

Abstract
Objective
Pharmacological inhibition of the cholesteryl ester transfer protein (CETP) in humans increases high-density lipoprotein (HDL) cholesterol (HDL-C) levels; however, its effects on apolipoprotein A-I (apoA-I) containing HDL subspecies, apoA-I turnover, and markers of reverse cholesterol transport are unknown. The present study was designed to address these issues.
Methods and Results
Nineteen subjects, 9 of whom were taking 20 mg of atorvastatin for hypercholesterolemia, received placebo for 4 weeks, followed by the CETP inhibitor torcetrapib (120 mg QD) for 4 weeks. In 6 subjects from the nonatorvastatin cohort, the everyday regimen was followed by a 4-week period of torcetrapib (120 mg BID). At the end of each phase, subjects underwent a primed-constant infusion of (5,5,5-2H3)-L-leucine to determine the kinetics of HDL apoA-I. The lipid data in this study have been reported previously. Relative to placebo, 120 mg daily torcetrapib increased the amount of apoA-I in α1-migrating HDL in the atorvastatin (136%; P<0.001) and nonatorvastatin (153%; P<0.01) cohorts, whereas an increase of 382% (P<0.01) was observed in the 120 mg twice daily group. HDL apoA-I pool size increased by 8±15% in the atorvastatin cohort (P=0.16) and by 16±7% (P<0.0001) and 34±8% (P<0.0001) in the nonatorvastatin 120 mg QD and BID cohorts, respectively. These changes were attributable to reductions in HDL apoA-I fractional catabolic rate (FCR), with torcetrapib reducing HDL apoA-I FCR by 7% (P<0.10) in the atorvastatin cohort, by 8% (P<0.001) in the nonatorvastatin 120 mg QD cohort, and by 21% (P=0.01) in the nonatorvastatin 120 mg BID cohort. Torcetrapib did not affect HDL apoA-I production rate. In addition, torcetrapib did not significantly change serum markers of cholesterol or bile acid synthesis or fecal sterol excretion.
Conclusions
These data indicate that partial inhibition of CETP via torcetrapib in patients with low HDL-C: (1) normalizes apoA-I levels within α1-migrating HDL, (2) increases plasma concentrations of HDL apoA-I by delaying apoA-I catabolism, and (3) does not significantly influence fecal sterol excretion.
Keywords: apolipoprotein A-I, bile acids, cholesteryl ester transfer protein, CETP inhibition, high-density lipoproteins, kinetics
Epidemiologic studies have consistently demonstrated that plasma concentrations of high-density lipoprotein (HDL) cholesterol and apolipoprotein A-I (apoA-I) are inversely correlated with the incidence of coronary heart disease (CHD).1–3 Clinical trial results indicate that even modest increases in HDL cholesterol (HDL-C) concentrations can significantly reduce CHD risk.4–5 However, 3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors, or statins, have only modest effects on HDL-C levels, raising them on average by 5% to 10%.6 Although fibrates and niacin can raise HDL-C levels, the increases in HDL-C are rarely >25%, and niacin is often not well tolerated.
Among the alternative HDL-raising strategies actively being explored is cholesteryl ester transfer protein (CETP) inhibition.7 CETP is a plasma glycoprotein that facilitates the transfer of cholesteryl esters (CEs) from HDL to apoB-containing lipoproteins.8,9 Humans with CETP deficiency attributable to molecular defect(s) in the CETP gene have markedly elevated plasma levels of HDL-C and apoA-I,10–12 the latter of which are the result of significantly delayed apoA-I catabolism.13 These observations have led to the hypothesis that pharmacological inhibition of CETP may increase HDL-C and apoA-I levels to a greater extent than currently available therapies. The concept of therapies targeted specifically toward HDL metabolism has gained support of late from the finding that infusions of apoA-I Milano–phospholipid (PL) complexes over a 5-week period induced regression of coronary atherosclerosis in a small clinical trial.14
We reported previously that pharmacological inhibition of CETP in humans significantly increased steady-state HDL-C levels in normolipidemic subjects15 and in patients with low HDL-C (<40 mg/dL).16 In the former subjects, 120 mg torcetrapib QD increased plasma concentrations of HDL-C and apoA-I by means of 73% and 24%,15 respectively, whereas corresponding increases of 53% and 15% were observed in subjects with low HDL-C.16 The present study was designed to assess the effects of CETP inhibition on dynamic aspects of HDL metabolism, specifically apoA-I kinetics and fecal sterol excretion, in patients with low HDL-C levels. Our data indicate that CETP inhibition significantly increased apoA-I pool size (PS) because of delayed catabolism but did not alter fecal sterol excretion.
Methods
Subjects
Subjects were recruited at 2 medical centers: Tufts-New England Medical Center, in Boston, Mass, and the University of Pennsylvania School of Medicine, in Philadelphia. Patients were eligible for this trial if they met the following criteria: an age of 18 to 70 years, an HDL-C of <40 mg/dL, triglycerides (TGs) of <400 mg/dL, a low-density lipoprotein cholesterol (LDL-C) level of <160 mg/dL, and a body mass index between 18 and 35 kg/m2. Subjects having an LDL-C of >160 mg/dL were considered for the atorvastatin arm of this study provided that they met all other criteria, including that of an LDL-C of <160 mg/dL once stabilized on 20 mg atorvastatin. Exclusion criteria were described previously in detail.16 The study protocol was approved by the human investigation review committee of each institution, and informed written consent was obtained from each participant.
Experimental Design
This was a single-blind, placebo-controlled, fixed-sequence study designed to examine the effects of torcetrapib on lipoprotein metabolism in subjects with low HDL-C. A total of 19 subjects were enrolled in this trial, with 9 subjects in the atorvastatin arm and 10 in the nonatorvastatin arm. A detailed description of the study design has been reported previously.16 Briefly, the study consisted of an introductory period of 2 to 4 weeks, during which time subjects were screened and, if necessary (LDL-C >160 mg/dL), stabilized on 20 mg atorvastatin. All subjects next received placebo for 4 weeks, followed by 120 mg torcetrapib QD for an additional 4 weeks. Six subjects from the nonatorvastatin cohort also participated in a third phase, in which they received 120 mg torcetrapib BID for 4 weeks.
At the end of each 4-week phase, subjects underwent a primed-constant infusion of deuterated leucine while fed a small meal each hour for 20 hours to determine the kinetics of HDL apoA-I.17 At 11 AM (0 hour), (5,5,5-2H3)-L-leucine (10 μmol/kg body weight) was injected intravenously as a bolus over 1 minute and then by continuous infusion 10 μmol/L · kg body weight−1 · hours−1 over a 15-hour period. Blood samples (20 mL) were collected at 0, 30, 35, and 45 minutes and at 1, 1.5, 2, 3, 4, 6, 9, 10, 12, 14, and 15 hours, and HDL particles were isolated by sequential density ultracentrifugation.
At a visit 1 week before the metabolic study, subjects were provided with detailed instructions and containers for complete stool collection. Stools were collected for the entire 3-day period before the metabolic study at the end of each phase.
Plasma Lipid and Lipoprotein Determinations
On the day of infusion, blood samples were collected from subjects after a 12-to 14-hour fast into tubes containing 0.1% EDTA. Plasma was isolated by centrifugation at 2500 rpm, 4°C, for 20 minutes. Plasma HDL-C levels were determined subsequent to dextran sulfate–magnesium precipitation of apoB-containing lipoproteins.18 Levels of total cholesterol (TC) and TG in HDL were determined using standard enzymatic methods.19 Free (unesterified) cholesterol (FC) and PL concentrations in HDL were determined with a Hitachi 911 autoanalyzer, using reagent kits from Wako Diagnostics. CE concentrations were calculated by subtracting FC from TC and multiplied by 1.68 to correct for fatty acid content. HDL total protein (PRO) was assessed using the Pierce Micro BCA kit, with the modification of adding 10% sodium dodecyl sulfate to denature lipoprotein TGs. Cholesterol, TG, and HDL-C assays have been standardized through the Centers for Disease Control (Atlanta, Ga).
Analysis of ApoA-I–Containing HDL Subpopulations by 2D Lipoprotein Electrophoresis
The distribution of apoA-I–containing HDL subpopulations in the plasma was determined using nondenaturing 2D agarose–polyacrylamide gel electrophoresis, as reported previously.20 With this method, absolute concentrations (in milligrams apoA-I per dL plasma) of apoA-I–containing HDL subpopulations are calculated by multiplying the plasma total apoA-I concentration for a given subject (mg/dL) by the percentile value of each subpopulation.
Quantitation and Isolation of Apolipoproteins
Plasma apoA-I levels at each kinetic time point were measured on a Hitachi 911 autoanalyzer (Hitachi, Inc.) using an immunoturbidimetric assay and reagents and calibrators from Wako Diagnostics. ApoA-I was isolated from HDL by SDS-PAGE using a Tris–glycine buffer system, as described previously.21
Determinations of Isotopic Enrichment
ApoA-I bands were excised from the polyacrylamide gels, hydrolyzed in 12 N HCl at 110°C for 24 hours, and amino acids converted to N-propyl ester, N-heptafluorobutyramide derivatives, as described previously in detail,22 before analysis on an Agilent Technologies 6890/5973 gas chromatography–mass spectrometry (GC/MS). To identify labeled and unlabeled leucine, amino acids were ionized by methane-negative chemical ionization. Selected ion monitoring at mass-to-charge ratios of 349 and 352 were used to determine the moles percent enrichment (labeled/unlabeled) of each sample. Enrichment was calculated from the moles percent enrichment and then converted to a tracer/tracee ratio according to the following formula: tracer/tracee=e(t)/e(I)−e(t), where e(t) is the enrichment at time t, and e(I) represents the isotopic abundance of the infusate determined by GC/MS.23
Kinetic Analysis
The kinetics of HDL apoA-I were assessed using a multicompartmental model as described previously.24 The SAAMII program was used to fit the model to the observed tracer data using a weighted least-squares approach to determine the best fit. The very LDL apoB-100 plateau was used for liver-derived apoB-100 as described previously.25 ApoA-I PS was defined as plasma apoA-I concentration (mg/dL) multiplied by plasma volume (0.45 dL/kg body weight). ApoA-I production rate (PR) was calculated using the formula PR (mg · kg−1 · day−1)=[fractional catabolic rate (FCR; pools per day)×apoA-I concentration (mg/dL)×plasma volume (0.45 dL/kg body weight)]/body weight (kg).
Determination of Serum Lathosterol and 7α-Hydroxy-4-Cholesten-3-One Concentrations
Serum levels of lathosterol (μg/mL), a cholesterol precursor, were assayed by mass spectrometry using an isotopedilution method, as previously described in detail.26,27 Serum concentrations of 7α-hydroxy-4-cholesten-3-one (C4; ng/mL), an intermediate in bile acid synthesis, were determined by high-performance liquid chromatography, as reported recently.28 A strong correlation has been shown between the level of C4 in serum or plasma and the enzymatic activity of hepatic cholesterol 7α-hydroxylase,28 the rate-limiting enzyme in the synthesis of bile acids.
Measurement of Fecal Bile Acid and Neutral Sterol Content
Stools were collected for the entire 3-day period before the metabolic study at the end of each phase, as described above. The neutral sterols cholesterol, coprostanol, and coprostanone, as well as bile acids and plant sterols, were measured in extracted stool samples using GC/MS.29–31 Because no dietary marker was used, the content of fecal neutral sterols and fecal bile acids is expressed as mg/mg plant sterol to correct for possible variation in fecal flow.
Statistical Analyses
The normality of end points was assessed by the Shapiro–Wilk goodness-of-fit test,32 in addition to visual examination of histograms and box plots. Paired t tests were used to assess differences between the placebo and drug phases within a given group, whereas 2-sample t tests were used to detect statistically significant differences between the atorvastatin and nonatorvastatin groups (SAS System for Windows, Release 8.02, 1999 to 2001; SAS Institute). Percent change relative to placebo was computed on an individual subject basis and summarized descriptively by cohort. All data in the text and tables are presented as mean±SD.
Results
Effects of Torcetrapib on HDL Composition
As reported previously,16 CETP inhibition with torcetrapib significantly increased plasma concentrations of HDL-C and apoA-I in this study (Figure). The effects of torcetrapib on HDL composition, expressed as percent weight/weight, are shown in Table 1. As expected, torcetrapib had its greatest effect on the CE and TG components of HDL. Relative to placebo, 120 mg torcetrapib QD increased HDL CE content from a mean of 12.2±1.1% to 15.9±1.1% (P<0.00001) in the atorvastatin cohort and from a mean of 11.2±2.3% to 15.6±1.8% (P<0.0001) in the nonatorvastatin cohort. A mean increase of 45±12%, from 11.9±1.3% to 17.1±1.4% (P<0.001), was seen in the 120 mg torcetrapib BID cohort. In contrast, HDL TG content was significantly reduced by torcetrapib. Torcetrapib reduced HDL TG from 7.4±1.7% to 5.4±1.6% (P<0.04) in the atorvastatin cohort and from 9.3±2.2% to 4.9±1.0% (P<0.0001) and 8.1±1.0% to 4.2±0.9% (P<0.01) in the 120 mg nonatorvastatin QD and BID cohorts, respectively.
Figure.
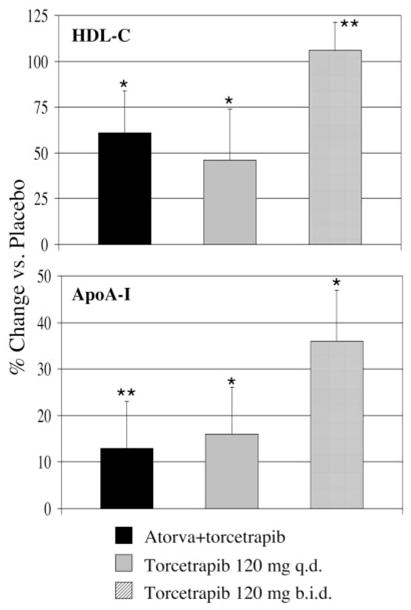
Effect of torcetrapib on plasma concentrations of HDL-C (top) and apoA-I (bottom) in subjects with low HDL-C. Relative to placebo, 120 mg torcetrapib daily increased plasma HDL-C levels by a mean of 61%, from 29±4 to 47±10 mg/dL, in the atorvastatin group, by 46%, from 32±7 to 46±14 mg/dL, in the 120 mg torcetrapib once daily group, and by 106%, from 34±5 to 70±15 mg/dL, in the 120 mg torcetrapib twice daily group. Plasma apoA-I concentrations were increased by 13%, 16%, and 36% in these groups, respectively. *P<0.001; **P<0.01 for comparison with placebo phase.
TABLE 1.
Effects of Torcetrapib on HDL Composition*
| Parameter | Atorvastatin+Torcetrapib | Torcetrapib
|
|
|---|---|---|---|
| 120 mg QD n=9 | 120 mg QD n=10 | 120 mg BID n=6 | |
| HDL-FC | |||
| Placebo | 2.3±0.4 | 1.9±0.4 | 2.0±0.5 |
| Torcetrapib | 2.8±0.8 | 2.3±0.4¶ | 2.6±0.6§ |
| % change | 22±35 | 19±12 | 34±19 |
| HDL-CE | |||
| Placebo | 12.2±1.1 | 11.2±2.3 | 11.9±0.7 |
| Torcetrapib | 15.9±1.1† | 15.6±1.8‡ | 17.1±1.4¶ |
| % change | 31±10 | 46±39 | 45±12 |
| HDL-TG | |||
| Placebo | 7.3±1.7 | 9.3±2.2 | 8.1±0.9 |
| Torcetrapib | 5.4±1.6|| | 4.9±1.0‡ | 4.2±0.8§ |
| % change | −22±34 | −46±14 | −48±10 |
| HDL-PL | |||
| Placebo | 17.8±1.5 | 16.4±1.6 | 16.8±1.4 |
| Torcetrapib | 19.7±1.8§ | 19.0±1.0§ | 19.8±1.0§ |
| % change | 11±8 | 17±14 | 19±9 |
| HDL-PRO | |||
| Placebo | 60.4±2.2 | 61.1±2.4 | 61.4±1.8 |
| Torcetrapib | 56.2±3.2¶ | 58.2±2.4§ | 56.4±2.3§ |
| % change | −7±4 | −5±4 | −8±3 |
Data are expressed as % weight/weight, mean±SD.
The absolute concentration (mean±SD, mg/dL) of each HDL component is provided in a data supplement (available online at http://atvb.ahajournals.org).
P<0.00001;
P<0.0001;
P<0.001;
P<0.01;
P<0.04 for comparison with placebo phase.
The PL and total PRO content of HDL were also significantly altered by torcetrapib. Torcetrapib increased HDL PL content from 17.8±1.5% to 19.7±1.8% (P<0.01) in the atorvastatin cohort, from 16.4±1.6% to 19.0±1.0% (P<0.01) in the 120 mg nonatorvastatin QD cohort, and from 17.1±1.2% to 19.9±1.0% (P<0.01) in the 120 mg nonatorvastatin BID cohort. HDL PRO was significantly reduced by torcetrapib, with mean reductions of 7.0±3.7% (P<0.001), 4.7±4.2% (P<0.01), and 7.9±3.6% (P<0.01) versus placebo observed for the atorvastatin, 120 mg nonatorvastatin QD, and 120 mg nonatorvastatin BID cohorts, respectively.
Effects of Torcetrapib on ApoA-I–Containing HDL Subpopulations
The effects of torcetrapib on concentrations of apoA-I–containing HDL subpopulations are provided in Table 2. Neither concentrations of apoA-I within pre–β-1 HDL nor pre–β-2 HDL were significantly affected by 120 mg torcetrapib QD or BID. In contrast, torcetrapib had striking effects on concentrations of apoA-I within the largest α-migrating subpopulation of HDL: α-1. Relative to placebo, 120 mg torcetrapib QD increased the amount of apoA-I in α-1 in the atorvastatin (136%; P<0.001) and nonatorvastatin (153%; P<0.01) groups, whereas an increase of 382% (P<0.01) was observed in the 120 mg BID group. The latter increase was also significantly different relative to the 120 mg QD phase (64%; P<0.02). In contrast, torcetrapib reduced rather than increased levels of apoA-I in α-3–migrating HDL, with mean reductions of 20% (P<0.01), 14% (P<0.11), and 29% (P<0.01) noted during the 120 mg atorvastatin and nonatorvastatin QD and BID phases, respectively. Concentrations of apoA-I in α-2–migrating HDL were significantly increased by torcetrapib only in the atorvastatin cohort (19%; P<0.01). Finally, relative to placebo, torcetrapib increased concentrations of apoA-I in preα-1–migrating HDL in the atorvastatin cohort (142%; P<0.01), as well as in the 120 mg nonatorvastatin torcetrapib QD (111%; P<0.01) and BID (713%; P<0.05) cohorts. No significant differences were observed in apoA-I–containing subpopulations on placebo or drug when the atorvastatin and nonatorvastatin cohorts were compared.
TABLE 2.
Effects of Torcetrapib on Concentrations of ApoA-I–Containing HDL Subpopulations*
| Parameter | Atorvastatin+Torcetrapib | Torcetrapib
|
|
|---|---|---|---|
| 120 mg QD n=9 | 120 mg QD n=10 | 120 mg BID n=6 | |
| Pre–β-1, mg/dL | |||
| Placebo | 7.1±3.7 | 10.4±4.2 | 8.5±3.4 |
| Torcetrapib | 6.2±5.1 | 9.8±3.8 | 11.2±5.4 |
| % change | −16±48 | 16±70 | 47±89 |
| Pre–β-2, mg/dL | |||
| Placebo | 1.8±0.7 | 2.1±1.3 | 2.0±1.3 |
| Torcetrapib | 1.7±0.9 | 2.1±1.0 | 2.0±1.2 |
| % change | −2±46 | 14±44 | 4±29 |
| α-1, mg/dL | |||
| Placebo | 9.0±3.4 | 9.5±5.5 | 10.0±6.3 |
| Torcetrapib | 20.1±7.5† | 23.1±16.4‡ | 39.8±17.0‡ |
| % change | 136±70 | 153±161 | 382±230 |
| α-2, mg/dL | |||
| Placebo | 35.9±7.2 | 33.6±7.3 | 37.1±7.1 |
| Torcetrapib | 43.2±11.5‡ | 40.7±10.1 | 45.8±9.2 |
| % change | 19±14 | 23±33 | 30±44 |
| α-3, mg/dL | |||
| Placebo | 41.4±5.3 | 42.7±7.9 | 42.5±9.7 |
| Torcetrapib | 33.1±7.4‡ | 37.3±12.8 | 29.9±7.4‡ |
| % change | −20±15 | −14±21 | −29±11 |
| Pre–α-1, mg/dL | |||
| Placebo | 2.6±1.5 | 2.9±2.6 | 3.0±3.1 |
| Torcetrapib | 5.4±3.0‡ | 5.6±4.5‡ | 11.3±8.1¶ |
| % change | 142±100 | 111±89 | 713±114 |
| Pre–α-2, mg/dL | |||
| Placebo | 4.5±1.6 | 4.3±2.8 | 4.5±3.2 |
| Torcetrapib | 6.5±3.2¶ | 5.5±3.3 | 7.4±3.8 |
| % change | 42±34 | 46±80 | 104±117 |
| Pre–α-3, mg/dL | |||
| Placebo | 3.5±1.2 | 3.9±2.1 | 4.1±2.0 |
| Torcetrapib | 3.5±1.5 | 3.0±2.0 | 3.0±1.4 |
| % change | 3±26 | −9±57 | −20±34 |
Data are presented as mean±SD.
P<0.001;
P<0.01;
P<0.05 for comparison with placebo phase.
Effects of Torcetrapib on HDL ApoA-I Kinetic Parameters
HDL apoA-I kinetic parameters at the end of the placebo and drug phases are presented in Table 3. Relative to placebo, 120 mg torcetrapib QD increased HDL apoA-I PS by 8±15% (P=0.16) in the atorvastatin cohort and by 16±7% (P<0.0001) in the nonatorvastatin cohort. When these 2 cohorts were compared, the relatively greater percent increase versus placebo seen in the nonatorvastatin cohort was not significantly different from that of the atorvastatin cohort (P=0.14). The 120 mg BID dose of torcetrapib increased HDL apoA-I PS to the greatest extent (34±8%). Analysis of the HDL apoA-I kinetic data revealed that the increases in HDL apoA-I PS were primarily attributable to reductions in HDL apoA-I FCR. A dose of 120 mg torcetrapib QD reduced HDL apoA-I FCR (pools per day) by 7±13%, from 0.237±0.042 to 0.218±0.030 (P<0.10), in the atorvastatin cohort and by 8±5% in the nonatorvastatin cohort, from 0.252±0.071 to 0.237±0.074 (P<0.001). In the 120 mg torcetrapib BID cohort, HDL apoA-I FCR was reduced by 21%, from a mean of 0.228±0.033 on placebo to a mean of 0.179±0.020 on drug (P<0.01). HDL apoA-I PR was not significantly altered by torcetrapib.
TABLE 3.
Effects of Torcetrapib on HDL ApoA-I Kinetic Parameters*
| Parameter | Atorvastatin+Torcetrapib | Torcetrapib
|
|
|---|---|---|---|
| 120 mg QD n=9 | 120 mg QD n=10 | 120 mg BID n=6 | |
| ApoA-I PS, mg | |||
| Placebo | 3485±681 | 3728±479 | 3593±279 |
| Torcetrapib | 3773±932 | 4331±553† | 4807±197† |
| % change | 8±15 | 16±7 | 34±8 |
| ApoA-I FCR, pools per day | |||
| Placebo | 0.237±0.042 | 0.252±0.071 | 0.228±0.033 |
| Torcetrapib | 0.218±0.030 | 0.237±0.074‡ | 0.179±0.020¶ |
| % change | −7±13 | −8±5 | −21±10 |
| ApoA-I PR, mg ·kg ·d−1 | |||
| Placebo | 9.5±2.4 | 10.7±2.0 | 10.1±0.9 |
| Torcetrapib | 9.4±2.0 | 11.5±2.4 | 10.5±0.9 |
| % change | 2±23 | 8±11 | 5±10 |
Data are presented as mean±SD.
P<0.0001;
P<0.001;
P<0.01 for comparison with placebo phase.
Effects of Torcetrapib on Serum and Fecal Concentrations of Neutral Sterols and Bile Acids
The effects of torcetrapib on serum concentrations of 7α-hydroxy-4-cholesten-3-one (C4), an intermediate in bile acid synthesis, and serum concentrations of lathosterol, an intermediate in cholesterol synthesis, are shown in Table 4. Neither the 120 mg QD nor BID doses of torcetrapib resulted in statistically significant changes in C4 concentrations relative to placebo. In terms of serum lathosterol concentrations, subjects in the atorvastatin cohort had significantly lower levels relative to the nonatorvastatin cohort. However, no significant differences in serum lathosterol levels were observed between the placebo and torcetrapib phases in any of the cohorts.
TABLE 4.
Effects of Torcetrapib on Precursors of Bile Acid and Cholesterol Synthesis and Fecal Bile Acid and Neutral Sterol Content
| Parameter | Atorvastatin+Torcetrapib | Torcetrapib
|
|
|---|---|---|---|
| 120 mg QD n=9 | 120 mg QD n=10 | 120 mg BID n=6 | |
| Serum 7α-hydroxy-4-cholesten-3-one, ng/mL | |||
| Placebo | 23±20 | 38±29 | 29±18 |
| Torcetrapib | 22±13 | 36±27 | 38±22 |
| % change | 16±66 | 17±52 | 35±49 |
| Serum lathosterol, μg/mL | |||
| Placebo | 0.6±0.3† | 2.0±0.8 | 1.7±0.5 |
| Torcetrapib | 0.5±0.2† | 2.4±0.9 | 2.3±1.0 |
| % change | −18±28¶ | 16±20 | 32±43 |
| Fecal bile acids, mg/mg plant sterols | |||
| Placebo | 1.4±0.4 | 2.4±1.5 | 1.9±0.7 |
| Torcetrapib | 1.2±0.4*|| | 2.2±1.2 | 2.2±1.1 |
| % change | −16±20 | 4±50 | 13±46 |
| Fecal neutral sterols, mg/mg plant sterols | |||
| Placebo | 1.9±0.7‡ | 3.3±1.0 | 2.9±0.5 |
| Torcetrapib | 1.7±0.4† | 3.3±0.6 | 2.9±1.0 |
| % change | −8±22 | 5±28 | −1±32 |
P<0.04 for comparison with placebo phase;
P<0.0001;
P<0.01;
P<0.02;
P<0.05 for comparison with 120 mg nonatorvastatin torcetrapib QD cohort.
As shown in Table 4, 120 mg torcetrapib QD reduced fecal bile acid content, expressed in relation to plant sterol content, in the atorvastatin cohort by 16±20%, from 1.4 to 1.2 mg/mg (P<0.04). However, torcetrapib did not significantly alter fecal bile acid concentrations in the nonatorvastatin cohorts, nor did it significantly change fecal sterol content in any of the cohorts.
Discussion
Because of its critical role in HDL metabolism, CETP represents an attractive target for HDL-raising therapies. Two inhibitors of CETP (JTT-70533 and torcetrapib)15,16 have been shown to significantly increase plasma HDL-C concentrations in humans. The main goal of the present study was to assess the effect of CETP inhibition with torcetrapib on HDL apoA-I metabolism in patients with low levels of HDL-C.
Analysis of the HDL apoA-I kinetic data revealed that the increases in apoA-I PS observed in subjects treated with torcetrapib were primarily attributable to reductions in HDL apoA-I clearance rate. Torcetrapib reduced HDL apoA-I FCR by 7% (P=0.10) in the atorvastatin cohort and by 8% (P<0.001) and 21% (P<0.01) in the 120 mg nonatorvastatin QD and BID cohorts, respectively. Torcetrapib did not significantly alter apoA-I PR in any of the cohorts. Thus, torcetrapib modulates plasma HDL apoA-I concentrations primarily via its effects on apoA-I catabolism. This finding is consistent with that of Ikewaki et al,13 who reported that the elevated concentrations of apoA-I observed in patients with CETP deficiency were due solely to delayed apoA-I catabolism.
The major role of CETP is to mediate the transfer of CE from HDL to apoB-containing lipoproteins and, in turn, of TG from apoB-containing lipoproteins to HDL.8,9 Therefore, it is not surprising that torcetrapib significantly influenced the lipid composition of HDL particles. Specifically, torcetrapib increased the CE and PL content of HDL while decreasing HDL TG content. These changes in HDL composition led to significant increases in mean HDL particle diameter in these subjects on torcetrapib.16 It has been demonstrated consistently that HDL particle size can influence the metabolism of apoA-I.34–37 In fact, one study has reported that as much as 70% of the variability in apoA-I FCR is attributable to variability in estimates of HDL size or density.37 Thus, the differences in apoA-I FCR observed in our study were likely attributable to torcetrapib-induced changes in HDL composition and, ultimately, HDL particle size.
We hypothesize that the relationship between increased particle size and decreased apoA-I FCR is not one of cause and effect, but rather, that the alterations in catabolism may be secondary to changes in apoA-I conformation. In vivo and in vitro work support this concept. Horowitz et al38 clearly demonstrated that HDL composition and size can affect the metabolic fate of apoA-I. Their data suggest that increased renal clearance of apoA-I occurs when HDL are cholesterol depleted and relatively TG enriched because of the dissociation of apoA-I from HDL. The in vitro experiments of Liang et al39 have further shown that the dissociation of apoA-I from HDL is a concentration-dependent phenomenon, such that increasing the level of HDL decreases the reduction in HDL particle size and the dissociation of apoA-I.
Torcetrapib also significantly influenced the distribution of apoA-I among HDL subpopulations. The most dramatic changes were observed in concentrations of the largest apoA-I–containing subpopulation of HDL: α-1. Relative to placebo, 120 mg torcetrapib QD increased levels of apoA-I in the α-1 subpopulation of HDL from 9.0 to 20.1 mg/dL in the atorvastatin cohort and from 9.5 to 23.1 mg/dL in the nonatorvastatin cohort, whereas the twice per day dose increased α-1 from 10.0 to 39.8 mg/dL. In comparison, we reported previously a mean apoA-I concentration in α-1 of 19.6 mg/dL for 79 healthy, normolipidemic subjects having a mean age of 53±13 years.40 Thus, in our subjects with low HDL-C, the once-daily dose of torcetrapib normalized the amount of apoA-I in α-1. We believe that this is a key observation in light of the fact that we have shown previously that levels of α-1 are significantly reduced in patients with CHD.40 Moreover, we reported that increased concentrations of α-1 observed during simvastatin–niacin combination therapy were inversely associated with progression of coronary stenosis in patients from the HDL-Atherosclerosis Treatment Study.41 Under normal metabolic conditions, the α-1 sub-population of HDL does not contain apoA-II.42 With this in mind, it is important to note that torcetrapib did not alter the distribution of apoA-II among α-migrating HDL subspecies (data not shown). In contrast, we reported recently that apoA-II was localized to the α-1 HDL subpopulation in patients with heterozygous and homozygous CETP deficiency.43
A crucial question is whether CETP inhibition influences the rate of reverse cholesterol transport (RCT). Schwartz et al44 reported recently that in normolipidemic humans, most HDL CE excreted into the bile gets there via apoB-containing lipoproteins and, therefore, presumably via CETP-mediated transfer. Hence, there is theoretical concern that CETP inhibition may impair RCT and, ultimately, be proatherogenic. Preliminary studies with CP-456,643, a close analog of torcetrapib, have shown that the removal of HDL CE from the plasma of rabbits is not reduced,45 suggesting that the RCT pathway is not compromised by CETP inhibition. Additional support for the latter concept comes from the work of Morehouse et al,46 which showed that the percentage of aortic surface covered with lesions was 60% lower in rabbits treated with torcetrapib relative to controls. Statistical analysis revealed that the reduction of aortic atherosclerosis in the torcetrapib-treated rabbits was significantly associated with the increase in HDL-C concentrations. Although the results of these studies are promising, whether or not the marked increases in HDL-C seen in humans treated with torcetrapib will translate into reduced cardiovascular disease risk remains to be determined.
In the present study, RCT was assessed indirectly, using fecal concentrations of neutral sterols and bile acids as surrogate measures. Torcetrapib did not substantially alter concentrations of either fecal sterols or bile acids, indicating that it did not influence fecal sterol excretion in these subjects. In the atorvastatin cohort, it should be noted that the dominant effect observed on these parameters was that associated with statin treatment because patients on atorvastatin had significantly lower serum concentrations of lathosterol (reduced cholesterol synthesis) and, in turn, decreased fecal sterol content, on placebo than did subjects in the nonatorvastatin cohorts. Although torcetrapib did not increase fecal sterol excretion, as did high doses of recombinant apoA-I in the study of Eriksson et al,47 increased flux of cholesterol from the periphery to the liver may not increase fecal sterol secretion or plasma lathosterol levels if it leads to a decrease in hepatic cholesterol synthesis and a proportional increase in cholesterol synthesis in the peripheral tissues. Moreover, important methodological differences prevent a direct comparison of these 2 studies. Included among these differences are: (1) modes of therapeutic delivery (oral versus intravenous infusion); (2) dosages of therapeutic agents (small daily doses of torcetrapib versus large doses of apoA-I, in effect equivalent to a dramatic increase in apoA-I synthesis); and (3) the duration of stool collection (3 days after 4 weeks of therapy versus 9 days before and 9 days after therapy).
To summarize, our data indicate that the CETP inhibitor torcetrapib increases plasma concentrations of HDL apoA-I by delaying apoA-I catabolism. The delayed apoA-I catabolism is likely attributable to CE enrichment of HDL which, in turn, leads to increased HDL particle size. Torcetrapib markedly increased concentrations of α1-migrating HDL, which we have consistently shown to be inversely associated with atherosclerotic risk.40,41 When given either as monotherapy or in combination with atorvastatin, torcetrapib did not significantly alter fecal sterol excretion or bile acid synthesis. In conclusion, CETP inhibition has multiple effects on the dynamic aspects of HDL metabolism that may be relevant to its ultimate effects on atherosclerotic cardiovascular disease in humans.
Acknowledgments
This work was supported in part by the Department of Clinical Research, Medicinal Products Research and Development, Pfizer, Inc., Groton, Conn. Support was also provided by the General Clinical Research Center of New England Medical Center, which is funded by the National Center for Research Resources of the National Institutes of Health (NIH; M01-RR00054), and the General Clinical Research Center of the University of Pennsylvania (M01-RR00040). M.E.B. and E.J.S. were supported in part by NIH R01-HL60935 from the National Heart, Lung, and Blood Institute, and B.A. and M.R. by the Swedish Research Council (03X-7137). The authors wish to thank the nursing and dietary staff of each clinical research center, as well as Rodrigo Ferreira, Aisha Faruqi, Jennifer Dykhouse, Rose Giordano, Katalin Horvath, Judith R. McNamara, Anna Lillethun, Linda Morrell, Anita Löfgren, and Ingela Arvidsson for technical assistance. We are also grateful to Drs Gregory G. Dolnikowski and P. Hugh Barrett for their expert advice.
References
- 1.Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 2.Gordon DJ, Rifkind BM. High-density lipoprotein—the clinical implications of recent studies. N Engl J Med. 1989;321:1311–1316. doi: 10.1056/NEJM198911093211907. [DOI] [PubMed] [Google Scholar]
- 3.Miller NE. Associations of high-density lipoprotein subclasses and apolipoproteins with ischemic heart disease and coronary atherosclerosis. Am Heart J. 1987;113:589–597. doi: 10.1016/0002-8703(87)90638-7. [DOI] [PubMed] [Google Scholar]
- 4.Manninen V, Elo MO, Frick MH, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P. Lipid alterations and decline in the incidence of coronary heart disease in the Helsinki Heart Study. J Am Med Assoc. 1988;260:641–651. [PubMed] [Google Scholar]
- 5.Robins SJ, Collins D, Wittes JT, Papademetriou V, Deedwania PC, Schaefer EJ, McNamara JR, Kashyap ML, Hershman JM, Wexler LF, Rubins HB. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. J Am Med Assoc. 2001;285:1585–1591. doi: 10.1001/jama.285.12.1585. [DOI] [PubMed] [Google Scholar]
- 6.Asztalos BF, Horvath KV, McNamara JR, Roheim PS, Rubinstein JJ, Schaefer EJ. Comparing the effects of five different statins on the HDL subpopulation profiles of coronary heart disease patients. Atherosclerosis. 2002;164:361–369. doi: 10.1016/s0021-9150(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 7.Barter PJ, Brewer HB, Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 8.Hesler CB, Swenson TL, Tall AR. Purification and characterization of a human plasma cholesteryl ester transfer protein. J Biol Chem. 1987;262:2275–2282. [PubMed] [Google Scholar]
- 9.Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34:1255–1274. [PubMed] [Google Scholar]
- 10.Brown ML, Inazu A, Hesler CB, Agellon LB, Mann C, Whitlock ME, Marcel YL, Milne RW, Koizumi J, Mabuchi H, Takeda R, Tall AR. 1989. Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature. 1989;342:448–451. doi: 10.1038/342448a0. [DOI] [PubMed] [Google Scholar]
- 11.Koizumi J, Mabuchi H, Yoshimura A, Michishita I, Takeda M, Itoh H, Sakai Y, Sakai T, Ueda K, Takeda R. Deficiency of serum cholesterylester transfer activity in patients with familial hyperalphalipoproteinemia. Atherosclerosis. 1985;58:175–186. doi: 10.1016/0021-9150(85)90064-4. [DOI] [PubMed] [Google Scholar]
- 12.Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, Maruhama Y, Mabuchi H, Tall AR. Increased high-density lipoprotein levels caused by a common cholesterylester transfer protein gene mutation. N Engl J Med. 1990;323:1234–1238. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- 13.Ikewaki K, Rader DJ, Sakamoto T, Nishiwaki M, Wakimoto N, Schaefer JR, Ishikawa T, Fairwell T, Zech LA, Nakamura H, Nagano M, Brewer HB., Jr Delayed catabolism of high density lipoprotein apolipoproteins A-I and A-II in human cholesteryl ester transfer protein deficiency. J Clin Invest. 1993;92:1650–1658. doi: 10.1172/JCI116750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. J Am Med Assoc. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 15.Clark RW, Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, Cosgrove PG, Sand TM, Wester RT, Williams JA, Perlman ME, Bamberger MJ. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib. Arterioscler Thromb Vasc Biol. 2004;24:490–497. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- 16.Brousseau ME, Schaefer EJ, Wolfe ML, Bloedon LT, Digenio AG, Clark RW, Mancuso JP, Rader DJ. Effects of a potent inhibitor of cholesteryl ester transfer protein on plasma lipoproteins in patients with low HDL cholesterol. N Engl J Med. 2004;350:1505–1515. doi: 10.1056/NEJMoa031766. [DOI] [PubMed] [Google Scholar]
- 17.Lichtenstein AH, Hachey DL, Millar JS, Jenner JL, Booth L, Ordovas JM, Schaefer EJ. Measurement of human apolipoprotein B-48 and B-100 kinetics in triglyceride-rich lipoproteins using [5,5,5–2H3]leucine. J Lipid Res. 1992;33:907–914. [PubMed] [Google Scholar]
- 18.Warnick GR, Benderson J, Albers JJ. Dextran sulfate-Mg2+ precipitation procedure for quantitation of high-density lipoprotein cholesterol. Clin Chem. 1982;28:1379–1388. [PubMed] [Google Scholar]
- 19.McNamara JR, Schaefer EJ. Automated enzymatic standardized lipid analyses for plasma and lipoprotein fractions. Clin Chim Acta. 1987;166:1–8. doi: 10.1016/0009-8981(87)90188-4. [DOI] [PubMed] [Google Scholar]
- 20.Asztalos BF, Sloop CH, Wong L, Roheim PS. Two-dimensional electrophoresis of plasma lipoproteins: recognition of new apoA-I-containing subpopulations. Biochim Biophys Acta. 1993;1169:291–300. doi: 10.1016/0005-2760(93)90253-6. [DOI] [PubMed] [Google Scholar]
- 21.Cohn JS, Wagner DA, Cohn SD, Millar JS, Schaefer EJ. Measurement of very low density and low density lipoprotein apolipoprotein (Apo) B-100 and high density lipoprotein Apo A-I production in human subjects using deuterated leucine: effect of fasting and feeding. J Clin Invest. 1990;85:804–811. doi: 10.1172/JCI114507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welty FK, Lichtenstein AH, Barrett PH, Dolnikowski GG, Ordovas JM, Schaefer EJ. Production of apolipoprotein B-67 in apolipoprotein B-67/B-100 heterozygotes: technical problems associated with leucine contamination in stable isotope studies. J Lipid Res. 1997;38:1535–1543. [PubMed] [Google Scholar]
- 23.Cobelli C, Toffolo G, Bier DM, Nosadini R. Models to interpret kinetic data in stable isotope tracer studies. Am J Physiol. 1987;253:E551–E564. doi: 10.1152/ajpendo.1987.253.5.E551. [DOI] [PubMed] [Google Scholar]
- 24.Batista MC, Welty FK, Diffenderfer MR, Sarnak MJ, Schaefer EJ, Lamon-Fava S, Asztalos BF, Dolnikowski GG, Brousseau ME, Marsh JB. Apolipoprotein A-I, B-100, and B-48 metabolism in subjects with chronic kidney disease, obesity, and the metabolic syndrome. Metabolism. 2004;53:1255–1261. doi: 10.1016/j.metabol.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Welty FK, Lichtenstein AH, Barrett PHR, Dolnikowski GG, Schaefer EJ. Human apolipoprotein (apo) B-48 and apoB-100 kinetics with stable isotopes. Arterioscler Thromb Vasc Biol. 1999;19:2966–2974. doi: 10.1161/01.atv.19.12.2966. [DOI] [PubMed] [Google Scholar]
- 26.Lund E, Sisfontes L, Reihner E, Bjorkhem I. Determination of serum levels of unesterified lathosterol by isotope dilution-mass spectrometry. Scand J Clin Lab Invest. 1989;49:165–171. doi: 10.3109/00365518909105417. [DOI] [PubMed] [Google Scholar]
- 27.Reihner E, Rudling M, Stahlberg D, Berglund L, Ewerth S, Bjorkhem I, Einarsson K, Angelin B. Influence of pravastatin, a specific inhibitor of HMG-CoA reductase, on hepatic metabolism of cholesterol. N Engl J Med. 1990;323:224–228. doi: 10.1056/NEJM199007263230403. [DOI] [PubMed] [Google Scholar]
- 28.Galman C, Arvidsson I, Angelin B, Rudling M. Monitoring hepatic cholesterol 7α-hydroxylase activity by assay of the stable bile acid intermediate 7α-hydroxy-4-cholesten-3-one in peripheral blood. J Lipid Res. 2003;44:859–865. doi: 10.1194/jlr.D200043-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Miettinen TA, Ahrens EH, Jr, Grundy SM. Quantitative isolation and gas-liquid chromatographic analysis of total dietary and fecal neutral steroids. J Lipid Res. 1965;6:411–424. [PubMed] [Google Scholar]
- 30.Grundy SM, Ahrens EH, Jr, Miettinen TA. 1965. Quantitative isolation and gas-liquid chromatographic analysis of total dietary and fecal bile acids. J Lipid Res. 1965;6:397–410. [PubMed] [Google Scholar]
- 31.Miettinen TA. Gas-liquid chromatographic determination of fecal neutral steroids using a capillary column. Clin Chim Acta. 1982;124:245–248. doi: 10.1016/0009-8981(82)90393-x. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro SS, Wilk MB. An analysis of variance test for normality (complete samples) Biometrika. 1965;52:591–611. [Google Scholar]
- 33.de Grooth GJ, Kuivenhoven JA, Stalenhoef AFH, de Graaf J, Zwinderman AH, Posma JL, van Tol A, Kastelein JJ. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response study. Circulation. 2002;105:2159–2165. doi: 10.1161/01.cir.0000015857.31889.7b. [DOI] [PubMed] [Google Scholar]
- 34.Schaefer EJ, Zech LA, Jenkins LL, Bronzert TJ, Rubalcaba EA, Lindgren RT, Aamodt RL, Brewer HB., Jr Human apolipoprotein A-I and A-II metabolism. J Lipid Res. 1982;23:850–862. [PubMed] [Google Scholar]
- 35.Brinton EA, Eisenberg S, Breslow JL. Elevated high-density lipoprotein cholesterol levels correlate with decreased apolipoprotein A-I and A-II fractional catabolic rate in women. J Clin Invest. 1989;84:262–269. doi: 10.1172/JCI114149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brinton EA, Eisenberg S, Breslow JL. Human HDL cholesterol levels are determined by apoA-I fractional catabolic rate, which correlates inversely with estimates of HDL particle size: effects of gender, hepatic and lipoprotein lipases, triglyceride and insulin levels, and body fat distribution. Arterioscler Thromb. 1994;14:707–720. doi: 10.1161/01.atv.14.5.707. [DOI] [PubMed] [Google Scholar]
- 37.Melchior GW, Castle CK, Vidmar TJ, Polites HG, Marotti KR. ApoA-I metabolism in cynomolgus monkeys: male-female differences. Biochim Biophys Acta. 1990;1043:97–105. doi: 10.1016/0005-2760(90)90115-e. [DOI] [PubMed] [Google Scholar]
- 38.Horowitz BS, Goldberg IJ, Merab J, Vanni T, Ramakrishnan R, Ginsberg HN. Increased plasma and renal clearance of an exchangeable pool of apolipoprotein A-I in subjects with low levels of high density lipoprotein cholesterol. J Clin Invest. 1991;91:1743–1752. doi: 10.1172/JCI116384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang H-Q, Rye K-A, Barter PJ. Dissociation of lipid-free apolipoprotein A-I from high density lipoproteins. J Lipid Res. 1994;35:1187–1199. [PubMed] [Google Scholar]
- 40.Asztalos BF, Roheim PS, Milani RL, Lefevre M, McNamara JR, Horvath KV, Schaefer EJ. Distribution of ApoA-I-containing HDL subpopulations in patients with coronary heart disease. Arterioscler Thromb Vasc Biol. 2000;20:2670–2676. doi: 10.1161/01.atv.20.12.2670. [DOI] [PubMed] [Google Scholar]
- 41.Asztalos BF, Batista M, Horvath KV, Cox CE, Dallal GE, Morse JS, Brown GB, Schaefer EJ. Change in alpha-1 HDL concentration predicts progression in coronary artery stenosis. Arterioscler Thromb Vasc Biol. 2003;23:847–852. doi: 10.1161/01.ATV.0000066133.32063.BB. [DOI] [PubMed] [Google Scholar]
- 42.Asztalos BF, Lefevre M, Foster TA, Tulley R, Windhauser M, Wong L, Roheim PS. Normolipidemic subjects with low HDL cholesterol levels have altered HDL subpopulations. Arterioscler Thromb Vasc Biol. 1997;17:1885–1893. doi: 10.1161/01.atv.17.10.1885. [DOI] [PubMed] [Google Scholar]
- 43.Asztalos BF, Horvath KV, Kojinami K, Nartsupha C, Cox CE, Batista M, Schaefer EJ, Inazu A, Mabuchi H. Apolipoprotein composition of HDL in cholesteryl ester transfer protein deficiency. J Lipid Res. 2004;45:448–455. doi: 10.1194/jlr.M300198-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer and output in vivo in humans. J Lipid Res. 2004;45:1594–1607. doi: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Kee P, Rye K-A, Morehouse L, Barrett PHR, Barter PJ. The effects of cholesteryl ester transfer protein (CETP) inhibition on the metabolism of cholesteryl esters in high density lipoproteins: insights into reverse cholesterol transport. 52nd Annual Scientific Meeting of the Cardiac Society of Australia and New Zealand; 8–11 August 2004; Brisbane, Queensland, Australia. [Google Scholar]
- 46.Morehouse LA, Sugarman ED, Bourassa P-A, Milici AJ. HDL elevation by the CETP-inhibitor torcetrapib prevents aortic atherosclerosis in rabbits. The Am Heart Association Scientific Sessions; 7–10 November 2004; New Orleans, La, United States. 2004. p. Abstract 1168. [Google Scholar]
- 47.Eriksson M, Carlson LA, Miettinen TA, Angelin B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I: potential reverse cholesterol transport in humans. Circulation. 1999;100:594–598. doi: 10.1161/01.cir.100.6.594. [DOI] [PubMed] [Google Scholar]