

Abstract
von Hippel–Lindau (VHL) disease is a hereditary cancer syndrome caused by inherited mutations that inactivate the VHL tumour suppressor gene. The VHL locus encodes pVHL, whose best studied function is to bind to and down-regulate the hypoxia-inducible factor (HIF) family of oxygen-dependent transcription factors. Early efforts have established the fundamental role of HIF in VHL-defective tumorigenesis and in particular renal cell carcinoma. However, recent findings have revealed an alternate side to the story, the HIF-independenttumour suppressor functions of pVHL. These include pVHL's ability to regulate apoptosis and senescence as well as its role in the maintenance of primary cilium and orchestrating the deposition of the extracellular matrix. To what extent these HIF-dependent and HIF-independent functions cooperate in VHL-defective tumorigenesis remains to be determined.
Keywords: von Hippel–Lindau, pVHL, HIF, hypoxia-inducible factor, tumorigenesis, renal cell carcinoma, pheochromocytoma, senescence, cilia
Introduction
VHL disease was first described in the medical literature in the late 19th century by the British surgeon and ophthalmologist, Treacher Collins, who reported on the occurrence of bilateral retinal haemangiomas in a pair of siblings [1]. Subsequent observations by Eugen von Hippel and Arvid Lindau linked the occurrence of retinal haemangiomas to central nervous system haemangioblastomas [2]. The term VHL disease was later coined by the neurosurgeon Harvey Cushing.
Patients with VHL disease are at increased risk for a variety of cancers, including renal cell carcinoma (RCC) of the clear cell histology, central nervous system haemangioblastomas (especially of the cerebellum and spinal cord), retinal haemangiomas and pheochromocytomas [2]. Other manifestations include visceral cysts of the kidney and pancreas, pancreatic islet cell tumours and epididymal or broad ligament papillary cystadenomas (in men and women, respectively) (Fig. 1). In affected families, cancer risk is transmitted in an autosomal-dominant manner.
Fig 1.
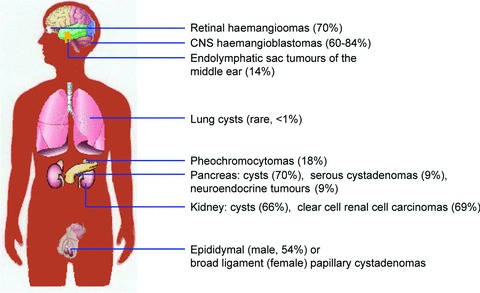
Clinical manifestations of VHL disease. Summary of the spectrum of benign and malignant tumours seen in association with VHL disease. Special notes: Retinal haemangiomas are found in up to 70% of VHL patients who survive to age 60 years. Haemangioblastomas are the most common lesions associated with VHL disease and are found most frequently in the cerebellum and spinal cord. Lung cysts appear to be rare, occurring in <1% of VHL patients. Pancreatic neuroendocrine tumours are relatively rare (9%) and are typically nonfunctional. The incidence of broad ligament papillary cystadenomas in women is not known secondary to their asymptomatic nature.
Genetic linkage studies performed in the 1980s indicated that the VHL gene (VHL) resides on chromosome 3p25, which is a region of the genome that is commonly deleted in sporadic kidney cancers [3]. This information was used to successfully isolate the VHL gene in 1993 [4]. Although RCC and haemangioblastomas are the leading cause of death in patients with VHL disease, retinal haemangiomas have the potential to cause significant morbidity (blindness) because of their association with posterior retinal detachment. Over the past century, studies focusing on the structure and function of the VHL tumour suppressor gene and its protein product, pVHL, have been highly informative with respect to the pathogenesis of clear cell renal carcinoma as well as the molecular mechanisms of oxygen sensing.
The VHL protein, pVHL
The VHL gene consists of three exons and is ubiquitously expressed. Translation of the VHL mRNA gives rise to two different protein products secondary to the presence of two distinct in-frame ATG codons (codon 1 and 54), which can both serve as translational initiation sites [5–7]. In most biochemical and functional assays, the two proteins (pVHL30 and pVHL19) behave similarly and unless otherwise noted are referred to generically as pVHL. pVHL is primarily a cytoplasmic protein but can also be found elsewhere, including the nucleus, the mitochondria and in association with the endoplasmic reticulum [8]. In fact, pVHL shuttles back and forth between the nucleus and the cytoplasm, and pVHL cannot suppress tumour growth when artificially restrained from doing so [9, 10].
HIF-dependent pVHL functions
Many functions have been attributed to pVHL; however, the one best characterized and most clearly linked to the development of pVHL-defective tumours, is targeting of the hypoxia-inducible factor (HIF) transcription factor for proteolytic degradation (Fig. 2). HIF is a heterodimeric transcription factor consisting of an unstable α subunit and a stable β subunit. Three HIFα genes (HIF1α, HIF2α and HIF3α) have been identified in the human genome [11]. Both HIF1α and HIF2α have two transcriptional activation domains, the N-terminal transactivation domain (NTAD) and the C-terminal transactivation domain (CTAD), which activate target genes upon DNA binding [12].
Fig 2.
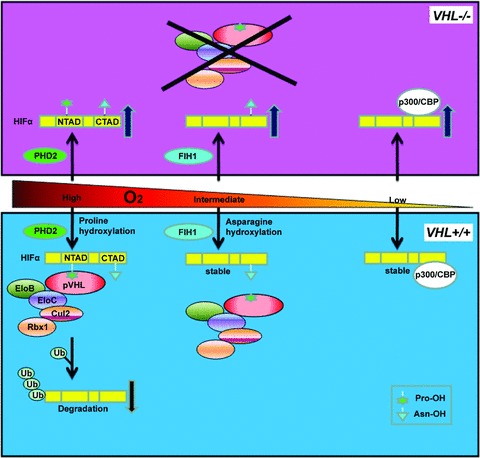
pVHL controls HIF via oxygen-sensitive hydroxylation. HIFα subunits have both an NTAD and a CTAD transactivation domain. When O2 levels are high, HIFα is hydroxylated on one or both conserved prolyl (Pro) residues located within the NTAD by the oxygen-dependent PHD2. This prolyl hydroxylation event generates a high affinity binding site for the pVHL E3 ubiquitin ligase complex composed of Cullin 2 (Cul2), Elongin B (EloB), Elongin C (EloC) and Rbx1. The pVHL complex polyubiquitinates HIFα, leading to its destruction by the proteasome. When O2 levels are intermediate, HIFα is hydroxylated by factor inhibiting HIF (FIH1) at a conserved asparaginyl (Asn) residue located in the CTAD, inhibiting HIF's interaction with the transcriptional co-activators p300/CBP. When O2 levels are low, HIFα subunits are stabilized, able to heterodimerize with the constitutively stable HIFβ, interact with p300/CBP and promote the transcription of downstream target genes. In cells lacking VHL, PHD2 and FIH1 remain active but HIFα subunits are not polyubquitinated and therefore allowed to accumulate.
HIF1α and HIF2α do not appear to be fully redundant in function. Although germline knock-out of HIF1α and HIF2α results in embryonic lethality the timing and cause of death appear to differ [13–15]. Moreover, post-natal inactivation of HIF1α and HIF2α leads to differing phenotypes as well [16]. Finally, the global gene expression changes induced by HIF1 and HIF2 show that they produce overlapping yet distinct gene expression profiles in both cells and in mice [17–21]. The role of HIF3α, which possesses a NTAD but lacks a CTAD, in transcriptional regulation is less well defined, and some HIF3α splice variants appear to inhibit HIF-dependent transcriptional activation in vitro and in vivo[22–25].
Hydroxylation of HIF
When oxygen levels are high (normoxia), HIFα subunits are enzymatically hydroxylated on one or both prolyl residues that reside near the NTAD by members of the oxygen- and 2-oxoglutarate-dependent prolyl hyroxylase (PHD) family [26–29]. There are at least three PHDs identified to date: PHD1 (EGLN2), PHD2 (EGLN1) and PHD3 (EGLN3) [30]. Although PHD2 is believed to be the primary hydroxylase for both HIF1α and HIF2α, other studies indicate that PHD3 may be mainly responsible for HIF2α hydroxylation [31, 32]. Hydroxylation of one or both proline residues within HIF1α and HIF2α creates a high affinity pVHL binding site. pVHL is part of a multi-subunit ubiquitin ligase complex composed of elongin-B, elongin-C, Cullin-2 and ring-box 1 (Rbx1) [33]. pVHL serves as a substrate recognition component that brings the ubiquitin conjugating machinery into proximity of its substrate, HIFα subunits, and leads to HIFα polyubiquitylation and destruction [26, 27, 34, 35].
When oxygen availability is limiting (hypoxia), the PHDs are enzymatically inactive, HIFα is therefore not hydroxylated, and does not interact with the pVHL complex. HIFα subunits therefore accumulate, translocate to the nucleus, heterodimerize with HIFα (also called ARNT [aryl hydrocarbon nuclear translocator] and activate transcription of numerous target genes involved in cell proliferation, angiogenesis, glucose metabolism, apoptosis and other cellular processes. Similarly, in the setting of VHL inactivation, although HIFα subunits are prolyl hydroxylated, they are not degraded, and similar to hypoxia, are free to transactivate HIF target genes (Fig. 2).
Soon after the discovery that HIFα subunits were prolyl hydroxylated, they were noted to be post-translationally hydroxy-lated in an oxygen-dependent manner on a conserved asparaginyl residue located in the CTAD by the asparaginyl hydroxylase, factor-inhibiting HIF (FIH1) (Fig. 2) [36, 37]. Asparaginyl hydroxy-lation of HIF prevents recruitment of the transcriptional co-activators p300 and CREB (cAMP response element binding protein) binding protein (CBP), and disrupts HIF-mediated transactivation [37]. In contrast to the PHD family, FIH1 remains active even under conditions of moderate hypoxia suggesting that in this setting it may act as a secondary mechanism to inhibit HIF transcriptional activity [38]. Interestingly, the CTAD of HIF2α appears to be relatively more resistant to inhibition of FIH1 under normoxia than the HIF1α CTAD [39].
HIF is a key mediator of VHL defective tumorigenesis
Given the early age of onset of VHL-associated tumours such as retinal haemangiomas, VHL inactivation is likely to be sufficient for their development. Other VHL associated tumours, such as RCC however, have a longer latency and more variable penetrance, suggesting that VHL loss alone is insufficient for their tumorigenesis. Nonetheless, it is clear that VHL inactivation is necessary for their development and that HIF dysregulation plays an important role in this process. Indeed there are several lines of evidence that implicate HIFα, and in particular HIF2α, as playing an active role in VHL−/− renal cell carcinogenesis. First and foremost, RCC-associated pVHL mutants are invariably defective with respect to HIFα polyubiquitination and therefore all VHL defective RCCs produce either HIF1α and HIF2α or solely HIF2α[2, 40–42]. This would suggest that there may be selective pressure to maintain HIF2α expression, but not HIF1α. Indeed, whole exosome sequencing from sporadic RCCs detected a significant, but low frequency of truncating mutations in HIF1α suggesting that in RCC it may function as a tumour suppressor [43]. Second, an apparent switch from HIF1α to HIF2α expression occurs in preneoplastic lesions arising in human VHL+/− kidneys in association with increasing dysplasia and cellular atypia [42]. Furthermore, HIF2α activation in mice appears to induce gene expression changes similar to mice with VHL inactivation, whereas HIF1α activation does so to a much less significant degree [19]. Finally, in VHL−/− RCC cell lines, HIF2α, but not HIF1α, appears to be necessary and sufficient for tumour growth [44–47].
Germline inactivation of Vhl in mice is embryonic lethal and whereas Vhl+/−mice do not have a cancer prone phenotype, they do develop liver haemangiomas as a result of loss of the remaining wild-type Vhl allele [48]. Similarly, conditional inactivation of Vhl in hepatocytes results in vascular liver lesions accompanied by hepatic steatosis [48]. HIF expression is both necessary and sufficient to recapitulate the hepatic phenotypes seen in mice as inactivation of Anrt or dual activation of HIF1α and HIF2α were able to abrogate or induce the hepatic phenotypes seen in Vhl loss, respectively [19, 49]. Unfortunately at this time there are no autochthonous mouse models of Vhl−/− RCC that can be used to investigate HIF's role in that setting.
Several clues exist as to why HIF2α may be more oncogenic than HIF1α. First, HIF2α is less sensitive than HIF1α to the inhibition by FIH-1 and is therefore more transcriptionally active under normoxia [50]. Second, HIF1α more than HIF2α, remains susceptible to proteasomal degradation in VHL−/− cell lines [19]. Third, HIF2α appears to cooperate with MYC to activate MYC transcriptional targets whereas HIF1α antagonizes MYC transcriptional activation [51]. Interestingly, a recent genome-wide analysis of copy number alterations noted that a region of chromosome 8q encoding MYC is often amplified in both sporadic and VHL disease associated tumours [52]. Whether or not HIF2α activation in concert with MYC overexpression cooperate in vivo has yet to be determined.
HIF responsive genes
More than 100 direct HIF-responsive genes have been described with a number of these genes active in carcinogenesis (Fig. 3) [53]. These include genes that encode proteins responsible for cell proliferation (transforming growth factor [TGFα] and epidermal growth factor receptor [EGFR]); angiogenesis (vascular endothelial growth factor [VEGF], platelet-derived growth factor B [PDGF-B] and interleukin-8 [IL-8]); glucose uptake and metabolism (glucose transporter 1 [GLUT1], 6-phosphofructokinase 1 [PFK1]); and chemotaxis (stromal cell-derived factor [SDF1] and its receptor C-X-C chemokine receptor 4 [CXCR4]). A number of gene products that are expected to have effects on the tumour microenvironment such as extracellular matrix (ECM) formation and turnover (membrane type 1 matrix metalloproteinase [MMP1] and lysyl oxidase [LOX]) are HIF responsive. Moreover, epithelial to mesenchymal transition (EMT) related genes (Twist [TWIST1 and TWIST2] and hepatocyte growth factor receptor [HGFR]) are known HIF target genes as well [53].
Fig 3.
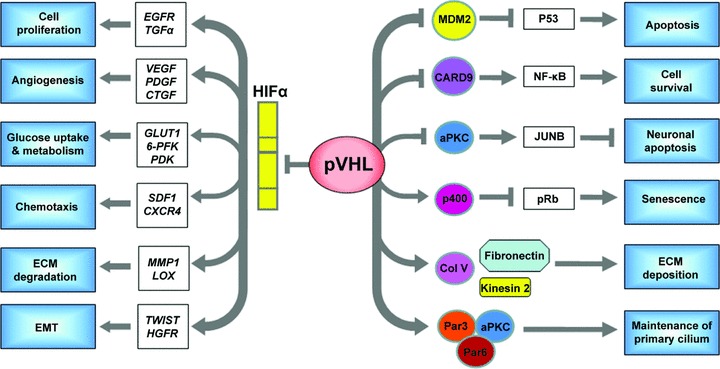
pVHL inactivation mediates both HIFdependent and HIF-independent pathways. HIF-responsive gene products play important roles in tumorigenesis. EGFR and TGFα promote cell proliferation and survival. VEGF, PDGF and connective tissue growth factor (CTGF) stimulate angiogenesis. Some proteins encoded by HIF-targeted gene products are responsible for regulating glucose uptake and metabolism, such as GLUT1, 6-PFK and pyruvate dehydrogenase kinase (PDK). CXCR4 and its ligand SDF1 stimulate chemotaxis and may also contribute to tumour cell invasion and metastases. MMP1 and lysyl oxidase (encoded by LOX) are implicated in ECM breakdown and tumour cell invasion/migration. Finally, dysregulation of TWIST (TWIST1 and TWIST2) and activation of HGFR (encoded by c-MET) are involved in EMT. pVHL has a number of HIF-independent functions as well. pVHL interacts with MDM2 and suppresses its ability to ubiqutinate p53, resulting in p53 accumulation and apoptosis. It can also act as an adaptor to bind CK2, which inactivates the NF-kB agonist CARD9, leads to inhibition of NF-kB signalling and overall inhibits cell survival. pVHL also down-regulates atypical protein kinase C (aPKC), which secondarily results in decreased levels of JUNB (an antagonist of JUN) thus permitting JUN-dependent neuronal apoptosis. Acute pVHL loss causes a senescent-like phenotype. It appears that pVHL increases p400 activity, which results in inactivation (hypophosphorylation) of the retinoblastoma protein (pRb) and prevents senescence. pVHL also interacts with collagen IV (Col IV), Kinesin 2 and fibronectin to ensure proper ECM deposition. Finally, pVHL plays an important role in primary cilium function by promoting microtubule stabilization and binding with aPKC and the polarity proteins Par3 and Par6.
HIF-independent pVHL functions
Recent evidence has accrued to indicate that pVHL has functions other than regulation of HIF-related pathways. The majority of these alternate functions have been discovered through biochemical interactions. However, gene expression studies also support the notion that there are HIF-independent gene expression changes induced by VHL loss [54, 55]. To what extent the HIF-independent functions of pVHL cooperate with HIF dysregulation in VHL defective tumorigenesis remains to be delineated.
Regulation of apoptosis
RCCs are notable for their insensitivity to conventional cytotoxic chemotherapies. The efficacy of chemotherapy is tightly linked to p53-mediated apoptosis [56]. However, most RCCs do not appear to harbour p53 mutations or loss suggesting either functional modulation of p53 activity or activation of alternative anti-apoptotic pathways [57, 58]. Both HIF and pVHL appear to be able to influence p53 function. Previous reports have shown that HIF can directly bind to and modulate p53 activity [59–61]. In addition, pVHL is able to regulate p53 function in a HIF-independent manner through suppression of MDM2-mediated ubiquitination and nuclear export resulting in an increase in its transcriptional activity [62]. Therefore pVHL loss appears to result in p53 inactivation by both HIF-dependent and HIF-independent effects.
The nuclear factor κB (NF-κB) pathway can mediate resistance to chemotherapy-induced apoptosis as well. pVHL deficient cells have been noted to have heightened NF-κB activity at least partially dependent upon HIF signalling [63–66]. In addition, pVHL can modulate NF-κB activity directly by binding with casein kinase 2 (CK2) and promoting the inhibitory phosphorylation of the NF-κB agonist CARD9 [67]. It seems possible that the ability of pVHL loss to both activate NF-κB and inactivate p53 may contribute to its profound chemoresistant phenotype.
Although sporadic RCC and haemangioblastomas harbour a high percentage of VHL mutations, VHL mutations are uncommon in truly sporadic pheochromocytomas [2, 68]. Indeed, 11% of apparently sporadic pheochromocytomas (defined by a lack of a family history or a spectrum of tumours suggestive of VHL disease) are actually due to occult germline, not sporadic, mutation of VHL [69]. This peculiarity, along with the knowledge that some VHL mutations that are associated with the development of pheochromocytoma (without an increased risk of RCC or haeman-gioblastoma) retain their ability to downregulate HIF, suggests that the development of VHL-associated pheochromocytomas is related to a HIF-independent function of pVHL [70, 71].
Insight into these apparent discrepancies has been recently elucidated by Kaelin and colleagues. It has been known for some time that during development there is an excess number of cells destined to become sympathetic neurons and that these cells’ survival is dependent upon nerve growth factor (NGF). As NGF becomes limiting, these cells undergo JUN-dependent apoptosis. VHL mutations that are linked to pheochromocytoma development result in the HIF-independent accumulation of JUNB, which is known to antagonize the pro-apoptotic function of JUN during NGF withdrawal [72]. Thus, patients inheriting pheochromocy-toma associated VHL mutations, presumably have an excess number of sympathetic neurons due to a relative insensitivity to NGF withdrawal induced apoptosis. However, whether the increased risk of pheochromocytoma development in patients with VHL disease is merely a reflection of an increased number of cells susceptible of forming pheochromocytomas or a distinct oncogenic mechanism associated with these mutations is unclear.
Control of cell senescence
Cellular senescence is the phenomenon of irreversible growth arrest in response to DNA damage (including shortened telomeres) but is also an important in vivo tumour suppressor mechanism [73, 74]. Interestingly, it has been recognized that physiological oxygenation can extend the replicative lifespan of cells in culture, which has typically been attributed to a relative decrease in the amount of oxidative stress [75]. Several reports have now confirmed that this phenomenon is at least in part due to stabilization of HIF [76, 77]. Interestingly, acute pVHL inactivation (with resultant HIF stabilization) was observed to induce senescence both in vitro and in vivo[78]. In this setting however, senescence appeared to be independent of both HIF and p53 function but primarily relied on activation of the retinoblastoma protein (pRb) and down-regulation of the SWItch/Sucrose NonFermentable (SWI2/SNF2) chromatin remodelling protein, p400. Recent work showing that induction of senescence by VHL loss is highly dependent upon oxygenation along with the differences in the senescence assays examined (i.e. replicative versus oncogene-induced senescence) may begin to explain the contrasting results [79].
Microtubule stabilization and maintenance of the primary cilium
pVHL associates with and is able to stabilize microtubules. This function of pVHL appears to be independent of its ability to either down-regulate HIF and its ubiquitin ligase function. Moreover, pVHL's ability to stabilize microtubules is lost in VHL mutations that predispose to the development of haemangioblastomas and pheochromocytomas, but not those associated with the development of RCC [80]. The primary cilium is a specialized structure on the cell surface that serves an antenna of the cell, and regulates the transduction of both chemical and mechanical signals [81]. The ciliary axoneme is composed of microtubules arranged in nine peripheral doublets that are templated from the basal body or mother centriole. Thus microtubule dynamics and formation and maintenance of the primary cilium are intimately linked.
Preneoplastic renal cysts are a common feature of VHL disease. Immunohistochemical and laser capture microdissection studies have demonstrated that the renal tubular epithelial cells lining these cysts have lost expression of VHL[42, 82, 83]. Other inherited familial syndromes are characterized by the development of renal cystic diseases of the kidney (e.g. autosomal dominant and recessive polycystic kidney disease [ADPKD and ARPKD, respectively], and Bardet Biedl syndrome) and despite being phenotypically diverse and having distinct extrarenal manifestations, these disorders are intriguingly unified by genetic defects that converge on the regulation of ciliogenesis and function [84].
pVHL's affects on microtubule dynamics is negatively regulated by its phosphorylation by glycogen synthase kinase 3β (GSK-3β) and appears to be HIF-independent, although some studies suggest that HIF dysregulation may play at least a partial role in the loss of microtubule stability imparted by pVHL inactivation [85–87]. Interestingly, active GSK-3β itself can promote microtubule stability and cilium maintenance in a pVHL-independent manner. When GSK-3β is inactive, for example following activation of the PI3Kinase-Akt pathway, microtubule stability and cilium maintenance appear to rely on pVHL once again. In keeping with the notion that GSK-3β and pVHL redundantly maintain primary cilia, it appears that the combined loss of VHL and PTEN in a genetically engineered mouse model cooperate to promote renal and genital tract cysts [88].
It is an apparent paradox that VHL mutants predisposing to RCC maintain the ability to regulate microtubule dynamics. One possibility is that the development of renal cysts secondary to loss of primary cilia on renal tubular cells lack significant malignant potential. In this scenario, the majority of RCCs associated with VHL disease would be expected to arise without an antecedent cystic phase. To some degree, this is in keeping with the observation that patients with polycystic kidney disease, despite having a high renal cystic burden are not clearly at a significantly higher risk for RCC [89].
Regulation of extracellular matrix formation and cell – cell adhesion
The ECM, a physical barrier to cancer cell migration and invasion, can provide survival signals to cancer cells and aide in the maintenance of cell polarity in concert with intercellular junctions [90]. pVHL can bind directly to both fibronectin and hydorxylated collagen IV, and interestingly all pVHL mutants studied to date are defective in this capacity [71, 91]. The inability of VHL deficient cells to bind ECM components results in ineffective ECM organization that is not mediated by HIF [92–94]. Moreover, pVHL's ability to orchestrate proper ECM deposition does not require binding to the other components of the pVHL complex such as Cullin2 and Elongins B and C and is regulated at least partially by the post-translational modification of pVHL by the ubiquitin-like molecule, NEDD8 [95, 96]. Similarly, cell polarity and assembly of intercellular junctions (i.e. adherens junctions and tight junctions) are defective in cells lacking VHL in a HIF-independent process [97]. How an intracellular protein such as pVHL modulates the assembly of the extracellular ECM components remains to be fully elucidated.
pVHL and synthetic lethality
Synthetic lethality occurs when two non-allelic mutations, which by themselves are not lethal, result in cell death when they exist simultaneously [98]. Synthetic lethality provides a framework to discover drugs that might preferentially kill cancer cells harbouring a cancer-relevant gene, yet leave normal cells unharmed. Two screens have been performed in attempt to target VHL deficient cells. A cell based small molecule synthetic lethality screen identified a compound, STF-62247, that selectively induces autophagic cell death in VHL-deficient RCC cells but not in those expressing wild-type VHL [99]. In addition, an shRNA screen targeting a select group of kinases identified and validated that silencing of CDK6, MET and MAP2K1 (MEK1) preferentially inhibited the growth of VHL−/− cells compared with their isogenic VHL wild-type counterparts [100]. Interestingly, in both screens the selective killing of cells lacking VHL was HIF-independent leaving open the possibility that therapies targeting these pathways might cooperate with those targeting HIF.
Conclusions
The VHL tumour suppressor gene is mutated or silenced in the majority of clear cell RCC. Loss of pVHL function results in the stabilization of HIFα and activation of HIF responsive genes. Many of these gene products have been shown to be oncogenic in the context of RCC. In recent years, our understanding of pVHL function has broadened to include several HIF-independent functions and it seems likely that more will be uncovered. Despite this broadened understanding of the consequences of VHL loss, the therapies in clinical use for RCC to date are primarily focused on dampening of HIF signalling and although effective have not achieved remarkable results. It will be interesting to determine whether targeting of HIF-independent pVHL functions either separately or in concert with HIF will lead to improved results.
Acknowledgments
W.Y.K. is a DAMON Runyon Merck Clinical Investigator. This work was supported by NIH grants R01CA142794 (W.Y.K.) and T32CA128590 (M.L.) and the UNC University Cancer Research Fund (W.Y.K.).
Conflict of interest
The authors state that there is no conflict of interest.
References
- 1.Collins E. Intra-ocular growths (two cases, brother and sister, with peculiar vascular new growth, probably retinal, affecting both eyes) Trans Ophthalmol Soc UK. 1894;14:141–9. [Google Scholar]
- 2.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 3.Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988;332:268–9. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- 4.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 5.Schoenfeld A, Davidowitz EJ, Burk RD. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci USA. 1998;95:8817–22. doi: 10.1073/pnas.95.15.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iliopoulos O, Ohh M, Kaelin WG., Jr pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci USA. 1998;95:11661–6. doi: 10.1073/pnas.95.20.11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blankenship C, Naglich JG, Whaley JM, et al. Alternate choice of initiation codon produces a biologically active product of the von Hippel Lindau gene with tumor suppressor activity. Oncogene. 1999;18:1529–35. doi: 10.1038/sj.onc.1202473. [DOI] [PubMed] [Google Scholar]
- 8.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nature Rev. 2002;2:673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 9.Lee S, Neumann M, Stearman R, et al. Transcription-dependent nuclear-cytoplas-mic trafficking is required for the function of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol. 1999;19:1486–97. doi: 10.1128/mcb.19.2.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S, Chen DY, Humphrey JS, et al. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc Natl Acad Sci USA. 1996;93:1770–5. doi: 10.1073/pnas.93.5.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001;13:167–71. doi: 10.1016/s0955-0674(00)00194-0. [DOI] [PubMed] [Google Scholar]
- 12.Sang N, Fang J, Srinivas V, et al. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1 alpha is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol Cell Biol. 2002;22:2984–92. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tian H, Hammer RE, Matsumoto AM, et al. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–4. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Compernolle V, Brusselmans K, Acker T, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. 2002;8:702–10. doi: 10.1038/nm721. [DOI] [PubMed] [Google Scholar]
- 16.Gruber M, Hu CJ, Johnson RS, et al. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA. 2007;104:2301–6. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang V, Davis DA, Haque M, et al. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005;65:3299–306. doi: 10.1158/0008-5472.CAN-04-4130. [DOI] [PubMed] [Google Scholar]
- 18.Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–86. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim WY, Safran M, Buckley MR, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006;25:4650–62. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu CJ, Sataur A, Wang L, et al. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18:4528–42. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu CJ, Wang LY, Chodosh LA, et al. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–74. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maynard MA, Qi H, Chung J, et al. Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem. 2003;278:11032–40. doi: 10.1074/jbc.M208681200. [DOI] [PubMed] [Google Scholar]
- 23.Maynard MA, Evans AJ, Hosomi T, et al. Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J. 2005;19:1396–406. doi: 10.1096/fj.05-3788com. [DOI] [PubMed] [Google Scholar]
- 24.Makino Y, Kanopka A, Wilson WJ, et al. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J Biol Chem. 2002;277:32405–8. doi: 10.1074/jbc.C200328200. [DOI] [PubMed] [Google Scholar]
- 25.Makino Y, Cao R, Svensson K, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–4. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- 26.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 27.Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 28.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–40. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 29.Yu F, White SB, Zhao Q, et al. Dynamic, site-specific interaction of hypoxia-inducible factor-1alpha with the von Hippel-Lindau tumor suppressor protein. Cancer Res. 2001;61:4136–42. [PubMed] [Google Scholar]
- 30.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Berra E, Richard DE, Gothie E, et al. HIF-1-dependent transcriptional activity is required for oxygen- mediated HIF-1alpha degradation. FEBS Lett. 2001;491:85–90. doi: 10.1016/s0014-5793(01)02159-7. [DOI] [PubMed] [Google Scholar]
- 32.Bishop T, Gallagher D, Pascual A, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3-/-mice. Mol Cell Biol. 2008;28:3386–400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nature Rev. 2008;8:865–73. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 34.Yu F, White SB, Zhao Q, et al. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci USA. 2001;98:9630–5. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masson N, Willam C, Maxwell PH, et al. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hewitson KS, McNeill LA, Riordan MV, et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–5. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 37.Lando D, Peet DJ, Gorman JJ, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–71. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dayan F, Roux D, Brahimi-Horn MC, et al. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006;66:3688–98. doi: 10.1158/0008-5472.CAN-05-4564. [DOI] [PubMed] [Google Scholar]
- 39.Yan Q, Bartz S, Mao M, et al. The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol Cell Biol. 2007;27:2092–102. doi: 10.1128/MCB.01514-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 41.Gordan JD, Lal P, Dondeti VR, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandriota KT, Davies DR, Murray PG, et al. Hif activation identifies early lesion in vhl kidneys: evidence for site-specific tumor suppressor funciton in the nephron. Cancer Cell. 2002;1:459–68. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 43.Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmer M, Doucette D, Siddiqui N, et al. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL-/- tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]
- 45.Kondo K, Kim WY, Lechpammer M, et al. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kondo K, Klco J, Nakamura E, et al. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 47.Maranchie JK, Vasselli JR, Riss J, et al. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 48.Haase VH, Glickman JN, Socolovsky M, et al. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci USA. 2001;98:1583–8. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rankin EB, Higgins DF, Walisser JA, et al. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol Cell Biol. 2005;25:3163–72. doi: 10.1128/MCB.25.8.3163-3172.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bracken CP, Fedele AO, Linke S, et al. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J Biol Chem. 2006;281:22575–85. doi: 10.1074/jbc.M600288200. [DOI] [PubMed] [Google Scholar]
- 51.Gordan JD, Bertout JA, Hu CJ, et al. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beroukhim R, Brunet JP, Di Napoli A, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–81. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Semenza GL. Targeting HIF-1 for cancer therapy. Nature Rev. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 54.Jiang Y, Zhang W, Kondo K, et al. Gene expression profiling in a renal cell carcinoma cell line: dissecting VHL and hypoxia-dependent pathways. Mol Cancer Res. 2003;1:453–62. [PubMed] [Google Scholar]
- 55.Zatyka M, da Silva NF, Clifford SC, et al. Identification of cyclin D1 and other novel targets for the von Hippel-Lindau tumor suppressor gene by expression array analysis and investigation of cyclin D1 genotype as a modifier in von Hippel- Lindau disease. Cancer Res. 2002;62:3803–11. [PubMed] [Google Scholar]
- 56.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–64. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 57.Vasavada SP, Novick AC, Williams BR. P53, bcl-2, and Bax expression in renal cell carcinoma. Urology. 1998;51:1057–61. doi: 10.1016/s0090-4295(98)00132-0. [DOI] [PubMed] [Google Scholar]
- 58.Tomasino RM, Morello V, Tralongo V, et al. p53 expression in human renal cell carcinoma: an immunohistochemical study and a literature outline of the cytogenetic characterization. Pathologica. 1994;86:227–33. [PubMed] [Google Scholar]
- 59.Sanchez-Puig N, Veprintsev DB, Fersht AR. Binding of natively unfolded HIF-1alpha ODD domain to p53. Mol Cell. 2005;17:11–21. doi: 10.1016/j.molcel.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 60.An WG, Kanekal M, Simon MC, et al. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998;392:405–8. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- 61.Sendoel A, Kohler I, Fellmann C, et al. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature. 2010;465:577–83. doi: 10.1038/nature09141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roe JS, Kim H, Lee SM, et al. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol Cell. 2006;22:395–405. doi: 10.1016/j.molcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 63.Oya M, Ohtsubo M, Takayanagi A, et al. Constitutive activation of nuclear factor-kappaB prevents TRAIL-induced apoptosis in renal cancer cells. Oncogene. 2001;20:3888–96. doi: 10.1038/sj.onc.1204525. [DOI] [PubMed] [Google Scholar]
- 64.Qi H, Ohh M. The von Hippel-Lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappaB-dependent anti-apoptotic pathway. Cancer Res. 2003;63:7076–80. [PubMed] [Google Scholar]
- 65.An J, Rettig MB. Mechanism of von Hippel-Lindau protein-mediated suppression of nuclear factor kappa B activity. Mol Cell Biol. 2005;25:7546–56. doi: 10.1128/MCB.25.17.7546-7556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pantuck AJ, An J, Liu H, et al. NF-kappaB-dependent plasticity of the epithelial to mesenchymal transition induced by Von Hippel-Lindau inactivation in renal cell carcinomas. Cancer Res. 2010;70:752–61. doi: 10.1158/0008-5472.CAN-09-2211. [DOI] [PubMed] [Google Scholar]
- 67.Yang H, Minamishima YA, Yan Q, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell. 2007;28:15–27. doi: 10.1016/j.molcel.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim WY, Kaelin WG., Jr Molecular pathways in renal cell carcinoma–rationale for targeted treatment. Semin Oncol. 2006;33:588–95. doi: 10.1053/j.seminoncol.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–66. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 70.Clifford SC, Cockman ME, Smallwood AC, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10:1029–38. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- 71.Hoffman MA, Ohh M, Yang H, et al. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–27. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- 72.Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–67. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 73.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 74.Kim WY, Sharpless NE. VHL inactivation: a new road to senescence. Cancer Cell. 2008;13:295–7. doi: 10.1016/j.ccr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 75.Parrinello S, Samper E, Krtolica A, et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003;5:741–7. doi: 10.1038/ncb1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Welford SM, Bedogni B, Gradin K, et al. HIF1alpha delays premature senescence through the activation of MIF. Genes Dev. 2006;20:3366–71. doi: 10.1101/gad.1471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bell EL, Klimova TA, Eisenbart J, et al. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007;27:5737–45. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Young AP, Schlisio S, Minamishima YA, et al. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008;10:361–9. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- 79.Welford SM, Dorie MJ, Li X, et al. Renal oxygenation suppresses VHL-loss-induced senescence that is caused by increased sensitivity to oxidative stress. Mol Cell Biol. 2010;30:4595–603. doi: 10.1128/MCB.01618-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hergovich A, Lisztwan J, Barry R, et al. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5:64–70. doi: 10.1038/ncb899. [DOI] [PubMed] [Google Scholar]
- 81.Singla V, Reiter JF. The primary cilium as the cell's antenna: signaling at a sensory organelle. Science. 2006;313:629–33. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 82.Zhuang Z, Roth MJ, Emmert-Buck MR, et al. Detection of the von Hippel-Lindau gene deletion in cytologic specimens using microdissection and the polymerase chain reaction. Acta Cytol. 1994;38:671–5. [PubMed] [Google Scholar]
- 83.Lubensky IA, Gnarra JR, Bertheau P, et al. Allelic deletions of the VHL gene detected in multiple microscopic clear cell renal lesions in von Hippel-Lindau disease patients. Am J Pathol. 1996;149:2089–94. [PMC free article] [PubMed] [Google Scholar]
- 84.Siroky BJ, Guay-Woodford LM. Renal cystic disease: the role of the primary cilium/centrosome complex in pathogenesis. Adv Chronic Kidney Dis. 2006;13:131–7. doi: 10.1053/j.ackd.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 85.Hergovich A, Lisztwan J, Thoma CR, et al. Priming-dependent phosphorylation and regulation of the tumor suppressor pVHL by glycogen synthase kinase 3. Mol Cell Biol. 2006;26:5784–96. doi: 10.1128/MCB.00232-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lutz MS, Burk RD. Primary cilium formation requires von hippel-lindau gene function in renal-derived cells. Cancer Res. 2006;66:6903–7. doi: 10.1158/0008-5472.CAN-06-0501. [DOI] [PubMed] [Google Scholar]
- 87.Esteban MA, Tran MG, Harten SK, et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–75. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 88.Frew IJ, Thoma CR, Georgiev S, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008;27:1747–57. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bonsib SM. Renal cystic diseases and renal neoplasms: a mini-review. Clin J Am Soc Nephrol. 2009;4:1998–2007. doi: 10.2215/CJN.02020309. [DOI] [PubMed] [Google Scholar]
- 90.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 91.Kurban G, Duplan E, Ramlal N, et al. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene. 2008;27:1004–12. doi: 10.1038/sj.onc.1210709. [DOI] [PubMed] [Google Scholar]
- 92.Tang N, Mack F, Haase VH, et al. pVHL function is essential for endothelial extra-cellular matrix deposition. Mol Cell Biol. 2006;26:2519–30. doi: 10.1128/MCB.26.7.2519-2530.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ohh M, Yauch RL, Lonergan KM, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–68. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- 94.Bishop T, Lau KW, Epstein AC, et al. Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004;2:e289. doi: 10.1371/journal.pbio.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Russell RC, Ohh M. NEDD8 acts as a ‘molecular switch’ defining the functional selectivity of VHL. EMBO Rep. 2008;9:486–91. doi: 10.1038/embor.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stickle NH, Chung J, Klco JM, et al. pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol Cell Biol. 2004;24:3251–61. doi: 10.1128/MCB.24.8.3251-3261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Calzada MJ, Esteban MA, Feijoo-Cuaresma M, et al. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006;66:1553–60. doi: 10.1158/0008-5472.CAN-05-3236. [DOI] [PubMed] [Google Scholar]
- 98.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nature Rev. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 99.Turcotte S, Chan DA, Sutphin PD, et al. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14:90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bommi-Reddy A, Almeciga I, Sawyer J, et al. Kinase requirements in human cells: III. Altered kinase requirements in VHL-/-cancer cells detected in a pilot synthetic lethal screen. Proc Natl Acad Sci USA. 2008;105:16484–9. doi: 10.1073/pnas.0806574105. [DOI] [PMC free article] [PubMed] [Google Scholar]