

Abstract
A little more than 10 years ago, the completed sequencing of the human genome boldly promised to usher in an era of enhanced understanding and accelerated development of treatments for most human diseases. Ten years later, many of these therapeutic goals have not been reached, but genomic technologies have dramatically enhanced our understanding of how genes and gene networks contribute to the pathogenesis of disease. In this review, we describe how genomic technologies have shaped our study of idiopathic pulmonary fibrosis (IPF), a devastating, progressive scarring of the lung parenchyma, a disease without a known cause, or treatment. We frame the important genomic discoveries in IPF of the previous decade in the clinical context of establishing a diagnosis of IPF and predicting the prognosis. Gene expression profiling of peripheral blood will help identify potential biomarkers for assessing the clinical severity of IPF. We highlight the growth of epigenetic research in IPF, including the contribution of microRNAs to the pathogenesis of disease. We suggest that the full power of genomic discoveries in IPF will be realized when researchers apply these techniques prospectively in large collaborative studies across institutions, support the training of young investigators in genomics, and employ systems biology approaches to the interpretation of genomic data. Clin Trans Sci 2011; Volume 4: 372–379
Keywords: lung, genetics, genes
Introduction
Publication of this review article now comes perhaps at a fortuitous time, just 10 years after the enthusiastic announcement by President Bill Clinton that the sequencing of the entire human genome had been completed, portending a revolution in the “diagnosis, prevention and treatment of most, if not all, human diseases.” 1 Articles in the lay press have appeared lamenting that “…the stunning advances in scientific knowledge and DNA‐processing technologies.” have done “…relatively little to improve medical treatments and human health.” 1 While it is true that the explosive growth of genomic research directly impacts only few patients, has genomic research failed to reach the promised goals? Perhaps the questions that we should ask about the current impact of genomic research on clinical medicine should be more focused. High‐throughput technologies, such as gene expression profiling by microarray, which are part of the genomic research armamentarium, have provided a large amount of disease‐specific data in a short period of time and have revolutionized the way we think about human disease.
The study of idiopathic pulmonary fibrosis (IPF), a progressive, lethal disease, which is shaped by the complex interplay of genetic, 2 , 3 , 4 epigenetic, 5 , 6 , 7 and environmental forces, 8 , 9 is the focus of our work and will provide an excellent opportunity to reexamine the accomplishments of disease‐focused genomic research. Genomic approaches to IPF seek to identify genes and gene networks that promote or sustain the scarring of the lung parenchyma. The aim of this review is to highlight the work of genomic research in IPF and to define how this research has impacted our understanding of the pathogenesis of IPF. Toward that end, we will attempt to answer the following questions:
-
1
What are the sources of genomic data in IPF?
-
2
Do genomic approaches using lung tissue and so‐called “surrogate” tissues, such as peripheral blood, provide clinically relevant diagnostic and prognostic information on IPF?
-
3
How has genomic research enhanced our understanding of the pathogenesis of IPF?
-
4
What is the state of “epigenomic” research in IPF?
-
5
What is the future of genomic research in IPF?
These questions will serve as a framework for reviewing the current state of genomic research in IPF and for understanding how genomic research will impact the clinically important questions in IPF: diagnosis, therapeutics, and prognosis. As our focus is on the genomics of IPF, we will limit our discussion to genomic studies of the human disease only.
Brief Introduction to IPF
IPF is a progressive disorder characterized by the unrelenting accumulation of myofibroblasts and the deposition of collagens in the lower respiratory tract leading ultimately to breathlessness and respiratory failure. It is a rare disease, with an estimated prevalence of 14–43 per 100,000 people, depending on the case definition of IPF. 10 Prevalence increases with age. The disease is typically discovered late in its course, and the median survival is close to 3 years from the time of diagnosis. 11 Except for lung transplantation, there is no approved treatment for IPF in the United States.
Usual interstitial pneumonia (UIP) is the typical histopathologic finding of patients with IPF and is characterized by areas of inflammation and fibroblast proliferation (fibroblastic foci) adjacent to areas of normal, uninjured, and seemingly uninvolved lung (temporal heterogeneity). Since the 1980s, the mechanistic description of IPF has shifted from a disease characterized by “inflammation leading invariably to fibrosis” to a more complex view that posits repeated epithelial injury and aberrant wound healing leading to the accumulation of fibroblasts in the lung. 12 Roles for multiple new pathways and phenomena, such as coagulation, 13 apoptosis, 14 , 15 , 16 angiogenesis, 17 , 18 oxidative injury, 19 , 20 development, 21 and recently, epithelial mesenchymal transition (EMT), have been proposed. 22 , 23 , 24 Central to this model is the observation that both alveolar epithelial cells and fibroblasts exhibit distinct and profound changes in their phenotypes. Alveolar epithelial cells proliferate and elaborate inflammatory signals. In response, fibroblasts are activated and differentiate into myofibroblasts. In addition, some epithelial cells begin expressing markers of mesenchymal cells, and some may proceed by EMT to differentiate into myofibroblasts as well. 25 As in normal wound healing, these myofibroblasts secrete collagens and other extracellular matrix proteins. The myofibroblasts contract the lung parenchyma leading to the restrictive ventilatory defect that characterizes IPF. But while normal wound healing involves signals to terminate the accumulation of fibroblasts and the deposition of collagen, in IPF, this process fails. Alveolar epithelial cells, in various stages of activation and EMT, do not properly repair the injured alveolar lining. The ongoing injury stimulates fibroblast accumulation and deposition of collagens. What causes the initial injury in IPF and why the process fails to terminate is the subject of intense investigation.
What are the sources of genomic data in IPF?
The most common source of genomic data in IPF is lung tissue from patients. IPF lung tissue used for diagnosis and genomic analysis is available at the time of biopsy, explant, or at autopsy. The inherent invasiveness in all of these procedures limits the number of samples that are readily available to individual researchers. The lack of tissue may be, perhaps, the most serious challenge facing IPF research. Larger sources of material are available when the research is done at a large center or in collaboration with multiple centers. The Lung Tissue Research Consortium (LTRC) funded by the National Institutes of Health (NIH) and discussed below is a major effort to overcome this obstacle. The invasiveness of the procedure prevents obtaining more than one sample per patient and quite often those samples are obtained during periods of relative disease stability, thus limiting our understanding of the molecular events leading to change in disease course and eventually to the patient’s death. Traditional autopsies are limited in their value because the time from death to harvest of tissues is long, and there is significant degradation of proteins, nucleic acids, and death of lung resident cells. To address this issue, our group is undertaking a program of “warm” autopsies 26 to help capture data from patients who die of IPF and especially in the context of the mostly lethal acute exacerbations of IPF. 27 In this program, lungs are obtained from IPF patients within 6 hours of death ensuring minimal degradation of nucleic acid and protein, as well as no changes to cell populations in the lung. Another challenge is the control samples—naturally, normal lungs are very rarely sampled. Thus, so‐called “control” lung tissue is either obtained from histologically normal lung distant from lung tumors, from cadaveric lung donors rejected for lung transplantation, or from the rare case of a biopsy for a granuloma or scar tissue. While concerns regarding effects of “cancer field effects” are often mentioned with regard to histologically normal lung distant from lung tumors, neither of the alternatives is ideal: the donor lungs frequently exhibit the effects of prolonged ventilation, the trauma that caused the death, and evidence of environmental damage such as cigarette smoke, and the benign biopsies are often inflamed, suggesting that there is no ideal control lung tissue. Despite these challenges, studies applying high‐throughput genomic technologies have been highly successful in obtaining valid insights regarding IPF.
Using easier‐to‐obtain materials, such as peripheral blood or broncho‐alveolar lavage, so‐called “surrogate tissues,” may permit the generation of even larger datasets and yield insights into the pathogenesis of disease. The tissues that can be studied are presented schematically in Figure 1 . In addition to direct analysis of tissue, cells, most commonly fibroblasts, can be cultured from the lung ex vivo under a variety of conditions and in response to various stimuli. Inflammatory cells from broncho‐alveolar lavage fluid can be profiled. 28 From peripheral blood, one can study gene expression in peripheral blood mononuclear cells (PBMC). These methods are most pertinent to biomarker discovery in IPF. In addition, fibrocytes, bone marrow‐derived collagen‐producing cells can be purified from PBMC and analyzed directly or following culture. 29 Data suggest that the concentration of fibrocytes in the peripheral blood correlates with disease severity and may be an excellent biomarker 30 (reviewed in 31 ). Circulating microRNAs can be detected free in the plasma residing in apoptotic bodies. 32 , 33 Boon et al. note that many of the genes associated with clinical progression in IPF have been detected in surrogate tissues in different diseases. 34 Genomic techniques have been applied to sputum and exhaled breath condensate, but there are no current studies in IPF. 35 , 36
Figure 1.
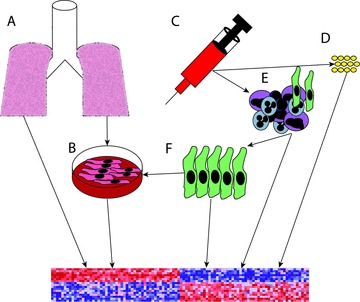
Sources of material for genomic analysis in pulmonary fibrosis. Genomic analysis can be performed directly on lung samples (A) or on primary cells cultured from the lung (B). Peripheral blood (C) can be drawn and genomic information can be obtained from free RNA residing in the plasma or in apoptotic bodies (D). Peripheral blood mononuclear cells (PBMC) (E) can be studied directly. Fibrocytes (F), bone marrow‐derived, collagen producing cells can be isolated from PBMC and analyzed directly or following stimulation in cell culture.
Do genomic approaches provide diagnostic and prognostic information in IPF?
The diagnosis of IPF is typically based on a surgical lung biopsy, an invasive procedure that is obviously accompanied by risk of serious complications. In many cases, a biopsy is not even possible because of the risk to the patient. It should be noted that according to American Thoracic Society/European Respiratory Society guidelines, a biopsy is not always necessary for diagnosis. 37 And while considered the gold standard for diagnosis of IPF, histopathology alone may fail in establishing a definitive diagnosis, even when reviewed by experienced pathologists. 38 These questions are of significant importance, since making the correct diagnosis has implications for prognosis and the response to therapies. Genomics approaches to diagnosis in IPF are not in clinical practice, but currently available data suggest that such assays may be clinically relevant in the near future. While gene expression profiling is probably not necessary to clinically differentiate IPF from normal patients, it may be important in identifying disease “subphenotypes” and potentially disease relevant markers for determining the rate of progression and outcomes. Genomic approaches to diagnosis raise an important question: is IPF a “single disease” with a unique genomic profile that distinguishes it from other forms of pulmonary fibrosis?
Since our group published the first microarray experiments comparing IPF lungs to uninjured controls in 2002, 39 the information gleaned from these experiments has helped future studies identify and prioritize biomarker discovery, 40 below is a description of the impact of these studies on diagnosis and prognosis prediction.
Diagnosis
Several studies have been published that explore disease‐specific gene expression signatures that distinguish UIP from other forms of diffuse parenchymal lung disease. These important works all support the hypothesis that disease‐specific gene expression signatures can be studied in the context of diagnosis. Selman et al. reported gene expression profiling experiments of lung samples from patients with IPF, hypersensitivity pneumonitis (HP), and nonspecific interstitial pneumonia (NSIP). 41 The genes enriched in IPF code for extracellular matrix proteins and cell growth and differentiation. In contrast, HP samples showed increased expression of genes coding for inflammatory proteins. When the set of genes that distinguished IPF from HP were applied to cases of NSIP, several cases of NSIP were misclassified as either IPF or HP, but the remaining samples did not classify as either IPF or HP suggesting that there may be a distinct gene signature for idiopathic NSIP. 41 Perhaps what is most notable about this study is the wealth of insight obtained by global analysis of gene expression. Profiling sporadic idiopathic interstitial pneumonias from familial interstitial pneumonias revealed distinctive signatures, but only minor gene expression changes were detected between UIP and NSIP. 42
A recently published article by Hsu and colleagues attempted to identify both distinctive and shared patterns of gene expression between patients with pulmonary fibrosis from systemic sclerosis (SSc‐PF) and IPF. 43 The lung tissues studied were obtained from patients with SSc‐PF exhibiting a UIP pattern. The authors found that nearly 70% of the genes that distinguish IPF and SSc‐PF from normal lungs are shared. This important work suggests that while IPF and SSc are unique clinical syndromes, the genomic profiles of IPF and SSc‐PF are quite similar and only a very small group of genes differentiates one from the other. This small group of genes may shed light on the nature of inciting injury and the genetic predisposition of the host that leads to the development of a particular phenotype of pulmonary fibrosis.
The major limitation of these data is clinical validation—does a particular pattern of gene expression diagnose UIP or HP or NSIP or SSc when routine histopathology or clinical criteria cannot? Can a gene expression signature identify patients who will respond to immunosuppressive therapy? Thus, while there is significant support to the notion that at least the decision to try antiinflammatory therapy could be based on gene expression patterns, the necessary studies validating this approach have not yet been performed.
Prognosis
There is no clinical staging system in IPF to grade severity of disease. There are several clinical predictors of increased mortality in IPF including the rate of decline in spirometry, 44 the degree of exercise limitation, 45 and the presence of pulmonary hypertension 46 or a clinical exacerbation. 47 Genomic approaches have been employed to study patients with IPF who exhibit clinical predictors of mortality compared to those who do not. All these studies are retrospective. Using gene expression signatures to predict clinical outcomes has not been evaluated prospectively in IPF, which is in contrast to a published prospective study in lung adenocarcinoma, for example. 48 IPF patients with pulmonary hypertension (World Health Organization Class III) have a distinctive gene expression signature when compared to patients with pulmonary arterial hypertension and normal subjects. 49 The patients with IPF and pulmonary hypertension exhibited profiles enriched with genes that function in transforming growth factor (TGF)‐β and platelet‐derived growth factor signaling pathways. 49 These networks are likely highly important to the pathogenesis of pulmonary hypertension in IPF.
Serial analysis of gene expression (SAGE) is a genomic technique by which short sequence (10–14 nucleotides) is identified by sequencing. The number of times a sequence appears reveals the abundance of each gene (reviewed in 50 ). Boon et al. sought to understand genomic predictors of progression in IPF. 34 SAGE studies in IPF revealed transcripts that distinguished stable (<10% reduction in forced vital capacity [FVC] over a 1‐year period following biopsy) versus progressive IPF (>10% reduction in FVC over a 1‐year period following biopsy. 34 A distinct molecular signature was identified by Selman and colleagues using microarray, analyzing patients with IPF who experience a rapid course (<6 months of symptoms vs. >24 months of symptoms prior to diagnosis). 51 What are the implications of such work? A gene expression signature that identifies rapid progressors from stable disease may, if validated in a larger dataset, identify potential biomarkers of IPF and stratify patients according to their need for lung transplant. Is there a distinct gene signature of short‐term versus long‐term survivors in IPF? The answers to these questions remain unknown.
How has genomic research enhanced our understanding of the pathogenesis of IPF?
Is there biologic relevance to the genes that are identified by genomic approaches? Grouping these genes into families based on similar functions has become easier as the analytic programs have become more sophisticated. The most common approach to analyzing the results of genomic data in IPF is the single‐gene approach. We have called this approach “cherry picking.” 52 These studies have focused on single genes and have used in vitro and in vivo techniques to determine the relevance of these genes to pulmonary fibrosis. These studies have helped shape our understanding of IPF and have identified potential drug targets. The majority of these studies have described factors that promote the accumulation of fibroblasts in the lung. The fibroblasts in IPF are excessively proliferative and resistant to apoptosis. 53 , 54 The first study using gene expression profiling of IPF lungs was published by Zuo et al. 39 The authors found significantly enriched expression of matrilysin (matrix metalloproteinase 7, MMP‐7) and that MMP‐7−/‐ mice were protected from bleomycin‐induced pulmonary fibrosis. While the mechanism is unclear, the upregulation of MMP‐7 in IPF has been repeatedly confirmed, and MMP‐7 55 has been proposed as a peripheral blood biomarker for IPF 40 .
Based on gene expression profiling of IPF lungs compared to normal controls, Bridges et al. identified significantly enriched expression of twist1, a transcription factor that has been implicated in tumor metastasis and in epithelial‐mesenchymal transition (EMT). 56 , 57 The authors determined that twist1 is necessary for fibroblast survival in vitro when fibroblasts are exposed to proapoptotic stimuli present in IPF lungs. The role of twist1 is even more notable because another group independently published on the role of twist1 as a necessary mediator of EMT in pulmonary fibrosis. 56 Other studies have focused on secreted factors in IPF. Osteopontin, also identified by gene expression profiling of IPF, was found to increase proliferation and migration in human lung fibroblasts and in the type II alveolar epithelial cell‐like A549 cell line. 58 Increased MMP‐7 and osteopontin gene expression were independently confirmed by quantitative real‐time PCR using laser‐capture microdissection of lung tissue from patients with UIP. 55
Aberrant angiogenesis has been hypothesized as a necessary process for driving pulmonary fibrosis. 18 , 59 , 60 , 61 To further study angiogenesis using genomic techniques, Cosgrove and colleagues, using gene expression profiling made the surprising observation that pigment epithelium‐derived factor, a protein with angiostatic properties is present in the fibroblastic foci in IPF and is induced by the profibrotic growth factor TGF‐β1. 62
Another genomic approach to studying IPF is to study isolated primary cells from the lungs in culture. Cell biology techniques using fibroblasts from patients with IPF support the hypothesis that these fibroblasts are inherently proproliferative and antiapoptotic. 53 , 54 Are genome‐wide mechanisms at work to explain the differences between IPF and normal fibroblasts? Our group has identified significantly increased expression of Wnt5a in IPF and that Wnt5a acts as a proproliferative and antiapoptotic signal in human lung fibroblasts. 63 Renzoni et al. stimulated normal and IPF human lung fibroblasts with TGF‐β1 and performed gene expression profiling. 64 While TGF‐β1 dramatically changes the gene expression profile of these fibroblasts, there were no substantial changes between IPF and normal human lung fibroblasts. Larsson and colleagues provide evidence in contrast that IPF fibroblasts are transcriptionally and translationally distinct. 65 After nine passages, the fibroblasts from IPF patients maintained gene expression pathways that were significantly different from controls. 65 When the cells were cultured in collagen matrices that differed by stiffness, IPF fibroblasts showed significant alterations in ribosome recruitment patterns in contrast to the normal controls. 65 The authors also found enriched expression of EMT genes in the IPF myofibroblasts providing further evidence of EMT as a source of myofibroblasts in IPF.
The studies mentioned above have all focused on individual genes. A more complex, but probably more informative, systems biology approach is to study global patterns of gene expression and to identify trends and correlations with other physiologic or pathophysiologic processes. 66 Studer and Kaminski have described this approach as “…a global quantitative analysis of how all components in a biological system interact to determine its phenotype.” 66 An important example of a systems biology approach to gene expression can be seen in a study from our group in collaboration with Drs. Selman and Pardo. Global analysis of the IPF gene expression dataset has revealed significantly enriched expression of genes in IPF that are associated with lung development. 67 Pathways that appear active in IPF include the Wnt and TGF‐β signaling cascades. 67 These data suggest that IPF may be an pathologic recapitulation of embryonic development, a process that is normally repressed in adult life. 67
Other Approaches
To date, two genome‐wide association studies in IPF has been published. 68 , 69 After genotyping 159 Japanese IPF patients and 934 controls, a single‐nucleotide polymorphism (SNP) in intron 2 of the telomerase reverse transcriptase gene was found to be significantly associated with IPF. Telomere length has been found in many prior studies to be associated with both human and experimental pulmonary fibrosis. 5 , 70 , 71 , 72 In the second study published by Hodgson, cases of familial pulmonary fibrosis were analyzed and a common haplotype was identified in patients versus normal controls. The haplotype harbored two functionally uncharacterized genes ELMOD2 and LOC152586. In situ hybridization revealed nearly absent expression of ELMOD2 compared to normal controls and may reflect a susceptibility gene in familial pulmonary fibrosis. 69 Further studies in vitro suggest that ELMOD2 expression in lung epithelial cells is necessary for innate antiviral responses. 73
Perhaps the most important contribution to the genetic understanding of IPF is forthcoming. Work by Seibold et al. 74 and confirmed by Zhang et al. 75 from our group identified a highly significant association of the rs35705950 SNP in the putative promoter of MUC5B with IPF. 74 , 75 The odds ratios for IPF for subjects heterozygous or homozygous for the minor allele of rs35705950 were 5.9 (95% CI 4.4–7.8) and 9.7 (95% CI 4.7–19.9), respectively. 75 These provocative data suggest that contrary to prior estimates, IPF may exhibit a more Mendelian pattern of inheritance.
By a variety of genomic approaches, several research groups have studied the genetic association of IPF with candidate genes that have been implicated in prior animal and human studies of pulmonary fibrosis. 76 , 77 , 78 , 79 , 80 , 81 , 82
What is the state of “epigenomic” research in IPF?
“Epigenomics” refers to the study of epigenetic mechanisms using many of the same techniques employed in genomic studies. Do epigenetic mechanisms, which are the regulation of phenotype or gene expression caused by changes independent of the underlying DNA sequence, explain the pathogenesis of IPF? 83 A few publications have reported on epigenetic regulation of gene expression in IPF. 5 , 6 , 7 , 84 Recent advances in genomic techniques have permitted the high‐throughput analysis of epigenetic regulation of several diseases including IPF (reviewed in 85 ). Pandit and colleagues have reported profiling of IPF lungs for microRNAs, small RNA molecular regulators of gene expression, and have identified the microRNA let7d as decreased in IPF. 86 Biologic validation found that let7d binds to the TGF‐β signaling intermediate smad3 and that in vivo inhibition of let7d increased collagen gene expression and EMT in murine lungs. Based on microRNA profiling, Liu et al. recently published that expression of microRNA 21 (miR‐21) is increased in IPF and in primary lung fibroblasts stimulated with TGF‐β. 87 Suppression of miR‐21 in vivo by antisense probes attenuated bleomycin‐induced lung injury in mice. 87 These lines of evidence support the hypothesis that altered microRNA expression in IPF may in part drive pulmonary fibrosis in humans.
The importance of epigenetic regulation has been increasingly recognized in IPF. Epigenetic analysis of single genes 6 , 7 , 88 and preliminary data from our group and others demonstrate that there are phenotypically significant changes in global methylation patterns in IPF tissue and fibroblasts. 89 , 90 , 91 , 92
What is the future of genomic research in IPF
What lessons have been learned after nearly a decade of experience with gene expression profiling experiments in specimens from human lungs 93 , 94 and 15 years after the publication of the first microarray experiments? 95 We believe that several obstacles need to be overcome in order to realize the full power of genomic analysis in IPF. First, for many tissues, availability is limited and many of the published studies use only small datasets. Second, there has been no central mechanism for sharing data across centers. Third, because of the potential clinical impact of this work, it is essential that important observations are replicated independently. Fourth, there needs to be adequate training for new genomics investigators. The currently funded projects in genomic approaches to IPF ( Table 1 ) help address some of these challenges. First, the funding of the LTRC in January 2004 by the NIH created a large repository of lung tissue, blood, imaging, and clinical data on patients with lung diseases including chronic obstructive pulmonary disease (COPD) and IPF. Use of LTRC specimens has resulted in the funding of several grants 96 including the Lung Genomic research Consortium (LGRC 97 ). The LGRC was established through an 11 million dollar grant from the National Heart, Lung, and Blood Institute (NHLBI) through funds from the American Recovery and Reinvestment Act (ARRA). 97 The multidisciplinary team across six centers, using samples from the LTRC, will assemble the largest database yet of clinical and genomic data in many diseases including IPF. 97 The database will be accessible to the research community and will be designed to be easily searchable. As of 2010, nearly 800 lung tissues from the LTRC have been received for genomic analysis. 97 From these samples, data will be obtained from deep sequencing, methylation studies, and gene and microRNA expression studies. We believe that the institution of the LTRC and the LGRC will greatly expand tissue availability and permit data sharing. We also need to ensure that there is adequate funding for training in translational genomics of lung disease by expanding and enhancing the K12 programs for career development awards in lung genomics.
Table 1.
Studies funded by the National Institutes of Health employing genomic techniques to study pulmonary fibrosis.
Another critical obstacle to unlocking the potential of genomic analysis in IPF has been reliance on the “single gene” approach. Single‐gene studies have proven to be very useful for identifying certain pathophysiologic pathways in IPF. However, we believe that the single‐gene approach provides only limited insight into the pathogenesis of a complex disease such as IPF. The emergence of epigenomic techniques will herald we believe, a new paradigm for understanding IPF. This shift ( Figure 2 ) will be characterized by a move from a linear gene‐centric view in which organ phenotype is determined by single‐gene responses that result from a snapshot response to injury to a global‐genome view that focuses on the global responsiveness of the genome, as measured by global methylation patterns and changes in microRNA expression. While the former view usually provides efficient explanations of the initial response to injury, the latter provides a working model of how abnormal organ phenotypes are maintained over time, years after the inciting injury has passed. Single‐gene studies have predominated over the past 10 years and have identified multiple targets for therapeutic intervention, but still no drugs are available for IPF that target these single‐gene products. The failure of single‐gene studies to bring new drugs to market is most easily explained by the complicated nature of IPF. Ten years of IPF genomic research has shown that the disease is not driven by one gene alone but by a network of thousands of genes acting together. To truly understand how the expression of all these genes together causes pulmonary fibrosis, a systems biology approach is needed. At the most basic level, systems biology techniques can unlock the full potential of genomic analysis by using the entire gene expression dataset. Genomic analysis using systems biology will be interdisciplinary, bridging clinicians with biologists, and computer scientists. Systems biology views pulmonary fibrosis as dynamic, modeling changes of gene expression over time. 66
Figure 2.
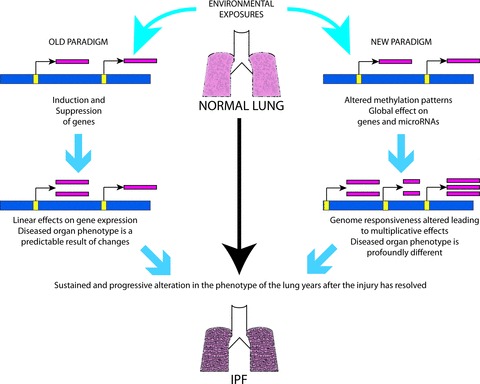
A paradigm shift. The role of epigenetics in IPF.
The final question is how genomic discoveries in IPF will impact patients. The increased availability of patients will more adequately power our studies so that results will be representative of the IPF population across the United States. The devastating nature of IPF demands that the new drugs that come to clinical trial have a strong likelihood of success. In this context, application of a robust systems biology model of a drug in a preclinical model, for example, could predict the chances for a drug’s success and save significant cost and effort. In addition, genomic profiles of the lung or surrogate tissues may provide significant information about drug effects, response to therapy, and disease pathogenesis. Thus, we propose that all new drug studies will collect samples for genomic analysis and include approaches for data analysis and sharing as the data collected will be extremely valuable even if the drug fails. As of 2011, there are no clinical trials in IPF targeting any of the molecules that have been reviewed here (MMP‐7, Twist, IGFBP5, osteopontin, Wnt5a, Let‐7d, or miR‐21). Of the trials that are currently recruiting patients or have recently been completed, three trials have been designed to target genes that may exhibit increased expression in IPF (LOXL2, TGF‐β3, and Collagen V). 98 Several trials target molecules that have been identified by more classical in vitro and in vivo studies. These include TGF‐β1, angiotensin II, JNK, CTGF, and CCL2. But a review of publicly available IPF datasets does not show significantly increased expression of these genes.
Finally, critical to translation would be the application of high‐throughput genomic technologies to surrogate tissues. The profiling of peripheral blood will allow sampling during disease progression and identification of biomarkers for disease progression and outcome. These biomarkers, when correlated with clinical characteristics, will stratify patients according to disease severity in clinical studies, identify those who can benefit from available therapies, and serve to prioritize patients for lung transplantation.
Conclusion
We think most biomedical researchers would agree that the publication of the human genome was unlikely to provide a “quick fix” to any human disease, especially a disease as complex as IPF. Genomic research had a significant impact on our understanding of IPF. Genomic profiling led to identification of multiple novel molecules that are mechanistically involved in the disease, as well as to some very significant global insights such as the aberrant activation of developmental pathways. In the more translational arena, genomic profiling demonstrated that IPF has a distinctive gene signature that may predict the rate of IPF disease progression as well as acute exacerbations. The clinical impact of these findings has been limited because of the lack of availability of tissue and little replication. We believe that the significant investment of NHLBI in creating tissue resources such as the LTRC as well as in large projects that provide genomic information will allow the pulmonary community to realize many of the bold predictions made 10 years ago when the human genome was first published. The discoveries derived from using these resources will transform our approach to the diagnosis and management of IPF and thus have a dramatic beneficial effect on the life of our patients.
Acknowledgments
This work was supported by the NIH: HL083085 (D.J.K.) and HL095397, LM009657, HL101715 (N.K.), and the Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease.
References
- 1. Editorial . The Genome, 10 Years Later. The New York Times. New York Edition ed. New York , 2010. [Google Scholar]
- 2. Nogee LM, Dunbar AE, 3rd , Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001; 344: 573–579. [DOI] [PubMed] [Google Scholar]
- 3. Thomas AQ, Lane K, Phillips J, 3rd , Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002; 165: 1322–1328. [DOI] [PubMed] [Google Scholar]
- 4. Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA 3rd, Sporn TA, et al Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005; 172: 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diaz de Leon A, Cronkhite JT, Katzenstein AL, Godwin JD, Raghu G, Glazer CS, Rosenblatt RL, Girod CE, Garrity ER, Xing C, Garcia CK. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. 5: e10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coward WR, Watts K, Feghali‐Bostwick CA, Jenkins G, Pang L. Repression of IP‐10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol Cell Biol. 30: 2874–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, Hagood JS. Thy‐1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008; 39: 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, Colby TV, Waldron JA. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case‐control study. Collaborating Centers Am J Epidemiol. 2000; 152: 307–315. [DOI] [PubMed] [Google Scholar]
- 9. Schwartz DA, Van Fossen DS, Davis CS, Helmers RA, Dayton CS, Burmeister LF, Hunninghake GW. Determinants of progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994; 149: 444–449. [DOI] [PubMed] [Google Scholar]
- 10. Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006; 174: 810–816. [DOI] [PubMed] [Google Scholar]
- 11. Fernandez Perez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, Yi ES, Ryu JH. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population‐based study. Chest. 137: 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001; 134: 136–151. [DOI] [PubMed] [Google Scholar]
- 13. Scotton CJ, Krupiczojc MA, Konigshoff M, Mercer PF, Lee YC, Kaminski N, Morser J, Post JM, Maher TM, Nicholson AG, et al Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J Clin Invest. 2009; 119: 2550–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, et al Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008; 294: L1119–L1126. [DOI] [PubMed] [Google Scholar]
- 15. Drakopanagiotakis F, Xifteri A, Polychronopoulos V, Bouros D. Apoptosis in lung injury and fibrosis. Eur Respir J. 2008; 32: 1631–1638. [DOI] [PubMed] [Google Scholar]
- 16. Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008; 178: 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keane MP, Arenberg DA, Lynch JP, 3rd , Whyte RI, Iannettoni MD, Burdick MD, Wilke CA, Morris SB, Glass MC, DiGiovine B, et al The CXC chemokines, IL‐8 and IP‐10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol. 1997; 159: 1437–1443. [PubMed] [Google Scholar]
- 18. Keane MP, Belperio JA, Burdick MD, Lynch JP, Fishbein MC, Strieter RM. ENA‐78 is an important angiogenic factor in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001; 164: 2239–2242. [DOI] [PubMed] [Google Scholar]
- 19. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase‐4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009; 15: 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kliment CR, Englert JM, Gochuico BR, Yu G, Kaminski N, Rosas I, Oury TD. Oxidative stress alters syndecan‐1 distribution in lungs with pulmonary fibrosis. J Biol Chem. 2009; 284: 3537–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol. 2009; 175: 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy. 65: 537–553. [DOI] [PubMed] [Google Scholar]
- 23. Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008; 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willis BC, Liebler JM, Luby‐Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial‐mesenchymal transition in alveolar epithelial cells by transforming growth factor‐beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005; 166: 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006; 3: 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindell KO, Erlen JA, Kaminski N. Lessons from our patients: development of a warm autopsy program. PLoS Med. 2006; 3: e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr., Lasky JA, Loyd JE, Noth I, Olman MA, et al Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007; 176: 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol. 2002; 26: 549–557. [DOI] [PubMed] [Google Scholar]
- 29. Curnow SJ, Fairclough M, Schmutz C, Kissane S, Denniston AK, Nash K, Buckley CD, Lord JM, Salmon M. Distinct types of fibrocyte can differentiate from mononuclear cells in the presence and absence of serum. PLoS One. 5: e9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, et al Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009; 179: 588–594. [DOI] [PubMed] [Google Scholar]
- 31. Strieter RM, Keeley EC, Burdick MD, Mehrad B. The role of circulating mesenchymal progenitor cells, fibrocytes, in promoting pulmonary fibrosis. Trans Am Clin Climatol Assoc. 2009; 120: 49–59. [PMC free article] [PubMed] [Google Scholar]
- 32. Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U. Plasma microRNA profiling reveals loss of endothelial miR‐126 and other microRNAs in type 2 diabetes. Circ Res. 107: 810–817. [DOI] [PubMed] [Google Scholar]
- 33. Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 101: 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, Brown KK, Schwarz MI, Schwartz DA. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS One. 2009; 4: e5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, Bleecker ER. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol. 125: 1028–1036 e1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carpagnano GE, Palladino GP, Gramiccioni C, Foschino Barbaro MP, Martinelli D. Exhaled ERCC‐1 and ERCC‐2 microsatellite alterations in NSCLC patients. Lung Cancer. 68: 305–307. [DOI] [PubMed] [Google Scholar]
- 37. American Thoracic Society . Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000; 161: 646–664. [DOI] [PubMed] [Google Scholar]
- 38. Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 2000; 162: 2213–2217. [DOI] [PubMed] [Google Scholar]
- 39. Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben‐Dor A, Lollini L, Morris D, Kim Y, DeLustro B, et al Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA. 2002; 99: 6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, et al MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008; 5: e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2006; 173: 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang IV, Burch LH, Steele MP, Savov JD, Hollingsworth JW, McElvania‐Tekippe E, Berman KG, Speer MC, Sporn TA, Brown KK, et al Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med. 2007; 175: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hsu E, Shi H, Jordan RM, Lyons‐Weiler J, Pilewski JM, Feghali‐Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 63: 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3: 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lederer DJ, Arcasoy SM, Wilt JS, D’Ovidio F, Sonett JR, Kawut SM. Six‐minute‐walk distance predicts waiting list survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174: 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005; 128: 2393–2399. [DOI] [PubMed] [Google Scholar]
- 47. Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors, and outcome. Eur Respir J. 2011; 37: 356–363. [DOI] [PubMed] [Google Scholar]
- 48. Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, et al Gene expression‐based survival prediction in lung adenocarcinoma: a multi‐site, blinded validation study. Nat Med. 2008; 14: 822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, Ahmad F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 298: H1235–H1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Velculescu VE, Vogelstein B, Kinzler KW. Analysing uncharted transcriptomes with SAGE. Trends Genet. 2000; 16: 423–425. [DOI] [PubMed] [Google Scholar]
- 51. Selman M, Carrillo G, Estrada A, Mejia M, Becerril C, Cisneros J, Gaxiola M, Perez‐Padilla R, Navarro C, Richards T, et al Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One. 2007; 2: e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaminski N, Rosas IO. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: can we identify the right target genes? Proc Am Thorac Soc. 2006; 3: 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moodley YP, Scaffidi AK, Misso NL, Keerthisingam C, McAnulty RJ, Laurent GJ, Mutsaers SE, Thompson PJ, Knight DA. Fibroblasts isolated from normal lungs and those with idiopathic pulmonary fibrosis differ in interleukin‐6/gp130‐mediated cell signaling and proliferation. Am J Pathol. 2003; 163: 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moodley YP, Misso NL, Scaffidi AK, Fogel‐Petrovic M, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA. Inverse effects of interleukin‐6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol. 2003; 29: 490–498. [DOI] [PubMed] [Google Scholar]
- 55. Kelly MM, Leigh R, Gilpin SE, Cheng E, Martin GE, Laurent GJ, Thompson PJ, Knight DA. Cell‐specific gene expression in patients with usual interstitial pneumonia. Am J Respir Crit Care Med. 2006; 174: 557–565. [DOI] [PubMed] [Google Scholar]
- 56. Pozharskaya V, Torres‐Gonzalez E, Rojas M, Gal A, Amin M, Dollard S, Roman J, Stecenko AA, Mora AL. Twist: a regulator of epithelial‐mesenchymal transition in lung fibrosis. PLoS One. 2009; 4: e7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bridges RS, Kass D, Loh K, Glackin C, Borczuk AC, Greenberg S. Gene expression profiling of pulmonary fibrosis identifies Twist1 as an antiapoptotic molecular “rectifier” of growth factor signaling. Am J Pathol. 2009; 175: 2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M, et al Up‐regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005; 2: e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Strieter RM, Gomperts BN, Keane MP. The role of CXC chemokines in pulmonary fibrosis. J Clin Invest. 2007; 117: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Burdick MD, Murray LA, Keane MP, Xue YY, Zisman DA, Belperio JA, Strieter RM. CXCL11 attenuates bleomycin‐induced pulmonary fibrosis via inhibition of vascular remodeling. Am J Respir Crit Care Med. 2005; 171: 261–268. [DOI] [PubMed] [Google Scholar]
- 61. Keane MP, Belperio JA, Arenberg DA, Burdick MD, Xu ZJ, Xue YY, Strieter RM. IFN‐gamma‐inducible protein‐10 attenuates bleomycin‐induced pulmonary fibrosis via inhibition of angiogenesis. J Immunol. 1999; 163: 5686–5692. [PubMed] [Google Scholar]
- 62. Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, Schwarz MI, Cool CD, Worthen GS. Pigment epithelium‐derived factor in idiopathic pulmonary fibrosis: a role in aberrant angiogenesis. Am J Respir Crit Care Med. 2004; 170: 242–251. [DOI] [PubMed] [Google Scholar]
- 63. Vuga LJ, Ben‐Yehudah A, Kovkarova‐Naumovski E, Oriss T, Gibson KF, Feghali‐Bostwick C, Kaminski N. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol. 2009; 41: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Renzoni EA, Abraham DJ, Howat S, Shi‐Wen X, Sestini P, Bou‐Gharios G, Wells AU, Veeraraghavan S, Nicholson AG, Denton CP, et al Gene expression profiling reveals novel TGFbeta targets in adult lung fibroblasts. Respir Res. 2004; 5: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Larsson O, Diebold D, Fan D, Peterson M, Nho RS, Bitterman PB, Henke CA. Fibrotic myofibroblasts manifest genome‐wide derangements of translational control. PLoS One. 2008; 3: e3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Studer SM, Kaminski N. Towards systems biology of human pulmonary fibrosis. Proc Am Thorac Soc. 2007; 4: 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med. 2008; 5: e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, Taniguchi H, Kubo M, Kamatani N, Nakamura Y. A genome‐wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008; 45: 654–656. [DOI] [PubMed] [Google Scholar]
- 69. Hodgson U, Pulkkinen V, Dixon M, Peyrard‐Janvid M, Rehn M, Lahermo P, Ollikainen V, Salmenkivi K, Kinnula V, Kere J, et al ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006; 79: 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008; 105: 13051–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fridlender ZG, Cohen PY, Golan O, Arish N, Wallach‐Dayan S, Breuer R. Telomerase activity in bleomycin‐induced epithelial cell apoptosis and lung fibrosis. Eur Respir J. 2007; 30: 205–213. [DOI] [PubMed] [Google Scholar]
- 72. Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, et al Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007; 356: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 73. Pulkkinen V, Bruce S, Rintahaka J, Hodgson U, Laitinen T, Alenius H, Kinnula VL, Myllarniemi M, Matikainen S, Kere J. ELMOD2, a candidate gene for idiopathic pulmonary fibrosis, regulates antiviral responses. FASEB J. 24: 1167–1177. [DOI] [PubMed] [Google Scholar]
- 74. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, et al A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 364: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 364: 1576–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Checa M, Ruiz V, Montano M, Velazquez‐Cruz R, Selman M, Pardo A. MMP‐1 polymorphisms and the risk of idiopathic pulmonary fibrosis. Hum Genet. 2008; 124: 465–472. [DOI] [PubMed] [Google Scholar]
- 77. Zorzetto M, Ferrarotti I, Trisolini R, Lazzari Agli L, Scabini R, Novo M, De Silvestri A, Patelli M, Martinetti M, Cuccia M, et al Complement receptor 1 gene polymorphisms are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003; 168: 330–334. [DOI] [PubMed] [Google Scholar]
- 78. Latsi P, Pantelidis P, Vassilakis D, Sato H, Welsh KI, du Bois RM. Analysis of IL‐12 p40 subunit gene and IFN‐gamma G5644A polymorphisms in idiopathic pulmonary fibrosis. Respir Res. 2003; 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu L, Dai HP, Xiao B, Zhang S, Ban CJ, Xin P. Association of ENA‐78, IP‐10 and VEGF gene polymorphism with idiopathic pulmonary fibrosis. Zhonghua Yi Xue Za Zhi. 2009l; 89: 2690–2694. [PubMed] [Google Scholar]
- 80. Zorzetto M, Ferrarotti I, Campo I, Trisolini R, Poletti V, Scabini R, Ceruti M, Mazzola P, Crippa E, Ottaviani S, et al NOD2/CARD15 gene polymorphisms in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005; 22: 180–185. [PubMed] [Google Scholar]
- 81. Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, Markin C, Renzoni E, Lympany P, Thomas AQ, et al Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004; 59: 977–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hutyrova B, Pantelidis P, Drabek J, Zurkova M, Kolek V, Lenhart K, Welsh KI, Du Bois RM, Petrek M. Interleukin‐1 gene cluster polymorphisms in sarcoidosis and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002; 165: 148–151. [DOI] [PubMed] [Google Scholar]
- 83. Bird A. Perceptions of epigenetics. Nature. 2007; 447: 396–398. [DOI] [PubMed] [Google Scholar]
- 84. Coward WR, Watts K, Feghali‐Bostwick CA, Knox A, Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol. 2009; 29: 4325–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis Translational Research 2011; 157: 191–199. [DOI] [PubMed] [Google Scholar]
- 86. Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, Konishi K, Yousem SA, Singh M, Handley D, et al Inhibition and role of let‐7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 182: 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E. miR‐21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 207: 1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sanders YY, Tollefsbol TO, Varisco BM, Hagood JS. Epigenetic Regulation of thy‐1 by histone deacetylase inhibitor in rat lung fibroblasts. Am J Respir Cell Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dakhlallah DPM, Marsh C. miR‐17 92 cluster as a prognostic marker in human Idiopathic Pulmonary Fibrosis (IPF). Am J Respir Crit Care Med. [Abstract] 2010; 181: A2018. [Google Scholar]
- 90. Sanders YY, Hagood JS. The effects of histone deacetylase inhibitors on fibrotic lung fibroblasts survival and apoptosis. Am J Respir Crit Care Med. 2010; 181: A2015. [Google Scholar]
- 91. Milosevic J, Pandit K, Magister M, Bais AS, Benos PV, Rabinovich EI, Kaminski N. The role and regulation of miR‐154 microRNA family in lung fibrosis. Am J Respir Crit Care Med. [Abstract] 2010; 181: A2019. [Google Scholar]
- 92. Rabinovich EI, Yakhini Z, Benos PV, Pandit KV, Milosevic J, Kapetanaki M, Richards TJ, Chensny LJ, Kaminski N. The human CpG islands arrays reveal changes in global methylation patterns in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010; 181: A2017. [Google Scholar]
- 93. Wang T, Hopkins D, Schmidt C, Silva S, Houghton R, Takita H, Repasky E, Reed SG, et al Identification of genes differentially over‐expressed in lung squamous cell carcinoma using combination of cDNA subtraction and microarray analysis. Oncogene. 2000; 19: 1519–1528. [DOI] [PubMed] [Google Scholar]
- 94. Hong TM, Yang PC, Peck K, Chen JJ, Yang SC, Chen YC, Wu CW. Profiling the downstream genes of tumor suppressor PTEN in lung cancer cells by complementary DNA microarray. Am J Respir Cell Mol Biol. 2000; 23: 355–363. [DOI] [PubMed] [Google Scholar]
- 95. Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995; 270: 467–470. [DOI] [PubMed] [Google Scholar]
- 96. RePORT N . Available at: http://projectreporter.nih.gov/reporter.cfm. [Accessed May 4, 2011].
- 97. Lung Genomics Research Consortium . Available at: http://www.lung‐genomics.org/index.html. (Accessed May 4, 2011).
- 98. National Institutes of Health . ClinicalTrials.gove: A service of the U.S. National Institutes of Health. Available at: http://www.clinicaltrials.gov. Accessed March 30, 2011.