

Abstract
JC virus (JCV) encodes a small basic phosphoprotein from the late coding region called agnoprotein, which has been shown to play important regulatory roles in the viral replication cycle. In this study, we report that agnoprotein forms highly stable dimers and higher order oligomer complexes. This was confirmed by immunoblotting and mass spectrometry studies. These complexes are extremely resistant to strong denaturing agents, including urea and SDS. Central portion of the protein, amino acids spanning from 17 to 42 is important for dimer/oligomer formation. Removal of 17 to 42 aa region from the viral background severely affected the efficiency of the JCV replication. Extracts prepared from JCV-infected cells showed a double banding pattern for agnoprotein in vivo. Collectively, these findings suggest that agnoprotein forms functionally active homodimer/oligomer complexes and these may be important for its function during viral propagation and thus for the progression of PML.
Keywords: JC virus, SV40, BKV, PML, agnoprotein, stable dimer, oligomer, transcription, replication
INTRODUCTION
Dimer and higher order oligomer formation of proteins is common, and is frequently a necessary phenomenon for biological systems. Subunit interactions are essential processes for regulation of the functions of many proteins including enzymes, ion channels, receptors and transcription factors (Marianayagam, Sunde, and Matthews, 2004). Dimer/oligomer formation can also help to minimize the genome size of the organisms by allowing multiple combinations of active protein molecules. On the contrary, such formations may have harmful consequences for a biological system when non-native oligomers associated with pathogenic states are generated. For example, β-amyloid deposition in the brains of affected individuals results in Alzheimer’s disease. This protein forms fibrillar structures through the self-association of the monomers (Serpell, Sunde, and Blake, 1997; Sunde and Blake, 1997; Sunde et al., 1997).
There are many examples of the self-association and oligomerization of viral proteins. Examples include polyomavirus large T antigen, (LT-Ag) and human deficiency virus 1 (HIV-1) small proteins, Rev and vpr. LT-Ag binds to its target sequences as a double hexamer in the origin of replication and initiates the viral DNA replication (Auborn et al., 1988; Lynch and Frisque, 1990; Simmons, Loeber, and Tegtmeyer, 1990). Rev, a small HIV-1 protein, is involved in the transport of incomplete spliced RNA molecules from nucleus to cytoplasm. This protein binds to Rev response elements present in the intron region of the viral transcripts and was found to function as both stable dimers and oligomers as evidenced by 3D structural studies (Daugherty et al., 2010; Daugherty, D’Orso, and Frankel, 2008; Daugherty, Liu, and Frankel, 2010; DiMattia et al., 2010). Like Rev, Vpr also forms stable dimers, arrests cells at G2/M phase transition and induces apoptosis (Bolton and Lenardo, 2007; Cui et al., 2006; Fritz et al., 2008; Fritz et al., 2010; Godet et al., 2010; Iordanskiy et al., 2004; Poon, Chang, and Chen, 2007). Vpr 3D structure has been determined by NMR, showing that the carboxy-terminal helix is responsible for its dimer formation (Bourbigot et al., 2005). An Ebola virus-specific transcription factor, VP30 was also found to form homodimers. A region within this protein spanning amino acid residues 94-112 is essential for oligomerization, where a cluster of four leucine residues was shown to have a critical importance. Mutation of only one of these leucine residues resulted in a molecule that no longer forms oligomers and therefore supports EBOV-specific transcription (Hartlieb et al., 2003). Some viral proteins undergo nucleic acid-induced polymerization process in a sequence specific manner. One example of those is the Borna disease virus (BDV) nucleoprotein which requires the presence of 5’-specific BDV RNA for its oligomerization process (Hock et al., 2010)
JC virus (JCV), a member of the polyomaviridae family of viruses and is known to be the etiologic agent of fatal disease, progressive multifocal leukoencephalopathy (PML) (Frisque, 1992; Major et al., 1992). The viral genome encodes several regulatory proteins, including large T antigen (LT-Ag), small t antigen (Sm t-Ag) T’165, T’136, T’135 and agnoprotein (Bollag et al., 2010; Bollag et al., 2006; Khalili, Sariyer, and Safak, 2008; Khalili et al., 2005; Major et al., 1992; Safak et al., 2001). Agnoprotein is a small basic protein, expressed by all three JCV, BK virus (BKV) and simian virus 40 (SV40) polyomaviruses. Its expression during the viral lytic cycle has been demonstrated by biochemical and immunocytochemical methods in infected cells (Gilbert et al., 1981; Rinaldo, Traavik, and Hey, 1998; Safak et al., 2002) and in tissue sections (Okada et al., 2002). It is a predominantly cytoplasmic protein localized to the perinuclear region of infected cells, however, a small fraction of it was also found to be localized to the nucleus. This is supported by a recent report by Unterstab et al., where it was found that agnoprotein becomes nuclear when both amino acids 25 (Ala) and 39 (Phe) are mutated to aspartic acid (Asp) and glutamic acid (Glu) respectively (Unterstab et al., 2010). Amino acid sequence alignment of the agnoproteins for JCV, BKV and SV40 shows about 70% sequence identity among these functionally related proteins (Khalili et al., 2005; Safak et al., 2001). While the amino-terminal and central regions of each agnoprotein exhibit considerable sequence identity with one another, the sequences toward the carboxy-terminal region are more divergent.
It has been recently shown that amino terminus of agnoprotein is targeted for phosphorylation by a well-characterized protein kinase, protein kinase C (PKC). This modification plays a significant role in the function of this protein during the viral replication cycle of BK virus and JC virus (Johannessen et al., 2008; Sariyer et al., 2006). SV40 agnoprotein was also previously reported to be phosphorylated but no function was assigned to it (Nomura, Khoury, and Jay, 1983). Agnoprotein has also been previously found to functionally interact with viral and cellular proteins, including LT-Ag (Safak et al., 2001) and Sm t-Ag (Sariyer, Khalili, and Safak, 2008), Y-box binding protein, Yb-1 (Safak et al., 2002), p53 (Darbinyan et al., 2002) and HP1α (Suzuki et al., 2005). Mutational analysis of agnoprotein from the closely related virus SV40 suggested that it may have effects on various aspects of the viral lytic cycle including transcription, translation, virion production and maturation of the viral particles (Alwine, 1982; Haggerty, Walker, and Frisque, 1989; Hay, Skolnik-David, and Aloni, 1982; Hou-Jong, Larsen, and Roman, 1987; Margolskee and Nathans, 1983; Ng, Behm, and Bina, 1985; Ng et al., 1985)
Based on amino acid sequence, the predicted molecular weight of JCV agnoprotein is ~8.00 kDa. However, this protein has been previously detected as two discrete bands in extracts prepared from both transfected and infected cells (Del Valle et al., 2002; Merabova et al., 2008). These findings are also consistent with recent observations by Suzuki et al., (Suzuki et al., 2010), who demonstrated the homodimer and homooligomer formation of agnoprotein by intermolecular fluorescence resonance energy transfer (FRET) analysis and chemical crosslinking studies. It was also previously observed that bacterially produced GST-fusion protein of agno was found to induce multiple higher molecular weight protein complexes (Safak et al., 2001). Those complexes were initially thought to be bacterial proteins that strongly interact and co-purify with agnoprotein during the affinity purification process (Safak et al., 2001). Previously, it was not clear that they result from the dimer/oligomer formation property of agnoprotein itself. In this report, we have shown that bacterially produced MBP and GST fusion agnoproteins form highly stable homodimers and oligomers in vitro, mapped the dimerization domain of agnoprotein to amino acids 17-42 and investigated the functional consequences of the deletion of the dimerization domain from the viral background, where deletion of this domain severely affected the replication cycle of JCV. Finally, analysis of agnoprotein, expressed in infected cells, showed two discrete bands on immunoblots suggesting that agnoprotein may also form dimers and perhaps oligomers in vivo and functions accordingly during the JCV infection cycle.
MATERIALS AND METHODS
Cell lines
SVG-A is a subclonal population of a human glial cell line which was established by transformation of human fetal glial cell line with an origin-defective SV40 mutant and has been described previously (Major et al., 1985). These cells do not express either SV40 viral capsid proteins (VPs) or agnoprotein. Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin, 100 μg/ml). They were maintained at 37°C in a humidified atmosphere with 7% CO2.
Plasmid constructs
Full length (FL) JCV agnoprotein open reading frame with three stop codons at the end of the gene was directionally subcloned into Nco I / Eco RI sites of pMAL-c5x bacterial expression vector to produce MBP-Agno fusion protein by PCR-based cloning. Bluescript KS-JCV-Mad-1 WT plasmid was used as a template for PCR amplification by using the following primers: 5’ primer (5’-CCT CTA TCG CAG CCC ATC CC ATG GGC GTT CTT CGC CAG CTG TCA CGT AAG 3’); 3’primer (5’-CCT CTA TCG CAG CCC ATC GAA TTC CTA TTA TTA TGT AGC TTT TGG TTC AGG CAA AGC ACT -3). PCR product was ethanol-precipitated, digested with Nco I and Eco RI restriction enzymes (underlined sequences indicate the restriction sites for Nco I and Eco RI respectively), gel purified and ligated into pMAL-c5x bacterial expression vector and the resulting plasmid was designated as pMAL-c5x JCV-Agno-FL. The following fragments of agnoprotein, encompassing amino acids, 1-16, 1-42, 17-71 and 43-71, were also cloned into pMAL-c5x vector at Nco I and Eco RI sites by PCR-based cloning as described for FL agnoprotein. They were designated as pMAL-c5x JCV-Agno (1-16), pMAL-c5x JCV-Agno (1-42), pMAL-c5x JCV-Agno (17-71) and pMAL-c5x JCV-Agno (43-71). These truncation mutants of agnoprotein also contained a triple stop codon at the end of the truncated gene. Full length of BKV and SV40 agnoprotein coding sequences was also cloned into pMAL-c5x vector by PCR-based cloning at Nco I and Eco RI sites; and named pMAL-c5x-BKV Agno-FL and pMAL-c5x-SV40 Agno-FL. FL JCV agnoprotein genome with three stop codon at the end of the gene was also directionally subcloned into pGEX 1λT at Bam HI/Eco RI sites by PCR-based cloning as described above to produce the GST-Agno fusion protein and designated as pGEX 1λT-JCV Agno FL. Agnoprotein genome was PCR-amplified from Bluescript KS-JCV-Mad-1 WT plasmid, using the following primers: 5’ primer (5’-CCT CTA TCG CAG CCC GGA TTC ATG GGC GTT CTT CGC CAG CTG TCA CGT AAG 3’); 3’primer (5’-CCT CTA TCG CAG CCC ATC GAA TTC CTA TTA TTA TGT AGC TTT TGG TTC AGG CAA AGC ACT -3). Amino acids 17 to 42 were deleted from the virus background by deletion mutagenesis using appropriate primers and this plasmid was designated as Bluescript KS-JCV Mad-1 Agno (Δ17-42). The integrity of the agnoprotein genome in all constructs was verified by DNA sequencing.
Expression and affinity purification of Agno-fusion proteins
For the expression of the different MBP-Agno protein fusions, pMAL-c5x-JCV-Agno-FL and pMAL-c5x vector containing the deletion mutants of agnoprotein were transformed into E. coli DH5α cells. Bacterial cells were grown in 0.5-1 L LB media supplemented with ampicillin (100 μg/mL and glucose (2 g/L) at 37°C until OD595 = 0.4. Protein production was induced by the addition of and 0.3 mM IPTG and the incubation temperature was lowered to 28°C and maintained at that temperature for 2h. Bacterial cells were harvested by centrifugation (8000 rpm, Sorval-SLA-3000 rotor) and the resulting pellet from the various MBP-Agno fusions were each resuspended in 20 mM Tris-HCl pH 7.4 containing 200 mM NaCl and 1 mM EDTA [Amylose fast flow (FF) column buffer]. The cell suspension was then incubated on ice for 30 min in the presence of lysozyme and protease inhibitor cocktail, after which the cells were lysed by sonication. Clear lysates were obtained after a high speed centrifugation at 15,000 rpm (Thermo Scientific, F21-8x50y rotor). The lysates were then incubated with 125 μL of amylose FF resin (New England Biolabs) overnight at 4°C to ensure efficient binding of the proteins to the resin beads. After incubation, the protein bound amylose resins for each MBP-Agno protein fusion (FL and mutants) were washed with five column volumes of amylose column binding buffer (20 mM Tris pH 7.4, 200 mM NaCl, 1 mM EDTA). Target proteins were either stored on beads at 4°C or eluted with 10 mM maltose prepared in the same buffer and stored at -30°C until use.
The expression and protein extraction of the pGEX 1λT JCV-Agno (GST-Agno FL) was carried out following a similar procedure as described above for the MBP-Agno fusion proteins with a few modifications. Protein expression was induced by the addition of 0.3 mM IPTG. The bacterial pellet resulting from the cell growth was resuspended in 40 mL PENT buffer (20 mM Tris HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 % NP-40 and 1% N-L-Sarcosyl), and sonicated followed by a high speed centrifugation at 15000 rpm (Thermo Scientific, F21-8x50y rotor). GST-Agno FL lysate was incubated with 150 μL bed volume of GSH-Sepharose resin (GE HealthCare) overnight at 4°C. GST-Agno beads were washed three times with PENT buffer and resuspended in 1 × PBS and were either kept in bound form at 4°C or eluted from the GSH-Sepharose 4B beads using 20 mM glutathione in 50 mM Tris-HCl buffer, pH 8.0. The eluted GST-Agno protein was stored at -70°C until use. The partner affinity tags MBP and GST were also each expressed and purified as a single protein following the methods used for MBP-Agno and GST-Agno fusion proteins, respectively.
Western blotting
For detection of agnoprotein in fusion protein complex, one microgram of MBP alone and MBP-Agno proteins (FL and mutants of agnoprotein) were analyzed on a 10% SDS-polyacrylamide gel after treating the samples with 1 × SDS loading dye containing β-mercaptoethanol and heating them at 95°C for various amounts of time as indicated in the respective figure legends. Proteins were transferred to a nitrocellulose membrane for 3h and detected with anti-Agno polyclonal antibody and the detection was performed using ECL system following the manufacturer’s guidelines (Amersham, UK). For detection of agnoprotein in extracts prepared from the transfected/infected cells, 40 μg of whole cell extracts prepared from SVG-A cells either untransfected or transfected with Bluescript KS-JCV-Mad-1 WT plasmid (Sariyer et al., 2006) were treated with SDS sample buffer and heated at 95°C for 5 min. In addition, 100 μg whole cell extract was immunoprecipitated (IP) with either normal rabbit serum (5 μl) or with a polyclonal anti-agno antibody (Del Valle et al., 2002; Safak et al., 2002) (5 μl), treated in 1 × SDS loading dye and heated at 95°C for 5 min. Both immunoprecipitated and normal protein samples were then separated on a 15% SDS-polyacrylamide gel and transferred onto nitrocellulose filter (Bio-Rad) for 10 min at 250 mA current. The blotted membranes were probed with a polyclonal anti-agnoprotein antibody as described earlier (Safak et al., 2002; Sariyer et al., 2006). The detection was performed using ECL system following the manufacturer’s guidelines (Amersham, UK). For detection of VP1 in nuclear extracts prepared from the transfected/infected cells, 40 μg of protein was separated on a 10% SDS-polyacrylamide gel, transferred onto cellulose membrane for 3h at 250 mA and probed with anti-VP1 monoclonal antibody (PAB597) using ECL detection kit.
Factor Xa cleavage
Purified MBP-Agno was dialyzed against the reaction buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl) and incubated with Factor Xa (NEB) in the presence of 2 mM CaCl2 for 3 hrs at room temperature (RT). The samples were then analyzed on a 10% SDS-polyacrylamide gel. In addition, cleavage products were also analyzed by Western blotting, for which samples were first separated on a 15% SDS-polyacrylamide gel, transferred onto nitrocellulose filter (Bio-Rad) and probed with a polyclonal anti-agno antibody (Safak et al., 2002).
LC-MS analysis
LC-MS analysis was performed by Wistar Institute Proteomic Facility, Philadelphia, PA (see supplement 1).
MALDI-TOF analysis
MALDI-TOF analysis of the MBP-Agnoprotein complexes was performed by Wistar Institute Proteomic Facility, Philadelphia, PA (see supplement 2).
Mapping homodimer and oligomer formation domain of agnoprotein
Eluted MBP alone or MBP-Agno FL or MBP-Agno deletion mutants (1-16, 1-41, 17-71 and 43-71) (5μg each) were separated on a 10% SDS-polyacrylamide gel and analyzed by coomassie staining to monitor dimer/oligomer formation.
Treatment of MBP-Agno dimer and oligomers with strong denaturing agents
The stability of dimer and oligomers of agnoprotein was tested by employing a prolonged heat treatment at 95°C for various amount of time as indicated in the respective figure legend. Ten micrograms of MBP-Agno was mixed with 1 × SDS sample buffer (final volume) and heated at 95°C for 5, 10, 20, 30, 45 and 60 minutes. Five micrograms of each sample was fractioned on a 8% SDS-polyacrylamide gel and analyzed by coomassie blue staining. The protein bands were quantified using NIH ImageJ software. A hundred nanogram of the sample were analyzed by Western blotting using a polyclonal anti-agno antibody (Safak et al., 2002). Similarly, 5 μg of MBP-Agno protein was subjected to treatment with an increasing concentration of SDS (1, 2, 5, 10, 15 and 20%) for 30 min at room temperature. 1 × SDS sample buffer was added to the samples and samples were then heated at 95°C for 5 min, separated on a 10% SDS-polyacrylamide gel and analyzed by coomassie blue staining. The protein bands were then quantified using NIH ImageJ software. The stability of agnoprotein dimers and oligomers was also tested by treating the samples with high concentration of urea. Five micrograms of MBP-Agno were treated with 8M Urea (final concentration) at room temperature for different periods of time as indicated in the respective figure, followed by addition of SDS sample loading dye and heating at 95°C for 5 minutes. The samples were then separated on a 8% SDS-polyacrylamide gel and analyzed by coomassie blue staining.
Transfection/infection
SVG-A cells were transfected with either Bluescript KS-JCV Mad-1 WT or Bluescript KS-JCV Mad-1 Agno (Δ17-42) plasmids (8μg/2×106cells/75cm2 flask) using lipofectin 2000 reagent (Sariyer et al., 2006). Complete genome of JCV was liberated from Bluescript KS vector by Bam HI digestion and used in transfections. Whole cell and nuclear extracts were prepared at indicated time points from infected and uninfected cells as described previously (Akan et al., 2006; Sariyer et al., 2006) and analyzed by Western blotting using anti-VP1 antibody (pAB597).
Anion exchange chromatography
MBP-Agno was purified in large scale using 10 ml amylose FF columns, as described above, and pooled fractions of MBP-Agno fusion protein (~17 mg) eluted from the amylose resin were first exchanged against 25 mM Tris-HCl, pH 7.5, 50 mM NaCl and 1 mM EDTA (Buffer A) using a PD10 gel filtration column. This sample was then loaded onto a 25 mL Q Sepharose anion exchange column (Amersham Biosciences) preequilibrated with the same buffer. The column was initially washed with 50 mL of Buffer A to remove unbound proteins. The fusion protein was eluted using a linear gradient of 50-1000 mM NaCl collecting 8 mL fractions. Aliquots (15 μl) of each fraction from the anion-exchange step were analyzed on a 4-12% SDS-polyacrylamide gel, followed by coomassie staining.
Gel filtration chromatography
In an attempt to separate the different oligomeric forms of the fusion protein, the Q Sepharose fractions containing the MBP-Agno dimers and monomers were pooled and concentrated using an Amicon spin concentrator (10 kDa MW cut off) to obtain a total of 12 mg of protein. Gel filtration chromatography was carried out using a high resolution resin (Sephacryl HR200, exclusion range of 5 to 250 kDa) equilibrated with 25 mM Tris-HCl, 50 mM NaCl and 1 mM EDTA. Fractions were collected using 1.5x column volume buffer (~180 mL) to elute the proteins. Fractions were analyzed on a 4-12% SDS-polyacrylamide gel followed by coomassie staining.
Replication assay
Replication assays were carried out as previously described (Safak et al., 2001). Briefly, SVG-A cells (2×106 cells) grown in 75 cm2 flasks were transfected either with JCV Mad-1 WT or JCV Mad-1 Agno (Δ17-42) viral genomes (8 μg/2×106 cells/75 cm2 flask) by lipofectin-2000 transfection method, as described under the transfection section. At indicated time points posttransfection, low-molecular-weight DNA containing both input and replicated viral DNA was isolated by the Hirt method (Hirt, 1967), digested with Bam HI and Dpn I enzymes, resolved on a 1% agarose gel and analyzed by Southern blotting.
Viral particle release assay
JCV Mad-1 WT genome or JCV Mad-1 Agno (Δ17-42) genomes were separately transfected into SVG-A cells (8μg DNA/2×106 cells/75cm2 flask), as described under transfection/infection section. Supernatants from infected cells were collected at indicated time points, centrifuged at 16,000 × g to clear cell debris and were subjected to immunoprecipitation using an anti-VP1 monoclonal antibody (PAB597) (4μl). Half of the samples were analyzed by Western blotting using anti-VP1 antibody (PAB597), and the other half was analyzed by Southern blotting for detection of the encapsidated viral DNA. The viral DNA from capsids was purified employing Qiagen spin columns (Ziegler et al., 2004), digested with Bam HI and Dpn I enzymes, resolved on an 1% agarose gel and analyzed by Southern blotting using probes prepared from whole Mad-1 genome.
RESULTS
Detection of agnoprotein-induced high molecular weight complexes in affinity purified fusion protein fractions
Our previous analysis of GST-agno fusion protein by SDS-polyacrylamide gel consistently revealed the presence of additional agnoprotein-induced higher molecular weight complexes (Safak et al., 2001; Sariyer et al., 2006). These complexes were originally presumed to be bacterial proteins that strongly interact and thereby co-purify with agnoprotein during affinity purification process (Safak et al., 2001; Sariyer et al., 2006). In this report, we sought to further investigate the nature of these complexes, by determining whether complex formation only occurs when it is fused to GST (glutahione-S-transferase). To address this question, agnoprotein was fused to a maltose binding protein (MBP), then expressed in Escherichia. coli and purified by affinity chromatography. As shown in Fig. 1A and 1B, the expected size of monomeric GST-Agno is near 32 kDa and that of MBP-Agno is 51 kDa. However, in both cases, additional bands were observed in the respective lanes, running at significantly higher levels than the expected sizes of each agno fusion protein (GST and MBP). This eliminates the possibility that the observed phenomenon is a fusion protein-specific case, but rather occurs regardless of the type of the backbone of the fusion protein. It was also our experience working with MBP-Agno protein construct that it was found to be expressed in higher amounts in E. coli than GST-Agno and readily eluted from the affinity resin compared to GST-Agno. Due to these reasons, MBP-Agno construct was chosen for further protein characterization studies.
Fig. 1. Expression of agnoprotein as GST and MBP fusion protein.
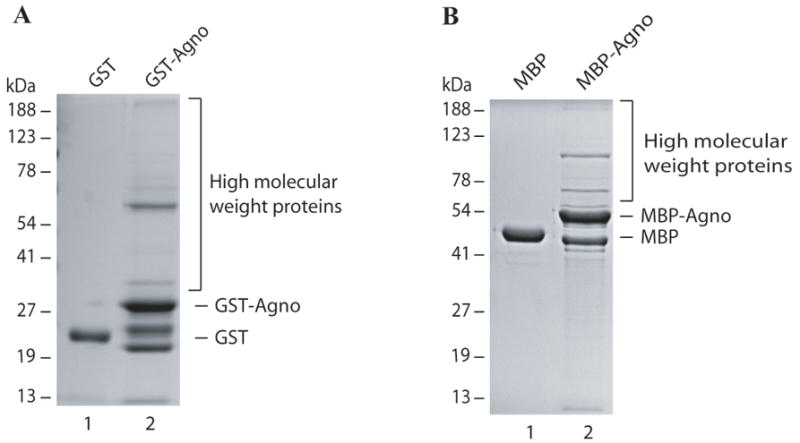
(A) GST alone (5μg) and GST-Agno (5μg) were expressed in E. coli, affinity purified using GSH-Sepharose 4B resin, separated on a 10% SDS-polyacrylamide gel and analyzed by coomassie staining. (B) MBP alone (5μg) and MBP-Agno (5μg) were expressed in E. coli, affinity purified using amylose FF resin, separated on a 10% SDS-polyacrylamide gel and analyzed by coomassie staining.
Detection of high molecular weight complexes by anti-agno antibody
Next, the identity of the proteins participating in these complexes was investigated to determine if these proteins are in fact bacterial proteins that co-purify with agnoprotein during affinity purification or, alternatively, whether agnoprotein itself can form stable homodimers and oligomers, resulting in this pattern of higher molecular weight bands on SDS-polyacrylamide gel. To distinguish between these two possibilities, affinity purified MBP alone and MBP-Agno were separated on a 10% SDS-polyacrylamide gel (Fig. 2A), then transferred on a nitrocellulose membrane and probed with an anti-agnoprotein antibody (Fig. 2B). As expected, MBP-Agno (monomer) was detected on the membrane at about 51 kDa (See MALDI-TOF analysis, supplement 2). However, the anti-agnoprotein antibody also detected all of the higher molecular weight complexes that were observed on a SDS-polyacrylamide gel, while no higher molecular weight bands were detected in the lane that contained MBP alone (Fig. 2B, lane 1). These findings support a case that the higher molecular weight complexes result from a stable homodimer and oligomer formation property of agnoprotein itself. Next, the identity of each high molecular weight complex was determined by mass spectrometry analysis (see Supplement 1). Results showed that these complexes mainly consists of MBP and agnoprotein with little impurities, strongly suggesting that high molecular weight bands result from the homodimer and oligomer formation property of agnoprotein, but did not exclude a possibility that a bacterial protein or proteins could be a part of these complexes. To exclude this possibility, we investigated the content of agnoprotein dimers with a highly sensitive MALDI-TOF analysis (see Supplement 2). It turned out that agnoprotein dimer complexes do not contain any other bacterial protein, clearly confirming the case that stable dimer formation results from the intrinsic homodimer formation property of agnoprotein itself.
Fig. 2. Analysis of agnoprotein-associated high molecular weight proteins by immunoblotting and Factor Xa cleavage.
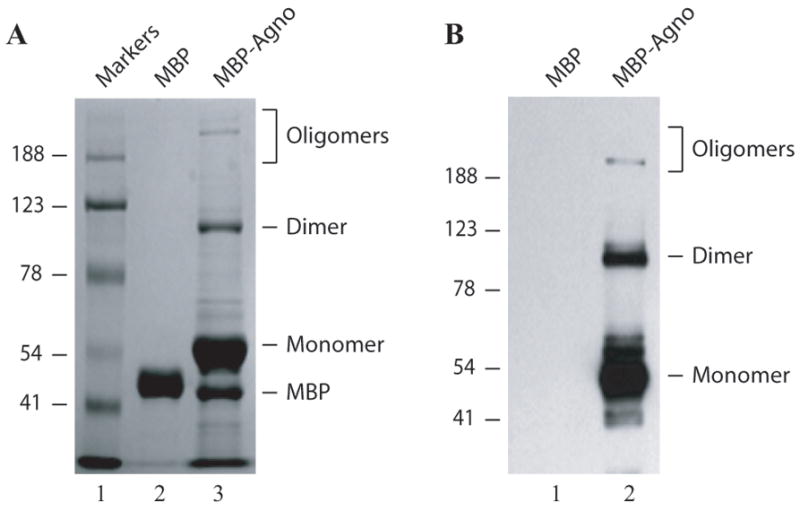
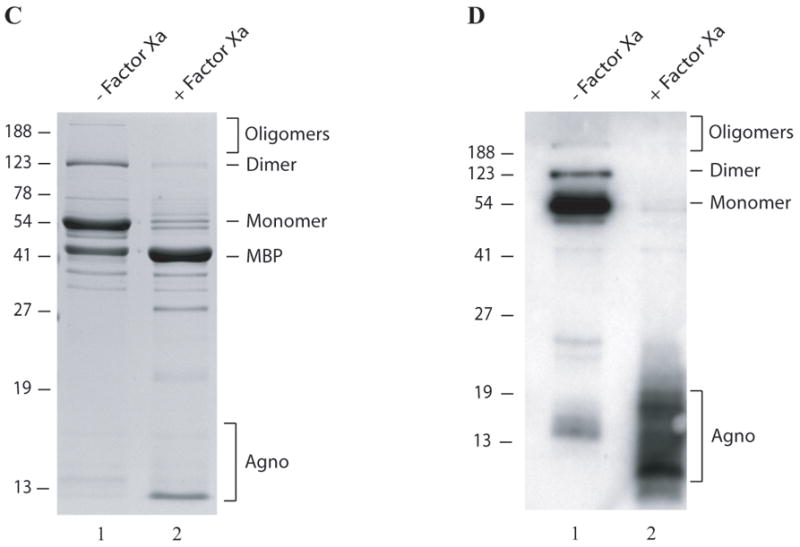
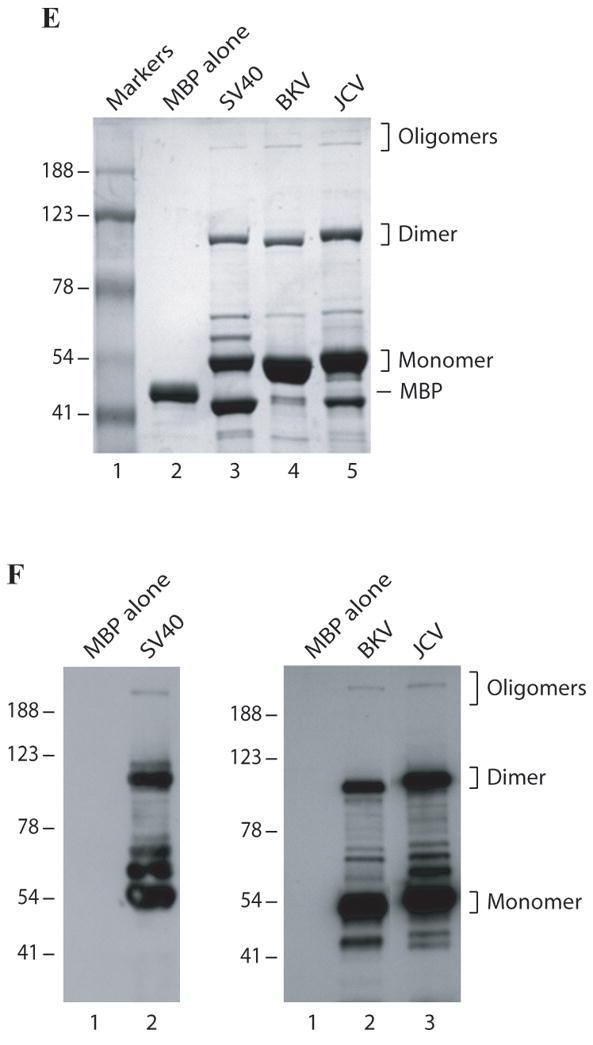
(A) MBP alone (5μg) and MBP-Agno (5μg) were separated on a 8% SDS-polyacrylamide gel and analyzed by coomassie staining. (B) Immunoblot analysis of agnoprotein-associated complexes. Affinity purified MBP alone (5μg) (lane 1) and MBP-Agno (100 ng) were separated on a 8% SDS-polyacrylamide gel, transferred onto nitrocellulose membrane, probed with an anti-agnoprotein antibody (Del Valle et al., 2002) and detected with ECL. (C) Intact MBP-Agno (5μg) (lane 1) and cleaved form by Factor Xa as described in materials and methods, separated on a 12% SDS-polyacrylamide gel and analyzed by coomassie staining. A bracket points to the cleavage products of MBP-Agno. (D) Intact MBP-Agno (5μg) (lane 1) and cleaved by Factor Xa as described in materials and methods, separated on a 15% SDS-polyacrylamide gel and analyzed by Western blotting using an anti-agno antibody (Del Valle et al., 2002; Sariyer et al., 2006). (E) MBP alone, MBP-JCV Agno, MBP-BKV Agno and MBP-SV40 Agno (5μg each) were separated on a 8% SDS-polyacrylamide gel and analyzed by coomassie staining. (F) Immunoblot analysis of MBP fusion protein of JCV, BKV and SV40 agnoproteins. Affinity purified MBP alone (5μg), MBP-JCV Agno (150 ng), MBP-BKV Agno (0.5μg) and MBP-SV40 Agno (10μg) were separated on a 8% SDS-polyacrylamide gel, transferred onto nitrocellulose membrane, probed with an anti-agnoprotein antibody and detected with ECL.
Factor Xa cleavage products were detected by anti-agno antibody running as multiple bands
In order to investigate whether MBP protein alone contributes to dimer and oligomer formation of MBP-Agno, agnoprotein was cleaved from MBP by using Factor Xa and the resulting products were analyzed by a SDS-polyacrylamide gel (Fig. 2C) followed by Western blotting (Fig. 2D). The majority of the higher molecular weight complexes disappeared upon cleavage by Factor Xa, although a small amount of uncleaved products remained intact, likely due to incomplete cleavage reaction (Fig. 2C, lane 2). Detection of the cleavage products by anti-agnoprotein antibody as multiple bands on a Western blot further confirmed that agnoprotein itself is responsible for such complexes but not MBP alone (Fig. 2D). The multiple banding patterns of the cleaved products of agnoprotein also support the conclusion that agnoprotein not only forms homodimers and oligomers, but that those complexes are highly stable under denaturing conditions.
Since JCV, BKV and SV40 agnoproteins show high degree of homology (>70%) with one another, we reasoned that BKV and SV40 agnoproteins also form dimer/oligomers. SDS-polyacrylamide gel analysis of MBP-fusion protein of BKV and SV40 agnoproteins followed by coomassie staining clearly showed that both proteins also form similar stable dimer/oligomers (Fig. 2E) and immunoblotting studies confirmed these findings (Fig. 2F).
Mapping of the agnoprotein region(s) responsible for dimer and oligomer formation
In order to map the region(s) of agnoprotein responsible for dimer and oligomer formation, different deletion mutants of agnoprotein were expressed in E. coli as MBP-Agno fusion and then affinity purified. The ability of dimer/oligomer formation of these mutants was analyzed by SDS-polyacrylamide gel followed by coomassie staining (Fig. 3C). As expected, FL agnoprotein forms a stable dimer and oligomers (Fig. 3C, lane 3). Deletion of a 29 aa region from the carboxy-terminus (1-42 construct) and a 16 aa region from the amino-terminus (17-71 construct) of agnoprotein did not have a noticeable effect on the ability to form higher molecular weight complexes (lanes 4 and 5, respectively). However, deletion of a 55 aa region from the carboxy-terminus (1-16 construct) and a 42 aa region from the amino-terminus (43-71 construct) abrogated dimer/oligomer formation ability of agnoprotein (Fig. 3C, lanes 6 and 7 respectively). From this data, it was deduced that the central portion of agnoprotein, encompassing the aa 17-42 region, is critical for the formation of dimers and oligomers. According to a computer-generated protein structural model, agnoprotein adopts an α-helical structure within 17-42 amino acid region (Fig. 3B). Future studies are required to further delineate the amino acids responsible for dimer/oligomer formation of agnoprotein. The dimer/oligomer formation ability of FL agnoprotein and the various truncation mutants are summarized in Fig. 3D. The FL of the primary sequences (WT agno) and its truncated version (Agno Δ17-42) are shown in Fig. 3A.
Fig. 3. Mapping of the region of agnoprotein responsible for dimer and oligomer formation.
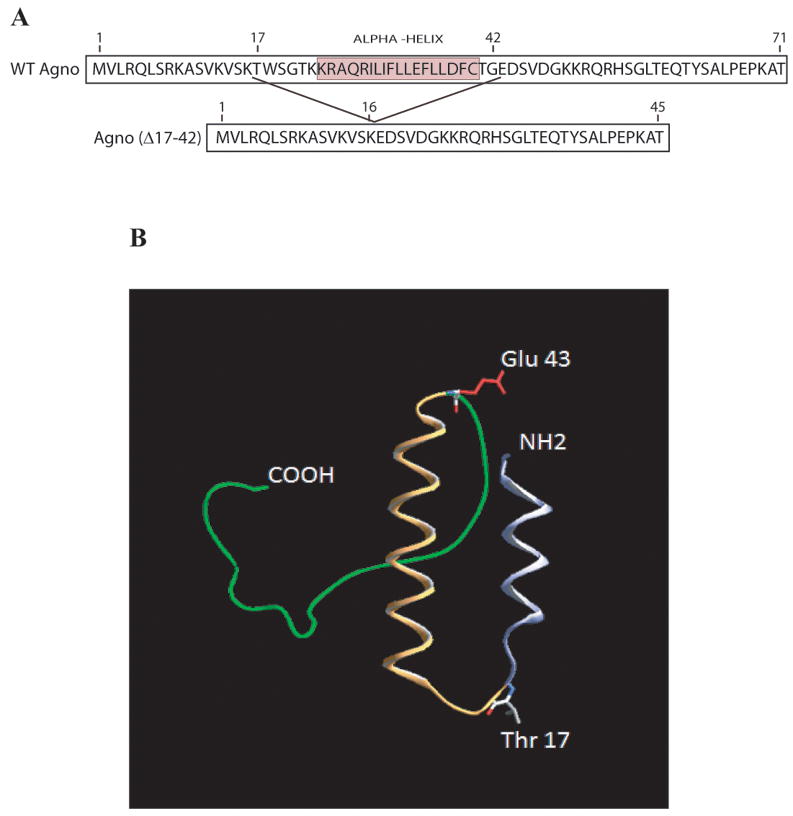
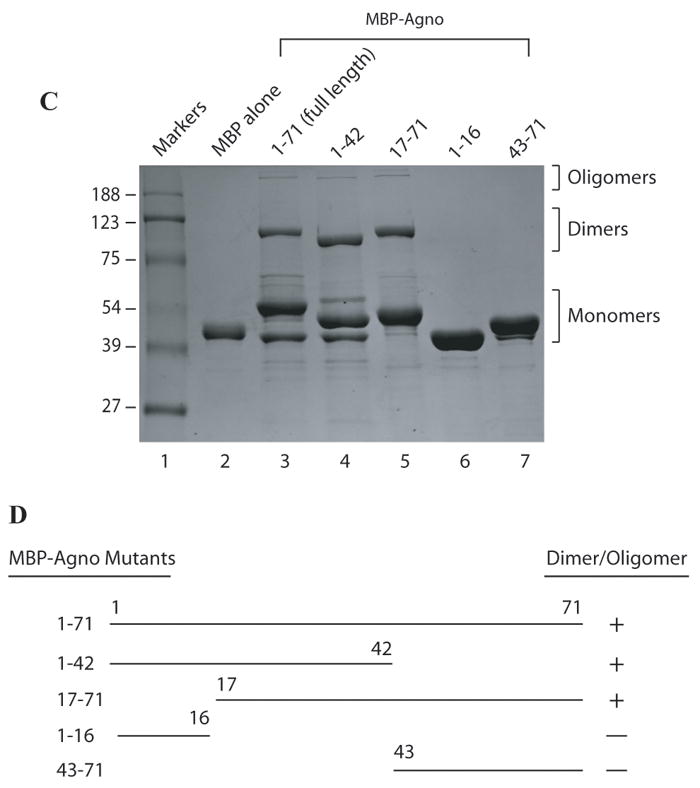
(A) Primary sequence of agnoprotein (Agno-WT). Highlighted region spanning amino acids (17-42) was deleted from the viral background (Agno-Δ17-42). (B) Predicted three dimensional structure of agnoprotein was generated by I-Tasser program (Roy et al 2010). (C) FL agnoprotein and its deletion mutants (aa 1-42, 1-16, 17-71, and 43-71) were fused to MBP, expressed in E. coli, separated on a 8% SDS-polyacrylamide gel and analyzed by coomassie staining. (D) Map of agnoprotein depicting the regions critical for dimer/oligomer formation. “+” and “−” indicate presence and absence of dimer/oligomer formation respectively.
Dimers and oligomers of agnoprotein are resistant to strong denaturing conditions
One of the interesting characteristics of the dimers and oligomers of agnoprotein is being resistant to denaturing agents under normal SDS-polyacrylamide gel conditions. To test their ability to resist even stronger denaturing agents, these complexes were examined in the presence of high concentration of urea and SDS. To this end, 5 μg of MBP-Agno was treated with 8M urea (final concentration) for different times at room temperature. SDS-sample buffer was then added to the protein, heated at 95°C for increasing times from 5 to 120 min, followed by separation on a 10% SDS-polyacrylamide gel and coomassie staining. Unexpectedly, the treatment with 8M urea did not have a significant effect on the integrity of homodimers and oligomers (Fig. 4A, lanes 4, 6, 8 and 10). The behavior of the homodimers and oligomers of agnoprotein was also analyzed in the presence of high concentration of SDS, ranging from 4% to 20%. Even the addition of increasing levels of SDS had only a minor effect on the integrity of the homodimers and oligomers of agnoprotein, even at the highest levels of SDS tested (Fig. 4B and C).
Fig. 4. Exposure of agnoprotein to high concentration of SDS and Urea.
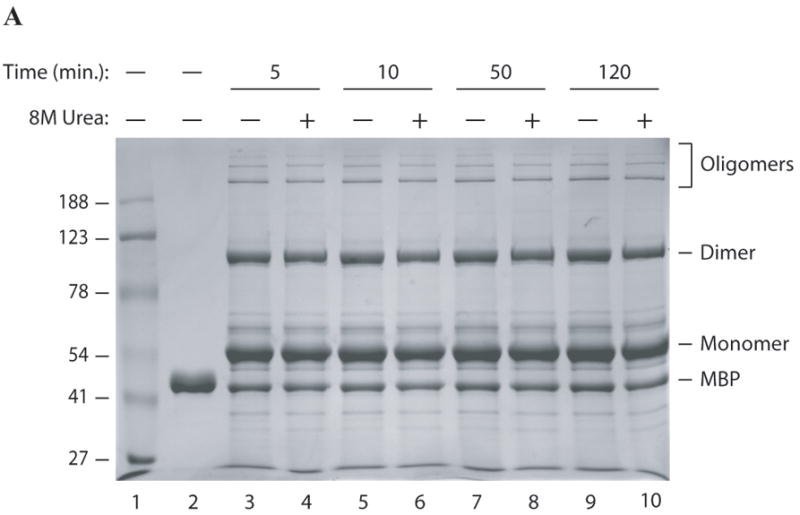
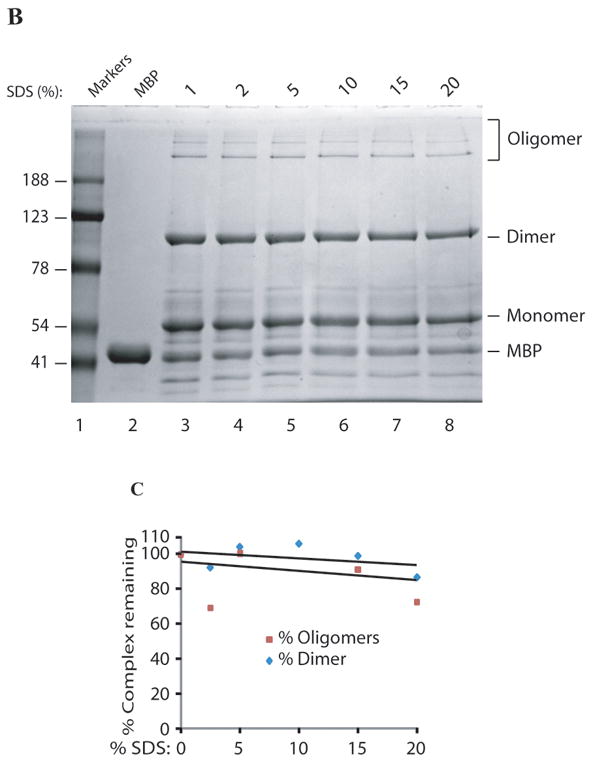
MBP-Agnoprotein samples (10 μg each) were treated with high concentration of urea (8M, final) for a a period of times as indicated before resuspension of the samples in 1 × SDS Laemmli sample buffer. Samples were then heated at 95°C for 5 min, separated on a 8% SDS-polyacrylamide gel and analyzed by coomassie staining. (B) Aliquots of MBP-Agno protein (10 μg) were resuspended in 1 × SDS Laemmli sample buffer after pretreatment of increasing concentration of SDS as indicated, heated at 95°C for 5 min, separated on a 8% SDS-polyacrylamide gel and analyzed by coomassie staining. Lane 2: MBP control. Lane 3: No prior SDS treatment. (C) Protein bands corresponding dimers and oligomers were quantified by gel densitometry using ImageJ program. Percent values were calculated relative to the control sample without pre-SDS treatment.
Prolonged heat treatment shows a considerable effect on the destabilization of agnoprotein dimers and oligomers
The extreme stability of agnoprotein dimers and oligomers under different strong denaturing conditions could be either the result of covalent bond formation or strong non-covalent interactions, such as salt bridges and hydrophobic interactions, between the agnoprotein monomers. Prolonged heat treatment of the protein samples would test these two possibilities since non-covalent interactions will be less resistant to thermal denaturation. MBP alone or MBP-Agno protein samples were exposed to prolonged heat treatment ranging from 5 min to 60 min at 95°C. Following heat treatment, the samples were divided into two portions, one portion separated by a SDS-polyacrylamide gel, while the other portion was analyzed by Western blotting using an anti-agno polyclonal antibody. Approximately 60% of the homodimers and 85% of the homo-oligomers were destroyed upon 1h heat treatment and consequently MBP-Agno monomer band intensity increased (Fig 5A, B and C), suggesting that the high resistance of the dimers and oligomers of agnoprotein against strong denaturing agents is not the result of covalent cross-linking, but likely from numerous strong ionic and hydrophobic attractions between the monomers.
Fig. 5. Prolonged heat treatment of agnoprotein at 95°C.
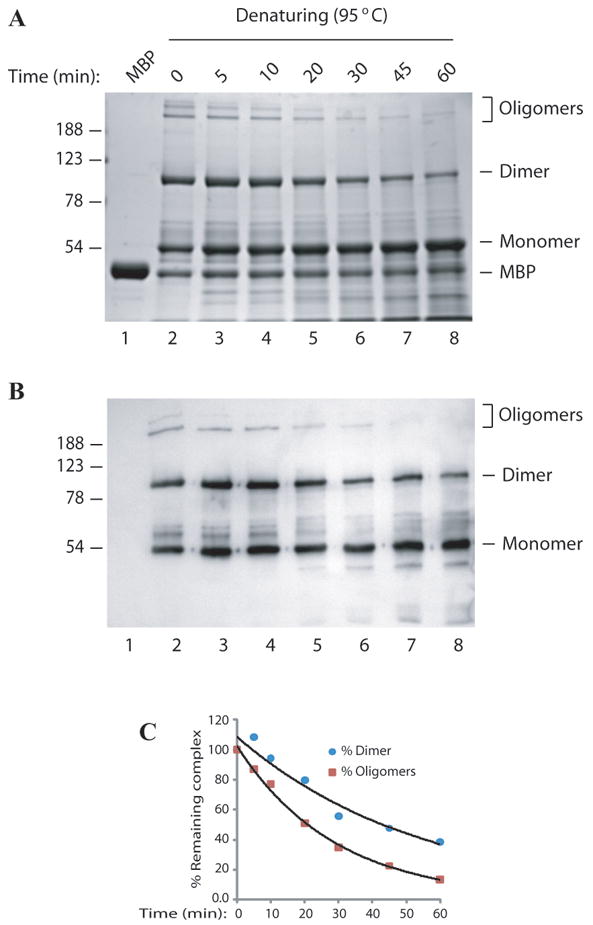
MBP-Agnoprotein samples (10 ug) were resuspended in 1 × SDS Laemmli sample buffer, heated for different lengths of time at 95°C as indicated. Samples were then analyzed on a 10% SDS-polyacrylamide gel (A) and by Western blotting using an anti-agno antibody (Del Valle et al., 2002; Sariyer et al., 2006) (B). (C) Densitometry analysis was performed using Image J software to determine signal levels on the bands corresponding to the dimer and oligomers. Signal levels were plotted against corresponding temperatures.
The level of dimer and oligomer formation of agnoprotein increases in a time- and temperature-dependent manner
During the initial characterization of agnoprotein dimers and oligomers, it was observed that the levels of dimer/oligomers significantly increased over time, seen upon storages at temperatures above 0°C. It was reasoned that temperatures above the freezing point may have a positive effect on the rate of dimer and oligomer formation of agnoprotein in vitro. To investigate the effect of temperature, affinity column purified fresh protein samples were stored at -30°C, 4°C, 25°C and 37°C for 24h and then were analyzed by a SDS-polyacrylamide gel. The level of dimer and oligomer formation increased in a temperature-dependent manner, with higher levels seen in samples kept at 37°C for 24h (Fig. 6A, lane 5) compared to those stored at -30°C (Fig. 6A, lane 2). These findings confirm the effect of temperature on dimer and oligomer formation of agnoprotein. It was also observed that the level of the intensity of a band, indicated by an arrow, remained virtually unchanged under the same conditions, even though this band also contained agnoprotein by mass spectrometry analysis. This band likely resulted from a cleavage product of MBP-Agno causing a differential fractionation of the protein complex.
Fig. 6. The level of agnoprotein dimer and oligomer formation is time and temperature dependent.
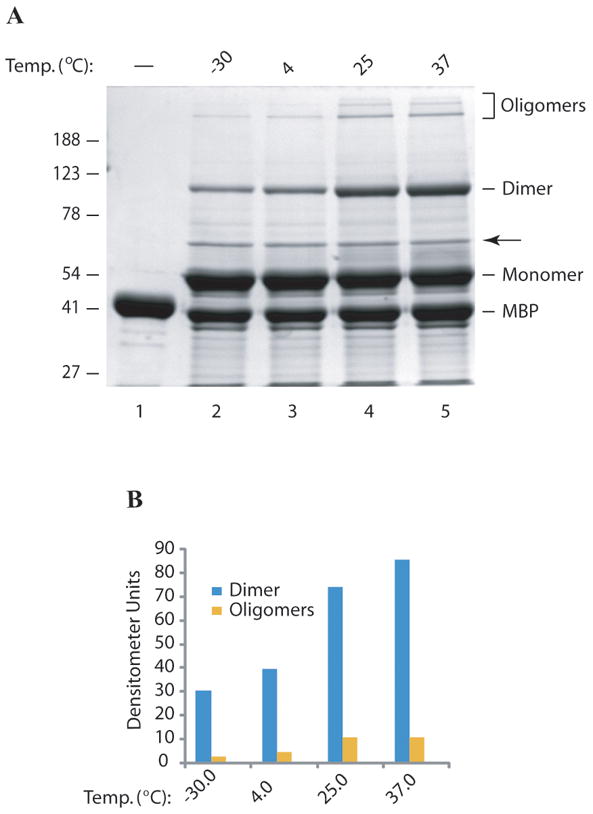
(A) MBP and MBP-Agno protein were expressed in E. coli and affinity purified using amylose column, eluted with maltose, frozen immediately and then stored at -30°C. Frozen samples (10 μg each) were thawed out and then incubated at different temperatures as indicated for 24h, treated with 1 × SDS Laemmli sample buffer and analyzed on a 10% SDS-polyacrylamide gel followed by coomassie blue staining. An arrow points to a cleavage product of MBP-Agno. (B) The signal levels on the bands corresponding to the dimers and oligomers were densitometrically determined using ImageJ software and plotted against corresponding temperatures.
Purification of MBP-Agno by ion exchange and gel filtration chromatography
Amylose affinity column purified MBP-Agnoprotein was further purified by ion-exchange chromatography, followed by gel filtration to characterize the behavior of the monomers, dimers and oligomers in solution. As shown in Fig. 7A, any free MBP can easily be separated from the rest of MBP-agnoprotein (Fig. 7A, fractions 1-6). However, the agnoprotein complexes (monomers, dimers and oligomers) were not separated from each other on an ion-exchange column (fractions 5-13). Each fraction contained all forms of agnoprotein (monomers, dimers, and oligomers). This suggests that either each form of agnoprotein exhibits a similar affinity to the column or they remain as a single complex in the chromatography solution that was used. To further investigate these questions, fractions 7 through 14 were pooled and run through a gel filtration column (Fig. 7B). It turned out that all of the agnoprotein complexes were found to co-elute from this column, even though each complex has a different size by SDS-polyacrylamide gel analysis, suggesting that some complexes tightly and some others loosely bound one other under chromatography buffer conditions.
Fig. 7. Purification of MBP-Agno by ion-exchange and gel exclusion chromatography.
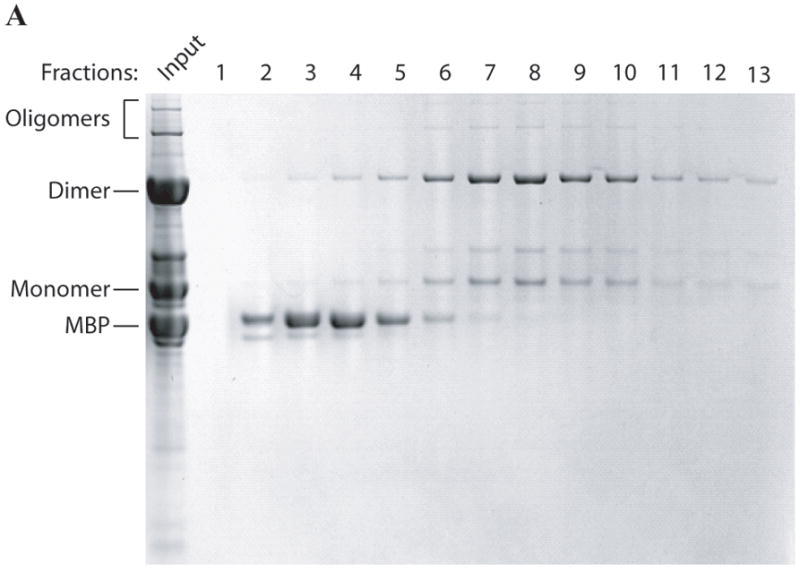
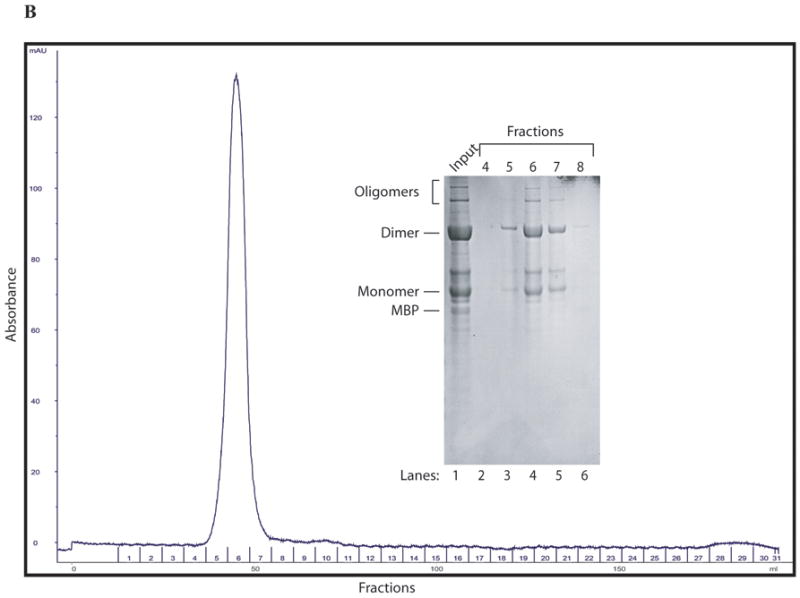
(A) After amylose FF affinity purification, the fractions containing MBP-Agno was pooled and dialyzed as described in the methods and loaded onto a Q Sepharose anion exchange column (25 ml). The column washed with a buffer A (see materials and methods and proteins were eluted using a linear NaCl gradient (50-1000 mM). The fractions were collected and protein samples were analyzed on a 4-20 % SDS-polyacrylamide gel followed by coomassie blue staining. (B) Purification of agnoprotein by gel filtration column. After Q Sepharose purification step fractions containing MBP-Agno were pooled and applied to a gel filtration column (Sephacryl HR 2000, see materials and methods) equilibrated with a buffer A. The fractions were collected and analyzed on a 4-20% SDS-polyacrylamide gel followed by coomassie blue staining. The gel filtration column elution profile and the coomassie stained SDS-polyacrylamide gel are shown.
Detection of agnoprotein as two distinct bands in extracts from infected cells
The final issue to be resolved is whether agnoprotein can also forms stable dimer/oligomer structures in infected cells, and perhaps these higher order structures may have functional roles in vivo. To address this question, SVG-A cells, derived from primary human glial cells, (Major et al., 1985) were infected and whole cell extracts prepared at 15th day of post infection were analyzed either by direct Western blotting or by immunoprecipitation (IP):Western using an anti-agnoprotein antibody. As seen in Fig. 8, both in direct Western (lanes 1 and 2) and in IP:Western cases (lanes 5 and 6), agnoprotein was detected as two distinct bands, with the upper band as the most prominent one. The presence of multiple forms of agnoprotein in these infected cells supports the findings from in vitro studies, and is also consistent with previous reports in the literature by Del Valle et al., (Del Valle et al., 2002) and Merabova et al., (Merabova et al., 2008). Each reported the detection of agnoprotein by Western blotting as two distinct bands under denaturing conditions, however, no explanation was provided for their findings. More recently, Suzuki et al., also reported the dimerization and oligomerization of agnoprotein in transfection assays by fluorescence resonance energy transfer (FRET) technique and cross-linking respectively (Suzuki et al., 2010). The findings in this current study, combined with those from previous studies show that agnoprotein forms, and perhaps even functions, as a dimer or higher order oligomer during viral replication cycle.
Fig. 8. Agnoprotein is present as two distinct bands in extracts from infected cells.
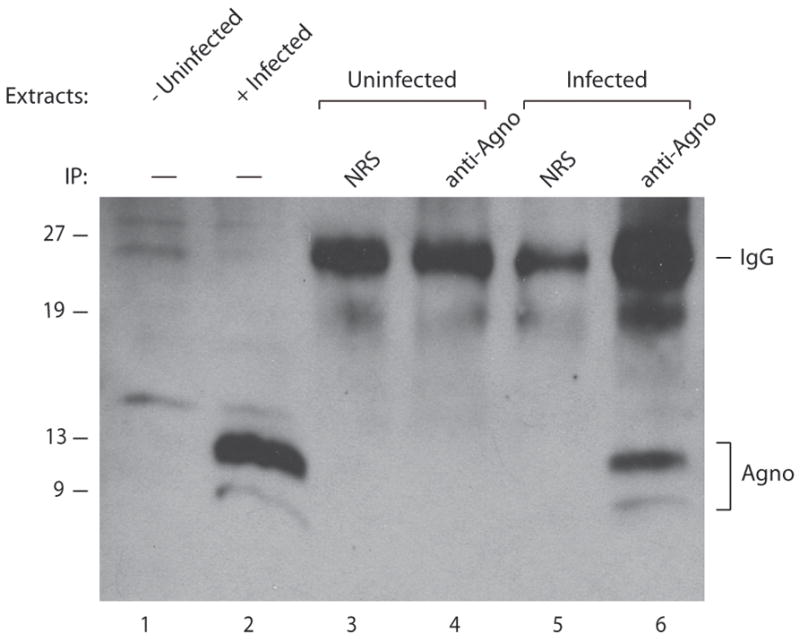
SVG-A cells were infected with JCV Mad-1 strain as described in materials and methods. Whole cell extracts were prepared from either uninfected or infected cells at 15th day of postinfection. Extracts from infected or uninfected cells (150 μg each) were immunoprecipitated (IP) using either normal rabbit serum (NRS, 5 μg) or anti-agnoprotein antibody (anti-agno, 5 μg) (Del Valle et al., 2002; Sariyer et al., 2006) as indicated, and were analyzed by Western blotting using anti-agnoprotein antibody (Del Valle et al., 2002; Sariyer et al., 2006). Whole cell extracts prepared from either uninfected (lane 1) or infected cells (lane 2), (40 μg each) were directly loaded on a 15% SDS-polyacrylamide gel and analyzed.
Deletion of amino acids from 17 to 42 region of agnoprotein severely affects the viral replication cycle
Mapping of the dimerization/oligomerization domain of agnoprotein demonstrated that, in the absence of amino acids 17-42 region, the ability of dimer/oligomer formation of agnoprotein was abolished. We next wanted to test the functional consequences of the deletion of this region on the level of the JCV protein expression and replication. JCV Mad-1 genome lacking the amino acids 17-42 region of agnoprotein and JCV WT was transfected into SVG-A cells separately and nuclear extracts were prepared at 5th and 15th day of posttransfection and analyzed for the expression level of VP1 by Western blotting. As shown in Fig. 9A, JCV capsid protein VP1 is readily detectable at the 5th day of posttransfection in extracts prepared from the cells transfected with either WT (lane 4) or mutant viral genome (lane 2). Even the level of VP1 expression for mutant virus was higher than WT at 5th day posttransfection. Surprisingly, while the level of expression of VP1 increased significantly in the extracts prepared at 15th day time point, as expected, from the cells transfected with WT genome, the mutant virus was not able to sustain its replication cycle until 15th day posttransfection. The level of VP1 expression was undetectable at this time point. In parallel, the ability of the replication of the mutant virus was also analyzed by Dpn I assay. This assay detects only the newly replicated viral DNA while digesting away the transfected DNA (input) by Southern blot analysis as described under material and methods. Using this method, one can distinguish the newly replicated DNA from the input DNA (transfected into the cells). Consistent with our findings from protein expression studies in Fig. 9A, the mutant virus was found to replicate as efficiently as WT by 5th day posttransfection (compare lane 2 to lane 4). However, while the level of replication of WT significantly increased at 15d posttransfection, as expected, that of mutant dropped to the undetectable levels (compare lane 3 to 5). Taken together, these findings suggest that deletion of the dimerization/oligomerization domain of agnoprotein severely affects both the viral protein expression, VP1, and replication in long-term infection cycles of JCV.
Fig. 9. Effect of deletion of amino acids from 17 to 42 region of agnoprotein on viral replication cycle.
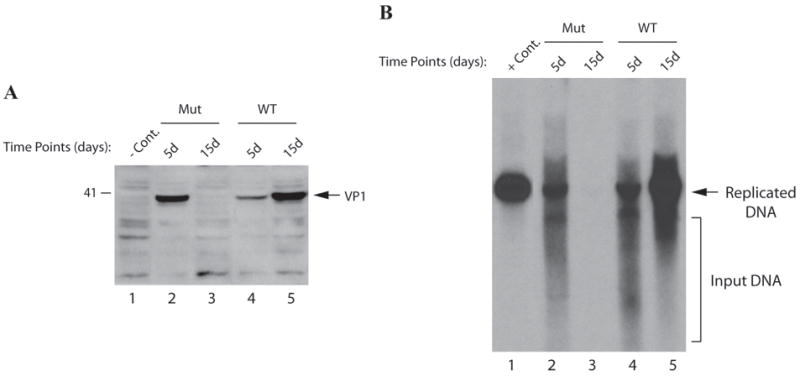
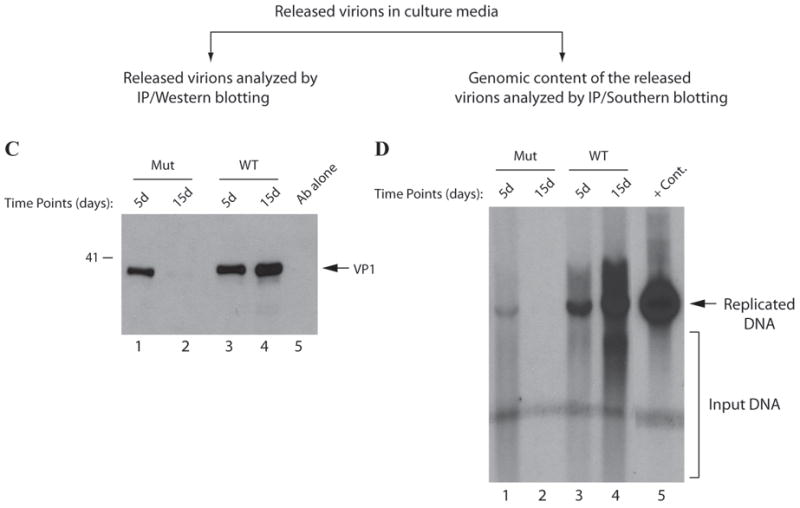
(A) Western blot analysis of VP1 from infected cells. Nuclear extracts were prepared at indicated time points from SVG-A cells transfected/infected with either JCV Mad-1 WT or JCV Mad-1 Agno (Δ17-42) deletion mutant and analyzed by Western blotting using anti-VP1 monoclonal antibody (pAB597). In lane 1, nuclear extracts prepared from the uninfected SVG-A cells were loaded as a negative control (- Cont.). (B) Southern blot analysis of the replicated DNA. In parallel to the Western blot analysis in panel A, viral DNA (newly replicated DNA from transfected/infected cells) was isolated by Hirt’s method as described in materials and methods, digested with Bam HI and Dpn I restriction enzymes. Bam HI linearizes the viral DNA and Dpn I digests the transfected DNA (bacterially-produced and methylated) but leaves unmethylated and newly-replicated DNA remaining intact. Digested DNA was separated on a 1% agarose gel, transferred onto a synthetic membrane and probed for detection of the newly replicated DNA using a probe prepared from the JCV Mad-1 WT as a template. In lane 1, 2 ng of JCV Mad-1 WT linearized by Bam HI digestion was loaded as positive control (+ Cont.). (C) Western blot analysis of the released virions. In parallel to the Western blot analysis of VP1 of the nuclear extracts in panel A, supernatants from the infected cells were collected at indicated time points, subjected to immunoprecipitation using anti-VP1 antibody (PAB597) (10 ml supernatant). Immunoprecipitants were divided into two equal portions. One portion was analyzed by Western blotting (C) using the same antibody. (D) As described for panel C, the other portion of the samples was processed for DNA isolation as described previously (Ziegler et al., 2004) and isolated DNA was analyzed by Southern blotting as described for panel B to determine the level of the genomic content of the released virions.
Analysis of the virions released from the cells infected with Mad-1 agno (Δ17-42) mutant
The inability of agnoprotein deletion mutant to sustain viral replication cycle suggested a possibility that this mutant virus was either defective in release of the infectious viral particles or that the released particles were deficient in DNA content. To distinguish between these possibilities, a viral release assay was performed, where the released viral particles were immunoprecipitated from the culture media and analyzed by both Western (Fig. 9C) and Southern blottings (Fig. 9D). The culture media was collected from the cells transfected either with WT or mutant at different time points and viral particles were subjected to immunoprecipitation using an anti-VP1 antibody. The samples were then splitted into two equal portions. One portion was analyzed by Western blotting using anti-VP1 antibody. The other portion was analyzed by Southern blotting for detection of encapsidated viral DNA. As shown in Fig. 9C, VP1 was readily detectable for WT at every time point taken posttransfection (lanes 3 and 4) by Western blotting. We also observed comparable levels of virion release for the mutant virus at 5th day posttransfection (lanes 1). However, the observed signal declined to undetectable levels 15thth day data point for the mutant virus compared to WT (lanes 2 and 4 respectively). These results demonstrated that the mutant virus was capable of releasing the viral particles at least as efficiently as WT. In light of these findings, we then reasoned that perhaps the released viral particles were deficient in viral DNA and were therefore unable to successfully propagate. To investigate this possibility, we analyzed the viral DNA content of the other half of the immunoprecipitated samples by Southern blotting. As demonstrated in Fig. 9D, we detected, as expected, a relatively strong signal for encapsidated WT viral DNA, isolated at 5th day posttransfection (lane 3) and the intensity of the signal gradually increased for the later time point (lane 4). In contrast, we detected a relatively weak signal for the mutant virus at 5th day posttransfection and did not observe a detectable level of signal for the 15th day time point, which strongly supports the hypothesis that the viral particles, are efficiently released from the cells transfected with the mutant virus but were mostly deficient in DNA content. It is conceivable that this results in a defective propagation cycle of the mutant virus at the subsequent rounds of infection cycles. This finding suggests that agnoprotein may be involved in viral encapsidation process and dimerization/oligomerization property of agnoprotein perhaps plays a critical role in this and other processes that agnoprotein associated with during the viral replication cycle.
DISCUSSION
Evidence has been presented to show that agnoprotein forms highly stable dimers and oligomers when it is fused to GST and MBP and expressed in E. coli. More significantly, analysis of agnoprotein in cell extracts prepared from the infected cells also showed distinct bands indicating that agnoprotein may form and function as dimer/oligomers during viral infection cycle in vivo, correlating with the previously published data where homodimer formation of agnoprotein was demonstrated by intermolecular fluorescence resonance energy transfer (FRET) analysis as well as high order oligomers by biochemical cross linking (Suzuki et al., 2010). In addition, the agnoprotein region which is responsible for the stable dimerization/oligomerization was found to encompass one of the predicted helical domains of the protein, amino acids spanning 17-42. Deletion of this region from the viral background resulted in a phenotype that is unable to sustain viral replication cycle, highlighting the importance of the dimerization domain of agnoprotein in viral replication cycle. Our current studies in this report clearly show the importance of the roughly mapped dimer/oligomer formation domain of agnoprotein for its function, however it does not tell us much about the exact mechanism(s) through which agnoprotein fulfills its functions. Further mutational analysis of the region is required to gain insight into such a mechanism(s). It should, however, be mentioned here that previously published mapping studies almost always pointed this dimer/oligomer formation domain of the protein as the “protein-protein interaction domain” with other proteins, including LT-Ag, Sm t-Ag, YB-1, p53 (Darbinyan et al., 2002; Safak et al., 2001; Safak et al., 2002; Sariyer, Khalili, and Safak, 2008). Collectively, it appears that this region not only plays a role in forming dimers/oligomers but also mediates interaction with other proteins. In addition, N-terminus of agnoprotein, aa 1-18, was shown to interact with heterochromatin protein 1 (HP1) (Okada et al., 2005).
At the beginning of these studies, it was initially assumed that the high molecular weight complexes that had been observed resulted from bacterial proteins that strongly interact and co-purify with agnoprotein. Alternatively, these higher molecular weight proteins may have been produced by read-through of the stop codon. However, the latter possibility was eliminated by demonstrating that the addition of three consecutive stop codons at the end of the agnogene did not prevent the formation of these protein complexes (data not shown). In addition, detection of high molecular weight complexes with an antibody, specific for agnoprotein and further analysis of these complexes by LC-Mass Spectrometry (see Supplement 1) and MALDI-TOF spectrometry (see Supplement 2) confirmed that higher molecular weight complexes are generated by the ability of agnoprotein to form stable dimer and oligomers. Particularly MALDI-TOF spectrometry analysis of the purified MBP-Agno clearly demonstrated that no bacterial protein is present in the agnoprotein dimer complexes (see supplement 2), which also eliminates that the possibility that dimer formation is mediated by a bacterial protein.
One of the interesting characteristics of these complexes is their stability in the presence of a variety of denaturing agents, including high concentration of SDS and 8M urea; conditions that would completely denature most proteins. Only prolonged heat treatment at 95°C had a significant effect on the integrity of these complexes, suggesting that these assemblies are not formed by covalent bonds between the monomers, but are most likely stabilized by a network of salt bridges and/or hydrophobic interactions. Another interesting observation was that the level of dimers and oligomers increased in both a time-dependent and a temperature-dependent manner. Mapping studies showed that the amino acids in the central core region appear to be critical for dimer/oligomer formation. Three dimensional structure prediction studies using the I-Tasser program (Roy, Kucukural, and Zhang, 2010) indicated that the majority of the sequences of dimer/oligomer formation domain of agnoprotein most likely participate in formation of an α-helical structure (Fig. 3B). Based on this computer generated structure, the agnoprotein appears to possess an isoleucine-, phenylalaine- and leucine-rich domain spanning amino acids 28-37 in this amphipathic helix (Fig. 3A). The amphipathic nature of the α-helical region of agnoprotein was predicted by different computer-aided prediction studies (http://heliquest.ipmc.cnrs.fr). In addition, in a recent study by Unterstab et al., it was reported that a roughly corresponding region from BKV agnoprotein also exhibits an amphipathic nature (Unterstab et al., 2010). Prediction studies also suggest that this region contains a nuclear export-like signal (NES-like) similar to those characterized for HIV Rev protein (Fig. 3A). It is likely that these isoleucine- and leucine-rich sequences present within the dimerization play critical roles for the formation of stable dimer and oligomers.
In literature, a number of viral and eukaryotic proteins that form SDS-resistant dimers or oligomers have also been reported. NOS (nitric oxide synthetase) was, for example, shown to form highly stable dimers in cells that are resistant to heat and denaturing reagents (Kolodziejski, Rashid, and Eissa, 2003), supporting the idea that the active form of this enzyme is a stable dimer. Several small regulatory proteins of HIV, including vpr, Rev, Nef, vif, and tat were also reported to form stable dimers and oligomers (Cullen, 1998a; Frankel, Bredt, and Pabo, 1988; Frankel et al., 1988; Kwak et al.; Yang, Sun, and Zhang, 2001; Zhao et al., 1994). Vpr forms SDS-resistant dimers and oligomers when expressed in E. coli and mammalian cells and dimer structure has been determined indicating that leu-like zipper interaction between two helixes (Bourbigot et al., 2005; Fritz et al., 2008; Venkatachari et al., 2010; Zhao et al., 1994). This protein is a multifunctional protein involved in different steps of viral infection cycle, including increased transcription from the long terminal repeats, enhances viral replication, inducing G2 cell cycle arrest, regulating apoptosis and facilitating the entry of the HIV pre-integration complex through the nuclear pore (Bourbigot et al., 2005; Fritz et al., 2008; Venkatachari et al., 2010; Zhao et al., 1994). One of the implications that Vpr dimerization is important for its function was provided by Fritz et al (Fritz et al., 2010) who showed that vpr oligomerization is crucial for HIV gag protein recognition and the accumulation of gag at the plasma membrane (Fritz et al., 2010). Another HIV regulatory protein Rev also forms stable dimers and higher order oligomers evidenced by 3D structure determination. Rev is a 116 amino acid protein that binds to the ~350 nt Rev response element (RRE) RNA found in the introns of partially spliced and unspliced HIV RNA, and its role is to direct HIV RNA to the cytoplasm. The transported RNA molecules are then either translated into the viral proteins or are packaged as genomic RNA into the virions (Cullen, 1998a; Cullen, 1998b; Cullen, 2003a; Cullen, 2003b; Pollard and Malim, 1998). In order to export its bound RNA, oligomeric form of Rev binds to the human nuclear export receptor, CRM1, forming a ternary complex stabilized by GTP-bound Ran (Cullen, 2003b; Fornerod et al., 1997) and upon unloading its cargo, Rev shuttles back into the nucleus. Ebola virus transcription factor VP30 was also shown to function as a homodimer during the viral transcription. Specific mutations within the dimerization region abrogated the VP30-dependent transcription (Hartlieb et al., 2003). In JCV case, it is possible that agnoprotein may also bind to JCV-specific viral transcripts and transport them to the translation centers in cytoplasm. In such a scenario, the dimeric and oligomeric structures of agnoprotein would be crucial for their binding to the viral transcripts and transporting them to cytoplasm for translation.
The study of the functional properties of dimer/oligomer formation of agnoprotein is important for the understanding of its role in JCV replication cycle. These studies will help to establish the significance of this protein for JCV life cycle. We had previously reported that this protein is a target for phosphorylation by protein kinase C, where Ser7, Ser11 and Thr21 were shown to be phosphorylated by PKC. Single or combinatorial mutations at these sites converted JCV in an essentially nonfunctional virus after the first round of replication cycle (Sariyer et al., 2006). Similar work by Johannessen et al., demonstrated the functional importance of the phosphorylated forms of agnoprotein for BKV replication cycle (Johannessen et al., 2008). Analysis of a deletion mutant of BKV agnoprotein by Myhre et al., also showed that BKV has a restricted life cycle in the absence of agnoprotein, but viral function can be rescued when native agnoprotein is provided in trans (Myhre et al., 2010). In this paper, we also attempted to further characterize the functional consequences of the deletion of the agnoprotein dimer formation domain from the viral background (Fig. 9A&B). Close examination of the levels of the viral protein expression and viral DNA replication at different time points after infections showed that in the absence of amino acids spanning 17-42, mutant virus cannot sustain its replication cycle in SVG-A cells. Although the level of viral protein expression VP1 and that of replication are readily comparable between WT and mutant at 5th day postransfection, these levels decreased to the undetectable levels at 15th day time point for the mutant virus (Fig. 9A&B). These findings suggest that the 17-42 amino acid region, which is involved in agnoprotein dimerization and oligomerization, plays an essential role in the function of this protein during the viral replication cycle. It should be noted here that it is difficult to detect the expression of the mutant agnoprotein in extracts prepared from transfected/infected cells by Western. It appears that this mutant protein after removal of the amino acids 17-42 becomes unstable and degrades rapidly in vivo. Bacterial expression studies with the mutant protein (MBP-Agno (Δ17-42) also demonstrated that it degrades quickly as soon as bacterial cells are lysed for protein purification, supporting our findings from transfection/infection studies (data not shown). Another interesting point is that although SVG-A cells constitutively express SV40 large T antigen, which was previously shown to cross-regulate the replication of JCV (Lynch, Haggerty, and Frisque, 1994), the expression of LT-Ag alone is not sufficient to maintain productive viral replication cycle in the absence of WT agnoprotein. This observation is consistent with our recent findings, where it was observed that in the absence of agnoprotein, neither JCV nor SV40 can replicate as efficiently as WT (Sariyer et al., 2011)
JCV has a relatively long replication cycle. It takes 6-7 days to complete its first cycle although viral release can occur before the completion of the infection cycle in SVG-A cells. Infection cycle for SV40, however, takes 1-2 days (Sariyer et al., 2011). Since the mutant virus failed to sustain its replication cycle during the second round of propagation, we reasoned that the mutant virus was defective in the process of viral release from the infected cells. To address this question, a viral release assay was employed (Fig. 9C&D) (Sariyer et al., 2006). It was found that mutant viral particles were efficiently released from the infected cells compared to WT, evidenced by detection of comparable level of VP1 in the supernatant of the cells infected with either WT or mutant (Fig. 9C). We then investigated whether the released mutant viral particles were in fact deficient in viral DNA content and therefore defective in propagation. Analysis of such particles by Southern blotting showed that in deed the released virions are deficient in DNA content compared to WT. This, in part, explains why the mutant virus was defective in propagation but does not explain the mechanisms that render the mutant defective in viral maturation. Experiments are underway to investigate why the mutant virus cannot continue its replication cycle. In conclusion, our experiments suggest that agnoprotein may form dimers/oligomers in vivo and such structures may play critical roles for its function during the replication cycle of JCV.
Supplementary Material
Acknowledgments
We would like to thank past and present members of the Department of Neuroscience and Center for Neurovirology for their insightful discussion and sharing of ideas and reagents. This work was made possible by grants awarded by NIH to MS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology. 2006;349(1):66–78. doi: 10.1016/j.virol.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Alwine JC. Evidence for simian virus 40 late transcriptional control: mixed infections of wild-type simian virus 40 and a late leader deletion mutant exhibit trans effects on late viral RNA synthesis. J Virol. 1982;42(3):798–803. doi: 10.1128/jvi.42.3.798-803.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auborn KJ, Markowitz RB, Wang E, Yu YT, Prives C. Simian virus 40 (SV40) T antigen binds specifically to double-stranded DNA but not to single-stranded DNA or DNA/RNA hybrids containing the SV40 regulatory sequences. J Virol. 1988;62(6):2204–8. doi: 10.1128/jvi.62.6.2204-2208.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag B, Hofstetter CA, Reviriego-Mendoza MM, Frisque RJ. JC virus small T antigen binds phosphatase PP2A and Rb family proteins and is required for efficient viral DNA replication activity. PLoS One. 2010;5(5):e10606. doi: 10.1371/journal.pone.0010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag B, Kilpatrick LH, Tyagarajan SK, Tevethia MJ, Frisque RJ. JC virus T’135, T’136 and T’165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol. 2006;12(6):428–42. doi: 10.1080/13550280601009553. [DOI] [PubMed] [Google Scholar]
- Bolton DL, Lenardo MJ. Vpr cytopathicity independent of G2/M cell cycle arrest in human immunodeficiency virus type 1-infected CD4+ T cells. J Virol. 2007;81(17):8878–90. doi: 10.1128/JVI.00122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbigot S, Beltz H, Denis J, Morellet N, Roques BP, Mely Y, Bouaziz S. The C-terminal domain of the HIV-1 regulatory protein Vpr adopts an antiparallel dimeric structure in solution via its leucine-zipper-like domain. Biochem J. 2005;387(Pt 2):333–41. doi: 10.1042/BJ20041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Tungaturthi PK, Ayyavoo V, Ghafouri M, Ariga H, Khalili K, Srinivasan A, Amini S, Sawaya BE. The role of Vpr in the regulation of HIV-1 gene expression. Cell Cycle. 2006;5(22):2626–38. doi: 10.4161/cc.5.22.3442. [DOI] [PubMed] [Google Scholar]
- Cullen B. HIV-1 auxiliary proteins:making connections in dying cell. Cell. 1998a;93:685–692. doi: 10.1016/s0092-8674(00)81431-2. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Retroviruses as model systems for the study of nuclear RNA export pathways. Virology. 1998b;249(2):203–10. doi: 10.1006/viro.1998.9331. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Nuclear mRNA export: insights from virology. Trends Biochem Sci. 2003a;28(8):419–24. doi: 10.1016/S0968-0004(03)00142-7. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Nuclear RNA export. J Cell Sci. 2003b;116(Pt 4):587–97. doi: 10.1242/jcs.00268. [DOI] [PubMed] [Google Scholar]
- Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21(36):5574–81. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci U S A. 2010;107(28):12481–6. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, D’Orso I, Frankel AD. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell. 2008;31(6):824–34. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010;17(11):1337–42. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radhakrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Natl Cancer Inst. 2002;94(4):267–73. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- DiMattia MA, Watts NR, Stahl SJ, Rader C, Wingfield PT, Stuart DI, Steven AC, Grimes JM. Implications of the HIV-1 Rev dimer structure at 3.2 A resolution for multimeric binding to the Rev response element. Proc Natl Acad Sci U S A. 2010;107(13):5810–4. doi: 10.1073/pnas.0914946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducret A, Bartone N, Haynes PA, Blanchard A, Aebersold R. A simplified gradient solvent delivery system for capillary liquid chromatography-electrospray ionization mass spectrometry. Anal Biochem. 1998;265(1):129–38. doi: 10.1006/abio.1998.2873. [DOI] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90(6):1051–60. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Bredt DS, Pabo CO. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science. 1988;240(4848):70–3. doi: 10.1126/science.2832944. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Chen L, Cotter RJ, Pabo CO. Dimerization of the tat protein from human immunodeficiency virus: a cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc Natl Acad Sci U S A. 1988;85(17):6297–300. doi: 10.1073/pnas.85.17.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ, White FA. The molecular biology of JC virus, causative agent of progressive multifocal leukoencophalopathy. In: Roose RP, editor. Molecular Neurovirology, pathogenesis of viral CNS infections. Humana Press Inc.; Totowa, NJ: 1992. pp. 25–158. [Google Scholar]
- Fritz JV, Didier P, Clamme JP, Schaub E, Muriaux D, Cabanne C, Morellet N, Bouaziz S, Darlix JL, Mely Y, de Rocquigny H. Direct Vpr-Vpr interaction in cells monitored by two photon fluorescence correlation spectroscopy and fluorescence lifetime imaging. Retrovirology. 2008;5:87. doi: 10.1186/1742-4690-5-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz JV, Dujardin D, Godet J, Didier P, De Mey J, Darlix JL, Mely Y, de Rocquigny H. HIV-1 Vpr oligomerization but not that of Gag directs the interaction between Vpr and Gag. J Virol. 2010;84(3):1585–96. doi: 10.1128/JVI.01691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Nomura S, Anderson CW, George K. Identification of the SV40 agnoproduct: a DNA binding protein. Nature. 1981;291:346–349. doi: 10.1038/291346a0. [DOI] [PubMed] [Google Scholar]
- Godet AN, Guergnon J, Croset A, Cayla X, Falanga PB, Colle JH, Garcia A. PP2A1 binding, cell transducing and apoptotic properties of Vpr(77-92): a new functional domain of HIV-1 Vpr proteins. PLoS One. 2010;5(11):e13760. doi: 10.1371/journal.pone.0013760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty S, Walker DL, Frisque RJ. JC virus-simian virus 40 genomes containing heterologous regulatory signals and chimeric early regions: identification of regions restricting transformation by JC virus. J Virol. 1989;63(5):2180–90. doi: 10.1128/jvi.63.5.2180-2190.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlieb B, Modrof J, Muhlberger E, Klenk HD, Becker S. Oligomerization of Ebola virus VP30 is essential for viral transcription and can be inhibited by a synthetic peptide. J Biol Chem. 2003;278(43):41830–6. doi: 10.1074/jbc.M307036200. [DOI] [PubMed] [Google Scholar]
- Hay N, Skolnik-David H, Aloni Y. Attenuation in the control of SV40 gene expression. Cell. 1982;29(1):183–93. doi: 10.1016/0092-8674(82)90102-7. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26(2):365–9. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Hock M, Kraus I, Schoehn G, Jamin M, Andrei-Selmer C, Garten W, Weissenhorn W. RNA induced polymerization of the Borna disease virus nucleoprotein. Virology. 2010;397(1):64–72. doi: 10.1016/j.virol.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Hou-Jong MH, Larsen SH, Roman A. Role of the agnoprotein in regulation of simian virus 40 replication and maturation pathways. J Virol. 1987;61(3):937–9. doi: 10.1128/jvi.61.3.937-939.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanskiy S, Zhao Y, DiMarzio P, Agostini I, Dubrovsky L, Bukrinsky M. Heat-shock protein 70 exerts opposing effects on Vpr-dependent and Vpr-independent HIV-1 replication in macrophages. Blood. 2004;104(6):1867–72. doi: 10.1182/blood-2004-01-0081. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Myhre MR, Dragset M, Tummler C, Moens U. Phosphorylation of human polyomavirus BK agnoprotein at Ser-11 is mediated by PKC and has an important regulative function. Virology. 2008;379(1):97–109. doi: 10.1016/j.virol.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Khalili K, Sariyer IK, Safak M. Small tumor antigen of polyomaviruses: role in viral life cycle and cell transformation. J Cell Physiol. 2008;215(2):309–19. doi: 10.1002/jcp.21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili K, White MK, Sawa H, Nagashima K, Safak M. The agnoprotein of polyomaviruses: a multifunctional auxiliary protein. J Cell Physiol. 2005;204(1):1–7. doi: 10.1002/jcp.20266. [DOI] [PubMed] [Google Scholar]
- Kolodziejski PJ, Rashid MB, Eissa NT. Intracellular formation of “undisruptable” dimers of inducible nitric oxide synthase. Proc Natl Acad Sci U S A. 2003;100(24):14263–8. doi: 10.1073/pnas.2435290100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YT, Raney A, Kuo LS, Denial SJ, Temple BR, Garcia JV, Foster JL. Self-association of the Lentivirus protein. Nef Retrovirology. 2010;7:77. doi: 10.1186/1742-4690-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KJ, Frisque RJ. Identification of critical elements within the JC virus DNA replication origin. J Virol. 1990;64(12):5812–22. doi: 10.1128/jvi.64.12.5812-5822.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KJ, Haggerty S, Frisque RJ. DNA replication of chimeric JC virus-simian virus 40 genomes. Virology. 1994;204(2):819–22. doi: 10.1006/viro.1994.1600. [DOI] [PubMed] [Google Scholar]
- Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5(1):49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A. 1985;82(4):1257–61. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolskee RF, Nathans D. Suppression of a VP1 mutant of simian virus 40 by missense mutations in serine codons of the viral agnogene. J Virol. 1983;48(2):405–9. doi: 10.1128/jvi.48.2.405-409.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29(11):618–25. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Merabova N, Kaniowska D, Kaminski R, Deshmane SL, White MK, Amini S, Darbinyan A, Khalili K. JC virus agnoprotein inhibits in vitro differentiation of oligodendrocytes and promotes apoptosis. J Virol. 2008;82(3):1558–69. doi: 10.1128/JVI.01680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre MR, Olsen GH, Gosert R, Hirsch HH, Rinaldo CH. Clinical polyomavirus BK variants with agnogene deletion are non-functional but rescued by trans-complementation. Virology. 2010;398(1):12–20. doi: 10.1016/j.virol.2009.11.029. [DOI] [PubMed] [Google Scholar]
- Ng SC, Behm M, Bina M. DNA sequence alterations responsible for the synthesis of thermosensitive VP1 in temperature-sensitive BC mutants of simian virus 40. J Virol. 1985;54(2):646–9. doi: 10.1128/jvi.54.2.646-649.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SC, Mertz JE, Sanden-Will S, Bina M. Simian virus 40 maturation in cells harboring mutants deleted in the agnogene. J Biol Chem. 1985;260(2):1127–32. [PubMed] [Google Scholar]
- Nomura S, Khoury G, Jay G. Subcellular localization of the simian virus 40 agnoprotein. J Virol. 1983;45(1):428–33. doi: 10.1128/jvi.45.1.428-433.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Sawa H, Endo S, Orba Y, Umemura T, Nishihara H, Stan AC, Tanaka S, Takahashi H, Nagashima K. Expression of JC virus agnoprotein in progressive multifocal leukoencephalopathy brain. Acta Neuropathol. 2002;104(2):130–6. doi: 10.1007/s00401-002-0526-8. [DOI] [PubMed] [Google Scholar]
- Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep. 2005;6(5):452–7. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard VW, Malim MH. The HIV-1 Rev protein. Annu Rev Microbiol. 1998;52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- Poon B, Chang MA, Chen IS. Vpr is required for efficient Nef expression from unintegrated human immunodeficiency virus type 1 DNA. J Virol. 2007;81(19):10515–23. doi: 10.1128/JVI.00947-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo CH, Traavik T, Hey A. The agnogene of the human polyomavirus BK is expressed. J Virol. 1998;72(7):6233–6. doi: 10.1128/jvi.72.7.6233-6236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75(3):1476–86. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Sadowska B, Barrucco R, Khalili K. Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J Virol. 2002;76(8):3828–38. doi: 10.1128/JVI.76.8.3828-3838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80(8):3893–903. doi: 10.1128/JVI.80.8.3893-3903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Khalili K, Safak M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: inhibition by small t antigen. Virology. 2008;375(2):464–79. doi: 10.1016/j.virol.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8(1):255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell LC, Sunde M, Blake CC. The molecular basis of amyloidosis. Cell Mol Life Sci. 1997;53(11-12):871–87. doi: 10.1007/s000180050107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DT, Loeber G, Tegtmeyer P. Four major sequence elements of simian virus 40 large T antigen coordinate its specific and nonspecific DNA binding. J Virol. 1990;64(5):1973–83. doi: 10.1128/jvi.64.5.1973-1983.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunde M, Blake C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv Protein Chem. 1997;50:123–59. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273(3):729–39. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Okada Y, Semba S, Orba Y, Yamanouchi S, Endo S, Tanaka S, Fujita T, Kuroda S, Nagashima K, Sawa H. Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules: role of agnoprotein-induced dissociation of FEZ1 from microtubules in viral propagation. J Biol Chem. 2005;280(26):24948–56. doi: 10.1074/jbc.M411499200. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 2010;6(3):e1000801. doi: 10.1371/journal.ppat.1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterstab G, Gosert R, Leuenberger D, Lorentz P, Rinaldo CH, Hirsch HH. The polyomavirus BK agnoprotein co-localizes with lipid droplets. Virology. 2010;399(2):322–31. doi: 10.1016/j.virol.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Venkatachari NJ, Walker LA, Tastan O, Le T, Dempsey TM, Li Y, Yanamala N, Srinivasan A, Klein-Seetharaman J, Montelaro RC, Ayyavoo V. Human immunodeficiency virus type 1 Vpr: oligomerization is an essential feature for its incorporation into virus particles. Virol J. 2010;7:119. doi: 10.1186/1743-422X-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Sun Y, Zhang H. The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J Biol Chem. 2001;276(7):4889–93. doi: 10.1074/jbc.M004895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LJ, Wang L, Mukherjee S, Narayan O. Biochemical mechanism of HIV-1 Vpr function. Oligomerization mediated by the N-terminal domain. J Biol Chem. 1994;269(51):32131–7. [PubMed] [Google Scholar]
- Ziegler K, Bui T, Frisque RJ, Grandinetti A, Nerurkar VR. A rapid in vitro polyomavirus DNA replication assay. J Virol Methods. 2004;122(1):123–7. doi: 10.1016/j.jviromet.2004.08.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.