandan N, Gennarelli TA. New mechanism for inducing closed head injury in the rat. Biomed Sci Instrum. 2005;41:86–91. [PubMed] [Google Scholar]
- Empey PE, McNamara PJ, Young B, Rosbolt MB, Hatton J. Cyclosporin A disposition following acute traumatic brain injury. J Neurotrauma. 2006;23:109–116. doi: 10.1089/neu.2006.23.109. [DOI] [PubMed] [Google Scholar]
- Enomoto T, Osugi T, Satoh H, McIntosh TK, Nabeshima T. Pre-injury magnesium treatment prevents traumatic brain injury-induced hippocampal ERK activation, neuronal loss, and cognitive dysfunction in the radial-arm maze test. J Neurotrauma. 2005;22:783–792. doi: 10.1089/neu.2005.22.783. [DOI] [PubMed] [Google Scholar]
- Esen F, Erdem T, Aktan D, Kalayci R, Cakar N, Kaya M, et al. Effects of magnesium administration on brain edema and blood-brain barrier breakdown after experimental traumatic brain injury in rats. J Neurosurg Anesthesiol. 2003;15:119–125. doi: 10.1097/00008506-200304000-00009. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Farace E, Alves WM. Do women fare worse? A metaanalysis of gender differences in outcome after traumatic brain injury. J Neurosurg. 2000;93:539–545. doi: 10.3171/jns.2000.93.4.0539. [DOI] [PubMed] [Google Scholar]
- Feldman Z, Gurevitch B, Artru AA, Oppenheim A, Shohami E, Reichenthal E, et al. Effect of magnesium given 1 hour after head trauma on brain edema and neurological outcome. J Neurosurg. 1996;85:131–137. doi: 10.3171/jns.1996.85.1.0131. [DOI] [PubMed] [Google Scholar]
- Ferrand-Drake M, Zhu C, Gido G, Hansen AJ, Karlsson JO, Bahr BA, et al. Cyclosporin A prevents calpain activation despite increased intracellular calcium concentrations, as well as translocation of apoptosis-inducing factor, cytochrome c and caspase-3 activation in neurons exposed to transient hypoglycemia. J Neurochem. 2003;85:1431–1442. doi: 10.1046/j.1471-4159.2003.01794.x. [DOI] [PubMed] [Google Scholar]
- Fijalkowski RJ, Stemper BD, Pintar FA, Yoganandan N, Crowe MJ, Gennarelli TA. New rat model for diffuse brain injury using coronal plane angular acceleration. J Neurotrauma. 2007;24:1387–1398. doi: 10.1089/neu.2007.0268. [DOI] [PubMed] [Google Scholar]
- Fijalkowski RJ, Yoganandan N, Zhang J, Pintar FA. A finite element model of region-specific response for mild diffuse brain injury. Stapp Car Crash J. 2009;53:193–213. doi: 10.4271/2009-22-0007. [DOI] [PubMed] [Google Scholar]
- Finnie JW, Blumbergs PC. Traumatic brain injury. Vet Pathol. 2002;39:679–689. doi: 10.1354/vp.39-6-679. [DOI] [PubMed] [Google Scholar]
- Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4:1328–1337. doi: 10.1038/nprot.2009.148. [DOI] [PubMed] [Google Scholar]
- Floyd CL, Golden KM, Black RT, Hamm RJ, Lyeth BG. Craniectomy position affects morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. J Neurotrauma. 2002;19:303–316. doi: 10.1089/089771502753594873. [DOI] [PubMed] [Google Scholar]
- Foda MA, Marmarou A. A new model of diffuse brain injury in rats. Part II: morphological characterization. J Neurosurg. 1994;80:301–313. doi: 10.3171/jns.1994.80.2.0301. [DOI] [PubMed] [Google Scholar]
- Fork M, Bartels C, Ebert AD, Grubich C, Synowitz H, Wallesch CW. Neuropsychological sequelae of diffuse traumatic brain injury. Brain Inj. 2005;19:101–108. doi: 10.1080/02699050410001726086. [DOI] [PubMed] [Google Scholar]
- Fromm L, Heath DL, Vink R, Nimmo AJ. Magnesium attenuates post-traumatic depression/anxiety following diffuse traumatic brain injury in rats. J Am Coll Nutr. 2004;23:529S–533S. doi: 10.1080/07315724.2004.10719396. [DOI] [PubMed] [Google Scholar]
- Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–378. doi: 10.1016/j.neubiorev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Fukui S, Signoretti S, Dunbar JG, Marmarou A. The effect of cyclosporin A on brain edema formation following experimental cortical contusion. Acta Neurochir Suppl. 2003;86:301–303. doi: 10.1007/978-3-7091-0651-8_65. [DOI] [PubMed] [Google Scholar]
- Ganz JC, Hall C, Zwetnow NN. Cerebral blood flow during experimental epidural bleeding in swine. Acta Neurochir (Wien) 1990;103:148–157. doi: 10.1007/BF01407522. [DOI] [PubMed] [Google Scholar]
- Gao WM, Chadha MS, Kline AE, Clark RS, Kochanek PM, Dixon CE, et al. Immunohistochemical analysis of histone H3 acetylation and methylation – evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res. 2006;1070:31–34. doi: 10.1016/j.brainres.2005.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Oda Y, Wei EP, Povlishock JT. The adverse pial arteriolar and axonal consequences of traumatic brain injury complicated by hypoxia and their therapeutic modulation with hypothermia in rat. J Cereb Blood Flow Metab. 2010;30:628–637. doi: 10.1038/jcbfm.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeraerts T, Friggeri A, Mazoit JX, Benhamou D, Duranteau J, Vigue B. Posttraumatic brain vulnerability to hypoxia-hypotension: the importance of the delay between brain trauma and secondary insult. Intensive Care Med. 2008;34:551–560. doi: 10.1007/s00134-007-0863-0. [DOI] [PubMed] [Google Scholar]
- Gennarelli TA. Animate models of human head injury. J Neurotrauma. 1994;11:357–368. doi: 10.1089/neu.1994.11.357. [DOI] [PubMed] [Google Scholar]
- Ghajar J. Traumatic brain injury. Lancet. 2000;356:923–929. doi: 10.1016/S0140-6736(00)02689-1. [DOI] [PubMed] [Google Scholar]
- Gilmer LK, Ansari MA, Roberts KN, Scheff SW. Age-related mitochondrial changes after traumatic brain injury. J Neurotrauma. 2010;27:939–950. doi: 10.1089/neu.2009.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg MD, Zhao W, Alonso OF, Loor-Estades JY, Dietrich WD, Busto R. Uncoupling of local cerebral glucose metabolism and blood flow after acute fluid-percussion injury in rats. Am J Physiol. 1997;272:H2859–H2868. doi: 10.1152/ajpheart.1997.272.6.H2859. [DOI] [PubMed] [Google Scholar]
- Giri BK, Krishnappa IK, Bryan RM, Jr, Robertson C, Watson J. Regional cerebral blood flow after cortical impact injury complicated by a secondary insult in rats. Stroke. 2000;31:961–967. doi: 10.1161/01.str.31.4.961. [DOI] [PubMed] [Google Scholar]
- Gonzenbach RR, Schwab ME. Disinhibition of neurite growth to repair the injured adult CNS: focusing on Nogo. Cell Mol Life Sci. 2008;65:161–176. doi: 10.1007/s00018-007-7170-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss CW, Hoffman SW, Stein DG. Behavioral effects and anatomic correlates after brain injury: a progesterone dose-response study. Pharmacol Biochem Behav. 2003;76:231–242. doi: 10.1016/j.pbb.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Graham DI, Adams JH, Doyle D. Ischaemic brain damage in fatal non-missile head injuries. J Neurol Sci. 1978;39:213–234. doi: 10.1016/0022-510x(78)90124-7. [DOI] [PubMed] [Google Scholar]
- Graham DI, Raghupathi R, Saatman KE, Meaney D, McIntosh TK. Tissue tears in the white matter after lateral fluid percussion brain injury in the rat: relevance to human brain injury. Acta Neuropathol. 2000;99:117–124. doi: 10.1007/pl00007414. [DOI] [PubMed] [Google Scholar]
- Graham DI, Adams JH, Murray LS, Jennett B. Neuropathology of the vegetative state after head injury. Neuropsychol Rehabil. 2005;15:198–213. doi: 10.1080/09602010443000452. [DOI] [PubMed] [Google Scholar]
- Greenberg G, Mikulis DJ, Ng K, DeSouza D, Green RE. Use of diffusion tensor imaging to examine subacute white matter injury progression in moderate to severe traumatic brain injury. Arch Phys Med Rehabil. 2008;89(12) Suppl:S45–S50. doi: 10.1016/j.apmr.2008.08.211. [DOI] [PubMed] [Google Scholar]
- Groswasser Z, Cohen M, Keren O. Female TBI patients recover better than males. Brain Inj. 1998;12:805–808. doi: 10.1080/026990598122197. [DOI] [PubMed] [Google Scholar]
- Guluma KZ, Saatman KE, Brown A, Raghupathi R, McIntosh TK. Sequential pharmacotherapy with magnesium chloride and basic fibroblast growth factor after fluid percussion brain injury results in less neuromotor efficacy than that achieved with magnesium alone. J Neurotrauma. 1999;16:311–321. doi: 10.1089/neu.1999.16.311. [DOI] [PubMed] [Google Scholar]
- Habgood MD, Bye N, Dziegielewska KM, Ek CJ, Lane MA, Potter A, et al. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci. 2007;25:231–238. doi: 10.1111/j.1460-9568.2006.05275.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RJ, White-Gbadebo DM, Lyeth BG, Jenkins LW, Hayes RL. The effect of age on motor and cognitive deficits after traumatic brain injury in rats. Neurosurgery. 1992;31:1072–1077. doi: 10.1227/00006123-199212000-00013. discussion 1078. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Temple MD, O'Dell DM, Pike BR, Lyeth BG. Exposure to environmental complexity promotes recovery of cognitive function after traumatic brain injury. J Neurotrauma. 1996;13:41–47. doi: 10.1089/neu.1996.13.41. [DOI] [PubMed] [Google Scholar]
- Hanell A, Clausen F, Bjork M, Jansson K, Philipson O, Nilsson LN, et al. Genetic deletion and pharmacological inhibition of Nogo-66 receptor impairs cognitive outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27:1297–1309. doi: 10.1089/neu.2009.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harford-Wright E, Thornton E, Vink R. Angiotensin-converting enzyme (ACE) inhibitors exacerbate histological damage and motor deficits after experimental traumatic brain injury. Neurosci Lett. 2010;481:26–29. doi: 10.1016/j.neulet.2010.06.044. [DOI] [PubMed] [Google Scholar]
- Hatton J, Rosbolt B, Empey P, Kryscio R, Young B. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg. 2008;109:699–707. doi: 10.3171/JNS/2008/109/10/0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath DL, Vink R. Magnesium sulphate improves neurologic outcome following severe closed head injury in rats. Neurosci Lett. 1997;228:175–178. doi: 10.1016/s0304-3940(97)00394-7. [DOI] [PubMed] [Google Scholar]
- Heath DL, Vink R. Neuroprotective effects of MgSO4 and MgCl2 in closed head injury: a comparative phosphorus NMR study. J Neurotrauma. 1998;15:183–189. doi: 10.1089/neu.1998.15.183. [DOI] [PubMed] [Google Scholar]
- Heath DL, Vink R. Concentration of brain free magnesium following severe brain injury correlates with neurologic motor outcome. J Clin Neurosci. 1999a;6:505–509. doi: 10.1016/s0967-5868(99)90011-5. [DOI] [PubMed] [Google Scholar]
- Heath DL, Vink R. Improved motor outcome in response to magnesium therapy received up to 24 hours after traumatic diffuse axonal brain injury in rats. J Neurosurg. 1999b;90:504–509. doi: 10.3171/jns.1999.90.3.0504. [DOI] [PubMed] [Google Scholar]
- Heath DL, Vink R. Optimization of magnesium therapy after severe diffuse axonal brain injury in rats. J Pharmacol Exp Ther. 1999c;288:1311–1316. [PubMed] [Google Scholar]
- Hellewell SC, Yan EB, Agyapomaa DA, Bye N, Morganti-Kossmann MC. Post-traumatic hypoxia exacerbates brain tissue damage: analysis of axonal injury and glial responses. J Neurotrauma. 2010;27:1997–2010. doi: 10.1089/neu.2009.1245. [DOI] [PubMed] [Google Scholar]
- Helmy A, Carpenter KL, Hutchinson PJ. Microdialysis in the human brain and its potential role in the development and clinical assessment of drugs. Curr Med Chem. 2007;14:1525–1537. doi: 10.2174/092986707780831113. [DOI] [PubMed] [Google Scholar]
- Helmy A, Carpenter KL, Skepper JN, Kirkpatrick PJ, Pickard JD, Hutchinson PJ. Microdialysis of cytokines: methodological considerations, scanning electron microscopy, and determination of relative recovery. J Neurotrauma. 2009;26:549–561. doi: 10.1089/neu.2008.0719. [DOI] [PubMed] [Google Scholar]
- Henninger N, Dutzmann S, Sicard KM, Kollmar R, Bardutzky J, Schwab S. Impaired spatial learning in a novel rat model of mild cerebral concussion injury. Exp Neurol. 2005;195:447–457. doi: 10.1016/j.expneurol.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Hillered L, Enblad P. Nonischemic energy metabolic crisis in acute brain injury. Crit Care Med. 2008;36:2952–2953. doi: 10.1097/CCM.0b013e3181872178. [DOI] [PubMed] [Google Scholar]
- Hillered L, Vespa PM, Hovda DA. Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. J Neurotrauma. 2005;22:3–41. doi: 10.1089/neu.2005.22.3. [DOI] [PubMed] [Google Scholar]
- Hillman J, Aneman O, Anderson C, Sjögren F, Säberg C, Mellergård P. A microdialysis technique for routine measurement of macromolecules in the injured human brain. Neurosurgery. 2005;56:1264–1268. doi: 10.1227/01.neu.0000159711.93592.8d. [DOI] [PubMed] [Google Scholar]
- Hlatky R, Valadka AB, Goodman CJ, Contant CF, Robertson CS. Patterns of energy substrates during ischemia measured in the brain by microdialysis. J Neurotrauma. 2004;21:894–906. doi: 10.1089/0897715041526195. [DOI] [PubMed] [Google Scholar]
- Hoane MR. Treatment with magnesium improves reference memory but not working memory while reducing GFAP expression following traumatic brain injury. Restor Neurol Neurosci. 2005;23:67–77. [PubMed] [Google Scholar]
- Hoane MR, Lasley LA, Akstulewicz SL. Middle age increases tissue vulnerability and impairs sensorimotor and cognitive recovery following traumatic brain injury in the rat. Behav Brain Res. 2004;153:189–197. doi: 10.1016/j.bbr.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Hoffman SW, Fulop Z, Stein DG. Bilateral frontal cortical contusion in rats: behavioral and anatomic consequences. J Neurotrauma. 1994;11:417–431. doi: 10.1089/neu.1994.11.417. [DOI] [PubMed] [Google Scholar]
- Hoffman AN, Malena RR, Westergom BP, Luthra P, Cheng JP, Aslam HA, et al. Environmental enrichment-mediated functional improvement after experimental traumatic brain injury is contingent on task-specific neurobehavioral experience. Neurosci Lett. 2008;431:226–230. doi: 10.1016/j.neulet.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukkelhoven CW, Steyerberg EW, Rampen AJ, Farace E, Habbema JD, Marshall LF, et al. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99:666–673. doi: 10.3171/jns.2003.99.4.0666. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol. 2009;215:243–252. doi: 10.1016/j.expneurol.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imer M, Omay B, Uzunkol A, Erdem T, Sabanci PA, Karasu A, et al. Effect of magnesium, MK-801 and combination of magnesium and MK-801 on blood-brain barrier permeability and brain edema after experimental traumatic diffuse brain injury. Neurol Res. 2009;31:977–981. doi: 10.1179/174313209X385617. [DOI] [PubMed] [Google Scholar]
- Inglese M, Makani S, Johnson G, Cohen BA, Silver JA, Gonen O, et al. Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg. 2005;103:298–303. doi: 10.3171/jns.2005.103.2.0298. [DOI] [PubMed] [Google Scholar]
- Ishige N, Pitts LH, Hashimoto T, Nishimura MC, Bartkowski HM. Effect of hypoxia on traumatic brain injury in rats: Part 1. Changes in neurological function, electroencephalograms, and histopathology. Neurosurgery. 1987;20:848–853. doi: 10.1227/00006123-198706000-00005. [DOI] [PubMed] [Google Scholar]
- Israelsson C, Bengtsson H, Kylberg A, Kullander K, Lewen A, Hillered L, et al. Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. J Neurotrauma. 2008;25:959–974. doi: 10.1089/neu.2008.0562. [DOI] [PubMed] [Google Scholar]
- Jones NC, Cardamone L, Williams JP, Salzberg MR, Myers D, O'Brien TJ. Experimental traumatic brain injury induces a pervasive hyperanxious phenotype in rats. J Neurotrauma. 2008;25:1367–1374. doi: 10.1089/neu.2008.0641. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Hilton GD, Stoica BA, Zapple DN, Faden AI. Fluid-percussion-induced traumatic brain injury model in rats. Nat Protoc. 2010;5:1552–1563. doi: 10.1038/nprot.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafadar AM, Sanus GZ, Is M, Coskun A, Tanriverdi T, Hanimoglu H, et al. Prolonged elevation of magnesium in the cerebrospinal fluid of patients with severe head injury. Neurol Res. 2007;29:824–829. doi: 10.1179/016164107X181879. [DOI] [PubMed] [Google Scholar]
- Kalsotra A, Turman CM, Dash PK, Strobel HW. Differential effects of traumatic brain injury on the cytochrome p450 system: a perspective into hepatic and renal drug metabolism. J Neurotrauma. 2003;20:1339–1350. doi: 10.1089/089771503322686139. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889–900. doi: 10.3171/jns.1990.73.6.0889. [DOI] [PubMed] [Google Scholar]
- Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140:685–697. doi: 10.1016/j.neuroscience.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Kharatishvili I, Pitkanen A. Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res. 2010;90:47–59. doi: 10.1016/j.eplepsyres.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Kilbourne M, Kuehn R, Tosun C, Caridi J, Keledjian K, Bochicchio G, et al. Novel model of frontal impact closed head injury in the rat. J Neurotrauma. 2009;26:2233–2243. doi: 10.1089/neu.2009.0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AE, Wagner AK, Westergom BP, Malena RR, Zafonte RD, Olsen AS, et al. Acute treatment with the 5-HT(1A) receptor agonist 8-OH-DPAT and chronic environmental enrichment confer neurobehavioral benefit after experimental brain trauma. Behav Brain Res. 2007;177:186–194. doi: 10.1016/j.bbr.2006.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotapka MJ, Graham DI, Adams JH, Gennarelli TA. Hippocampal pathology in fatal human head injury without high intracranial pressure. J Neurotrauma. 1994;11:317–324. doi: 10.1089/neu.1994.11.317. [DOI] [PubMed] [Google Scholar]
- Kroppenstedt SN, Kern M, Thomale UW, Schneider GH, Lanksch WR, Unterberg AW. Effect of cerebral perfusion pressure on contusion volume following impact injury. J Neurosurg. 1999;90:520–526. doi: 10.3171/jns.1999.90.3.0520. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW, et al. The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma. 2001;18:1229–1240. doi: 10.1089/089771501317095269. [DOI] [PubMed] [Google Scholar]
- Kupina NC, Detloff MR, Bobrowski WF, Snyder BJ, Hall ED. Cytoskeletal protein degradation and neurodegeneration evolves differently in males and females following experimental head injury. Exp Neurol. 2003;180:55–73. doi: 10.1016/s0014-4886(02)00048-1. [DOI] [PubMed] [Google Scholar]
- Lammie GA, Piper IR, Thomson D, Brannan F. Neuropathologic characterization of a rodent model of closed head injury – addition of clinically relevant secondary insults does not significantly potentiate brain damage. J Neurotrauma. 1999;16:603–615. doi: 10.1089/neu.1999.16.603. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, McKenna SE, Song P, Wang H, Durham L, Yeung N, et al. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–1107. doi: 10.1089/neu.2006.0192. [DOI] [PubMed] [Google Scholar]
- Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R, et al. Mild head injury increasing the brain's vulnerability to a second concussive impact. J Neurosurg. 2001;95:859–870. doi: 10.3171/jns.2001.95.5.0859. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol. 2001;24:169–181. doi: 10.1385/MN:24:1-3:169. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Shimizu S, Marklund N, Thompson HJ, Schwab ME, Saatman KE, et al. Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience. 2005;134:1047–1056. doi: 10.1016/j.neuroscience.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Lewen A, Li GL, Olsson Y, Hillered L. Changes in microtubule-associated protein 2 and amyloid precursor protein immunoreactivity following traumatic brain injury in rat: influence of MK-801 treatment. Brain Res. 1996;719:161–171. doi: 10.1016/0006-8993(96)00081-9. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- Li S, Kuroiwa T, Ishibashi S, Sun L, Endo S, Ohno K. Transient cognitive deficits are associated with the reversible accumulation of amyloid precursor protein after mild traumatic brain injury. Neurosci Lett. 2006;409:182–186. doi: 10.1016/j.neulet.2006.09.054. [DOI] [PubMed] [Google Scholar]
- Li XY, Li J, Feng DF, Gu L. Diffuse axonal injury induced by simultaneous moderate linear and angular head accelerations in rats. Neuroscience. 2010;169:357–369. doi: 10.1016/j.neuroscience.2010.04.075. [DOI] [PubMed] [Google Scholar]
- Lifshitz J, Sullivan PG, Hovda DA, Wieloch T, McIntosh TK. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion. 2004;4:705–713. doi: 10.1016/j.mito.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma. 1988;5:1–15. doi: 10.1089/neu.1988.5.1. [DOI] [PubMed] [Google Scholar]
- Lindgren SO. Experimental studies of mechanical effects in head injury. Acta Chir Scand Suppl. 1966;360:1–100. [PubMed] [Google Scholar]
- Lippert-Gruner M, Maegele M, Pokorny J, Angelov DN, Svestkova O, Wittner M, et al. Early rehabilitation model shows positive effects on neural degeneration and recovery from neuromotor deficits following traumatic brain injury. Physiol Res. 2007;56:359–368. doi: 10.33549/physiolres.930971. [DOI] [PubMed] [Google Scholar]
- Lo EH, Singhal AB, Torchilin VP, Abbott NJ. Drug delivery to damaged brain. Brain Res Brain Res Rev. 2001;38:140–148. doi: 10.1016/s0165-0173(01)00083-2. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF, Davis J, et al. Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery. 2005;56:364–374. doi: 10.1227/01.neu.0000149008.73513.44. discussion 364–374. [DOI] [PubMed] [Google Scholar]
- Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci. 2009a;14:3795–3813. doi: 10.2741/3489. [DOI] [PubMed] [Google Scholar]
- Lu XC, Chen RW, Yao C, Wei H, Yang X, Liao Z, et al. NNZ-2566, a glypromate analog, improves functional recovery and attenuates apoptosis and inflammation in a rat model of penetrating ballistic-type brain injury. J Neurotrauma. 2009b;26:141–154. doi: 10.1089/neu.2008.0629. [DOI] [PubMed] [Google Scholar]
- Luheshi NM, Rothwell NJ, Brough D. Dual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapy. Br J Pharmacol. 2009;157:1318–1329. doi: 10.1111/j.1476-5381.2009.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Steyerberg EW, Butcher I, Dammers R, Lu J, Marmarou A, et al. Prognostic value of computerized tomography scan characteristics in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:303–314. doi: 10.1089/neu.2006.0033. [DOI] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- McIntosh TK. Neurochemical sequelae of traumatic brain injury: therapeutic implications. Cerebrovasc Brain Metab Rev. 1994;6:109–162. [PubMed] [Google Scholar]
- McIntosh TK, Noble L, Andrews B, Faden AI. Traumatic brain injury in the rat: characterization of a midline fluid-percussion model. Cent Nerv Syst Trauma. 1987;4:119–134. doi: 10.1089/cns.1987.4.119. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Faden AI, Yamakami I, Vink R. Magnesium deficiency exacerbates and pretreatment improves outcome following traumatic brain injury in rats: 31P magnetic resonance spectroscopy and behavioral studies. J Neurotrauma. 1988;5:17–31. doi: 10.1089/neu.1988.5.17. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Yamakami I, Faden AI. Magnesium protects against neurological deficit after brain injury. Brain Res. 1989;482:252–260. doi: 10.1016/0006-8993(89)91188-8. [DOI] [PubMed] [Google Scholar]
- McKee JA, Brewer RP, Macy GE, Borel CO, Reynolds JD, Warner DS. Magnesium neuroprotection is limited in humans with acute brain injury. Neurocrit Care. 2005;2:342–351. doi: 10.1385/NCC:2:3:342. [DOI] [PubMed] [Google Scholar]
- Maegele M, Riess P, Sauerland S, Bouillon B, Hess S, McIntosh TK, et al. Characterization of a new rat model of experimental combined neurotrauma. Shock. 2005;23:476–481. doi: 10.1097/01.shk.0000159929.87737.5c. [DOI] [PubMed] [Google Scholar]
- Maegele M, Sauerland S, Bouillon B, Schafer U, Trubel H, Riess P, et al. Differential immunoresponses following experimental traumatic brain injury, bone fracture and ‘two-hit’-combined neurotrauma. Inflamm Res. 2007;56:318–323. doi: 10.1007/s00011-007-6141-3. [DOI] [PubMed] [Google Scholar]
- Margulies S, Hicks R. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26:925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion DW. Evidenced-based guidelines for traumatic brain injuries. Prog Neurol Surg. 2006;19:171–196. doi: 10.1159/000095191. [DOI] [PubMed] [Google Scholar]
- Marklund N, Lewander T, Clausen F, Hillered L. Effects of the nitrone radical scavengers PBN and S-PBN on in vivo trapping of reactive oxygen species after traumatic brain injury in rats. J Cereb Blood Flow Metab. 2001;21:1259–1267. doi: 10.1097/00004647-200111000-00002. [DOI] [PubMed] [Google Scholar]
- Marklund N, Sihver S, Langstrom B, Bergstrom M, Hillered L. Effect of traumatic brain injury and nitrone radical scavengers on relative changes in regional cerebral blood flow and glucose uptake in rats. J Neurotrauma. 2002;19:1139–1153. doi: 10.1089/08977150260337958. [DOI] [PubMed] [Google Scholar]
- Marklund N, Bakshi A, Castelbuono DJ, Conte V, McIntosh TK. Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr Pharm Des. 2006;12:1645–1680. doi: 10.2174/138161206776843340. [DOI] [PubMed] [Google Scholar]
- Marklund N, Bareyre FM, Royo NC, Thompson HJ, Mir AK, Grady MS, et al. Cognitive outcome following brain injury and treatment with an inhibitor of Nogo-A in association with an attenuated downregulation of hippocampal growth-associated protein-43 expression. J Neurosurg. 2007;107:844–853. doi: 10.3171/JNS-07/10/0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110:1227–1237. doi: 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- Marmarou CR, Povlishock JT. Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury. Exp Neurol. 2006;197:353–362. doi: 10.1016/j.expneurol.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K. A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- Maruichi K, Kuroda S, Chiba Y, Hokari M, Shichinohe H, Hida K, et al. Graded model of diffuse axonal injury for studying head injury-induced cognitive dysfunction in rats. Neuropathology. 2009;29:132–139. doi: 10.1111/j.1440-1789.2008.00956.x. [DOI] [PubMed] [Google Scholar]
- Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- Matsushita Y, Bramlett HM, Kuluz JW, Alonso O, Dietrich WD. Delayed hemorrhagic hypotension exacerbates the hemodynamic and histopathologic consequences of traumatic brain injury in rats. J Cereb Blood Flow Metab. 2001;21:847–856. doi: 10.1097/00004647-200107000-00010. [DOI] [PubMed] [Google Scholar]
- Mauritz W, Wilbacher I, Majdan M, Leitgeb J, Janciak I, Brazinova A, et al. Epidemiology, treatment and outcome of patients after severe traumatic brain injury in European regions with different economic status. Eur J Public Health. 2008;18:575–580. doi: 10.1093/eurpub/ckn079. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Kunene NK, Gilman CB, Hamm RJ, Hafez N, Bullock MR. Severe human traumatic brain injury, but not cyclosporin a treatment, depresses activated T lymphocytes early after injury. J Neurotrauma. 2006;23:962–975. doi: 10.1089/neu.2006.23.962. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Beat A, Singh A, Bullock MR. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. 2009a;218:363–370. doi: 10.1016/j.expneurol.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Mazzeo AT, Brophy GM, Gilman CB, Alves OL, Robles JR, Hayes RL, et al. Safety and tolerability of cyclosporin A in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009b;26:2195–2206. doi: 10.1089/neu.2009.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney DF, Ross DT, Winkelstein BA, Brasko J, Goldstein D, Bilston LB, et al. Modification of the cortical impact model to produce axonal injury in the rat cerebral cortex. J Neurotrauma. 1994;11:599–612. doi: 10.1089/neu.1994.11.599. [DOI] [PubMed] [Google Scholar]
- Menon DK. Unique challenges in clinical trials in traumatic brain injury. Crit Care Med. 2009;37(1) Suppl:S129–S135. doi: 10.1097/CCM.0b013e3181921225. [DOI] [PubMed] [Google Scholar]
- Meyer MJ, Megyesi J, Meythaler J, Murie-Fernandez M, Aubut JA, Foley N, et al. Acute management of acquired brain injury part I: an evidence-based review of non-pharmacological interventions. Brain Inj. 2010;24:694–705. doi: 10.3109/02699051003692118. [DOI] [PubMed] [Google Scholar]
- Miller JD, Bullock R, Graham DI, Chen MH, Teasdale GM. Ischemic brain damage in a model of acute subdural hematoma. Neurosurgery. 1990;27:433–439. doi: 10.1097/00006123-199009000-00016. [DOI] [PubMed] [Google Scholar]
- Missios S, Harris BT, Dodge CP, Simoni MK, Costine BA, Lee YL, et al. Scaled cortical impact in immature swine: effect of age and gender on lesion volume. J Neurotrauma. 2009;26:1943–1951. doi: 10.1089/neu.2009.0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, et al. Experimental models of traumatic brain injury: do we really need to build a better mousetrap. Neuroscience. 2005;136:971–989. doi: 10.1016/j.neuroscience.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Mosenthal AC, Lavery RF, Addis M, Kaul S, Ross S, Marburger R, et al. Isolated traumatic brain injury: age is an independent predictor of mortality and early outcome. J Trauma. 2002;52:907–911. doi: 10.1097/00005373-200205000-00015. [DOI] [PubMed] [Google Scholar]
- Muir JK, Boerschel M, Ellis EF. Continuous monitoring of posttraumatic cerebral blood flow using laser-Doppler flowmetry. J Neurotrauma. 1992;9:355–362. doi: 10.1089/neu.1992.9.355. [DOI] [PubMed] [Google Scholar]
- Napolitano E, Elovic EP, Qureshi AI. Pharmacological stimulant treatment of neurocognitive and functional deficits after traumatic and non-traumatic brain injury. Med Sci Monit. 2005;11:RA212–RA220. [PubMed] [Google Scholar]
- Ng K, Mikulis DJ, Glazer J, Kabani N, Till C, Greenberg G, et al. Magnetic resonance imaging evidence of progression of subacute brain atrophy in moderate to severe traumatic brain injury. Arch Phys Med Rehabil. 2008;89(12) Suppl:S35–S44. doi: 10.1016/j.apmr.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Nichol AD, Cooper DJ, POLAR Study Investigators on behalf of the ANZICS-Clinical Trials Group, EPO Study Investigators on behalf of the ANZICS-Clinical Trials Group Can we improve neurological outcomes in severe traumatic brain injury? Something old (early prophylactic hypothermia) and something new (erythropoietin) Injury. 2009;40:471–478. doi: 10.1016/j.injury.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Hillered L, Ponten U, Ungerstedt U. Changes in cortical extracellular levels of energy-related metabolites and amino acids following concussive brain injury in rats. J Cereb Blood Flow Metab. 1990;10:631–637. doi: 10.1038/jcbfm.1990.115. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Hillered L, Olsson Y, Sheardown MJ, Hansen AJ. Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J Cereb Blood Flow Metab. 1993;13:183–192. doi: 10.1038/jcbfm.1993.22. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Ronne-Engstrom E, Flink R, Ungerstedt U, Carlson H, Hillered L. Epileptic seizure activity in the acute phase following cortical impact trauma in rat. Brain Res. 1994;637:227–232. doi: 10.1016/0006-8993(94)91237-8. [DOI] [PubMed] [Google Scholar]
- Nilsson P, Gazelius B, Carlson H, Hillered L. Continuous measurement of changes in regional cerebral blood flow following cortical compression contusion trauma in the rat. J Neurotrauma. 1996;13:201–207. doi: 10.1089/neu.1996.13.201. [DOI] [PubMed] [Google Scholar]
- O'Connor C, Heath DL, Cernak I, Nimmo AJ, Vink R. Effects of daily versus weekly testing and pre-training on the assessment of neurologic impairment following diffuse traumatic brain injury in rats. J Neurotrauma. 2003;20:985–993. doi: 10.1089/089771503770195830. [DOI] [PubMed] [Google Scholar]
- O'Connor CA, Cernak I, Johnson F, Vink R. Effects of progesterone on neurologic and morphologic outcome following diffuse traumatic brain injury in rats. Exp Neurol. 2007;205:145–153. doi: 10.1016/j.expneurol.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Okiyama K, Smith DH, Gennarelli TA, Simon RP, Leach M, McIntosh TK. The sodium channel blocker and glutamate release inhibitor BW1003C87 and magnesium attenuate regional cerebral edema following experimental brain injury in the rat. J Neurochem. 1995;64:802–809. doi: 10.1046/j.1471-4159.1995.64020802.x. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- Passineau MJ, Green EJ, Dietrich WD. Therapeutic effects of environmental enrichment on cognitive function and tissue integrity following severe traumatic brain injury in rats. Exp Neurol. 2001;168:373–384. doi: 10.1006/exnr.2000.7623. [DOI] [PubMed] [Google Scholar]
- Piek J. Decompressive surgery in the treatment of traumatic brain injury. Curr Opin Crit Care. 2002;8:134–138. doi: 10.1097/00075198-200204000-00008. [DOI] [PubMed] [Google Scholar]
- Ponsford JL, Myles PS, Cooper DJ, McDermott FT, Murray LJ, Laidlaw J, et al. Gender differences in outcome in patients with hypotension and severe traumatic brain injury. Injury. 2008;39:67–76. doi: 10.1016/j.injury.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancan M, Otto VI, Hans VH, Gerlach I, Jork R, Trentz O, et al. Upregulation of ICAM-1 and MCP-1 but not of MIP-2 and sensorimotor deficit in response to traumatic axonal injury in rats. J Neurosci Res. 2001;63:438–446. doi: 10.1002/1097-4547(20010301)63:5<438::AID-JNR1039>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196:126–137. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Reid WM, Rolfe A, Register D, Levasseur JE, Churn SB, Sun D. Strain-related differences after experimental traumatic brain injury in rats. J Neurotrauma. 2010;27:1243–1253. doi: 10.1089/neu.2010.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riess P, Bareyre FM, Saatman KE, Cheney JA, Lifshitz J, Raghupathi R, et al. Effects of chronic, post-injury Cyclosporin A administration on motor and sensorimotor function following severe, experimental traumatic brain injury. Restor Neurol Neurosci. 2001;18:1–8. [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Risling M, Plantman S, Angeria M, Rostami E, Bellander BM, Kirkegaard M, et al. Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage. 2011;54S1:S89–S97. doi: 10.1016/j.neuroimage.2010.05.031. [DOI] [PubMed] [Google Scholar]
- Robertson CL, Clark RS, Dixon CE, Alexander HL, Graham SH, Wisniewski SR, et al. No long-term benefit from hypothermia after severe traumatic brain injury with secondary insult in rats. Crit Care Med. 2000;28:3218–3223. doi: 10.1097/00003246-200009000-00017. [DOI] [PubMed] [Google Scholar]
- Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17:367–388. doi: 10.1089/neu.2000.17.367. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bareyre FM, Grady MS, McIntosh TK. Acute cytoskeletal alterations and cell death induced by experimental brain injury are attenuated by magnesium treatment and exacerbated by magnesium deficiency. J Neuropathol Exp Neurol. 2001;60:183–194. doi: 10.1093/jnen/60.2.183. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT, Workshop Scientific Team and Advisory Panel Members Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saljo A, Bolouri H, Mayorga M, Svensson B, Hamberger A. Low-level blast raises intracranial pressure and impairs cognitive function in rats: prophylaxis with processed cereal feed. J Neurotrauma. 2010;27:383–389. doi: 10.1089/neu.2009.1053. [DOI] [PubMed] [Google Scholar]
- Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- Sarrafzadeh AS, Kiening KL, Unterberg AW. Neuromonitoring: brain oxygenation and microdialysis. Curr Neurol Neurosci Rep. 2003;3:517–523. doi: 10.1007/s11910-003-0057-2. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Dunn L. A model of acute subdural hematoma in the mouse. J Neurotrauma. 2001;18:1241–1246. doi: 10.1089/089771501317095278. [DOI] [PubMed] [Google Scholar]
- Sawauchi S, Marmarou A, Beaumont A, Signoretti S, Fukui S. Acute subdural hematoma associated with diffuse brain injury and hypoxemia in the rat: effect of surgical evacuation of the hematoma. J Neurotrauma. 2004;21:563–573. doi: 10.1089/089771504774129892. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury – an inflammatory disease. Brain Res Brain Res Rev. 2005;48:388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Seelig JM, Becker DP, Miller JD, Greenberg RP, Ward JD, Choi SC. Traumatic acute subdural hematoma: major mortality reduction in comatose patients treated within four hours. N Engl J Med. 1981;304:1511–1518. doi: 10.1056/NEJM198106183042503. [DOI] [PubMed] [Google Scholar]
- Shear DA, Williams AJ, Sharrow K, Lu XC, Tortella FC. Neuroprotective profile of dextromethorphan in an experimental model of penetrating ballistic-like brain injury. Pharmacol Biochem Behav. 2009;94:56–62. doi: 10.1016/j.pbb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Shimamura M, Garcia JM, Prough DS, Hellmich HL. Laser capture microdissection and analysis of amplified antisense RNA from distinct cell populations of the young and aged rat brain: effect of traumatic brain injury on hippocampal gene expression. Brain Res Mol Brain Res. 2004;122:47–61. doi: 10.1016/j.molbrainres.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Shohami E, Beit-Yannai E, Horowitz M, Kohen R. Oxidative stress in closed-head injury: brain antioxidant capacity as an indicator of functional outcome. J Cereb Blood Flow Metab. 1997;17:1007–1019. doi: 10.1097/00004647-199710000-00002. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT. The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma. 2001;18:607–614. doi: 10.1089/089771501750291846. [DOI] [PubMed] [Google Scholar]
- Smith DH, Okiyama K, Gennarelli TA, McIntosh TK. Magnesium and ketamine attenuate cognitive dysfunction following experimental brain injury. Neurosci Lett. 1993;157:211–214. doi: 10.1016/0304-3940(93)90739-8. [DOI] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, et al. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma. 1995;12:169–178. doi: 10.1089/neu.1995.12.169. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil. 2003;18:307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- Sozda CN, Hoffman AN, Olsen AS, Cheng JP, Zafonte RD, Kline AE. Empirical comparison of typical and atypical environmental enrichment paradigms on functional and histological outcome after experimental traumatic brain injury. J Neurotrauma. 2010;27:1047–1057. doi: 10.1089/neu.2010.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spain A, Daumas S, Lifshitz J, Rhodes J, Andrews PJ, Horsburgh K, et al. Mild fluid percussion injury in mice produces evolving selective axonal pathology and cognitive deficits relevant to human brain injury. J Neurotrauma. 2010;27:1429–1438. doi: 10.1089/neu.2010.1288. [DOI] [PubMed] [Google Scholar]
- Stein SC, Spettell C, Young G, Ross SE. Delayed and progressive brain injury in closed-head trauma: radiological demonstration. Neurosurgery. 1993;32:25–30. doi: 10.1227/00006123-199301000-00004. discussion 30–31. [DOI] [PubMed] [Google Scholar]
- Stiefel MF, Tomita Y, Marmarou A. Secondary ischemia impairing the restoration of ion homeostasis following traumatic brain injury. J Neurosurg. 2005;103:707–714. doi: 10.3171/jns.2005.103.4.0707. [DOI] [PubMed] [Google Scholar]
- Sullivan HG, Martinez J, Becker DP, Miller JD, Griffith R, Wist AO. Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg. 1976;45:521–534. [PubMed] [Google Scholar]
- Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol. 2000a;161:631–637. doi: 10.1006/exnr.1999.7282. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Hicks RR, Gibson TR, Fletcher-Turner A, Scheff SW. Dose-response curve and optimal dosing regimen of cyclosporin A after traumatic brain injury in rats. Neuroscience. 2000b;101:289–295. doi: 10.1016/s0306-4522(00)00380-8. [DOI] [PubMed] [Google Scholar]
- Svetlov SI, Prima V, Kirk DR, Gutierrez H, Curley KC, Hayes RL, et al. Morphologic and Biochemical Characterization of Brain Injury in a Model of Controlled Blast Overpressure Exposure. J Trauma. 2010;69:795–804. doi: 10.1097/TA.0b013e3181bbd885. [DOI] [PubMed] [Google Scholar]
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J. A systematic review of brain injury epidemiology in Europe. Acta Neurochir (Wien) 2006;148:255–268. doi: 10.1007/s00701-005-0651-y. discussion 268. [DOI] [PubMed] [Google Scholar]
- Tan AA, Quigley A, Smith DC, Hoane MR. Strain differences in response to traumatic brain injury in Long-Evans compared to Sprague-Dawley rats. J Neurotrauma. 2009;26:539–548. doi: 10.1089/neu.2008.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya K, Marmarou CR, Okuno K, Prieto R, Marmarou A. Effect of secondary insults upon aquaporin-4 water channels following experimental cortical contusion in rats. J Neurotrauma. 2010;27:229–239. doi: 10.1089/neu.2009.0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR, Anderson GD, Winn HR, Ellenbogen RG, Britz GW, Schuster J, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6:29–38. doi: 10.1016/S1474-4422(06)70630-5. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, et al. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Thornton E, Vink R, Blumbergs PC, Van Den Heuvel C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006;1094:38–46. doi: 10.1016/j.brainres.2006.03.107. [DOI] [PubMed] [Google Scholar]
- Tsuchida E, Bullock R. The effect of the glycine site-specific N-methyl-D-aspartate antagonist ACEA1021 on ischemic brain damage caused by acute subdural hematoma in the rat. J Neurotrauma. 1995;12:279–288. doi: 10.1089/neu.1995.12.279. [DOI] [PubMed] [Google Scholar]
- Tsuchida E, Harms JF, Woodward JJ, Bullock R. A use-dependent sodium channel antagonist, 619C89, in reduction of ischemic brain damage and glutamate release after acute subdural hematoma in the rat. J Neurosurg. 1996;85:104–111. doi: 10.3171/jns.1996.85.1.0104. [DOI] [PubMed] [Google Scholar]
- Turner RJ, Dasilva KW, O'Connor C, van den Heuvel C, Vink R. Magnesium gluconate offers no more protection than magnesium sulphate following diffuse traumatic brain injury in rats. J Am Coll Nutr. 2004;23:541S–544S. doi: 10.1080/07315724.2004.10719399. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Milman A, Holloway HW, Li Y, Harvey BK, Shen H, et al. Apoptotic and behavioral sequelae of mild brain trauma in mice. J Neurosci Res. 2007;85:805–815. doi: 10.1002/jnr.21160. [DOI] [PubMed] [Google Scholar]
- Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg. 2000;93:815–820. doi: 10.3171/jns.2000.93.5.0815. [DOI] [PubMed] [Google Scholar]
- Vespa P, Bergsneider M, Hattori N, Wu HM, Huang SC, Martin NA, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25:763–774. doi: 10.1038/sj.jcbfm.9600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, O'Connor CA, Nimmo AJ, Heath DL. Magnesium attenuates persistent functional deficits following diffuse traumatic brain injury in rats. Neurosci Lett. 2003;336:41–44. doi: 10.1016/s0304-3940(02)01244-2. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Willard LA, Kline AE, Wenger MK, Bolinger BD, Ren D, et al. Evaluation of estrous cycle stage and gender on behavioral outcome after experimental traumatic brain injury. Brain Res. 2004;998:113–121. doi: 10.1016/j.brainres.2003.11.027. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Kline AE, Ren D, Willard LA, Wenger MK, Zafonte RD, et al. Gender associations with chronic methylphenidate treatment and behavioral performance following experimental traumatic brain injury. Behav Brain Res. 2007;181:200–209. doi: 10.1016/j.bbr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley AR, Mir AK. Targeting the Nogo-A signalling pathway to promote recovery following acute CNS injury. Curr Pharm Des. 2007;13:2470–2484. doi: 10.2174/138161207781368611. [DOI] [PubMed] [Google Scholar]
- Wang D, Jiang R, Liu L, Dong JF, Zhang JN. Membrane neovascularization and drainage of subdural hematoma in a rat model. J Neurotrauma. 2010;27:1489–1498. doi: 10.1089/neu.2009.1057. [DOI] [PubMed] [Google Scholar]
- Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Hartings JA, Lu XC, Rolli ML, Dave JR, Tortella FC. Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma. 2005;22:313–331. doi: 10.1089/neu.2005.22.313. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Hartings JA, Lu XC, Rolli ML, Tortella FC. Penetrating ballistic-like brain injury in the rat: differential time courses of hemorrhage, cell death, inflammation, and remote degeneration. J Neurotrauma. 2006a;23:1828–1846. doi: 10.1089/neu.2006.23.1828. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Ling GS, Tortella FC. Severity level and injury track determine outcome following a penetrating ballistic-like brain injury in the rat. Neurosci Lett. 2006b;408:183–188. doi: 10.1016/j.neulet.2006.08.086. [DOI] [PubMed] [Google Scholar]
- Wright DW, Kellermann AL, Hertzberg VS, Clark PL, Frankel M, Goldstein FC, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391–402. doi: 10.1016/j.annemergmed.2006.07.932. [DOI] [PubMed] [Google Scholar]
- Xiao-Sheng H, Sheng-Yu Y, Xiang Z, Zhou F, Jian-ning Z. Diffuse axonal injury due to lateral head rotation in a rat model. J Neurosurg. 2000;93:626–633. doi: 10.3171/jns.2000.93.4.0626. [DOI] [PubMed] [Google Scholar]
- Yang L, Afroz S, Michelson HB, Goodman JH, Valsamis HA, Ling DS. Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury. J Neurotrauma. 2010;27:1541–1548. doi: 10.1089/neu.2009.1244. [DOI] [PubMed] [Google Scholar]
- Zafonte R, Friedewald WT, Lee SM, Levin B, Diaz-Arrastia R, Ansel B, et al. The citicoline brain injury treatment (COBRIT) trial: design and methods. J Neurotrauma. 2009;26:2207–2216. doi: 10.1089/neu.2009.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Groff RF, 4th, Chen XH, Browne KD, Huang J, Schwartz ED, et al. Hemostatic and neuroprotective effects of human recombinant activated factor VII therapy after traumatic brain injury in pigs. Exp Neurol. 2008;210:645–655. doi: 10.1016/j.expneurol.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweckberger K, Eros C, Zimmermann R, Kim SW, Engel D, Plesnila N. Effect of early and delayed decompressive craniectomy on secondary brain damage after controlled cortical impact in mice. J Neurotrauma. 2006;23:1083–1093. doi: 10.1089/neu.2006.23.1083. [DOI] [PubMed] [Google Scholar]