

Abstract
Background
Dimerization is an important regulatory mechanism of single membrane-spanning receptors. For instance, activation of receptor protein-tyrosine kinases (RPTKs) involves dimerization. Structural, functional and biochemical studies suggested that the enzymatic counterparts of RPTKs, the receptor protein-tyrosine phosphatases (RPTPs), are inhibited by dimerization, but whether RPTPs actually dimerize in living cells remained to be determined.
Results
In order to assess RPTP dimerization, we have assayed Fluorescence Resonance Energy Transfer (FRET) between chimeric proteins of cyan- and yellow-emitting derivatives of green fluorescent protein, fused to RPTPα, using three different techniques: dual wavelength excitation, spectral imaging and fluorescence lifetime imaging. All three techniques suggested that FRET occurred between RPTPα -CFP and -YFP fusion proteins, and thus that RPTPα dimerized in living cells. RPTPα dimerization was constitutive, extensive and specific. RPTPα dimerization was consistent with cross-linking experiments, using a non-cell-permeable chemical cross-linker. Using a panel of deletion mutants, we found that the transmembrane domain was required and sufficient for dimerization.
Conclusions
We demonstrate here that RPTPα dimerized constitutively in living cells, which may be mediated by the transmembrane domain, providing strong support for the model that dimerization is involved in regulation of RPTPs.
Introduction
Protein phosphorylation on tyrosine residues is one of the most important eukaryotic cell signalling mechanisms, and cellular protein phosphotyrosine (pTyr) levels are regulated by the antagonistic activities of the protein-tyrosine kinases (PTKs) and protein-tyrosine phosphatases (PTPs) [1]. Our insight into the function of PTPs - in contrast to the PTKs - is limited. However, in recent years evidence is emerging that PTPs play important and specific roles in biological processes [2,3,4]. Transmembrane PTPs, tentatively called receptor PTPs, RPTPs, are interesting, because their diverse extracellular domains may function as ligand binding domains. Recently, Pleiotrophin was identified as a ligand of RPTPβ/ζ, and binding of Pleiotrophin led to inactivation of RPTPβ/ζ activity, resulting in increased tyrosine phosphorylation of its substrate, β-Catenin [5]. The mechanism underlying ligand-induced modulation of RPTP activity remains to be determined.
Dimerization is a well-established regulatory mechanism for single membrane-spanning receptors [6,7], which may be involved in regulation of RPTPs. The crystal structure of RPTPα-D1, which is highly homologous to other PTPs [8,9,10,11,12,13,14], showed that an amino-terminal helix-turn-helix wedge-like segment interacts with the dyad-related monomer, in such a manner that both catalytic sites in the dimer are occluded [9]. It should be noted that the crystal structures of RPTPμ-D1 and of the complete cytoplasmic domain of LAR do not show dimers like RPTPα [11,12]. Functional experiments with EGFR/CD45, a chimeric protein, consisting of the extracellular domain of the Epidermal Growth Factor Receptor (EGFR) and the intracellular domain of the RPTP, CD45, demonstrated that ligand-induced dimerization leads to functional inactivation of CD45 [15]. Mutation of a single residue in the wedge of CD45 abolishes dimerization-induced functional inactivation [16]. The importance of tight regulation of CD45 was demonstrated by the introduction of a targeted point mutation in the wedge in mice. The CD45 E613R mutation causes lymphoproliferation and autoimmunity in mice, resulting in death [17]. RPTPα dephosphorylates and activates the PTK c-Src [18,19,20,21] via a phosphotyrosine displacement mechanism, involving Src pTyr529 and RPTPα pTyr789 [22]. Recently, we demonstrated that forced dimerization of RPTPα by introduction of a disulfide bridge in the extracellular domain leads to inactivation of RPTPα as assessed by analysis of c-Src Tyr529 phosphorylation and activity. Similar to CD45 [16], dimerization-induced inactivation of RPTPα was dependent on an intact wedge [23]. Therefore, it is clear that RPTPs can be regulated by dimerization. Experiments with chemical cross-linkers indicated that RPTPα forms dimers [24]. The use of chemical cross-linkers to analyze protein-protein interactions has many disadvantages due to the fact that detection of the interaction is indirect. Here we report the analysis of RPTPα dimerization in living cells using a non-invasive, non-destructive method, Fluorescence Resonance Energy Transfer (FRET).
FRET is a method to assess distances at the molecular level. FRET is a direct radiation-less transfer of energy from the excited donor fluorophore to the acceptor fluorophore, which occurs if the two fluorophores are in very near vicinity of each other, usually less than 60Å [25,26,27]. Recently, FRET has been applied to analyse interactions between proteins, fused to derivatives of Green Fluorescent Protein (GFP) [28,29]. For instance, GFP-FRET has been used to assay changes in cytosolic Ca2+-concentrations using "cameleons" [30,31], to probe interactions between Bcl-2 and Bax in mitochondria [32], to visualize nuclear interactions of Pit-1 with other transcription factors [33], to analyze interactions between nuclear import factors [34], to assess dimerization of G protein coupled receptors [35], and to follow the interaction between GRB2 and the Epidermal Growth Factor Receptor (EGFR) in vivo [36].
We have assayed FRET between fusion proteins of RPTPα, fused to CFP and YFP, by dual wavelength excitation, spectral imaging microscopy (SPIM) and fluorescence lifetime imaging microscopy (FLIM), and found that FRET occurred, using all three methods, suggesting that RPTPα dimerized constitutively. The FRET results were consistent with experiments using chemical cross-linkers. Finally, using deletion mutants, we mapped a site of interaction to the transmembrane domain, and demonstrate that the transmembrane domain is sufficient to drive dimerization. Our results provide strong support for the model that RPTPs are regulated by dimerization, and may be useful for analysis of factors that modulate RPTPalpha dimerization.
Results
FRET between RPTPalpha-GFP fusion proteins
In order to determine whether RPTPα formed dimers in living cells, we generated RPTPα-fusion proteins by substitution of RPTPα-D2 with CFP or YFP, a good donor-acceptor fluorophore pair for FRET [30,31] (Fig. 1A). RPTPα-516-CFP and RPTPα-516-YFP (individually, or in combination) were transiently transfected into SK-N-MC neuroepithelioma cells. RPTPα-516-YFP localized to the cell membrane as assessed by confocal microscopy (Fig. 1B). Similarly, RPTPα-516-CFP localized to the cell membrane (data not shown). The excitation spectra were determined in single living cells, using two filter sets, confirming that the excitation maxima are 452 and 514 nm for CFP and YFP, respectively [30,31], and that there was little overlap between the two excitation spectra (Fig. 1C). The excitation spectrum of a cell that coexpressed RPTPα-516-CFP and RPTPα-516-YFP suggested that FRET occurred, since a strong signal was detected at the YFP emission wavelength (curve 4, Fig. 1C) upon excitation below 440 nm (predominantly exciting CFP), whereas this signal was nearly absent in single RPTPα-516-CFP or RPTPα-516-YFP transfected cells (compare with curves 2 and 3). FRET between RPTPα-516-CFP and RPTPα-516-YFP suggested that RPTPα dimerized under these conditions.
Figure 1.

Expression and spectral properties of chimeric fluorescent RPTPα proteins. (A) Schematic representation of wild type RPTPα (RPTPα and RPTPα-516-XFP, a construct in which either CFP or YFP (indicated by XFP) was inserted at position 516, thereby replacing the membrane distal PTP domain, D2. The extracellular domain (ED), transmembrane domain (black box), membrane-proximal domain (D1) and membrane-distal domain (D2) are indicated. (B) Confocal micrograph of a single SK-N-MC neuroepithelioma cell transfected with an expression vector for RPTPα-516-YFP, illustrating membrane localization of RPTPα-516-YFP. (C) SK-N-MC cells were transfected with RPTPα-516-CFP (traces 1 and 2), RPTPα-516-YFP (trace 3), or both (trace 4). Excitation spectra of single, live cells were corrected for background and are depicted using filter #1 (longpass 490 nm)(trace 1), allowing detection of CFP, and using filter #2 (bandpass 530-560 nm)(traces 2 - 4), optimal for detection of YFP. Filter #2 is the FRET filter, allowing detection of FRET when CFP and YFP are in a protein complex.
Routinely, we determined fluorescence intensities in single living cells following excitation at 440 nm and 490 nm, using the FRET filter. Initially, we used RPTPα-516-CFP/-YFP, human EGFR-CFP/-YFP as a negative control, and constitutively dimeric RPTPα-P137C-CFP/-YFP as a positive control (Fig. 2A). The F(440)/F(490) ratio consists of three terms: direct YFP emission, direct CFP emission, and sensitized emission (directly proportional to FRET efficiency (E) and dimerization (α), two terms that cannot be separated from each other (see Materials and Methods). We have calculated the theoretical values of F(440)/F(490) under conditions of no dimerization (E*α = 0), and substantial dimerization (E*α = 0.5), and plotted these values against the ratio of the CFP- and YFP-concentrations R(CFP/YFP) (Fig. 2B). It is clear from Fig. 2B that F(440)/F(490) is substantially higher under conditions of dimerization and FRET (E*α = 0.5) than under conditions of no FRET and/or dimerization (E*α = 0). In addition, it is clear that changes in the CFP:YFP expression ratio affect F(440)/F(490). We have indicated the theoretical F(440)/F(490) levels under conditions of no dimerization and/or FRET with R(CFP/YFP) = 0.5 (F(440)/F(490) = 0.25) and R(CFP/YFP) = 1.5 (F(440)/F(490) = 0.37) (Fig. 2B, bottom and top dashed lines, respectively). We have analysed protein expression of the CFP- and YFP-fusion proteins by immunoblotting and found roughly similar expression levels for all the fusion proteins that we used, which was expected since the only difference between these proteins is a limited number of point mutations (six) that affect the fluorophore. The ratio of concentrations of CFP and YFP was close to 1.0 for all fusion protein pairs, and certainly 0.5<R(CFP/YFP) <1.5. Hence, if the experimental F(440)/F(490) for any CFP/YFP fusion protein pair is 0.25 or less, there is no dimerization and/or FRET. If F(440)/F(490) > 0.37, our simulations (Fig. 2B) indicate that FRET and hence a physical interaction (dimerization) occurs. If 0.25 <F(440)/F(490) < 0.37, one cannot distinguish whether the increased value is caused by a high CFP:YFP expression ratio in the cells or by dimerization-induced CFP to YFP FRET with this method. We postulate that under our experimental conditions with similar amounts of CFP and YFP fusion proteins expressed in the total cell population, an average F(440)/F(490) ratio value >0.37 for a large group of individual cell measurements is the result of FRET and dimerization.
Figure 2.

Specific and constitutive dimerization of RPTPα, as assessed by FRET. (A) Schematic representation of the constructs that were used. RPTPα-D2 was replaced by CFP or YFP in RPTPα-516-XFP. Pro137 was mutated to Cys in RPTPα-P137C-XFP and RPTPα-P137C, leading to the formation of a disulfide bridge, and thus to constitutive dimerization. In EGFR-XFP, the C-terminus of the EGFR was replaced by CFP or YFP. (B) Theoretical values of F(440)/F(490), calculated as described in the Experimental Procedures section for no FRET and/or dimerization (bottom trace), or significant dimerization (E*α = 0.5, upper trace). The dashed lines indicate the F(440)/F(490) levels under conditions of no FRET and/or dimerization with R(CFP/YFP) = 0.5 (lower dashed line), or R(CFP/YFP) = 1.5 (upper dashed line). F(440)/F(490) levels close to the lower dashed line indicate that no FRET and/or dimerization occurs. F(440)/F(490) values above the upper dashed line are most likely due to FRET. F(440)/F(490) values between the two dashed lines are ambiguous, since F(440)/F(490) values may indicate high CFP:YFP expression ratios and no FRET and/or dimerization, or low CFP:YFP expression ratios and little FRET and dimerization. (C) SK-N-MC cells were transiently transfected with combinations of CFP- and YFP-fusion proteins, fused to RPTPα-516, to the EGFR, or to constitutively dimeric RPTPα-P137C. The F(440)/F(490) ratios were determined in at least 10 single living cells from at least two independent experiments, and the error bars indicate the standard deviation. The dashed lines indicate the theoretically minimal F(440)/F(490) ratio (bottom dashed line), and the FRET threshold level (upper dashed line). (D) Expression of the fusion proteins in (C) was monitored by immunoblotting to verify that similar levels of fusion proteins were expressed, using an anti-GFP antibody (left two panels), or an anti-HA-tag antibody (right panel). (E) Wild type RPTPα and RPTPα-P137C were transfected into SK-N-MC cells. Lysates of these cells were run on 5% SDS-PAGE gels under reducing (+β-mercaptoethanol, β-ME) or non-reducing conditions (-β-ME). Immunoblots, probed with anti-RPTPα antibodies are depicted with the molecular weight of marker proteins that were co-electrophoresed with the samples on the left (in kDa). The position of monomeric (M) and dimeric (D) RPTPα is depicted on the right.
The F(440)/F(490) ratio of coexpressed RPTPα-516-CFP and -YFP was 0.46±0.07 (Fig. 2C), while the expression levels were similar (Fig. 2D), suggesting constitutive dimerization and FRET. As a control, we determined the F(440)/F(490) ratio of EGFR-CFP and -YFP, which was 0.20±0.05, suggesting there was no dimerization and/or FRET (Fig. 2C, D). To induce constitutive dimerization, we substituted Pro137 in the extracellular domain of RPTPα with cysteine. RPTPα-P137C formed a disulfide bridge, thus inducing constitutive homodimerization, and more than 80% of RPTPα-P137C was detected in the dimeric form by SDS-PAGE under non-reducing conditions [23] (Fig. 2E). The F(440)/F(490) ratio of constitutively dimeric RPTPα- P137C-CFP/-YFP was 0.50±0.07 (Fig. 2C) with similar expression of the -CFP and - YFP fusion proteins (Fig. 2D). Since the F(440)/F(490) ratios of RPTPα-516-CFP/- YFP and RPTPα-P137C-CFP/-YFP were comparable and well above the FRET/dimerization threshold of 0.37, we conclude that dimerization of wild type RPTPα was constitutive and extensive.
RPTPalpha dimerization assessed by SPIM and FLIM
In order to validate the excitation-ratio FRET data and exclude artefacts due to aberrant CFP:YFP expression ratios, we performed additional FRET-experiments for the full length RPTPα-CFP and -YFP fusion proteins using other methods and equipment. With SPIM, fluorescence emission spectra can be determined from living cells with spatial resolution along one dimension [37,38]. Using this technique, several spectra were obtained from plasma membranes of single living SK-N-MC neuroepithelioma cells that were cotransfected with RPTPα-CFP and either RPTPα- YFP or EGFR-YFP (control). From Fig. 3A it can be inferred that the RPTPα-CFP + RPTPα-YFP coexpressing cells displayed a clearly increased relative fluorescence at wavelengths between 500 and 600 nm, with a maximum increase at 527 nm upon excitation at 435 nm. The increased relative fluorescence closely matched a YFP emission spectrum, which was almost absent in control cells where similar amounts of RPTPα-CFP and EGFR-YFP were expressed. These data suggest that the increased YFP fluorescence observed in RPTPα-CFP and RPTPα-YFP double transfected cells is due to sensitized YFP emission resulting from CFP to YFP FRET.
Figure 3.

SPIM and FLIM confirm FRET in full length RPTPα-CFP and -YFP expressing cells. (A) Normalized uncorrected fluorescence emission spectra of plasma membrane regions of single SK-N-MC cells expressing both RPTPα-CFP and RPTPα-YFP (closed circles) or SK-N-MC cells expressing both RPTPα-CFP and EGFR-YFP (control, open circles) as obtained by fluorescence spectral imaging microscopy (SPIM). The emission spectra were normalized to the first emission peak of the CFP spectrum (at 475 nm) for comparative reasons (note that due to this normalization procedure no 'quenching' of CFP fluorescence due to FRET seems apparent from the figure). (B) CFP-fluorescence lifetime images (τφ, calculated from the phase shift) of cells expressing both RPTPα-CFP and RPTPα-YFP (left) or RPTPα-CFP and EGFR-YFP (control, right). The histogram represents the distribution of fluorescence lifetimes throughout the image. (C) Correlation between FLIM and SPIM data of cells expressing both RPTPα-CFP and RPTPα-YFP (closed symbols) or RPTPα-CFP and EGFR-YFP (control, open symbols). Each point represents a combined SPIM and FLIM measurement of a single cell. On the horizontal axis the normalized intensity of the emission spectrum at 527 nm is indicated (which is proportional to the YFP/CFP fluorescence emission ratio). Lifetimes determined from phase (τφ) and modulation (τM) are plotted with circles and squares, respectively. Regression analysis indicated a -85% and -88% correlation coefficient for the respective lines. Error bars represent pixel-by-pixel standard deviations in fluorescence lifetimes or fluorescence emission spectra.
To exclude the possibility that the increased YFP emission was caused by a high YFP to CFP expression ratio, we performed FLIM experiments on the very same cells by employing the other detector port of the SPIM-FLIM imaging microscope. FRET causes a decrease in the donor (i.e. CFP) fluorescence lifetime [25,38]. As shown in Fig. 3B, the CFP-fluorescence lifetimes (τφ) were lower in case of the RPTPα-CFP and RPTPα-YFP coexpressing cells than in control cells, coexpressing RPTPα-CFP and EGFR-YFP. The decrease in the CFP-fluorescence lifetime from 2.6 ns to 2.2 ns clearly demonstrated CFP to YFP FRET due to close proximity of the two fluorophores, which cannot be explained by variation in CFP:YFP expression ratios [38]. The control CFP-lifetime of 2.6 ns was identical to that of cells expressing cytosolic (unfused) CFP which has a τφ lifetime of 2.6 ns at the employed modulation frequency (data not shown, see also [38]). Because the reduction in CFP lifetime was rather low, we correlated the lifetime data with the SPIM data (see Fig. 3C). A clear negative correlation was observed between the emission ratios and the CFP fluorescence lifetimes (both τφ and τM). These data confirm the notion that the increased YFP fluorescence for the RPTPα-CFP and RPTPα-YFP coexpressing cells (Fig. 3A) indeed reflects sensitized emission resulting from FRET. Furthermore, the experiments in Fig. 3A and 3C also show that the FRET is not due to nonspecific RPTPα interactions as a result of high concentrations in the plasma membrane, since the control with EGFR-YFP did not show sensitized YFP emission. Hence these FRET experiments demonstrate a specific RPTPα-RPTPα interaction (i.e. dimerization) in the plasma membrane of living SK-N-MC neuroepithelioma cells.
Cross-linking of RPTPalpha using a chemical cross-linker
We used the non-cell-permeable irreversible chemical cross-linker, bis- [sulfosuccinimidyl]suberate (BS3), on intact cells to confirm that RPTPα-YFP dimerized. A considerable proportion of RPTPα-YFP, designated long form (L) (Fig. 4A), migrated with an apparent molecular weight that was consistent with dimerization following treatment of the cells with BS3 (indicated by LL) (Fig. 4B). The proportion of cross-linked dimers appeared to be low. It is noteworthy that - similar to RPTPα-YFP - less than 10% of the constitutively dimeric RPTPα-P137C was cross-linked by BS3 (data not shown), suggesting that the efficiency of cross-linking was low under these conditions. These results are similar to previously published results with wild type RPTPα, lacking GFP derivatives [24], indicating that the GFP-derivatives neither enhanced nor interfered with RPTPα-YFP dimerization. We tested dimerization of a deletion mutant of RPTPα, enabling us to investigate whether RPTPα homodimerized, or merely interacted with another protein of similar size. RPTPα-200-CFP, lacking almost the entire cytoplasmic domain, including the wedge, had an apparent molecular weight of approximately 110 kDa, and was designated short form (S) (Fig. 4A). RPTPα-200-CFP migrated as a dimer following treatment of the cells with BS3 (indicated by SS) (Fig. 4B). Dimerization of the short form appeared to be more efficient than of the long form (Fig. 4B). However, we cannot exclude that this is due - in part - to an artefact as a result of more efficient blotting of smaller proteins. Co-transfection of RPTPα-YFP (L) and RPTPα-200-CFP (S) followed by treatment with BS3 led to the formation of LL-dimers, SS-dimers, and a novel intermediate form, LS-heterodimers. Mixing of lysates from BS3-treated cells transfected with only RPTPα-YFP or RPTPα-200-CFP illustrated that the LS heterodimer was only detected after BS3-treatment of cells co-expressing RPTPα-YFP and RPTPα-200-CFP (Fig. 4B). Thus, BS3-mediated cross-linking confirmed the FRET measurements, and the heterodimerization results of the long and short forms demonstrated that RPTPα homodimerized specifically.
Figure 4.

Homodimerization of RPTPα as assessed using a chemical cross-linker. (A) Distinct RPTPα constructs with different molecular weights were used to be able to discriminate between them. The long form (L) is a full length RPTPα construct with YFP fused to the C-terminus, while the short form (S) lacks almost all cytoplasmic sequences and contains CFP fused to residue 200, close to the transmembrane domain. (B) SK-N-MC cells were transiently transfected with the constructs depicted in (A). The cells were treated with the non-cell-permeable cross-linker, bis [sulfosuccinimidyl]suberate (BS3), or left untreated, as indicated. Aliquots of the total cell lysates were run on a 5% SDS-polyacrylamide gel, the gel was blotted, and the immunoblot was probed with anti-HA-tag antibody. The positions of marker proteins that were co-electrophoresed with the samples are indicated on the left (in kDa). The position of monomeric RPTPα-YFP (the long form, L), dimeric RPTPα-YFP (LL), monomeric RPTPα-200-CFP (the short form, S), dimeric RPTPα-200-CFP (SS), and of the RPTPα-YFP/ RPTPα-200-CFP heterodimer (LS) are indicated, as well as a non-specific band (NS). Lysates from BS3-treated cells that had been transfected with either RPTPα-YFP or RPTPα-200-CFP were mixed (mix), clearly demonstrating that the heterodimer (LS) only formed in cells that had been co-transfected with RPTPα-YFP and RPTPα-200-CFP.
Mapping of the region involved in dimerization
In order to establish which regions in RPTPα were required for dimerization, we determined the F(440)/F(490) ratio of a panel of RPTPα mutants. Initially, we focussed on the region that is involved in dimerization in the crystal structure of RPTPα [9]. Subtle mutations and deletions of (part of) the wedge diminished RPTPα dimerization as assessed using the chemical cross-linker BS3 [24]. Surprisingly, neither subtle mutations, nor deletion of the entire wedge reduced FRET significantly (data not shown). Therefore, we generated additional deletion mutants and determined the F(440)/F(490) ratio (Fig. 5A, B). Neither deletion of D2 (RPTPα-516), D1 and D2 (RPTPα-250), nor deletion of the wedge, D1 and D2 (RPTPα-200) reduced the F(440)/F(490) ratio significantly. Expression of all the different CFP and YFP fusion protein pairs was similar (Fig. 5C). Some transmembrane domains are sufficient for dimerization, e.g. the transmembrane domain of glycophorin A [39]. To test whether the transmembrane domain of RPTPα was involved in dimerization, we deleted the extracellular domain of the RPTPα-200-CFP and -YFP constructs, rendering fusion proteins that consisted of little more than the RPTPα transmembrane domain and CFP or YFP (Fig. 5A). Analysis of the F(440)/F(490) ratio in cells transfected with RPTPα-200Δ ED-CFP and -YFP demonstrated that deletion of the extracellular domain did not reduce dimerization significantly (Fig. 5), indicating that the extracellular domain was not required for dimerization. To test the specificity of transmembrane domains in dimerization, we replaced RPTPα's transmembrane domain with the transmembrane domain of the EGFR (Fig. 5A) and found that the F(440)/F(490) ratio of cells coexpressing RPTPα-200-YFP and RPTPα-EGFR-200-CFP was close to the theoretical minimum (bottom dashed line) (Fig. 5B). The chimeric RPTPα EGFR-200-CFP and -YFP proteins did not homodimerize either (Fig. 5B). These results demonstrate that the transmembrane domain of RPTPα was sufficient for dimerization of RPTPα
Figure 5.
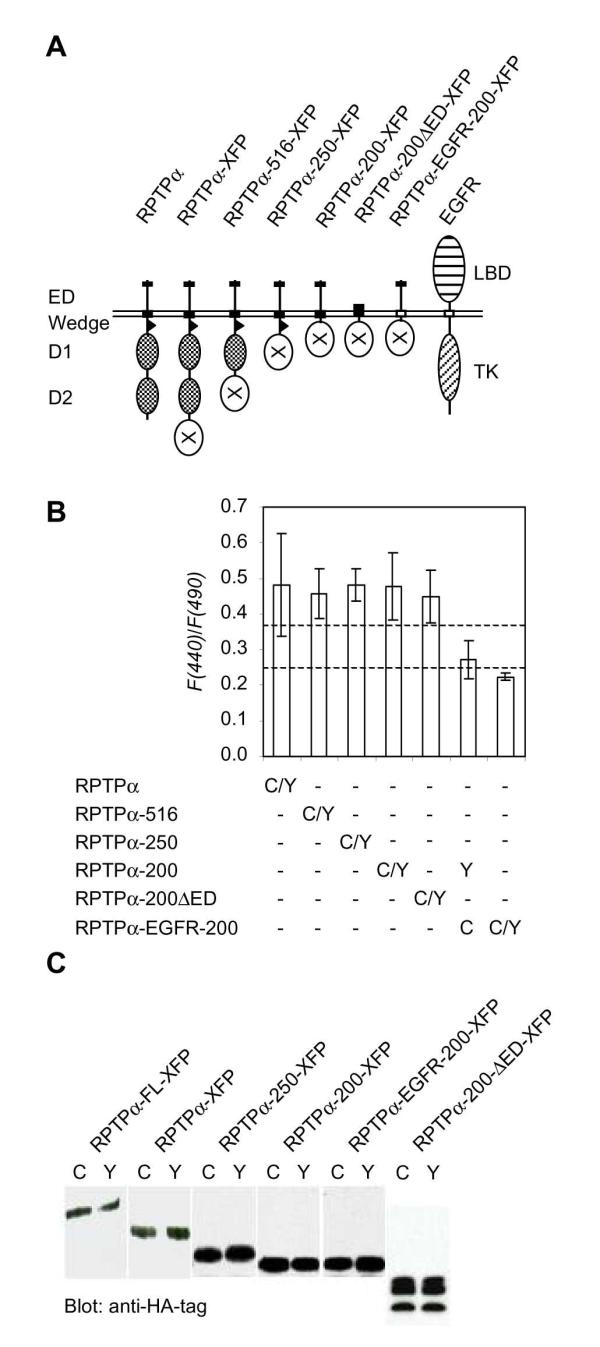
The transmembrane domain mediates RPTPα dimerization. (A) Schematic representation of RPTPα and deletion mutants. RPTPα fused to XFP (RPTPα-XFP) and deletion mutants lacking D2 (RPTPα-516-XFP), D1 and D2 (RPTPα-250-XFP), the entire cytoplasmic domain (RPTPα-200-XFP), or the cytoplasmic domain and the extracellular domain (RPTPα-200 ED-XFP) are depicted. RPTPα-EGFR-200-XFP is identical to RPTPα-200-XFP, except that the transmembrane and a short part of the intracellular domain were replaced by the corresponding regions of the human EGFR (depicted on the right). XFP (X) represents either YFP or CFP. The RPTPα extracellular domain (ED) is indicated, as well as the transmembrane domain (RPTPα, black box; EGFR, open box), the wedge (triangle) and the two PTP domains (D1 and D2). The EGFR ligand binding domain (LBD) and tyrosine kinase (TK) domain are indicated on the right. (B) Deletion mutants of RPTPα, fused to CFP and YFP (depicted in A), were co-transfected as indicated, and F(440)/F(490) was determined in single living cells. Averages of at least 10 individual cells from at least two independent experiments are depicted with error bars indicating the standard deviation. The dashed lines indicate theoretical minimal F(440)/F(490) ratio (bottom dashed line), and the FRET threshold level (upper dashed line). According to a two-tailed student's t-test, all values except for RPTPalpha-200-YFP/ RPTPalpha-EGFR-200-CFP are significantly different from the negative control, RPTPalpha-EGFR-200-CFP/ -YFP (p<0.003). (C) Expression of the constructs that were used in (B) was monitored by SDS-polyacrylamide gel electrophoresis using different gels (7.5 - 12.5%, depending on the size of the construct), and immunoblotting, using an anti-HA-tag antibody as a probe, to verify that CFP and YFP were expressed at similar levels. The molecular weights of the fusion proteins are: RPTPα-XFP, 160 kDa; RPTPα-516-XFP, 130 kDa; RPTPα-250-XFP, 110 kDa; RPTPα-200-XFP, 104 kDa; RPTPα-200Δ ED-XFP, 50 kDa; RPTPα-EGFR-200-XFP, 104 kDa.
Discussion
Here, we provide evidence that RPTPα dimerized in living cells. We have successfully used FRET to detect RPTPα dimers and dimerization was consistent with cross-linking experiments. Our results provide strong support for the model that dimerization is involved in regulation of RPTPs.
FRET was detected between RPTPα-CFP and RPTPα-YFP using three different techniques: dual wavelength excitation (Fig. 2), SPIM (Fig. 3A) and FLIM (Fig. 3B). The excitation-ratio method for determining FRET suffers from uncertainties when the CFP:YFP expression ratios, R(CFP/YFP), are not exactly known. Simulations show that high excitation ratios can be caused either by FRET or by an unfavorable (high) R(CFP/YFP) (Fig. 2B). Although for the entire cell population R(CFP/YFP) expression ratios were close to 1 (Fig. 2D), one cannot determine the expression ratio independently from FRET at the single cell level. Using the SPIM-method, FRET can be mimicked by unfavorable CFP/YFP expression ratios, but, in contrast to the excitation-ratio method, only when the R(CFP/YFP) is very low. The most reliable (but less sensitive) technique is FLIM. In conclusion, ratiometric analysis of FRET by dual wavelength excitation is a sensitive method for detection of FRET, which is reliable when similar levels of the two fluorophores are expressed.
We also demonstrated that RPTPα dimerized by chemical cross-linking, a technique that is fundamentally different from FRET analysis. BS3-mediated chemical cross- linking is a two-step chemical reaction, leading to a covalent bond, while FRET is only dependent on the distance between the two fluorophores. Dimerization, detected by chemical cross-linking appeared to be much less efficient than with FRET. However, this apparent difference is caused by the relatively low efficiency of the cross-linker, BS3. Only ∼ 10% of RPTPα-P137C was detected in dimeric form following BS3-mediated cross-linking, like wild type RPTPα (data not shown), while ∼ 80% of RPTPα-P137C dimerized according to non-denaturing gels (Fig. 2E). According to the FRET analysis dimerization was extensive, since similar FRET levels were detected in wild type RPTPα as in constitutively dimeric RPTPα-P137C (Fig. 2). Taken together, both FRET and cross-linking experiments indicate that dimerization of RPTPα is extensive. Subtle changes, for instance in the wedge, did not affect dimerization detected by FRET, but did lead to detection of reduced dimerization according to the cross-linking experiments [24]. This discrepancy may be explained by the difference in detection of dimerization. The extracellular domain may dimerize, without an interaction intracellularly. Therefore, extracellular cross-linkers may allow detection of these dimers, while FRET analysis does not, due to the topology of the two fluorophores in the intracellular domains. Importantly, the requirements for detection of dimerization are more stringent for cross-linkers than for FRET, since the linker between the two reactive groups in BS3 is only ∼ 12Å, while FRET allows distances up to ∼ 60Å between the two fluorophores. It is noteworthy that a distance of 60Å or less between the two fluorophores is only achieved when the two proteins are in a protein complex, not when they merely colocalize subcellularly. Taken together, both cross-linking and FRET analysis show that RPTPα dimerizes extensively.
FRET analysis demonstrated that the transmembrane domain was sufficient to drive dimerization of RPTPα which is consistent with cross-linking experiments [24] (Fig. 4B). Other regions in RPTPα may be involved in dimerization of the full length protein as well. For instance, cross-linking experiments demonstrated that the extracellular domain dimerized by itself [24]. By contrast, the extracellular domain of RPTPα fused to the EGFR transmembrane domain did not show FRET (Fig. 5B), suggesting that this fusion protein did not dimerize. This may be explained by the difference between cross-linking and FRET, as described above. Yet other regions in RPTPα may be involved in dimerization as well. We have evidence that RPTPα-D2 binds to RPTPα-D1 [40]. Similarly, CD45 and RPTPμ may be engaged in intra-or intermolecular interactions [41,42]. In addition, we and others found heterodimerization between RPTP-D1s and RPTP-D2s from different RPTPs, suggesting cross-talk between RPTPs [40,43]. It is noteworthy that the wedge is not required for dimerization, since deletion of the wedge did not abolish dimerization (Fig. 5, data not shown). Nevertheless, the wedge may be involved in stabilization of the dimer, since mutations in the wedge decreased the cross-linking efficiency [24]. Moreover, whereas the EGFR transmembrane domain by itself was not sufficient to mediate dimerization (Fig. 5), introduction of the juxtamembrane domain and D1 of RPTPα (residues 200-516) induced dimerization (data not shown). Taken together, multiple regions in RPTPα contribute to dimerization.
Here, we demonstrate for the first time that RPTPα dimerizes constitutively in living cells. Previously, indirect detection of protein-protein interactions using chemical cross-linkers demonstrated that CD45 homodimerizes [44]. In addition, RPTPα elutes from gel filtration columns as a large protein complex, suggesting dimerization or multimerization [45]. Regulation of dimerization is ill-understood. Like dimerization of RPTKs, dimerization of RPTPs may be regulated by ligand binding. RPTPs have diverse extracellular domains, and several RPTPs bind ligands [46,47,48,49]. Interestingly, GPI-linked Contactin binds laterally to the extracellular domain of RPTPα, i.e. in cis on the same cell [50]. Whether Contactin functions as a ligand, or as a ligand-binding moiety remains to be determined. Pleiotrophin, a ligand of RPTPβ/ζ inactivates RPTPβ/ζ activity [5]. Whether ligand binding affects dimerization of any of these RPTPs remains to be determined. RPTPα dimerization was constitutive and extensive. The transmembrane domain of RPTPα by itself was sufficient to drive dimerization, suggesting that ligands are not required for dimerization. However, ligand binding may modulate the extent of RPTPα dimerization. Analysis of RPTP dimerization using FRET may provide a powerful means to screen for factors that modulate dimerization or monomerization, and FRET may facilitate analysis of the dynamics of RPTP dimerization.
Dimerization of RPTPs may negatively regulate their activity. Ligand-induced dimerization of EGFR/CD45 led to functional inactivation [15]. Mutation of a single residue in the wedge of CD45 abolished ligand-induced inactivation [16], strongly supporting the model that dimerization leads to wedge-mediated occlusion of the catalytic sites. We have demonstrated that constitutively dimeric RPTPα-P137C was inactive, since it failed to dephosphorylate and activate c-Src in vivo. Mutation of the wedge rendered RPTPα-P137C active, while it did not affect dimerization, demonstrating that inactivation of RPTPs by dimerization was dependent on the wedge [23]. RPTPα-F135C with a disulfide bridge at position 135 dimerized constitutively, like RPTPα-P137C, but was still active, like wild type RPTPα. Apparently, dimerization-induced inactivation requires a specific rotational coupling, i.e. the monomers in the inhibited dimer need to be oriented in a specific geometry with respect to each other [23]. Ligand binding to the extracellular domain may induce rotation of the monomers, relative to each other, thus leading to activation or inactivation of RPTPα, similar to ligand-induced rotation that has been suggested to activate RPTKs [51,52].
Previously, we found that phorbol ester treatment of cells led to activation of RPTPα, and to phosphorylation of RPTPα Ser180 and Ser204 [53,54]. These serine phosphorylation sites are localized close to the wedge, suggesting that phosphorylation of these sites may interfere with interactions of the wedge. Phosphorylation of Ser180 and Ser204 may not affect dimerization per se, but still may lead to opening up of the catalytic site and thus to activation of RPTPα.
Since the crystal structures of RPTPμ-D1 and LAR did not show dimers like RPTPα-D1 [9,11,12], the model that RPTPs are regulated by dimerization has been the subject of debate. The transmembrane domain of RPTPα was sufficient to drive dimerization. It will be interesting to see whether the transmembrane domains of other RPTPs, including RPTPμ and LAR, drive dimerization as well. We propose that dimerization via the transmembrane domain of RPTPα provides a conformational basis for regulation by dimerization. Whether the dimer is inactive depends on the exact topology of the intracellular domain, which may be regulated by phosphorylation, by interaction with other proteins, or by rotational coupling for instance as a result of ligand binding to the extracellular domain. Here we demonstrate that RPTPα dimerized in living cells for the first time, providing strong support for the model that dimerization is involved in regulation of RPTP activity.
Materials and Methods
Cells, plasmids and transfections
SK-N-MC neuroepithelioma cells were grown in Dulbecco's Modified Eagle's Medium, supplemented with 10% Fetal Calf Serum. Expression vectors for chimeric RPTPα fusion proteins were generated by insertion of CFP and YFP [30] (kind gift of Roger Y. Tsien) on PCR-amplified BglII-fragments into pSG-HA- RPTPα [55] in which BglII sites had been introduced at the appropriate sites, position 792, 516, 250 or 200 (numbering according to [56]) by site-directed mutagenesis. In RPTPα-200Δ ED, the extracellular domain was deleted by deletion of an EcoNI-PstI fragment encompassing residues 30-129. RPTPα-EGFR-200 contains the extracellular domain of RPTPα (residues 1 - 142), fused to the transmembrane domain and 34 residues of the cytoplasmic domain of the EGFR (residues 646 - 702), comparable to RPTPα 200-XFP. Expression vectors for EGFR-XFP were generated by insertion of CFP and YFP into the endogenous BglII-site in the human EGFR (at position Ile923). SK-N-MC neuroepithelioma cells were grown on glass coverslips and transiently transfected with equal amounts (5-10 μg total plasmid DNA per 5 cm dish) of CFP- and YFP-fusion constructs by calcium phosphate precipitation exactly as described [55] and assayed 48 h after transfection.
Fluorescence Resonance Energy Transfer
For the FRET experiments, the cells were incubated at 220C in a HEPES buffered saline buffer (140 mM NaCl; 5 mM KCl; 2 mM CaCl2; 2 mM MgCl2; 10 mM HEPES; 10 mM glucose; 0.1% BSA; pH 7.50). We used a Leitz orthoplan upright microscope (Leitz GMBH, Wetzlar, Germany) and a SPEX Fluorolog fluorimeter (SPEX Industries, NJ) with two excitation monochromators (slit width: 8 nm). Two filter sets (Ploemopak) were used, the "CFP" filter set (filter #1) with an RKP510 dichroic mirror and a 490 nm long-pass emission filter, and the "FRET" filter (filter #2), equipped with a dichroic mirror RKP510 (reflection short-pass filter) and a BP530-560 (band-pass) emission filter (Leitz GMBH, Wetzlar, Germany). The fluorescence intensity was quantified with a Photon Counting Tube (type 9862, EMI Limited, Middlesex, England). The fluorescence intensities (obtained after excitation at 440 nm or 490 nm) were corrected for differences in excitation light intensities, using the reference photomultiplier. Fluorescence intensities were recorded from single living cells and corrected for background, using adjacent non-transfected cells. Routinely, we determined the F(440) and F(490) ratio in 5 or more individual cells that expressed both the CFP- and YFP-fusion proteins, using both filter sets. The ratio of F(440) and F(490) was determined using the FRET filter and consists of three terms: direct excitation of YFP, residual CFP fluorescence, and FRET, as indicated in the following formula:
![]()
In this formula, γ is a correction factor consisting of the relative transmission efficiencies of the microscope and the reflection efficiencies of the dichroic mirror at both excitation wavelengths. In our microscope γ was 1.0. The molar extinction coefficients, εCFP(440), εYFP(440), and εYFP(490) are respectively, 32.5, 10.5 and 55.3 x 103 M-1cm-1 [31]. QE(CFP/YFP) is the detection efficiency ratio for CFP to YFP detection, and we estimated the ratio QE(CFP/YFP) to be 0.2, based on the emission spectra of CFP and YFP, and on the filter set used. R(CFP/YFP) is the molar expression ratio in the cells of CFP and YFP-tagged fusion proteins. E is the FRET efficiency in a CFP and YFP labelled receptor dimer, which is dependent on the distance and orientation of the fluorophores. α is the degree of dimerization, ranging from 0 (no dimerization) to 1 (100% dimerization). The F(440)/F(490) ratio of cells expressing YFP fusion proteins only, was 0.16±0.04, consistent with the theoretical value (terms 2 and 3 of the formula are 0). In case there is no dimerization and/or FRET does not occur, term 3 in the formula above is 0 (α and/or E is 0), and F(440)/F(490) is directly proportional to the R(CFP/YFP) (see Fig. 2B). F(440)/F(490) ranges from 0.25 to 0.37 if R(CFP/YFP) ranges from 0.5 to 1.5, and E*α = 0 (no FRET and/or dimerization) (Fig. 2B). If F(440)/F(490)>0.37 and R(CFP/YFP) is close to 1.0, then term 3 in the formula above must be >0, indicating that FRET occurs between CFP and YFP in dimers of RPTPα fusion proteins. Therefore, we set the threshold level of F(440)/F(490) for FRET and dimerization at 0.37.
FLIM and SPIM
Both the SPIM and FLIM detector units were built on a Leica DMR upright epifluorescence microscope. Both detector systems are described in detail elsewhere [28,38]. Briefly, FLIM was implemented in the frequency domain using a homodyne detection scheme and a (wide field) RF-modulated image intensifier-CCD-coupled detector unit, and the SPIM detector unit consisted of an f/4 image spectrograph coupled to a back-illuminated chilled CCD camera.
For SPIM, excitation was provided by a 100 W Hg Arc lamp of which the 435 nm line was selected by inserting an Omega (Brattleboro, VT, USA) 435DF10 bandpass filter in the excitation light path. The excitation light was reflected onto the sample by an Omega 430DCLP dichroic mirror. A Leica 63x PL Apo water objective (NA = 1.2) was employed, using no coverslip, and immersing with HEPES buffered saline buffer (see above) at 20°C. Residual excitation light was rejected using a Schott (Mainz, Germany) GG455 longpass filter. In the spectrograph (Chromex 250 is (Chromex Inc., Albuquerque, NM, USA)) a 150 grooves/mm grating was used with a central wavelength of 500 nm. Single cells were positioned by aligning them across the entrance slit of the spectrograph (set at 200 μm width corresponding with a line of 3 μm width in the object plane). Acquisition time was 1-2 s. Regions of the image spectrum corresponding to the plasma membrane of labeled cells were distance averaged (typically 5-10 rows of pixels) and the resulting fluorescence spectra were corrected for background fluorescence and camera bias by background subtraction using an extracellular region just next to the plasma membrane region from the same spectral image.
For FLIM, the cells were excited with the 457 nm argon-ion laser line modulated at 60.116 MHz and the CFP fluorescence was selectively imaged using an Omega 470 DCLP dichroic mirror and an Omega 487RDF42 bandpass emission filter. 20 phase images (1-3 s each) were taken (10 with increasing and 10 with decreasing the phase allowing correction for photobleaching (which was less than 10% in all cases)). Reference phase settings and modulation were calibrated approximately every 30' by measuring a glass microcuvette filled with an erythrosine-B solution in water (single component fluorescence decay with a lifetime of 0.08 ns). The microscope setup, lifetime-image calculation and image processing are described in detail elsewhere [28].
Immunoblotting
Following transient transfection the cells were lysed in cell lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 10% glycerol, 10 u/ml aprotinin, 1 μM pMSF, 200 μM sodium orthovanadate), and the amount of protein in the lysates was determined. Subsequently, cell lysates containing equal amounts of protein were loaded on SDS-PAGE gels (7.5 - 12.5%, depending on the size of the construct). The material on the gels was blotted onto PVDF membranes by semi-dry blotting. The blots were blocked with 5% non-fat milk in TBS-T (50 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween-20) for 1 h, incubated with antibody in milk for 1.5 h, washed extensively and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h. Following extensive washing in TBS-T, the blots were developed using enhanced chemiluminescence. We used anti-GFP MAb (Transduction Labs, Lexington, KY), anti-HA-tag MAb (12CA5), and affinity purified anti-RPTPα antibodies, raised against the complete cytoplasmic domain (#5478) [57] as primary antibodies.
Chemical cross-linking
Prior to cross-linking, transiently transfected SK-N-MC neuroepithelioma cells were washed twice with ice cold phosphate-buffered saline (PBS). Subsequently, the cells were incubated with 2 mg/ml bis [sulfosuccinimidyl]suberate, BS3 (Pierce, Rockford, IL) in PBS for 1 h on ice. Following cross-linking, the cells were washed twice with ice cold PBS, and the cross-linker was quenched with 50 mM Tris for 15 min. The cells were lysed and aliquots of the lysates were run on a 5% SDS-PAGE gel, blotted and probed, using anti-HA-tag antibodies as described above.
Abbreviations
The abbreviations used are: RPTK, receptor protein-tyrosine kinase; RPTP, receptor protein-tyrosine phosphatase; FRET, fluorescence resonance energy transfer; GFP, green fluorescent protein; CFP, cyan fluorescent protein; YFP, yellow fluorescent protein; pTyr, phosphotyrosine; EGFR, epidermal growth factor receptor; SPIM, spectral imaging microscopy; FLIM, fluorescence lifetime imaging microscopy.
Acknowledgments
Acknowledgements
The authors would like to thank Roger Y. Tsien for the CFP- and YFP-cDNAs. G.J. is supported by a fellowship from the American Cancer Society. T.H. is a Frank and Else Schilling American Cancer Society Research Professor. This work was supported by grants from the National Cancer Institute (T.H.), the Royal Netherlands Academy of Arts and Sciences (T.W.J.G), and the Dutch Cancer Society (J.O. and J.d.H.).
Contributor Information
Leon GJ Tertoolen, Email: leon@niob.knaw.nl.
Christophe Blanchetot, Email: christof@niob.knaw.nl.
Guoqiang Jiang, Email: quoqiang_jiang@merck.com.
John Overvoorde, Email: johnov@niob.knaw.nl.
Theodorus WJ Gadella, Jr, Email: Dorus.Gadella@Laser.BC.WAU.NL.
Tony Hunter, Email: hunter@smtp.salk.edu.
Jeroen den Hertog, Email: hertog@niob.knaw.nl.
References
- Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- den Hertog J. Protein-tyrosine phosphatases in development. Mech Dev. 1999;85:3–14. doi: 10.1016/S0925-4773(99)00089-1. [DOI] [PubMed] [Google Scholar]
- Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/S0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- Van Vactor D, O'Reilly AM, Neel BG. Genetic analysis of protein tyrosine phosphatases. Curr Opin Genet Dev. 1998;8:112–126. doi: 10.1016/S0959-437X(98)80070-1. [DOI] [PubMed] [Google Scholar]
- Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci USA. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem Sci. 1994;19:459–463. doi: 10.1016/0968-0004(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Weiss A, Schlessinger J. Switching signals on or off by receptor dimerization. Cell. 1998;94:277–280. doi: 10.1016/S0092-8674(00)81469-5. [DOI] [PubMed] [Google Scholar]
- Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- Bilwes AM, den Hertog J, Hunter T, Noel JP. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature. 1996;382:555–559. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–450. doi: 10.1016/S0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Hoffmann KM, Tonks NK, Barford D. The crystal structure of domain 1 of receptor protein-tyrosine phosphatase mu. J Biol Chem. 1997;272:27505–27508. doi: 10.1074/jbc.272.44.27505. [DOI] [PubMed] [Google Scholar]
- Nam HJ, Poy F, Krueger NX, Saito H, Frederick CA. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97:449–457. doi: 10.1016/S0092-8674(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Stuckey JA, Schubert HL, Fauman EB, Zhang ZY, Dixon JE, Saper MA. Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature. 1994;370:571–575. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- Yang J, Liang X, Niu T, Meng W, Zhao Z, Zhou GW. Crystal structure of the catalytic domain of protein-tyrosine phosphatase SHP-1. J Biol Chem. 1998;273:28199–28207. doi: 10.1074/jbc.273.43.28199. [DOI] [PubMed] [Google Scholar]
- Desai DM, Sap J, Schlessinger J, Weiss A. Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase. Cell. 1993;73:541–554. doi: 10.1016/0092-8674(93)90141-c. [DOI] [PubMed] [Google Scholar]
- Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 1998;279:88–91. doi: 10.1126/science.279.5347.88. [DOI] [PubMed] [Google Scholar]
- Majeti R, Xu Z, Parslow TG, Olson JL, Daikh DI, Killeen N, Weiss A. An Inactivating Point Mutation in the Inhibitory Wedge of CD45 Causes Lymphoproliferation and Autoimmunity. Cell. 2000;103:1059–1070. doi: 10.1016/S0092-8674(00)00209-9. [DOI] [PubMed] [Google Scholar]
- den Hertog J, Pals CE, Peppelenbosch MP, Tertoolen LG, de Laat SW, Kruijer W. Receptor protein tyrosine phosphatase alpha activates pp60c-src and is involved in neuronal differentiation. EMBO J. 1993;12:3789–3798. doi: 10.1002/j.1460-2075.1993.tb06057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponniah S, Wang DZ, Lim KL, Pallen CJ. Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn. Curr Biol. 1999;9:535–538. doi: 10.1016/S0960-9822(99)80238-3. [DOI] [PubMed] [Google Scholar]
- Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr Biol. 1999;9:505–511. doi: 10.1016/S0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature. 1992;359:336–339. doi: 10.1038/359336a0. [DOI] [PubMed] [Google Scholar]
- Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. EMBO J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, den Hertog J, Su J, Noel J, Sap J, Hunter T. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature. 1999; 401:606–610. doi: 10.1038/44170. [DOI] [PubMed] [Google Scholar]
- Jiang G, den Hertog J, Hunter T. Receptor-like Protein Tyrosine Phosphatase alpha homodimerizes on the cell surface. Mol Cell Biol. 2000;20:5917–5929. doi: 10.1128/MCB.20.16.5917-5929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg RM. Fluorescence resonance energy transfer. 1996. pp. 179–252. [DOI] [PubMed]
- Selvin PR. Fluorescence resonance energy transfer. Meth Enzymol. 1995;246:300–334. doi: 10.1016/0076-6879(95)46015-2. [DOI] [PubMed] [Google Scholar]
- Wu P, Brand L. Resonance energy transfer: methods and applications. Anal Biochem. 1994;218:1–13. doi: 10.1006/abio.1994.1134. [DOI] [PubMed] [Google Scholar]
- Gadella TWJ. Fluorescence Lifetime Imaging Microscopy (FLIM): Instrumentation and Applications. 1999. pp. 467–479.
- Pollok BA, Heim R. Using GFP in FRET-based applications. Trends Cell Biol. 1999;9:57–60. doi: 10.1016/S0962-8924(98)01434-2. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Linder K, Berry G, Gordon GW, Heim R, Herman B. Bcl-2 and Bax interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer. Nat Biotechnol. 1998;16:547–552. doi: 10.1038/nbt0698-547. [DOI] [PubMed] [Google Scholar]
- Day RN. Visualization of Pit-1 transcription factor interactions in the living cell nucleus by fluorescence resonance energy transfer microscopy. Mol Endocrinol. 1998;12:1410–1419. doi: 10.1210/me.12.9.1410. [DOI] [PubMed] [Google Scholar]
- Damelin M, Silver PA. Mapping interactions between nuclear transport factors in living cells reveals pathways through the nuclear pore complex. Mol Cell. 2000;5:133–140. doi: 10.1016/s1097-2765(00)80409-8. [DOI] [PubMed] [Google Scholar]
- Overton MC, Blumer KJ. G-protein-coupled receptors function as oligomers in vivo. Curr Biol. 2000;10:341–344. doi: 10.1016/S0960-9822(00)00386-9. [DOI] [PubMed] [Google Scholar]
- Sorkin A, McClure M, Huang F, Carter R. Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr Biol. 2000;10:1395–1398. doi: 10.1016/S0960-9822(00)00785-5. [DOI] [PubMed] [Google Scholar]
- Gadella TWJ, Vereb GJ, Hadri AE, Rohrig H, Schmidt J, John M, Schell J, Bisseling T. Microspectroscopic imaging of nodulation factor-binding sites on living Vicia sativa roots using a novel bioactive fluorescent nodulation factor. Biophys J. 1997;72:1986–1996. doi: 10.1016/S0006-3495(97)78843-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadella TWJ, van der Krogt GN, Bisseling T. GFP-based FRET microscopy in living plant cells. Trends Plant Sci. 1999;4:287–291. doi: 10.1016/S1360-1385(99)01426-0. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Dempsey CE, Engelman DM. Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J Biol Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- Blanchetot C, den Hertog J. Multiple interactions between receptor protein-tyrosine phosphatase (RPTP) alpha and membrane-distal protein-tyrosine phosphatase domains of various RPTPs. J Biol Chem. 2000;275:12446–12452. doi: 10.1074/jbc.275.17.12446. [DOI] [PubMed] [Google Scholar]
- Feiken E, van EI, Gebbink MF, Moolenaar WH, Zondag GC. Intramolecular interactions between the juxtamembrane domain and phosphatase domains of receptor protein-tyrosine phosphatase RPTPmu. Regulation of catalytic activity. J Biol Chem. 2000;275:15350–15356. doi: 10.1074/jbc.275.20.15350. [DOI] [PubMed] [Google Scholar]
- Felberg J, Johnson P. Characterization of recombinant CD45 cytoplasmic domain proteins. Evidence for intramolecular and intermolecular interactions. J Biol Chem. 1998;273:17839–17845. doi: 10.1074/jbc.273.28.17839. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Fladd C, Batt J, Rotin D. The second catalytic domain of protein tyrosine phosphatase delta (PTP delta) binds to and inhibits the first catalytic domain of PTP sigma. Mol Cell Biol. 1998;18:2608–2616. doi: 10.1128/mcb.18.5.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Wu JJ, Maizel AL. Evidence for monomeric and dimeric forms of CD45 associated with a 30- kDa phosphorylated protein. J Biol Chem. 1992;267:16651–16659. [PubMed] [Google Scholar]
- Daum G, Regenass S, Sap J, Schlessinger J, Fischer EH. Multiple forms of the human tyrosine phosphatase RPTP alpha. Isozymes and differences in glycosylation. J Biol Chem. 1994;269:10524–10528. [PubMed] [Google Scholar]
- Brady-Kalnay SM, Flint AJ, Tonks NK. Homophilic binding of PTP mu, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol. 1993;122:961–972. doi: 10.1083/jcb.122.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebbink MF, Zondag GC, Wubbolts RW, Beijersbergen RL, van EI, Moolenaar WH. Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J Biol Chem. 1993;268:16101–16104. [PubMed] [Google Scholar]
- O'Grady P, Thai TC, Saito H. The laminin-nidogen complex is a ligand for a specific splice isoform of the transmembrane protein tyrosine phosphatase LAR. J Cell Biol. 1998;141:1675–1684. doi: 10.1083/jcb.141.7.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, Clary DO, Schilling J, Barnea G, Plowman GD. The carbonic anhydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal cell recognition molecule contactin. Cell. 1995;82:251–260. doi: 10.1016/0092-8674(95)90312-7. [DOI] [PubMed] [Google Scholar]
- Zeng L, D'Alessandri L, Kalousek MB, Vaughan L, Pallen CJ. Protein Tyrosine Phosphatase alpha (PTPalpha) and Contactin Form a Novel Neuronal Receptor Complex Linked to the Intracellular Tyrosine Kinase fyn. J Cell Biol. 1999;147:707–714. doi: 10.1083/jcb.147.4.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadella TWJ, Jovin TM. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J Cell Biol. 1995;129:1543–1558. doi: 10.1083/jcb.129.6.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Hunter T. When dimerization is not enough. Curr Biol. 1999;9:R568–R571. doi: 10.1016/S0960-9822(99)80357-1. [DOI] [PubMed] [Google Scholar]
- den Hertog J, Sap J, Pals CE, Schlessinger J, Kruijer W. Stimulation of receptor protein-tyrosine phosphatase alpha activity and phosphorylation by phorbol ester. Cell Growth Differ. 1995;6:303–307. [PubMed] [Google Scholar]
- Tracy S, van der Geer P, Hunter T. The receptor-like protein-tyrosine phosphatase, RPTP alpha, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J Biol Chem. 1995;270:10587–10594. doi: 10.1074/jbc.270.3.1408. [DOI] [PubMed] [Google Scholar]
- den Hertog J, Hunter T. Tight association of GRB2 with receptor protein-tyrosine phosphatase alpha is mediated by the SH2 and C-terminal SH3 domains. EMBO J. 1996;15:3016–3027. [PMC free article] [PubMed] [Google Scholar]
- Sap J, D'Eustachio P, Givol D, Schlessinger J. Cloning and expression of a widely expressed receptor tyrosine phosphatase. Proc Natl Acad Sci USA. 1990;87:6112–6116. doi: 10.1073/pnas.87.16.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hertog J, Tracy S, Hunter T. Phosphorylation of receptor protein-tyrosine phosphatase alpha on Tyr789, a binding site for the SH3-SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J. 1994;13:3020–3032. doi: 10.1002/j.1460-2075.1994.tb06601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]