

Abstract
The global incidence of obesity continues to rise and is a major driver of morbidity and mortality through cardiovascular and cerebrovascular diseases. Animal models used in the discovery of novel treatments for obesity range from straightforward measures of food intake in lean rodents to long-term studies in animals exhibiting obesity due to the continuous access to diets high in fat. The utility of these animal models can be extended to determine, for example, that weight loss is due to fat loss and/or assess whether beneficial changes in key plasma parameters (e.g. insulin) are evident. In addition, behavioural models such as the behavioural satiety sequence can be used to confirm that a drug treatment has a selective effect on food intake. Typically, animal models have excellent predictive validity whereby drug-induced weight loss in rodents subsequently translates to weight loss in man. However, despite this, at the time of writing orlistat (Europe; USA) remains the only drug currently marketed for the treatment of obesity, with sibutramine having recently been withdrawn from sale globally due to the increased incidence of serious, non-fatal cardiovascular events. While the utility of rodent models in predicting clinical weight loss is detailed, the review also discusses whether animals can be used to predict adverse events such as those seen with recent anti-obesity drugs in the clinic.
LINKED ARTICLES
This article is part of a themed issue on Translational Neuropharmacology. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2011.164.issue-4
Keywords: obesity, animal models, DIO, Qnexa, anti-obesity drug
Introduction
Among healthcare experts around the world, there is agreement that the global epidemic of obesity will be one of the leading causes of morbidity and mortality for current and future generations, unless the rise in the prevalence of this disorder is reversed. Indeed, the metabolic consequences of obesity are drivers of other life-threatening disorders including dyslipidaemia, hypertension, atherogenesis and Type 2 diabetes (for review see Heal et al., 2009). Once considered to be a problem mainly in Western cultures, developing nations have now joined the ranks of countries burdened by obesity. Indeed, a recent report from the World Health Organisation estimated that in 2008 approximately 500 million adults were obese and 1.5 billion were overweight worldwide (World Health Organisation, 2011).
In terms of pharmacotherapy for obesity, only four new drugs have been registered for the treatment of obesity over the last 15 years: dexfenfluramine (Redux), sibutramine (Meridia, Reductil), orlistat (Xenical) and rimonabant (Acomplia). Furthermore, due to the incidence of side effects, orlistat (Europe; USA) remains, at the time of writing, the only marketed drug, with sibutramine having recently been withdrawn in Europe (European Medicines Agency (EMEA, 2010) and the USA (voluntarily by Abbott) due to an increased risk of serious, non-fatal cardiovascular events, such as stroke or heart attack in the Sibutramine Cardiovascular OUTcome (SCOUT) trial. This situation appeared unlikely to change in the near future because both Qnexa (a proprietary formulation of topiramate and phentermine) and lorcaserin (a 5-HT2C receptor agonist) suffered setbacks when the Food and Drug Administration (FDA)-appointed expert advisory panel recommended that the products should not be approved for clinical use in the USA despite significant weight loss in clinical trials (Gadde and Allison, 2009; Smith et al., 2010). Importantly, the principal reason for drug withdrawals has been due to concerns over safety rather than the effect on body weight per se. For example, the CB1 receptor antagonist, rimonabant, significantly reduced body weight in clinical trials (Després et al., 2005; Van Gaal et al., 2005) but was not approved in the USA because treatment had been linked to severe psychiatric adverse events, e.g. anxiety, depression and suicidal ideation. Subsequently, the EMEA suspended the drug (EMEA, 2008). Interestingly, although some safety concerns remain, recently Contrave, a fixed dose combination therapy for obesity in a single tablet consisting of sustained release formulations of both naltrexone and bupropion, has been recommended for approval by the FDA advisory panel of experts. If this recommendation is subsequently endorsed by the FDA early in 2011, then this drug will be registered for the treatment of obesity.
Due to the poor track record of approved drugs in this therapeutic indication, there remains an enormous unmet need for the discovery of safer compounds delivering superior efficacy. As a result, the importance of animal models in not only detecting changes in body weight but also providing confidence that these changes are behaviourally specific and not a result of drug-induced side effects, is of critical importance. In addition to the key role in the screening of novel compounds for effects on food intake and/or body weight, animal models have utility in the identification of the physiological and genetic basis of obesity, which can result in the discovery and validation of new therapeutic targets [e.g. through the generation and characterization of transgenic animals (Powell, 2006)].
This review details a number of rodent models, focusing in particular on those with especial relevance to the discovery of new drugs for the treatment of obesity. While no single model is necessarily applicable to all drug research programmes, it is suggested that certain models have greater validity than others and, where possible, should be used in preference in order to achieve the best possible prediction of the outcome in clinical practice.
Animal models used in the discovery of novel ligands for the treatment of obesity
The regulation of body weight is dependent upon the interaction between food intake and energy expenditure. For example, if, over an extended period of time daily food intake increases with an insufficient increase in energy expenditure (the energy imbalance gap) then body weight will increase. This is typically the case in the development of obesity where the main driver is increased total energy intake (Swinburn et al., 2009). Specifically, access to cheap, highly palatable, calorie-dense foods has led to an increase in energy intake, which has been coupled with an increasingly sedentary lifestyle (e.g. increased use of cars, increased use of video games as a leisure activity, etc.). This relationship between food intake, energy expenditure and body weight leads not only to different mechanisms by which a drug may reduce body weight (i.e. through the reduction of food intake, stimulation of energy expenditure, or both), but is also of relevance in the selection and development of appropriate animal models for evaluating anti-obesity potential. For example, an acute model of food intake may have little utility in detecting compounds that may reduce body weight through the stimulation of energy expenditure.
Acute models of food intake
Rats and mice are the predominant models of human obesity used in drug discovery although phylogenetically, they are not closely related to man. However, like man, they are omnivores and have complex taste and digestive systems for identifying and consuming a variety of different foods. Furthermore, there are similarities neuroanatomically in those brain areas thought to play a role in the control of food intake and it is well established that a number of different neurotransmitters and peptides produce similar effects on food intake and energy homeostasis in laboratory rodents and man. Numerous pharmacological approaches for the treatment of obesity have focused on drug targets that reduce food intake. For example, the clinically effective drugs d-fenfluramine, sibutramine, rimonabant and lorcaserin all reduce food intake when given acutely to lean rats or mice (Neill and Cooper, 1989; Jackson et al., 1997; Colombo et al., 1998; Vickers et al., 1999; Verty et al., 2004; Smith et al., 2008; Thomsen et al., 2008). Hence, investigating the acute effect of candidate drugs on the food intake of a lean rat or mouse is not only a rapid screen, but provides information as to both the relative potencies of compounds to inhibit food intake and, also, the duration of action of compounds in vivo. Accordingly, superior compounds can be selected and studied further in, for example, chronic studies investigating effects on body weight.
Inter-laboratory variations in the methods used for detecting drug-induced changes in acute food intake are numerous. However, most if not all study paradigms tend to involve stimulating food intake in rats or mice to some extent and subsequently evaluating the effect of a treatment where baseline food intake is elevated. Male animals are usually used to avoid the influence of the oestrus cycle on food intake (Asarian and Geary, 2006). Perhaps the most straightforward method is to fast an animal before the test, e.g. overnight (Vickers et al., 2001; Hadcock et al., 2010). This method has advantages in that it is straightforward and induces a robust increase in the food intake of vehicle-treated controls. However, it could be argued that this approach is not physiological (e.g. an overnight fast would be a major stressor for a mouse) and there is evidence that the potency and duration of action of drug treatments can differ in such fasting paradigms compared with more refined approaches where baseline food intake may be stimulated either through the use of a palatable diet such as wet mash (e.g. Neill and Cooper, 1989; Vickers et al., 1996, 1999) or the use of reverse-phase lighting so that the initial part of the study is conducted during the dark period when rats in particular exhibit high levels of food intake (Jackson et al., 1997; Hadcock et al., 2010). Typically, in order to reduce food intake by the same amount (e.g. 50% of vehicle), higher doses are often needed in re-feeding models compared with freely-feeding models. For example, the CB1 receptor antagonist, CE-178253, more potently reduced food intake in a dark cycle induced feeding paradigm (i.e. spontaneous feeding) compared with an assay where animals were allowed to feed after an overnight fast (Hadcock et al., 2010). Such differences may be due to the large drive to eat of animals feeding after a fast, which needs to be overcome by higher doses of the drug.
One consideration when screening drugs in an acute food intake assay is that the test will be insensitive to drugs with a delayed onset of action. Such drugs include 5-HT6 receptor partial agonists (Heal et al., 2008) and MCH1 receptor antagonists (Shearman et al., 2003). On such occasions initial screens may require repeated drug administration, or, alternatively, in the case of MCH1 receptor antagonists, blockade of MCH-induced food intake (Shearman et al., 2003). Another disadvantage of acute food intake tests is that the assay is not relevant to all mechanisms of drug action. For example, an acute food intake model will prove of little value in the identification of either drugs that increase energy expenditure or lipase inhibitors, which may reduce body weight through the reduced absorption of fat from the gut.
In general, acute food intake studies do not need to be undertaken in obese animals. Furthermore, such studies can be run in rats or mice, because both species are sensitive to the acute effects of clinically effective compound classes such as serotonin and noradrenaline reuptake inhibitors (e.g. sibutramine), CB1 receptor antagonists (e.g. rimonabant) and 5-HT2C receptor agonists (e.g. lorcaserin) (Hewitt et al., 2002; Matsumoto and Iijima, 2003; Poncelet et al., 2003; Halford et al., 2010). At this stage of the discovery process the use of mice may prove advantageous because the amount of each compound required is much less. Accordingly, resource can be focused on the synthesis of novel ligands rather than scaling up existing candidates with suitable in vitro profiles for rat studies. That said, in some instances species selection is of critical importance. For example, in rats and humans 5-HT6 receptors are widely expressed and focused particularly in the basal ganglia (Hirst et al., 2003). In contrast, not only is the 5-HT6 receptor less widely expressed in the mouse CNS, but it has a different pharmacological profile to the other two species despite similar sequence homology (Hirst et al., 2003). Accordingly, where 5-HT6 receptor ligands are to be developed for the treatment of obesity (or any other disorder), then rat models should be used in preference to mouse models.
Long-term (chronic) models of food intake and body weight
Acute studies are typically undertaken in lean male animals in order to profile compounds rapidly and obtain information in regard to the potency, efficacy, duration of action, and potentially the side effect profile of a compound, in vivo. Such models can be used to select compounds that have a suitable profile for chronic testing (e.g. 28 days) because effects of a drug over a longer dosing period are usually required in order to demonstrate a maintained reduction of body weight. Sub-chronic or chronic feeding studies are sometimes performed in normal, lean rats and mice (e.g. Vickers et al., 2000; 2003a; Smith et al., 2008) or in animal models of ‘overweight’ such as rats maintained on a high-fat diet for a relatively short period (e.g. 2 weeks) before the start of the study (Thomas et al., 2006). These studies can be used as a screen to bridge the gap between acute feeding studies and chronic feeding studies in obese animals that are expensive.
The choice of sex in animal models of obesity is important. In humans, adipose tissue is distributed subcutaneously and in the abdomen (visceral fat). Females have relatively more subcutaneous fat than males whereas, in contrast, males have more visceral fat, which is associated with the complications of obesity (Wajchenberg, 2000). Recent studies have shown similar differences in the amount and distribution of adipose tissue in rats and in the circulating levels of the hormones related to adiposity. Thus, female rats have more fat and especially more subcutaneous fat than male rats and male rats have more visceral fat (Clegg et al., 2003a,b). In addition, although the levels of the adipose hormones, leptin and insulin, generally correlate with the amount of adipose tissue in both males and females, females are more sensitive to the inhibitory effects of leptin on food intake whereas male rats are more responsive to insulin (Clegg et al., 2003b). These differences argue that the effects of potential anti-obesity agents on body weight should be evaluated in both male and female animals.
Although rats and mice are the predominant models of human obesity, there are some important differences in physiology between rodents and man (e.g. rats do not have a vomit reflex or gall bladder). Furthermore, the standard environment for most rodent feeding studies is not analogous to the human situation as animals are typically individually housed (which restricts social interaction and therefore physical activity), in relatively small cages (which restrict physical activity) and are normally housed at temperatures of 20–23°C, which are several degrees lower than their thermoneutral temperature (29–32°C) such that animals will be expending energy to keep warm. In addition, food is readily available at all times, which does not always happen in man. Another limitation of these models is that they do not take into account the complex psychological factors that control food intake and can lead to overeating in man (Halford et al., 2010). That said, despite these weaknesses there are a number of strengths to obese animal models and these are discussed later.
Broadly speaking, obese animal models can be subdivided into two types: dietary-induced models of obesity and genetic models of obesity. An animal model of obesity will ideally mimic as closely as possible the human condition in terms of the causes of the disorder (construct validity) and the phenomenological similarity between the characteristics exhibited by the animal model and the specific symptoms of the human condition (face validity). Furthermore, the effects of pharmacological manipulation in the model should be identical to the clinical outcome in man (predictive validity). Although single gene mutations observed in some animal models have been linked to human obesity in the case of leptin (Montague et al., 1997), the leptin receptor (Clément et al., 1998) and others, such instances are rare as mentioned below and it is generally considered that an individual's susceptibility to obesity in the current environment of high calorie foods and reduced activity is determined by a polygenetic background (Bell et al., 2005; Mutch and Clément, 2006). Accordingly, polygenetic models such as the dietary-induced obese rat and mouse are of especial importance.
Dietary-induced obesity models
The first use of a ‘high-fat diet’ to induce obesity in rats was by Masek and Fabry (1959). Since then numerous methods have been published using the general approach whereby normal, lean rats or mice are provided free access to diets high in fat over a period of 3–4 months. With time animals exhibit increased weight gain characterized principally by a marked increase in body fat (e.g. Harrold et al., 2000; Jones et al., 2001; Naderali et al., 2001; Ravinet-Trillou et al., 2003; Li et al., 2008; Madsen et al., 2010). In addition, although the animals do not typically develop diabetes (hyperglycaemia) they exhibit insulin resistance, glucose intolerance, elevated plasma leptin and a mild dyslipidaemia with plasma cholesterol and triglyceride often elevated compared with appropriate controls on a standard diet (Dickinson et al., 1998; Harrold et al., 2000; Naderali et al., 2001; Ravinet-Trillou et al., 2003; Li et al., 2008; Madsen et al., 2010). Furthermore, there are some reports that blood pressure is altered in dietary-induced obese (DIO) rats (e.g. (Lobley et al., 2007). Indeed, some workers have developed models of dietary-induced obesity using spontaneously hypertensive rats (Miesel et al., 2010). Importantly, such changes in DIO mice and rats mimic the changes observed in obese patients (See Table 1).
Table 1.
Comparison of genetically-obese and DIO rodent models to human obesity
| Characteristic | ob/ob mouse | db/db mouse | fa/fa zucker rat | DIO rat/mouse | Common human obesity |
|---|---|---|---|---|---|
| Sex | Male or female | Male or female | Male or female | Male or female | Male or female1 |
| Polygenic basis | X | X | X | ✓ | ✓ |
| Access to calorie-dense high-fat diet | X | X | X | ✓ | ✓ |
| Marked visceral adiposity | ✓ | ✓ | ✓ | ✓ | ✓ |
| Hyperleptinaemia | X | ✓2 | ✓2 | ✓ | ✓ |
| Hyperinsulinaemia | ✓ | ✓ | ✓ | ✓ | ✓ |
| Hyperglycaemic | ✓ | ✓ | X | X/✓ | X/✓ |
| Hypertensive | ?/✓ | ?/✓ | ?/✓ | ?/✓ | ✓ |
| Sensitive to clinically effective weight loss agents | ?/✓ | ?/✓ | ?/✓ | ✓ | ✓ |
The table illustrates the similarities between the obesity and cardio-metabolic risk factors observed in DIO rats and mice, with humans.
However, greater than 70% of patients recruited into trials of novel anti-obesity drugs are female.
Increased plasma leptin levels are evident compared with age-matched controls. However, unlike DIO animals or most human patients with obesity, db/db mice and fa/fa Zucker rats have mutations of the leptin receptor such that leptin receptor function is impaired.
There are numerous differences in the methodology used throughout the literature, with some laboratories using commercially available high fat diets (e.g. Pierroz et al., 2002; Thornton-Jones et al., 2006) and others using cafeteria diets where animals have a choice of various palatable foods such as chocolate, peanuts, condensed milk, etc., which encourages overeating and is closer to the human situation than maintaining rats on a single food source (e.g. Naderali et al., 2001; Fisas et al., 2006). Although some diet-dependent differences in the phenotype of obese animals have been reported (Buettner et al., 2006), in general, despite across laboratory methodological variations, the metabolic profiles of rodents allowed long-term access to high-fat diets are reasonably uniform. Other differences across laboratories include the use only of animals that ‘respond’ to the high fat diet in terms of weight gain. Perhaps the most well-known example is the DIO and diet-resistant out-bred rat model of Levin and colleagues (Levin et al., 1997; Madsen et al., 2010).
Irrespective of the precise nature of the model used, the changes seen in DIO animals are remarkably consistent with those seen in obese patients in the clinic (for summary see Table 1). Indeed, functional genomic studies comparing gene expression changes in DIO rats and obese humans have demonstrated significant commonalities for various tissues and biological pathways affected in obesity (Li et al., 2008). For example, not only was the gene expression profile of adipose tissue similar in the DIO rat to that of obese Pima Indians, but genes in immune response and angiogenesis pathways were significantly up-regulated in obese rats and humans in comparison with non-obese controls (Li et al., 2008). Such data reinforce both the face and construct validity of the DIO rat as a model of human obesity. Using the functional genomics approach at least one key difference between the obese rat and human was identified. Hence, genes involved in lipid metabolism tended to be down-regulated in obese humans but not in the DIO rat model used in the publication (Li et al., 2008). While this finding illustrates that the DIO rat model is not a perfect match to the human situation, it also appears to be consistent with data suggesting that in the study of lipid metabolism, rats and mice are inappropriate models of the human situation and other rodent species such as the hamster are to be preferred (Kris-Etherton and Dietschy, 1997).
The ability of DIO animals to predict clinically meaningful weight loss is good. Hence, drugs such as d-fenfluramine (Bradbury et al., 2005), sibutramine (Jackson et al., 2004; 2005; Fisas et al., 2006; Madsen et al., 2010), orlistat (Hogan et al., 1987; Hauptman et al., 2000; Jackson et al., 2004) and rimonabant (Ravinet-Trillou et al., 2003; Jackson et al., 2005) all significantly reduce body weight in DIO rats or mice and are often used as reference agents in studies with novel ligands (e.g. Bradbury et al., 2005; Fisas et al., 2006; Hansen et al., 2010; Madsen et al., 2010). Accordingly, the model is widely used to gain insight into the potential weight loss a drug will deliver in the clinic. Because a placebo-subtracted weight loss of >5% maintained over 1 year is the primary efficacy end point for approval of anti-obesity drugs and sibutramine (for example) delivers a 5–10% reduction in the body weight of obese patients (Bray, 2001) then a dose of sibutramine that delivers approximately 10% weight loss in DIO animals would appear to be a sensible positive control to use in these studies (e.g. Fisas et al., 2006).
It could be argued that in rodent studies, reference compounds should be administered at doses reflecting their clinical steady-state exposure rather than the clinical % body weight loss. However, this is not practical. Many of the drugs that are identified using animal models have not been tested in man. Indeed, relatively little may be known about their pharmacokinetics in animals at the time of testing and they could not be given in comparable doses. Instead reference compounds are given at doses producing a clear endpoint, that is, a consistent degree of weight loss that matches their clinical efficacy as mentioned above.
Assessing the predictive validity of the DIO rat and mouse is a continuous process because it is only when compounds that are efficacious in an animal model are tested in the clinic, that the utility of the model is understood. Two novel drug applications recently reviewed by the FDA are the 5-HT2C receptor agonist, lorcaserin, and the proprietary combination of topiramate and phentermine, Qnexa. As one would expect, lorcaserin reduced body weight in DIO animals (e.g. the Levin rat model, Thomsen et al., 2008). Similarly, topiramate, phentermine and the combination were all observed to reduce body weight in the DIO rat (Jackson et al., 2007). In agreement with the clinical studies, the combination of phentermine and topiramate delivered greater effects on body weight compared with either treatment when dosed alone. Indeed, in the DIO rat the body weight loss with the combinations was substantial (15% cf. vehicle-treated controls) and greater than that typically observed with sibutramine in the model. The body weight and daily food intake data are shown in Figures 1 and 2 and illustrate that DIO animals are also useful in detecting the body weight efficacy of drug combinations and their overall effect on food intake. Such combination strategies are currently a popular approach in the development of novel anti-obesity agents. Indeed, the FDA advisory panel recently recommended that the buproprion/naltrexone fixed dose combination therapy, Contrave, be approved for the treatment of obesity. Accordingly, with the FDA expected to make a decision in January 2011, this proprietary combination may be the next anti-obesity drug registration. In preclinical studies, this combination reduces the acute food intake of DIO mice at doses of naltrexone and buproprion that do not significantly affect food intake when given alone (Greenway et al., 2009). To our knowledge this combination has not been tested long-term in a DIO rat model, and such data would be of especial interest. Further combination strategies being assessed in the clinic include pramlintide (synthetic amylin) and metreleptin (a human leptin analogue). This approach is not only effective in preclinical DIO rat studies using amylin and leptin (Roth et al., 2008) but the pramlintide/metreleptin combination also significantly reduced body weight in a 24 week study in obese or overweight patients (Ravussin et al., 2009).
Figure 1.
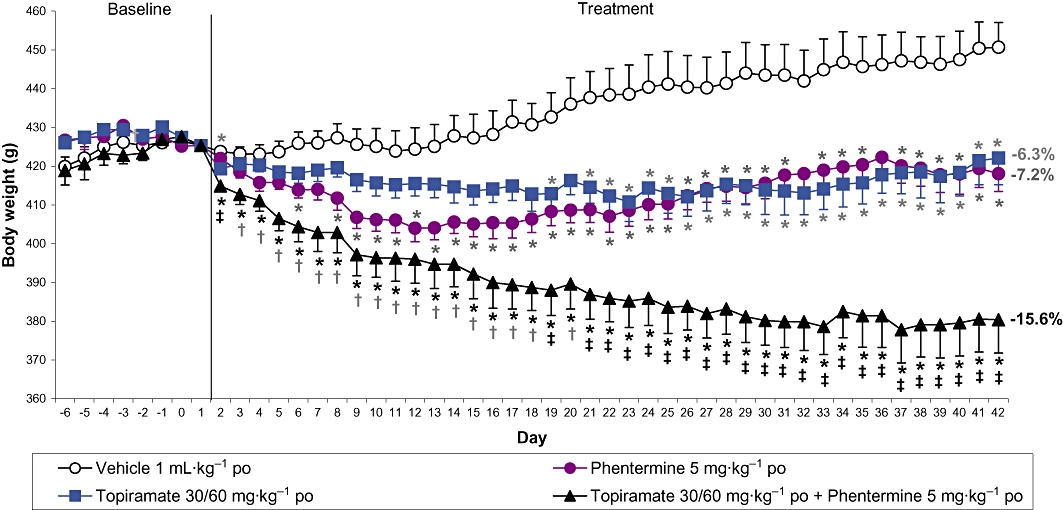
The effect of phentermine and topiramate alone and in combination on body weight in rats with dietary-induced obesity. Female Wistar rats were maintained on reverse-phase lighting (lights out 09.30–17.30 h) with free access to powdered high-fat chow, chocolate, peanuts and tap water during the induction of obesity (14 weeks) and throughout the feeding study. After a 7 day run-in period, during which rats were dosed orally with vehicle once daily, rats were dosed orally with vehicle, topiramate, phentermine or topiramate plus phentermine once daily for 41 days. Topiramate was given in a dose of 30 mg·kg−1 (increased to 60 mg·kg−1 on Day 15). Phentermine was given in a dose of 5 mg·kg−1. Rats, feeding jars and water bottles were weighed every day at the time of dosing, which was at the onset of the dark period. Topiramate and phentermine were dissolved in 1% Tylose MH50/0.1% poloxymer (in deionized water; dose volume 1 mL·kg−1). All doses are for the free base. Results are adjusted means ± SEM; n = 10. *P < 0.05 (vs. controls), †P < 0.05 (topiramate plus phentermine vs. topiramate alone), ‡P < 0.05 (topiramate plus phentermine vs. both topiramate and phentermine alone). Numbers represent % reduction in body weight compared with the control group on Day 42.
Figure 2.
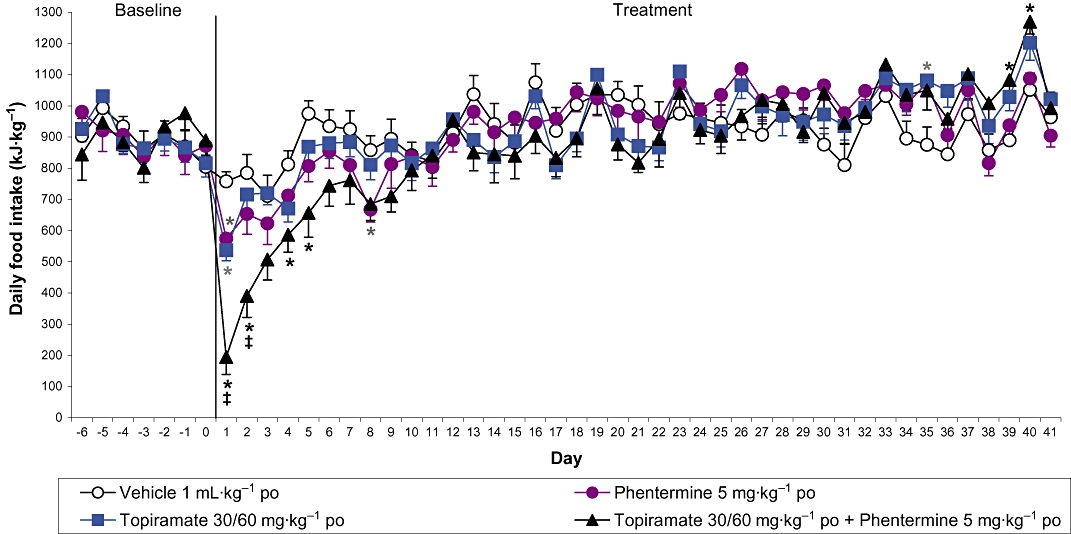
The effect of phentermine and topiramate alone and in combination on food intake in rats with dietary-induced obesity. Food intake data are expressed as kJ·kg−1 to take account of the different caloric values of the diet components (high fat chow, peanuts and chocolate). Results are adjusted means ± SEM; n = 9–10. *P < 0.05 (vs. controls), ‡P < 0.05 (topiramate plus phentermine vs. both topiramate and phentermine alone).
Statistical analysis can reveal whether the effects of two drugs on body weight, food intake and other factors are additive or synergistic in nature, that is, whether the response to the combination treatment is greater than predicted from the sum of the effects of the two drugs given alone. Such studies have shown that the reductions in body weight and food intake produced by the combination of topiramate and phentermine in DIO rats (Figures 1 and 2) are simply additive (unpublished data) whereas the reductions in body weight and food intake produced by the combination of amylin and leptin in DIO rats are synergistic as amylin restores leptin responsiveness in these animals (Trevaskis et al., 2008).
As discussed, the DIO rat model appears to have excellent predictive validity, even in regard to the modern trend towards the development of drug combinations for obesity. This is substantiated by Figure 3 where the weight loss induced by a number of drugs in the clinic is compared with the weight loss seen in the DIO rat.
Figure 3.
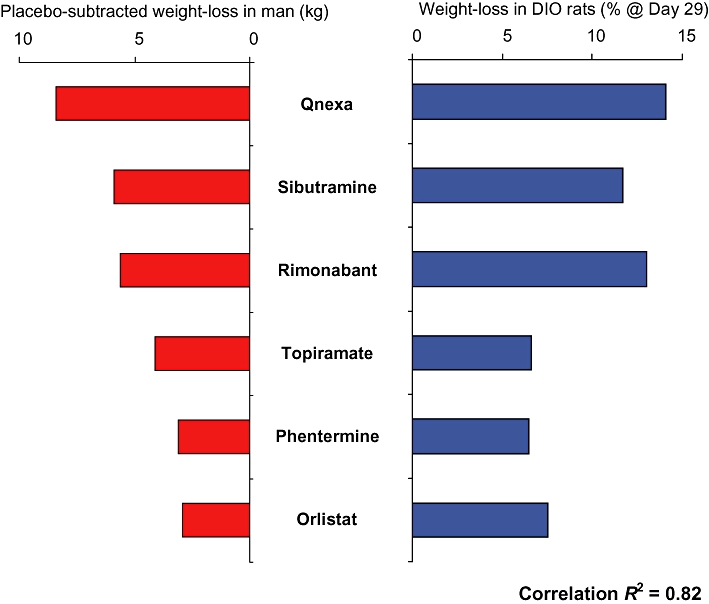
A comparison of the weight-loss produced by various anti-obesity drugs in man versus the body weight loss over a 28 day dosing period observed in the DIO rat. Correlation r2= 0.82. Figure adapted from Heal et al., 2011. DIO rat data taken from Jackson et al., 2004, Jackson et al., 2005, Jackson et al., 2007. Human data taken from Bray et al., 2003, http://www.vivus.com/pipeline/qnexa-obesity, Pi-Sunyer et al., 2006, Després et al., 2005, Apfelbaum et al., 1999, Hauptman et al., 2000, James et al., 2000.
Genetically modified models of obesity
Genetically obese animals that have been characterized following spontaneously occurring single gene mutations and are widely used as animal models of obesity include the ob/ob mouse, db/db mouse and the Zucker fa/fa rat (Ingalls et al., 1950; Zucker and Zucker, 1961; Coleman, 1978). The obesity in these animals becomes visible within several weeks of birth and they continue to put on weight and eventually become several times heavier than their lean counterparts with 50% of their body weight accounted for by fat. The phenotype of all these animals has now been well characterized (for review see Bailey and Flatt, 2003) and can depend on the background strain. All three models display hyperphagia, hyperinsulinaemia and hyperlipidaemia but differ in the degree of hyperglycaemia. Thus, db/db mice have markedly elevated plasma glucose levels and are often used as a model of diabetes whereas ob/ob mice display mild to moderate hyperglycaemia and Zucker fa/fa rats are not hyperglycaemic but are insulin-resistant (for review see Bailey and Flatt, 2003). As in obese patients, there are reports that genetically-obese animals such as the db/db mouse are hypertensive (e.g. Su et al., 2008), although such findings are not consistent across the literature and, unlike in obese humans (Kotsis et al., 2005), may be related to the incidence of sleeping behaviour and the light/dark cycle (Silvani et al., 2009). All three models also display a number of other abnormalities, notably in reproductive biology. Specifically, female ob/ob mice are infertile (Kennedy et al., 2010).
Each of these monogenetic models of obesity exhibits deficits in the signalling pathways involving the pleiotropic adipokine, leptin. In 1994, the obese gene and its protein, leptin, were discovered (Zhang et al., 1994). The mutation in ob/ob mice prevents the production of leptin whereas db/db mice and Zucker fa/fa rats have marked elevations in circulating leptin but have deficient leptin receptors (see Bailey and Flatt, 2003 and Kennedy et al., 2010 for reviews). The fact that the ob/ob mice are not hyperleptinaemic is a major discrepancy in the aetiology of the disease between ob/ob mice and common obesity in man where leptin levels are substantially increased (Correia and Haynes, 2007). Thus, in most mammals, including man, circulating leptin levels, correlate closely with the degree of adiposity (Maffei et al., 1995).
The discovery of leptin led to an extensive search for leptin or leptin receptor-deficient humans but it soon became apparent that only very small numbers of obese individuals had a loss-of-function mutation in this gene (12 people up to 2008; Farooqi and O'Rahilly, 2008). Furthermore, despite the finding that leptin-replacement therapy produced dramatic weight loss in leptin-deficient ob/ob mice and humans with congenital leptin deficiency (Farooqi and O'Rahilly, 2009), the results of clinical trials in normal obese patients using recombinant leptin were disappointing (Heymsfield et al., 1999). This was thought to be because polygenic common human obesity is associated with high circulating levels of leptin and leptin-resistance (Correia and Haynes, 2007). Polygenic DIO animals display hyperleptinaemia and are leptin-resistant and therefore mirror the normal obese state (Halaas et al., 1997; Dickinson et al., 1998; Roth et al., 2008; Madsen et al., 2010). It is clear that in contrast, monogenetic models such as ob/ob mice do not have as good construct or face validity and are an inferior model of common human obesity. While such models are routinely used in order to evaluate the effect of novel test compounds on body weight, caution should be taken when translating potential effects to man. For example, although the clinically effective CB1 receptor antagonist, rimonabant, reduces body weight in the fa/fa Zucker rat (Vickers et al., 2003b) and ob/ob mouse (Mohapatra et al., 2009), the effects of sibutramine in both these models, and others, appears to be modest (Day and Bailey, 1998; Matsumoto and Iijima, 2003) compared with the effects exhibited clinically. Indeed, the hypophagic effect of sibutramine is reported to be reduced in db/db mice, fa/fa Zucker rats and KK-Ay mice compared with wild-type animals (Matsumoto and Iijima, 2003). Furthermore, in our laboratory, doses of rimonabant that reduced body weight markedly in the DIO mouse did not affect the body weight of db/db mice (S. Vickers, unpubl. obs.). This contrasts with the clinical situation where, broadly speaking, sibutramine and rimonabant induce a similar weight loss in obese patients (for review see Vickers and Kennett, 2005).
Such examples indicate limitations in the predictive validity of these genetic models for assessing the effects of a drug on body weight loss and suggest that the use of DIO animals with their polygenetic basis and intact leptin system may provide a more robust approach for the screening of potential anti-obesity drugs. However, the relative sensitivity of genetically or dietary-induced obese animals (and appropriate wild-type or chow-fed controls) to reference anti-obesity drugs would ideally be tested more rigorously by the same laboratory using standardized conditions and matching drug exposure levels so that formal statistical analysis could be performed to confirm this hypothesis.
In addition to animals exhibiting an obese phenotype due to spontaneous gene mutations, molecular biology techniques have been used to produce obese animals by targeting specific genes. For example, mice with a targeted deletion of the 5-HT2C receptor exhibit mild obesity and Type 2 diabetes (Tecott et al., 1995). These models can provide useful information about the receptors and hormones that regulate the control of food intake and energy balance and can be used to provide proof of concept for novel anti-obesity targets. For example, the phenotype of the 5-HT2C receptor knockout mouse suggests that the 5-HT2C receptor is involved in the control of body weight and, in addition, suggests that 5-HT2C receptor agonists (such as the subsequently developed drug, lorcaserin) could be useful treatments for obesity. While such models may have value in further understanding the mechanisms involved in the control of food intake and body weight, and may provide useful tools in drug screening strategies for specific molecular targets, they do not reflect the human situation and, in general, are not used as models of obesity.
Long-term models of food intake and body weight: additional measurements
The evaluation of a test compound in an animal model over an extended dosing period such as 28 days is time-consuming and can be resource-intensive. For example, the animals are not only dosed and require husbandry on each day of the feeding study, but, in the case of dietary-induced obesity, require husbandry and handling during the induction of obesity, which can take several months. As a result, typically it is not only body weight and food intake that are measured during such studies because they also provide excellent opportunities to assess (for example) whether drug treatments have led to changes in water intake, improvements in glucose control or insulin sensitivity (e.g. by the performance of a glucose tolerance test), or to confirm that changes in body weight are attributable to fat loss (body composition analysis). In addition, blood samples can be taken to measure changes in key hormones and markers (e.g. leptin) or to assess drug exposure after (and during) chronic dosing.
In some instances (e.g. to investigate the potential role of a drug on energy expenditure) it is important to understand the contribution made by reduced food intake to the overall weight loss. Such studies usually involve pair-fed controls. Specifically, vehicle-treated animals are given the same amount of food as the drug treatment group and would be expected to lose the same amount of weight unless the reduction in body weight is due to factors other than, or in addition to, food intake. For example, if a compound acts to increase energy expenditure then the reduction in body weight of these animals will be greater than pair-fed controls. While such studies are routinely used (e.g. Vickers et al., 2003a; Thornton-Jones et al., 2006), one disadvantage of the approach is that the pair-fed control is likely to consume its daily food ration over a different time period. Specifically, an animal treated with an effective drug treatment may consume 10 g over a 24 h period whereas a pair-fed control is likely to be hungry and may consume this quantity over a much shorter time period (i.e. shortly after presentation of the food).
Water intake is often (although not always) measured during chronic feeding studies. Reductions in water intake are often secondary to reduced food intake. However, some drugs increase water intake while reducing food intake or having no effect on feeding behaviour at all demonstrating that these effects can be dissociated. Increased water consumption can be secondary to hyperactivity or diuresis. If changes in water intake are observed, body composition analysis can be used to examine the effects of the drugs on water content of the carcass.
Models used to assess the behavioural specificity of a drug treatment on food intake
Food intake in rats and mice can be decreased by a variety of factors including stress, sickness or drug-induced toxicity and not just by the enhancement of satiety or other specific mechanisms. For obvious reasons it is important to determine at an early stage of a research programme, that the role of a drug class in the regulation of food intake and body weight is specific in nature and not due to unwanted side effects. Ideally, a drug found to reduce gross food intake would be evaluated in other tests to examine whether the decrease in food intake was a specific effect on the mechanisms regulating food intake or due to non-specific disruption of normal feeding patterns (e.g. by hyperactivity or sedation) or drug-induced malaise.
The behavioural satiety sequence
In all feeding studies, animals should be observed every time the food is weighed. These gross behavioural observations should detect any overt behavioural effects such as increased or decreased locomotor activity or the induction of any other motor behaviours, which could interfere with the ability of the animal to eat. If required, the effects of the test drug on locomotor activity can be quantified using standard automated activity boxes. More rigorous methods couple behavioural observations with measurement of food intake (usually stimulated by mild food deprivation or the presentation of a palatable wet mash diet) to examine the effects of drugs on the behavioural satiety sequence (Antin et al., 1975). This characteristic sequence of behaviours (feeding followed by activity, grooming and resting) normally occurs when a rat or mouse eats until it is full (Antin et al., 1975; Kitchener and Dourish, 1994; Vickers et al., 1999). These studies, which can be performed in either rats or mice, have been used to show that anti-obesity agents such as d-fenfluramine and sibutramine inhibit food intake in a physiological manner by advancing the natural satiety sequence rather than by disrupting normal feeding behaviour (Halford et al., 1998; Vickers et al., 1999; Jackson et al., 2000; Tallett et al., 2009). Importantly, these drugs have also been reported to enhance satiety in man (Blundell and Hill, 1988; Hansen et al., 1999). On the other hand, comprehensive behavioural profiling revealed that the reduction in food intake produced by the CB1 antagonist, rimonabant, in rats may be due, at least in part, to response competition from compulsive scratching and grooming (Tallett et al., 2007), analogous to the well-documented ‘side effect’ of pruritis (itching) produced by this compound in man. For a comprehensive review of the value of behavioural satiety sequence analysis in the screening strategy for novel anti-obesity agents see Rodgers et al., 2010.
Models of drug-induced malaise and aversion
Some drugs may decrease food intake by producing gastrointestinal malaise, which can be difficult to detect as the animals may look behaviourally normal. Rats and mice lack the emetic response, which is a clear difference from man. However, the persistent eating of inert substances in rodents can be used to measure illness-response behaviour analogous to vomiting in other species (Takeda et al., 1993; 1995a,b; Yamamoto et al., 2002). This behaviour is called pica (after the magpie that is known for its voracious and indiscriminate appetite). The substance widely used to measure pica is kaolin or china clay. The hypothesis is that the animals eat kaolin, which is highly adsorbant. Toxins attach to the clay and are not absorbed into the circulation. Accordingly, this provides relief from the gastrointestinal distress. The model has been validated using motion sickness and a variety of agents known to produce sickness in man such as cisplatin, copper sulphate and apomorphine (Takeda et al., 1993). Furthermore, the increases in cisplatin-induced kaolin intake in rats and mice can be reduced by the 5-HT3 receptor antagonist, ondansetron, which prevents emesis in man (Takeda et al., 1993, 1995b; Yamamoto et al., 2002).
The technique has been used to show that the potential anti-obesity drugs, E-6837, exenatide (Byetta) and davalintide (AC2307), do not induce kaolin intake at doses reducing food intake (Fisas et al., 2006; Mack et al., 2006; 2010). Interestingly, despite these findings there is evidence in man that exenatide (an approved medicine for Type 2 diabetes that leads to some weight loss in patients) is associated with an increased incidence of nausea (Ellero et al., 2010). Hence, data from the assay should be treated with some caution and used in conjunction with other tests such as the behavioural satiety sequence and/or the conditioned taste aversion (CTA) assay.
The CTA assay is a paradigm where animals are allowed exposure (e.g. 3 h) to a novel palatable substance (typically a saccharin solution) and are then dosed with a test compound or vehicle. Subsequent to this conditioning (e.g. 72 h later), animals are re-exposed to the saccharin solution (often in the presence of an additional water bottle). Saccharin intake (or the preference for the saccharin solution over normal tap water) is recorded. A reduction in saccharin intake or saccharin preference ratio (i.e. the amount of saccharin solution drunk as a proportion of total fluid intake) compared with vehicle-treated controls is regarded as being indicative of a test compound, or dose, having aversive properties (Benoit et al., 2003; Fisas et al., 2006). In contrast to other paradigms this assay is not widely validated with anti-obesity agents and care should be taken in the interpretation of data because a number of drugs that are self-administered in humans (e.g. cocaine) are reported to support the development of a CTA in rodents (for review see Hunt and Amit, 1987). Hence, the test may be particularly sensitive to false positives. It does not identify the origin of the aversion that may be wholly unrelated to nausea or malaise and, accordingly, potential anti-obesity drugs should ideally be assessed in additional specificity tests in order to confirm the outcome of the CTA. Interestingly, not only does the peptide, GLP-1, support the development of a CTA (Thiele et al., 1997) but the CTA induced by lithium chloride is blocked by a GLP-1 receptor antagonist (Seeley et al., 2000). Such a finding is of relevance because the GLP-1 receptor agonist, exenatide, is associated with nausea in man as mentioned above (Ellero et al., 2010).
Recently, conditioned gaping in rats has been described as a new preclinical tool to examine whether drugs are likely to produce nausea in man. The test is based on observations that rats display a characteristic gaping reaction when intraorally infused with a flavoured solution that has previously been paired with an emetic drug (Parker and Limebeer, 2006). The advantage of this model is that is appears to be more selective than the CTA test as only emetic drugs produce conditioned gaping in rats. Furthermore, known antiemetic drugs prevent the development of conditioned gaping but not CTA (Parker and Limebeer, 2006).
Determining effects on energy expenditure
As discussed previously, pharmacological strategies to increase energy expenditure have implications in body weight control and may lead to the development of novel treatments for obesity. Increased energy output may be due to increased physical activity, basal metabolic rate and/or thermogenesis. Increased activity is normally detected by observation or the use of activity boxes as described earlier whereas increased basal metabolic rate and thermogenesis can typically be measured in animals using closed circuit calorimeters (indirect calorimetry). Indirect calorimetry is so-called because the expenditure of calories is calculated from a measurement of oxygen uptake and relies on the fact that burning 1 calorie (Kilocalorie) requires 208.06 mL of oxygen. Typically animals are placed in sealed chambers at a thermoneutral temperature (29°C in the case of rats so that they do not need to expend any energy to maintain their core temperature) and their resting oxygen consumption is recorded (Stock, 1975). Accordingly, this is used as a measure of energy expenditure.
This methodology has been used to demonstrate the thermogenic effects of thyroid hormones, β3-adrenoceptor agonists and the 5-HT and noradrenaline reuptake inhibitor, sibutramine, in rats (Silva, 1995; Connoley et al., 1999; Skill et al., 2000). Furthermore, chronic administration of sibutramine increased basal metabolic rate in these animals (Skill et al., 2000). A number of other thermogenic approaches have been explored as a strategy for the treatment of obesity including activation of growth hormone receptors; inhibition of glucocorticoid receptors and the modulation of transcription factors or enzymes that promote mitochondrial biogenesis and fatty acid oxidation (Clapham and Arch, 2007). However, most of these approaches have not been developed beyond the preclinical stage. This has been largely due to concerns that differences between the thermogenic capacity of rodents and man may give a misleading impression of the potential of a drug in the clinic. This notion was because it was believed for a long time that adult humans did not possess brown adipose tissue. Furthermore, although the weight loss induced by the uncoupling agent, dinitrophenol, demonstrates that pharmacological manipulation of energy expenditure could be used to a strategy to treat obesity, the compound led to fatal hyperthermia (see Elangbam, 2009) and it was unclear whether thermogenesis could be controlled to the extent that it produced weight loss but not discomfort or toxicity. Over recent years, however, important progress has been made in our understanding of brown adipose tissue biology and several independent research teams using a combination of positron-emission tomography and computed tomography (PET/CT) imaging, immunohistochemistry and gene and protein expression assays have shown that functional brown adipose tissue is present in man (Cypess and Kahn, 2010). These findings have renewed interest in the manipulation of energy output as a mechanism to produce weight loss. The presence of functional brown adipose tissue in man, which can be regulated by adrenergic stimulation, accords with reports that sibutramine induces thermogenesis in lean and obese humans and that it can decrease the decline in energy expenditure that accompanies weight loss (Hansen et al., 1998; 1999; Walsh et al., 1999; Saraçet al., 2006). These findings support the use of animals to investigate the effects of novel anti-obesity drugs on the energy output side of the energy balance equation, an aspect that is often ignored.
Side effects and safety of anti-obesity drugs
Animal models can be particularly useful to investigate the anti-obesity potential of both novel compounds and combinations of different compounds (which may or may not be novel). This later, drug combination, approach has become increasingly attractive as a possible method of overcoming some of the compensatory homeostatic mechanisms, which are called into play when weight loss occurs, and has the potential to produce more efficacious anti-obesity drugs (Kennett and Clifton, 2010). Existing animal models can be used to explore such combinations, and in the case of Qnexa and pramlintide/metreleptin for example, the DIO rat model in particular appears to have good predictive validity for therapeutic potential. However, despite animal models used in obesity drug research having, in some cases such as the DIO rat and mouse, excellent face, construct and predictive validity for efficacy to produce weight loss, the field has been beset by failures in the clinic in regard to unacceptable drug safety profiles. Anti-obesity drugs are likely to be taken for a long period, perhaps the lifetime of the patient. Accordingly, it is perhaps unsurprising that routine toxicologic screens did not detect the cardiovascular safety issues that emerged on repeated administration of anti-obesity drugs such as Aminorex and fenfluramine. Indeed, there are still no reliable, validated animal models, which can accurately predict whether drugs will produce pulmonary arterial hypertension and valvulopathy in man (Elangbam, 2009). This is of importance because fenfluramine was implicated in the induction of primary pulmonary hypertension (Brenot et al., 1993) and cardiac valvulopathy (Connolly et al., 1997). In addition, while animals can be used to assess the abuse liability of novel drugs (Ator and Griffiths, 2003) and should detect the abuse/dependence problems associated with the amphetamines, some psychiatric side effects are more difficult to evaluate in an animal model. For example, the CB1 receptor antagonist rimonabant was not recommended for clinical use in the USA and was suspended in Europe due to associations of the drug with severe psychiatric adverse events such as depression and suicidal ideation in patients. The results of animal experiments have been conflicting. Some studies clearly failed to predict the psychiatric side effects of rimonabant as CB1 receptor blockade was found to produce similar behavioural and neurochemical effects to established anxiolytics and antidepressants in rats and mice (Griebel et al., 2005; Witkin et al., 2005) whereas other workers reported that rimonabant was anxiogenic in rodent models (Navarro et al., 1997; Arévalo et al., 2001; Patel and Hillard, 2006) and produced a depression-like phenotype when given chronically to rats (Beyer et al., 2010), findings that are more in line with the clinical reports.
Rare and possibly human-specific adverse effects of novel drugs are by their very nature difficult to model in preclinical studies and such events will only emerge following extensive post-marketing surveillance. Accordingly, the successful development of anti-obesity drugs is likely to remain high risk because the bar of the regulatory hurdle has been raised with particularly onerous safety criteria in light of past compound failures (Heal et al., 2009). Indeed, psychiatric adverse events, abuse, dependence and withdrawal side effects are explicitly noted by the FDA and EMEA as potential safety issues to be investigated for centrally acting drug candidates.
Summary
This review has detailed the relative strengths and weaknesses of various animal models typically used during the research and development of novel anti-obesity drugs. The models available need to be selected according to the nature of the drug target and the mechanism of action, but in general they possess excellent utility in predicting weight loss in man, especially in the case of the DIO rat. Chronic studies can also examine whether the reductions in body weight are due to a specific loss of fat and accompanied by improvements in associated comorbidities and risk factors. Nevertheless, it should always be remembered that rodents are not humans and that species differences in pharmacokinetic disposition, efficacy and tolerability can occur. Furthermore, the doses used in rodent efficacy studies are likely to range beyond those tolerated in the clinic because of difficulties in assessing anything other than gross tolerability issues in rats and mice. Therefore, animal studies will always only have limited value in assessing the therapeutic index of novel anti-obesity agents in the clinic. Thus, over recent years novel anti-obesity drugs have significantly reduced body weight in clinical studies; however, the field has been beset with safety issues and limited efficacy and drugs have been withdrawn or have not been approved by the regulatory authorities because of negative risk/benefit profiles. Particular concerns are that anti-obesity drugs would have to be given chronically and that the expanding size of the anti-obesity market means that any approved drugs would be given to extremely large numbers of patients in a relatively short space of time. Accordingly, the development of anti-obesity drugs remains high-risk. That said, there remains an enormous unmet clinical need for safe and more efficacious treatments for obesity, and for any drug companies that succeed in developing such a product, the rewards are considerable.
Acknowledgments
None.
Glossary
Abbreviations
- 5-HT
5-hydroxytryptamine
- CTA
conditioned taste aversion
- DIO
dietary-induced obese
- EMEA
European Medicines Agency
- FDA
Food and Drug Administration
- GLP
glucagon-like-peptide
- MCH
melanin-concentrating hormone
Conflict of interest
The authors are employees of RenaSci Consultancy: a fee-for-service Contract Research Organisation.
References
- Antin J, Gibbs J, Holt J, Young RC, Smith GP. Cholecystokinin elicits the complete behavioral sequence of satiety in rats. J Comp Physiol Psychol. 1975;89:784–790. doi: 10.1037/h0077040. [DOI] [PubMed] [Google Scholar]
- Apfelbaum M, Vague P, Ziegler O, Hanotin C, Thomas F, Leutenegger E. Long-term maintenance of weight loss after a very-low-calorie diet: a randomized blinded trial of the efficacy and tolerability of sibutramine. Am J Med. 1999;106:179–184. doi: 10.1016/s0002-9343(98)00411-2. [DOI] [PubMed] [Google Scholar]
- Arévalo C, de Miguel R, Hernández-Tristán R. Cannabinoid effects on anxiety-related behaviours and hypothalamic neurotransmitters. Pharmacol Biochem Behav. 2001;70:123–131. doi: 10.1016/s0091-3057(01)00578-0. [DOI] [PubMed] [Google Scholar]
- Asarian L, Geary N. Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci. 2006;361:1251–1263. doi: 10.1098/rstb.2006.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ator NA, Griffiths RR. Principles of drug abuse liability assessment in laboratory animals. Drug Alcohol Depend. 2003;70(Suppl):S55–S72. doi: 10.1016/s0376-8716(03)00099-1. [DOI] [PubMed] [Google Scholar]
- Bailey CJ, Flatt PR. Animal syndromes resembling type 2 diabetes. In: Pickup JC, Williams G, editors. Textbook of Diabetes. 3rd edn. Oxford: Blackwell Publishing; 2003. pp. 25.1–25.30. [Google Scholar]
- Bell CG, Walley AJ, Froguel P. The genetics of human obesity. Nat Rev Genet. 2005;6:221–234. doi: 10.1038/nrg1556. [DOI] [PubMed] [Google Scholar]
- Benoit SC, Air EL, Wilmer K, Messerschmidt P, Hodge KM, Jones MB, et al. Two novel paradigms for the simultaneous assessment of conditioned taste aversion and food intake effects of anorexic agents. Physiol Behav. 2003;79:761–766. doi: 10.1016/s0031-9384(03)00189-6. [DOI] [PubMed] [Google Scholar]
- Beyer CE, Dwyer JM, Piesla MJ, Platt BJ, Shen R, Rahman Z, et al. Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol Dis. 2010;39:148–155. doi: 10.1016/j.nbd.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Blundell JE, Hill AJ. On the mechanism of action of dexfenfluramine: effect on alliesthesia and appetite motivation in lean and obese subjects. Clin Neuropharmacol. 1988;11(Suppl 1):S121–S134. [PubMed] [Google Scholar]
- Bradbury MJ, Campbell U, Giracello D, Chapman D, King C, Tehrani L, et al. Metabotropic glutamate receptor mGlu5 is a mediator of appetite and energy balance in rats and mice. J Pharmacol Exp Ther. 2005;313:395–402. doi: 10.1124/jpet.104.076406. [DOI] [PubMed] [Google Scholar]
- Bray GA. Drug treatment of obesity. Rev Endocr Metab Disord. 2001;2:403–418. doi: 10.1023/a:1011808701117. [DOI] [PubMed] [Google Scholar]
- Bray GA, Hollander P, Klein S, Kushner R, Levy B, Fitchet M, et al. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res. 2003;11:722–733. doi: 10.1038/oby.2003.102. [DOI] [PubMed] [Google Scholar]
- Brenot F, Herve P, Petitpretz P, Parent F, Duroux P, Simonneau G. Primary pulmonary hypertension and fenfluramine use. Br Heart J. 1993;70:537–541. doi: 10.1136/hrt.70.6.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner R, Parhofer KG, Woenckhaus M, Wrede CE, Kunz-Schughart LA, Schölmerich J, et al. Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J Mol Endocrinol. 2006;36:485–501. doi: 10.1677/jme.1.01909. [DOI] [PubMed] [Google Scholar]
- Clapham JC, Arch JRS. Thermogenic and metabolic antiobesity drugs: rationale and opportunities. Diabetes Obes Metab. 2007;9:259–275. doi: 10.1111/j.1463-1326.2006.00608.x. [DOI] [PubMed] [Google Scholar]
- Clegg DJ, Benoit SC, Fisher ME, Barrera JG, Seeley RJ, Woods SC. Sex hormones determine body fat distribution and sensitivity to adiposity signals. Appetite. 2003a;40:324. [Google Scholar]
- Clegg DJ, Riedy CA, Smith KA, Benoit SC, Woods SC. Differential sensitivity to central leptin and insulin in male and female rats. Diabetes. 2003b;52:682–687. doi: 10.2337/diabetes.52.3.682. [DOI] [PubMed] [Google Scholar]
- Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- Coleman DL. Diabetes and obesity: thrifty mutants? Nutr Rev. 1978;36:129–132. doi: 10.1111/j.1753-4887.1978.tb03726.x. [DOI] [PubMed] [Google Scholar]
- Colombo G, Agabio R, Diaz G, Lobina C, Reali R, Gessa GL. Appetite suppression and weight loss after the cannabinoid antagonist SR 141716. Life Sci. 1998;63:PL113–PL117. doi: 10.1016/s0024-3205(98)00322-1. [DOI] [PubMed] [Google Scholar]
- Connoley IP, Liu YL, Frost I, Reckless IP, Heal DJ, Stock MJ. Thermogenic effects of sibutramine and its metabolites. Br J Pharmacol. 1999;126:1487–1495. doi: 10.1038/sj.bjp.0702446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- Correia MLG, Haynes WG. Lessons from leptin's molecular biology: potential therapeutic actions of recombinat leptin and leptin-related compounds. Mini Rev Med Chem. 2007;7:31–38. doi: 10.2174/138955707779317858. [DOI] [PubMed] [Google Scholar]
- Cypess AM, Kahn CR. Brown fat as a therapy for obesity and diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:143–149. doi: 10.1097/MED.0b013e328337a81f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C, Bailey CJ. Effect of the antiobesity agent sibutramine in obese-diabetic ob/ob mice. Int J Obes Relat Metab Disord. 1998;22:619–623. doi: 10.1038/sj.ijo.0800636. [DOI] [PubMed] [Google Scholar]
- Després JP, Golay A, Sjöström L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–2134. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- Dickinson K, North TJ, Anthony DM, Jones RB, Heal DJ. Evaluation of a simplified cafeteria model for the induction of insulin resistant obesity in rats. Int J Obes. 1998;22(Suppl 3):S179. [Google Scholar]
- Elangbam CS. Review paper: current strategies in the development of anti-obesity drugs and their safety concerns. Vet Pathol. 2009;46:10–24. doi: 10.1354/vp.46-1-10. [DOI] [PubMed] [Google Scholar]
- Ellero C, Han J, Bhavsar S, Cirincione BB, Deyoung MB, Gray AL, et al. Prophylactic use of anti-emetic medications reduced nausea and vomiting associated with exenatide treatment: a retrospective analysis of an open-label, parallel-group, single-dose study in healthy subjects. Diabet Med. 2010;27:1168–1173. doi: 10.1111/j.1464-5491.2010.03085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMEA. The European Medicines Agency recommends suspension of the marketing authorisation of Acomplia. 2008. htttp://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2009/11/news_detail_000244.jsp&murl=menus/news_and_events/news_and_events.jsp&mid=WC0b01ac058004d5c1.
- EMEA. European Medicines Agency recommends suspension of marketing authorisation for sibutramine. 2010. http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/01/news_detail_000985.jsp.
- Farooqi IS, O'Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity. Nat Clin Pract Endocrinol Metab. 2008;4:569–577. doi: 10.1038/ncpendmet0966. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, O'Rahilly S. Leptin: a pivotal regulator of human energy homeostasis. Am J Clin Nutr. 2009;89:980S–984S. doi: 10.3945/ajcn.2008.26788C. [DOI] [PubMed] [Google Scholar]
- Fisas A, Codony X, Romero G, Dordal A, Giraldo J, Mercé R, et al. Chronic 5-HT6 receptor modulation by E-6837 induces hypophagia and sustained weight loss in diet-induced obese rats. Br J Pharmacol. 2006;148:973–983. doi: 10.1038/sj.bjp.0706807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadde KM, Allison DB. Combination therapy for obesity and metabolic disease. Curr Opin Endocrinol Diabetes Obes. 2009;16:353–358. doi: 10.1097/MED.0b013e3283304f90. [DOI] [PubMed] [Google Scholar]
- Greenway FL, Whitehouse MJ, Guttadauria M, Anderson JW, Atkinson RL, Fujioka K, et al. Rational design of a combination medication for the treatment of obesity. Obesity. 2009;17:30–39. doi: 10.1038/oby.2008.461. [DOI] [PubMed] [Google Scholar]
- Griebel G, Stemmelin J, Scatton B. Effects of the canabinoid CB1 receptor antagonist rimonabant in models of emotional reactivity in rodents. Biol Psychiatry. 2005;57:261–267. doi: 10.1016/j.biopsych.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Hadcock JR, Carpino PA, Iredale PA, Dow RL, Gautreau D, Thiede L, et al. Quantitative in vitro and in vivo pharmacological profile of CE-178253, a potent and selective cannabinoid type 1(CB1) receptor antagonist. BMC Pharmacol. 2010;10:9. doi: 10.1186/1471-2210-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci USA. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford JC, Wanninayake SC, Blundell JE. Behavioural satiety sequence (BSS) for the diagnosis of drug action on food intake. Pharmacol Biochem Behav. 1998;61:159–168. doi: 10.1016/s0091-3057(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Halford JCG, Boyland EJ, Blundell JE, Kirkham TC, Harrold JA. Pharmacological management of appetite expression in obesity. Nat Rev Endocrinol. 2010;6:255–269. doi: 10.1038/nrendo.2010.19. [DOI] [PubMed] [Google Scholar]
- Hansen DL, Toubro S, Stock MJ, Macdonald IA, Astrup A. Thermogenic effects of sibutramine in humans. Am J Clin Nutr. 1998;68:1180–1186. doi: 10.1093/ajcn/68.6.1180. [DOI] [PubMed] [Google Scholar]
- Hansen DL, Toubro S, Stock MJ, Macdonald IA, Astrup A. The effect of sibutramine on energy expenditure and appetite during chronic treatment without dietary restriction. Int J Obes Relat Metab Disord. 1999;23:1016–1024. doi: 10.1038/sj.ijo.0801059. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Hansen G, Tang-Christensen M, Larsen PJ, Axel AM, Raben A, et al. The novel triple monoamine reuptake inhibitor tesofensine induces sustained weight loss and improves glycemic control in the diet-induced obese rat: comparison to sibutramine and rimonabant. Eur J Pharmacol. 2010;636:88–95. doi: 10.1016/j.ejphar.2010.03.026. [DOI] [PubMed] [Google Scholar]
- Harrold JA, Widdowson PS, Clapham JC, Williams G. Individual severity of dietary obesity in unselected Wistar rats: relationship with hyperphagia. Am J Physiol Endocrinol Metab. 2000;279:E340–E347. doi: 10.1152/ajpendo.2000.279.2.E340. [DOI] [PubMed] [Google Scholar]
- Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9:160–167. doi: 10.1001/archfami.9.2.160. [DOI] [PubMed] [Google Scholar]
- Heal DJ, Smith SL, Fisas A, Codony X, Buschmann H. Selective 5-HT6 receptor ligands: progress in the development of a novel pharmacological approach to the treatment of obesity and related metabolic disorders. Pharmacol Ther. 2008;117:207–231. doi: 10.1016/j.pharmthera.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Heal DJ, Gosden J, Smith SL. Regulatory challenges for new drugs to treat obesity and comorbid metabolic disorders. Br J Clin Pharmacol. 2009;68:861–874. doi: 10.1111/j.1365-2125.2009.03549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal DJ, Gosden J, Smith SL. The 5-HT6 receptor as a target for developing novel anti-obesity drugs. In: Borsini F, editor. International Review of Neurology. Oxford: Academic Press; 2011. pp. 73–109. Vol 96. Pharmacology of 5-HT6 Receptors, Part II. [DOI] [PubMed] [Google Scholar]
- Hewitt KN, Lee MD, Dourish CT, Clifton PG. Serotonin 2C receptor agonists and the behavioural satiety sequence in mice. Pharmacol Biochem Behav. 2002;71:691–700. doi: 10.1016/s0091-3057(01)00709-2. [DOI] [PubMed] [Google Scholar]
- Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- Hirst WD, Abrahamsen B, Blaney FE, Calver AR, Aloj L, Price GW, et al. Differences in the central nervous system distribution and pharmacology of the mouse 5-hydroxytryptamine-6 receptor compared with rat and human receptors investigated by radioligand binding, site-directed mutagenesis, and molecular modelling. Mol Pharmacol. 2003;64:1295–1308. doi: 10.1124/mol.64.6.1295. [DOI] [PubMed] [Google Scholar]
- Hogan S, Fleury A, Hadvary P, Lengsfeld H, Meier MK, Triscari J, et al. Studies on the antiobesity activity of tetrahydrolipstatin, a potent and selective inhibitor of pancreatic lipase. Int J Obes. 1987;11(Suppl 3):35–42. [PubMed] [Google Scholar]
- Hunt T, Amit Z. Conditioned taste aversion induced by self-administered drugs: paradox revisited. Neurosci Biobehav Rev. 1987;11:107–130. doi: 10.1016/s0149-7634(87)80005-2. [DOI] [PubMed] [Google Scholar]
- Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered. 1950;41:317–318. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- Jackson HC, Needham AM, Hutchins LJ, Mazurkiewicz SE, Heal DJ. Comparison of the effects of sibutramine and other monoamine reuptake inhibitors on food intake in the rat. Br J Pharmacol. 1997;121:1758–1762. doi: 10.1038/sj.bjp.0701312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson HC, Pleasance IM, Mitchell JM, Heal DJ. Observational analysis of the effects of sibutramine, phentermine and aminorex on food intake in rats. Obes Res. 2000;8(Suppl 1):95S. [Google Scholar]
- Jackson HC, Dickinson K, Jones RB, Schumacher C, Jensen C. Comparison of the effects of sibutramine and orlistat on body weight, food and water intake and body composition in dietary-induced obese rats. Neuroscience. 2004 Program No 75.3 Neuroscience Meeting Planner, Society for Neuroscience: San Diego, CA. Online. [Google Scholar]
- Jackson HC, Cheetham SC, Dickinson K, Jones RB, Heal DJ, Gregory P, et al. Comparison of the effects of rimonabant and sibutramine in a rat model of dietary-induced obesity. Neuroscience. 2005 Program No 532.13 Neuroscience Meeting Planner, Society for Neuroscience: Washington, DC. Online. [Google Scholar]
- Jackson HC, Cheetham SC, Gregory PC, Antel J. Effect of chronic administration of topiramate and phentermine, alone and in combination, in an animal model of dietary-induced obesity. 629.15. Neuroscience. 2007 Program No 629.15 Neuroscience Meeting Planner, Society for Neuroscience: San Diego, CA. Online. [Google Scholar]
- James WP, Astrup A, Finer N, Hilsted J, Kopelman P, Rössner S, et al. Effect of sibutramine on weight maintenance after weight loss: a randomised trial. STORM Study Group. Sibutramine Trial of Obesity Reduction and Maintenance. Lancet. 2000;356:2119–2125. doi: 10.1016/s0140-6736(00)03491-7. [DOI] [PubMed] [Google Scholar]
- Jones RB, Dickinson K, Sands S, Skill MJ, Heal DJ, Kilpatrick IC. Effect of chronic administration of sibutramine on body composition analysis of lean and obese female rats. Obes Res. 2001;9:118S. [Google Scholar]
- Kennedy AJ, Ellacott KLJ, King VL, Hasty AH. Mouse models of the metabolic syndrome. Dis Model Mech. 2010;3:156–166. doi: 10.1242/dmm.003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennett GA, Clifton PG. New approaches to the pharmacological treatment of obesity: can they break through the efficacy barrier? Pharmacol Biochem Behav. 2010;97:63–83. doi: 10.1016/j.pbb.2010.07.020. [DOI] [PubMed] [Google Scholar]
- Kitchener SJ, Dourish CT. An examination of the behavioural specificity of hypophagia induced by 5-HT1B, 5-HT1C and 5-HT2 receptor agonists using the post-prandial satiety sequence in rats. Psychopharmacology (Berl) 1994;113:369–377. doi: 10.1007/BF02245211. [DOI] [PubMed] [Google Scholar]
- Kotsis V, Stabouli S, Bouldin M, Low A, Toumanidis S, Zakopoulos N. Impact of obesity on 24-hour ambulatory blood pressure and hypertension. Hypertension. 2005;45:602–607. doi: 10.1161/01.HYP.0000158261.86674.8e. [DOI] [PubMed] [Google Scholar]
- Kris-Etherton PM, Dietschy J. Design criteria for studies examining individual fatty acid effects on cardiovascular disease risk factors: human and animal studies. Am J Clin Nutr. 1997;65(Suppl):1590S–1596S. doi: 10.1093/ajcn/65.5.1590S. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol. 1997;273:R725–R730. doi: 10.1152/ajpregu.1997.273.2.R725. [DOI] [PubMed] [Google Scholar]
- Li S, Zhang HY, Hu CC, Lawrence F, Gallagher KE, Surapaneni A, et al. Assessment of diet-induced obese rats as an obesity model by comparative functional genomics. Obesity. 2008;16:811–818. doi: 10.1038/oby.2007.116. [DOI] [PubMed] [Google Scholar]
- Lobley GE, Bremner DM, Holtrop G, Johnstone AM, Maloney C. Impact of high-protein diets with either moderate or low carbohydrate on weight loss, body composition, blood pressure and glucose tolerance in rats. Br J Nutr. 2007;97:1099–1108. doi: 10.1017/S0007114507691934. [DOI] [PubMed] [Google Scholar]
- Mack CM, Moore CX, Jodka CM, Bhavsar S, Wilson JK, Hoyt JA, et al. Antiobesity action of peripheral exenatide (exendin-4) in rodents: effects on food intake, body weight, metabolic status and side-effect measures. Int J Obes. 2006;30:1332–1340. doi: 10.1038/sj.ijo.0803284. [DOI] [PubMed] [Google Scholar]
- Mack CM, Soares CJ, Wilson JK, Athanacio JR, Turek VF, Trevaskis JL, et al. Davalintide (AC2307), a novel amylin-mimetic peptide: enhanced pharmacological properties over native amylin to reduce food intake and body weight. Int J Obes. 2010;34:385–395. doi: 10.1038/ijo.2009.238. [DOI] [PubMed] [Google Scholar]
- Madsen AN, Hansen G, Paulsen SJ, Lykkegaard K, Tang-Christensen M, Hansen HS, et al. Long-term characterization of the diet-induced obese and diet-resistant rat model: a polygenetic rat model mimicking the human obesity syndrome. J Endocrinol. 2010;206:287–296. doi: 10.1677/JOE-10-0004. [DOI] [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- Masek J, Fabry P. High-fat diet and the development of obesity in albino rats. Experientia. 1959;15:444–445. doi: 10.1007/BF02157708. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Iijima H. Sibutramine sensitivity assay revealed a unique phenotype of bombesin BB3 receptor-deficient mice. Eur J Pharmacol. 2003;473:41–46. doi: 10.1016/s0014-2999(03)01908-3. [DOI] [PubMed] [Google Scholar]
- Miesel A, Müller H, Thermann M, Heidbreder M, Dominiak P, Raasch W. Overfeeding-induced obesity in spontaneously hypertensive rats: an animal model of the human metabolic syndrome. Ann Nutr Metab. 2010;56:127–142. doi: 10.1159/000278748. [DOI] [PubMed] [Google Scholar]
- Mohapatra J, Sharma M, Singh S, Pandya G, Chatterjee A, Balaraman R, et al. Involvement of adipokines in rimonabant-mediated insulin sensitivity in ob/ob mice. J Pharm Pharmacol. 2009;61:1493–1498. doi: 10.1211/jpp/61.11.0008. [DOI] [PubMed] [Google Scholar]
- Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- Mutch DM, Clément K. Unraveling the genetics of human obesity. PloS Genet. 2006;2:e188. doi: 10.1371/journal.pgen.0020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderali EK, Pickavance LC, Wilding JP, Williams G. Diet-induced endothelial dysfunction in the rat is independent of the degree of increase in total body weight. Clin Sci (Lond) 2001;100:635–641. doi: 10.1042/cs1000635. [DOI] [PubMed] [Google Scholar]
- Navarro M, Hernández E, Muñoz RM, del Arco I, Villanúa MA, Carrera MR, et al. Acute administration of the CB1 cannabinoid receptor antagonist SR 141716A induces anxiety-like responses in the rat. Neuroreport. 1997;8:491–496. doi: 10.1097/00001756-199701200-00023. [DOI] [PubMed] [Google Scholar]
- Neill JC, Cooper SJ. Evidence that d-fenfluramine anorexia is mediated by 5-HT1 receptors. Psychopharmacology (Berl) 1989;97:213–218. doi: 10.1007/BF00442252. [DOI] [PubMed] [Google Scholar]
- Parker LA, Limebeer CL. Conditioned gaping in rats: a selective measure of nausea. Auton Neurosci. 2006;129:36–41. doi: 10.1016/j.autneu.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Patel S, Hillard CJ. Pharmacological evaluation of cannabinoid receptor ligands in a mouse model of anxiety: further evidence for an anxiolytic role for endogenous cannabinoid signalling. J Pharmacol Exp Ther. 2006;318:304–311. doi: 10.1124/jpet.106.101287. [DOI] [PubMed] [Google Scholar]
- Pierroz DD, Ziotopoulou M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51:1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- Pi-Sunyer FX, Aronne LJ, Heshmati HM, Devin J, Rosenstock J. Effect of rimonabant, a cannabinoid-1 receptor blocker, on weight and cardiometabolic risk factors in overweight or obese patients: RIO-North America: a randomized controlled trial. JAMA. 2006;295:761–775. doi: 10.1001/jama.295.7.761. [DOI] [PubMed] [Google Scholar]
- Poncelet M, Maruani J, Calassi R, Soubrié P. Overeating, alcohol and sucrose consumption decrease in CB1 receptor deleted mice. Neurosci Lett. 2003;343:216–218. doi: 10.1016/s0304-3940(03)00397-5. [DOI] [PubMed] [Google Scholar]
- Powell DR. Obesity drugs and their targets: correlation of mouse knockout phenotypes with drug effects in vivo. Obes Rev. 2006;7:89–108. doi: 10.1111/j.1467-789X.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- Ravinet-Trillou C, Arnone M, Delgorge C, Gonalons N, Keane P, Maffrand JP, et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol. 2003;284:R345–R353. doi: 10.1152/ajpregu.00545.2002. [DOI] [PubMed] [Google Scholar]
- Ravussin E, Smith SR, Mitchell JA, Shringarpure R, Shan K, Maier H, et al. Enhanced weight-loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity. 2009;17:1736–1743. doi: 10.1038/oby.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers RJ, Holch P, Tallett AJ. Behavioural satiety sequence (BSS): separating wheat from chaff in the behavioural pharmacology of appetite. Pharmacol Biochem Behav. 2010;97:3–14. doi: 10.1016/j.pbb.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci USA. 2008;105:7257–7262. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraç F, Pehlivan M, Celebi G, Saygili F, Yilmaz C, Kabalak T. Effects of sibutramine on thermogenesis in obese patients assessed via immersion calorimetry. Adv Ther. 2006;23:1016–1029. doi: 10.1007/BF02850222. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, Blake K, Rushing PA, Benoit S, Eng J, Woods SC, et al. The role of CNS glucagon-like peptide-1 (7-36) amide receptors in mediating the visceral illness effects of lithium chloride. Neurosci. 2000;20:1616–1621. doi: 10.1523/JNEUROSCI.20-04-01616.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman LP, Camacho RE, Sloan Stribling D, Zhou D, Bednarek MA, Hreniuk DL, et al. Chronic MCH-1 receptor modulation alters appetite, body weight and adiposity in rats. Eur J Pharmacol. 2003;475:37–47. doi: 10.1016/s0014-2999(03)02146-0. [DOI] [PubMed] [Google Scholar]
- Silva JE. Thyroid hormone control of thermogenesis and energy balance. Thyroid. 1995;5:481–492. doi: 10.1089/thy.1995.5.481. [DOI] [PubMed] [Google Scholar]
- Silvani A, Bastianini S, Berteotti C, Franzini C, Lenzi P, Lo Martire V, et al. Sleep modulates hypertension in leptin-deficient obese mice. Hypertension. 2009;53:251–255. doi: 10.1161/HYPERTENSIONAHA.108.125542. [DOI] [PubMed] [Google Scholar]
- Skill MJ, Dickinson K, Jones RB, Heal DJ. Thermogenic effect of repeated sibutramine treatment in female Wistar rats. Int J Obes. 2000;24(Suppl):S135. [Google Scholar]
- Smith BM, Smith JM, Tsai JH, Schultz JA, Gilson CA, Estrada SA, et al. Discovery and structure-activity relationship of (1R)-chloro-2,3,4,5-tetrahydro-1-methyl-1H-3-benzazepine (Lorcaserin), a selective serotonin 5-HT2C receptor agonist for the treatment of obesity. J Med Chem. 2008;51:305–313. doi: 10.1021/jm0709034. [DOI] [PubMed] [Google Scholar]
- Smith SR, Weissman NJ, Anderson CM, Sanchez M, Chuang E, Stubbe S, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. N Engl J Med. 2010;363:245–256. doi: 10.1056/NEJMoa0909809. [DOI] [PubMed] [Google Scholar]
- Stock MJ. An automatic, closed-circuit oxygen consumption apparatus for small animals. J Appl Physiol. 1975;39:849–850. doi: 10.1152/jappl.1975.39.5.849. [DOI] [PubMed] [Google Scholar]
- Su W, Guo Z, Randall DC, Cassis L, Brown DR, Gong MC. Hypertension and disrupted blood pressure circadian rhythm in type 2 diabetic db/db mice. Am J Physiol Heart Circ Physiol. 2008;295:H1634–H1641. doi: 10.1152/ajpheart.00257.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinburn BA, Sacks G, Lo SK, Westerterp KR, Rush EC, Rosenbaum M, et al. Estimating the changes in energy flux that characterize the rise in obesity prevalence. Am J Clin Nutr. 2009;89:1723–1728. doi: 10.3945/ajcn.2008.27061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Hasegawa S, Morita M, Matsunaga T. Pica in rats is analogous to emesis: an animal model in emesis research. Pharmacol Biochem Behav. 1993;45:817–821. doi: 10.1016/0091-3057(93)90126-e. [DOI] [PubMed] [Google Scholar]
- Takeda N, Hasegawa S, Morita M, Horii A, Uno A, Yamatodani A, et al. Neuropharmacological mechanisms of emesis. I. Effects of antiemetic drugs on motion- and apomorphine-induced pica in rats. Methods Find Exp Clin Pharmacol. 1995a;17:589–596. [PubMed] [Google Scholar]
- Takeda N, Hasegawa S, Morita M, Horii A, Uno A, Yamatodani A, et al. Neuropharmacological mechanisms of emesis. II. Effects of antiemetic drugs on cisplatin-induced pica in rats. Methods Find Exp Clin Pharmacol. 1995b;17:647–652. [PubMed] [Google Scholar]
- Tallett AJ, Blundell JE, Rodgers RJ. Grooming, scratching and feeding: role of response competition in acute anorectic response to rimonabant in male rats. Psychopharmacology. 2007;195:27–39. doi: 10.1007/s00213-007-0880-2. [DOI] [PubMed] [Google Scholar]
- Tallett AJ, Blundell JE, Rodgers RJ. Sibutramine-induced anorexia: potent, dose-dependent and behaviourally-selective profile in male rats. Behav Brain Res. 2009;198:359–365. doi: 10.1016/j.bbr.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Van Dijk G, Campfield LA, Smith FJ, Burn P, Woods SC, et al. Central infusion of GLP-1, but not leptin, produces conditioned taste aversions in rats. Am J Physiol. 1997;272(Pt 2):R726–R730. doi: 10.1152/ajpregu.1997.272.2.R726. [DOI] [PubMed] [Google Scholar]
- Thomas GH, Babbs AJ, Jackson HC, Sørenson RV, Widdowson PS. PSN S1 & S2 are Novel 5-HT1A receptor agonists and norepinephrine reuptake inhibitors that reduce food intake and body weight in animal models of obesity. Progran No 62.4 Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience; 2006. Online. [Google Scholar]
- Thomsen WJ, Grottick AJ, Menzaghi F, Reyes-Saldana H, Espitia S, Yuskin D, et al. Lorcaserin, a novel selective human 5-hydroxytryptamine2C agonist: in vitro and in vivo pharmacological characterization. J Pharmacol Exp Ther. 2008;325:577–587. doi: 10.1124/jpet.107.133348. [DOI] [PubMed] [Google Scholar]
- Thornton-Jones ZD, Kennett GA, Benwell KR, Revell DF, Misra A, Sellwood DM, et al. The cannabinoid CB1 receptor inverse agonist, rimonabant, modifies body weight and adiponectin function in diet-induced obese rats as a consequence of reduced food intake. Pharmacol Biochem Behav. 2006;84:353–359. doi: 10.1016/j.pbb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Trevaskis JL, Coffey T, Cole R, Lei C, Wittmer C, Walsh B, et al. Amylin-mediated restoration of leptin responsiveness in diet-induced obesity: magnitude and mechanisms. Endocrinology. 2008;149:5679–5687. doi: 10.1210/en.2008-0770. [DOI] [PubMed] [Google Scholar]
- Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rössner S. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet. 2005;365:1389–1397. doi: 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- Verty AN, McGregor IS, Mallet PE. Consumption of high carbohydrate, high fat, and normal chow is equally suppressed by a cannabinoid receptor antagonist in non-deprived rats. Neurosci Lett. 2004;354:217–220. doi: 10.1016/j.neulet.2003.10.035. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Kennett GA. Cannabinoids and the regulation of ingestive behaviour. Curr Drug Targets. 2005;6:215–223. doi: 10.2174/1389450053174514. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Clifton PG, Dourish CT. Behavioural evidence that d-fenfluramine-induced anorexia in the rat is not mediated by the 5-HT1A receptor subtype. Psychopharmacology (Berl) 1996;125:168–175. doi: 10.1007/BF02249416. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Clifton PG, Dourish CT, Tecott LH. Reduced satiating effect of d-fenfluramine in serotonin 5-HT(2C) receptor mutant mice. Psychopharmacology (Berl) 1999;143:309–314. doi: 10.1007/s002130050952. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Benwell KR, Porter RH, Bickerdike MJ, Kennett GA, Dourish CT. Comparative effects of continuous infusion of mCPP, Ro 60-0175 and d-fenfluramine on food intake, water intake, body weight and locomotor activity in rats. Br J Pharmacol. 2000;130:1305–1314. doi: 10.1038/sj.bjp.0703443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers SP, Dourish CT, Kennett GA. Evidence that hypophagia induced by d-fenfluramine and d-norfenfluramine in the rat is mediated by 5-HT2C receptors. Neuropharmacology. 2001;41:200–209. doi: 10.1016/s0028-3908(01)00063-6. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Easton N, Webster LJ, Wyatt A, Bickerdike MJ, Dourish CT, et al. Oral administration of the 5-HT2C receptor agonist, mCPP, reduces body weight gain in rats over 28 days as a result of maintained hypophagia. Psychopharmacology (Berl) 2003a;167:274–280. doi: 10.1007/s00213-002-1378-6. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Webster LJ, Wyatt A, Dourish CT, Kennett GA. Preferential effects of the cannabinoid CB1 receptor antagonist, SR 141716, on food intake and body weight gain of obese (fa/fa) compared to lean Zucker rats. Psychopharmacology (Berl) 2003b;167:103–111. doi: 10.1007/s00213-002-1384-8. [DOI] [PubMed] [Google Scholar]
- Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocrinol Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- Walsh KM, Leen E, Lean ME. The effect of sibutramine on resting energy expenditure and adrenaline-induced thermogenesis in obese females. Int J Obes Relat Metab Disord. 1999;23:1009–1015. doi: 10.1038/sj.ijo.0801045. [DOI] [PubMed] [Google Scholar]
- Witkin JM, Tzavara ET, Nomikos GG. A role for cannabinoid CB1 receptors in mood and anxiety disorders. Behav Pharmacol. 2005;16:315–331. doi: 10.1097/00008877-200509000-00005. [DOI] [PubMed] [Google Scholar]
- World Health Organisation. Obesity and overweight: factsheet No 311. 2011. http://www.who.int/mediacentre/factsheets/fs311/en/index.html.
- Yamamoto K, Matsunaga S, Matsui M, Takeda N, Yamatodani A. Pica in mice as a new model for the study of emesis. Methods Find Exp Clin Pharmacol. 2002;24:135–138. doi: 10.1358/mf.2002.24.3.802297. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zucker LM, Zucker TF. Fatty. A new mutation in the rat. J Hered. 1961;52:275–278. [Google Scholar]