

Abstract
A unique mouse model was developed with elevated endogenous GH (2- to 3-fold) and IGF-I (1.2- to 1.4-fold), due to somatotrope-specific Cre-mediated inactivation of IGF-I receptor (IgfIr) and insulin receptor (Insr) genes (IgfIr,InsrrGHpCre, referred to as HiGH mice). We demonstrate that the metabolic phenotype of HiGH mice is diet dependent and differs from that observed in other mouse models of GH excess due to ectopic heterologous transgene expression or pituitary tumor formation. Elevated endogenous GH promotes lean mass and whole-body lipid oxidation but has minimal effects on adiposity, even in response to diet-induced obesity. When caloric intake is moderated, elevated GH improves glucose clearance, despite low/normal insulin sensitivity, which may be explained in part by enhanced IGF-I and insulin output. However, when caloric intake is in excess, elevated GH promotes hepatic lipid accumulation, insulin resistance, hyperglycemia, and ketosis. The HiGH mouse model represents a useful tool to study the role endogenous circulating GH levels play in regulating health and disease.
GH is considered to be antilipogenic, prolipolytic, and protein anabolic, whereas high levels of GH reduce insulin sensitivity and can lead to hyperglycemia. This information has been in part derived from studies examining the metabolic phenotype of normal or GH-deficient (GHD) subjects treated with GH (1–7), humans and animal models with GH-producing pituitary adenomas (8–12), mice with elevated GH levels due to inactivation of hepatic IGF-I expression (13), and mice with ectopic overexpression of heterologous GH transgenes (14–25). Although a wealth of information has been gathered from these clinical and basic studies, this may not serve to appropriately inform us regarding the impact of raising endogenous GH levels within the physiologic range, in the context of intact GH receptor signaling and IGF-I production. This information is important in that pharmacologic strategies are being considered to raise endogenous GH levels in hopes of improving body composition and metabolic function in aging and in disease states (26, 27).
Recently, we generated a mouse model with elevated endogenous GH levels by removal of the IGF-I negative feedback effect on pituitary somatotropes using Cre recombinase-mediated inactivation of the IGF-I receptor (IgfIr) gene in somatotropes (28). These mice displayed modest increases in IGF-I levels, without alterations in growth, lean mass, or glucose homeostasis. However, fat mass was significantly reduced. Given insulin can also act directly at the pituitary level to inhibit GH synthesis and release and is thought to contribute to suppressed GH levels observed in obesity (29), studies were conducted to inactivate both the IgfIr and insulin receptor (Insr) genes in pituitary somatotropes. In contrast to somatotrope-specific inactivation of IgfIr alone, inactivation of both IgfIr and Insr elevated endogenous GH (2- to 3-fold) and IGF-I (1.2- to 1.4-fold), sufficient to modestly increase linear growth (29). These initial observations confirm that both insulin and IGF-I are important in directly inhibiting GH output from pituitary somatotropes. In the present report, we have extended these findings to examine the impact of elevated endogenous GH and IGF-I, due to somatotrope-specific loss of IgfIr and Insr, on body composition and metabolic function in the context of moderate or excessive caloric intake.
Materials and Methods
Animals
All experimental procedures were approved by the animal care and use committee of Jesse Brown Veterans Affairs Medical Center. Somatotrope-specific inactivation of both IgfIr and Insr was accomplished by crossbreeding rat GH promoter driven Cre recombinase (rGHpCre) mice (30) to mice homozygous for the loxP-modified IgfIr (31) and Insr (32) alleles (IgfIr,Insrfl/fl), where all strains are in a C57BL/6J background. All experimental mice were obtained by crossbreeding IgfIr,Insrfl/fl female mice with IgfIr,Insrfl/fl rGHpCre+/− (IgfIr,InsrrGHpCre) male mice, to avoid the confounding effects of elevated maternal GH/IGF-I levels on pup development. Mice were grouped housed and maintained on a standard rodent chow diet (CHOW-fat, 17% kcal; carbohydrate, 56% kcal; protein 27% kcal; Formulab Diet, Purina Mills, Inc., Richmond, IN), or fed a low-fat (LF-fat, 10% kcal; carbohydrate, 70% kcal; protein, 20% kcal; Research Diets, New Brunswick, NJ) or a high-fat (HF-fat, 60% kcal; carbohydrate, 20% kcal; protein 20% kcal; Research Diets) diet, starting at 4 wk of age. It should be noted that the purified ingredients in the LF diet matches that of the HF diet (amino acid: L-cystine; protein: casein; carbohydrates: corn starch, maltodextrin, and sucrose; fat: soybean oil and lard), where saturated fat (lard) represents 90% of the kcal from fat in the HF diet and 44% of the kcal from fat in the LF diet.
Immunohistochemistry
For immunohistochemical staining, IgfIr,Insrfl/fl Cre+/− (HiGH) and IgfIr,Insrfl/fl Cre−/− (control) mice pituitaries were formalin fixed and paraffin embedded, and 5-μm sections were used. Samples were boiled under pressure for 20 min in 10 mm citric acid (pH 6.0) to retrieve antigen. To assess GH, INSR, and IGFIR presence, slides were incubated overnight at 4 C with guinea pig anti-GH (A. F. Parlow, National Hormone and Peptide Program, Torrance, CA) and rabbit anti-INSR (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) or anti-IGFIR (Cell Signaling, Beverly, MA) antibodies, respectively. Primary antibodies were immunostained with fluorescein isothiocyanate-conjugated goat antiguinea pig (Sigma, St. Louis, MO) or TexasRed-conjugated goat antirabbit (Vector Laboratories, Inc., Burlingame, CA) antibodies (1:500). Coverslips were mounted onto slides with Fluoroshield mounting medium with 4′,6-diamidino-2-phenylindole (Sigma), sealed, and processed immediately. Images were acquired with a DX43 clinical microscope and the CellSens digital imaging software (Olympus USA, Melville, NY). Analysis and merges were performed with ImageJ software (National Institutes of Health, Bethesda, MD).
In vivo evaluation of metabolic status
Glucose tolerance tests (GTT) were performed at 16 wk of age (12 wk of diet) after an overnight fast (2 g/kg glucose, ip) and insulin tolerance tests (ITT) were performed at 18 wk of age (14 wk of diet) under ad libitum-fed conditions (1 U/kg Novolin, ip), beginning between 0800 and 0900 h. Blood was collected at t0, for hormone and nutrient measurements, and at t30 during the GTT to assess glucose-stimulated insulin secretion (GSIS). The homeostatic model assessment of insulin resistance (HOMA-IR) was calculated using glucose and insulin levels obtained after overnight fasting, using the following formula: [fasting blood glucose (mg/dl) × fasting insulin (μU/ml)/22.5], as previously reported (33). Every 2 wk, whole-body composition (lean, fat, and extracellular water content) was assessed by NMR (MiniSpec LF50; Bruker Optics, Billerica, MA). Indirect calorimetry, activity level, and food and water intake were determined using the PHYSIOCAGE system (Harvard, Holliston, MA) and METABOLISM analysis software (Panlab Harvard Apparatus, Barcelona, Spain) as previously described (34), and volume of O2 and volume of CO2 were expressed as ml/min · kg of lean mass (35).
Circulating hormones and metabolites
GH (Millipore, Bedford, MA), IGF-I (Immunodiagnostic Systems, Fountain Hills, AZ), insulin (Millipore), corticosterone (Immunodiagnostic Systems), and prolactin (Calbiotech, Spring Valley, CA) levels were assessed using commercial ELISA kits. Triglycerides (TG) and nonesterified fatty acids (NEFA) were determined using reagents and microtiter plate procedures from Wako Diagnostics (Richmond, VA). Blood glucose was assessed by glucometer (AlphaTRACK blood glucose monitoring system; Abbott, Abbott Park, IL). Circulating IGF-I binding proteins (IGFBP) levels were assessed using a Mouse IGFBP Magnetic Bead Panel (Millipore). Performance parameters of the various assays used are provided in Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org.
mRNA analysis
Total RNA was extracted from tissues [pituitary, hypothalamus, liver, or adipose tissue] using the Absolutely RNA RT-PCR Miniprep Kit with deoxyribonuclease treatment (Stratagene, La Jolla, CA), reversed transcribed (cDNA First Strand Synthesis kit; MRI Fermentas, Hanover, MD) and amplified by RT-PCR. Primer sequences, GenBank accession nos., and product sizes are provided as Supplemental information (Supplemental Table 2).
Protein analysis
Pituitaries were homogenized in sodium dodecyl sulfate-dithiothreitol sample buffer (62.5 mm Tris-HCl, 2% sodium dodecyl sulfate, 20% glycerol, and 100 mm dithiothreitol) at 65 C, followed by sonication for 10 sec and heating for 5 min at 95 C. After quantification with Bradford assay (Bio-Rad, Hercules, CA), proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes (Millipore). Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline with 0.05% Tween 20 and incubated with the primary antibodies for IGFIR or INSR (Cell Signaling), washed, and incubated with goat antirabbit antibody conjugated to horseradish peroxidase (Sigma). Chemiluminescence Immun-Star WesternC kit (Bio-Rad) was used to visualize the signal, and the images were digitally recorded using Image Station 4000R Pro system (Kodak, New York, NY). Images were analyzed with the Molecular Imaging Software (Carestream, Rochester, NY).
Liver lipid analysis
Liver was homogenized in isopropanol (1 ml/100 mg of tissue) to extract the lipids as previously described (36). TG content was determined using reagents and microtiter plate procedures from Wako Diagnostics.
Statistics
Student's t tests were used to evaluate the expression of IGFIR and INSR in the pituitary, as well as the impact of endogenous GH elevation on GH, IGF-I, and prolactin plasma levels in CHOW-fed mice and on VO2, VCO2, EE, activity, food and water intake, and HOMA-IR in LF diet mice. Comparisons of the effect of GH status (HiGH vs. control) and diet (LF vs. HF) on body composition, tissue weights, circulating nutrient, hormone, and IGFBP levels was assessed by two-way ANOVA, using genotype and diet as fixed factors. Comparisons of the effect of GH status (HiGH vs. control) on calorimetric analysis, response to GTT and ITT, and GSIS was assessed by two-way ANOVA, using genotype and time as fixed factors. In all two-way ANOVA analysis, Newman-Keuls post hoc tests for multiple comparisons were performed.
Results
Validation and characterization of IgfIr,Insrfl/fl (control) and IgfIr,InsrrGHpCre (HiGH) mice
IgfIr,Insrfl/fl mice carry a IgfIr gene containing loxP sites flanking exon 3 and an Insr gene containing loxP sites flanking exon 4. To confirm the effectiveness and tissue specificity of the Cre recombinase in excising the floxed alleles, primers located in the exons that span the floxed alleles were used to amplify cDNA obtained from the pituitary, hypothalamus, liver, and adipose tissue. As shown in Fig. 1A, use of pituitary cDNA from IgfIr,Insrfl/fl Cre+/− or IgfIr,Insrfl/wt Cre+/− mice resulted in the amplification of two bands, the higher corresponding to the intact floxed allele (not recombined) and the lower corresponding to the recombined allele (when Cre recombinase is present). Only bands corresponding to the intact floxed allele were amplified using cDNA obtained from IgfIr,Insrfl/fl Cre−/− tissues and from hypothalamus, liver, or adipose tissue (fat), irrespective of genotype. It should be noted that Insr, but not IgfIr, cDNA was amplified in the liver, consistent with previous reports showing that although the liver is the primary source of circulating IGF-I, it is not a target for IGF-I actions. The recombination of the IgfIr and Insr genes translated into a significant reduction in pituitary receptor protein levels as determined by Western blot analysis (Fig. 1B), but the receptor protein was not eliminated. These results were fully anticipated based on the fact that 99% of somatotropes, which represent 30–50% of all pituitary cells, express Cre recombinase. Therefore, approximately half of the pituitary cell population would retain the ability to synthesize IGFIR and INSR. The somatotrope specificity of receptor knockout is further supported by immunohistochemical evidence showing loss of IGFIR and INSR immunostaining in GH immunopositive cells of IgfIr,Insrfl/fl Cre+/− pituitaries but not in GH-immunonegative cells (Supplemental Fig. 1).
Fig. 1.

Validation and characterization of the mouse model of elevated endogenous GH (IgfIr,InsrrGHpCre). A, Confirmation of pituitary specific, Cre-mediated IgfIr, and Insr gene recombination. Primers located in the exons that span the floxed alleles of IgfIr and Insr genes were used to amplify pituitary (Pit), hypothalamus (HPT), liver (Liv), and fat cDNA from 4-month-old male mice. In IgfIr,InsrrGHpCre pituitaries, two bands are amplified, the higher one corresponding to the wild-type allele (not recombined) and the lower one corresponding to the recombined allele (when Cre recombinase is present). Only the wild-type allele is amplified in all tissues of control (IgfIr,Insrfl/fl) mice and in HPT, liver, and fat of IgfIr,InsrrGHpCre mice. B, IGFIR and INSR protein levels in pituitaries from 4-month-old male and female IgfIr,InsrrGHpCre and IgfIr,Insrfl/fl, as assessed by Western blotting, using β-actin as a control. Plasma GH (C) and IGF-I (D) of 8-month-old male and female IgfIr,InsrrGHpCre and IgfIr,Insrfl/fl mice, under fed and fasted (18 h) conditions. E, Plasma PRL levels of 8-month-old male and female IgfIr,InsrrGHpCre and IgfIr,Insrfl/fl mice. B–E, Values are shown as mean ± sem (n = 6–12 mice/group). Asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001) indicate values that significantly differ from controls (IgfIr,Insrfl/fl) and letters (a and b) indicate values that significantly differ from respective fed groups. F, Physiologic endpoints of prolactin action. Virgin 4-month-old female IgfIr,Insrfl/fl (n = 13) and IgfIr,InsrrGHpCre (n = 13) mice were crossbred to male IgfIr,InsrrGHpCre mice, and the percent that became pregnant, the days to conception, the number of pups born, the litters survived until weaning, the pups survived per litter until weaning (d 21), and pup weights at d 28 were recorded.
Loss of IGF-I and insulin negative feedback in the somatotrope resulted in an increase in circulating GH levels (Fig. 1C), which was associated with a significant increase in total IGF-I (Fig. 1D), in male and female mice under both fed (CHOW) and fasted conditions. Rank plot analysis of GH values (37) suggests that GH is still released in a pulsatile fashion in IgfIr,InsrrGHpCre mice, but baseline, as well as pulse height, is increased relative to controls (Supplemental Fig. 2).
Because we have previously observed that approximately 10% of the pituitary lactotrope population in rGHpCre mice express Cre recombinase and IGF-I has been shown to be important in lactotrope development and function (30), we also examined endpoints of lactotrope function in IgfIr,InsrrGHpCre mice, including circulating prolactin levels (Fig. 1E), fertility (percent pregnant, days to conception, and number of pups per litter), and maternal behavior/milk production (pup weight and number of surviving litters and surviving pups per litter) (Fig. 1F), where all were found to not differ from controls (IgfIr,Insrfl/fl). These results indicate that prolactin output is sufficient to maintain luteal function, maternal behavior, and milk production and also confirms that gonadotrope function is not overtly altered. We also found there were no differences in circulating corticosterone levels (Table 1). This is in contrast to mouse models with ectopic expression of heterologous GH transgenes, where supraphysiologic levels of GH have been associated with impaired female fertility and elevated glucocorticoid levels (17, 18). Given the selective alteration in somatotrope function (increase in GH) in IgfIr,InsrrGHpCre mice, we will refer to this model as “HiGH ” mice in the remainder of the text and IgfIr,Insrfl/fl mice will be referred to as “controls.”
Table 1.
Plasma levels of various hormones and metabolites in male and female IgfIr,Insrfl/fl (controls) and IgfIr,InsrrGHpCre (HiGH)
| Male |
Female |
|||||||
|---|---|---|---|---|---|---|---|---|
| LF diet |
HF diet |
LF diet |
HF diet |
|||||
| Control | HiGH | Control | HiGH | Control | HiGH | Control | HiGH | |
| GH (ng/ml) | 2.8 ± 0.9 | 9.8 ± 2.7b | 1.4 ± 0.3 | 8.1 ± 2.1a | 11.0 ± 2.7 | 18.6 ± 2.1a | 5.7 ± 1.4 | 14.2 ± 2.2b |
| IGF-I (ng/ml) | 316 ± 21 | 376 ± 20 | 332 ± 20 | 413 ± 16 | 244 ± 23 | 296 ± 26 | 285 ± 14 | 379 ± 28a |
| Corticosterone (ng/ml) | 42.2 ± 5.5 | 40.4 ± 16.8 | 53.1 ± 7.5 | 61.9 ± 12.4 | 41.3 ± 26.5 | 76.7 ± 13.2 | 77.9 ± 13.2 | 85.2 ± 13.2 |
| Glucose (mg/dl) | 202 ± 5 | 205 ± 13 | 205 ± 9 | 246 ± 15 | 179 ± 26 | 187 ± 8 | 197 ± 8 | 216 ± 9 |
| TG (mg/dl) | 44.6 ± 5.6 | 35.7 ± 5.5 | 46.2 ± 3.9 | 26.0 ± 1.7c | 33.3 ± 7.1 | 24.73 ± 2.1a | 26.5 ± 2.7 | 29.3 ± 2.3 |
| NEFA (mEq/liter) | 0.49 ± 0.17 | 0.32 ± 0.13 | 0.23 ± 0.08 | 0.14 ± 0.08 | 1.28 ± 0.13 | 1.01 ± 0.07a | 1.19 ± 0.18 | 1.36 ± 0.18 |
| Ketones (nmol/liter) | 98.8 ± 21.2 | 66.6 ± 11.4 | 101.7 ± 10.5 | 176.7 ± 32.3b | 76.4 ± 29.4 | 77.94 ± 49.9 | 145.3 ± 27.2 | 157.6 ± 23.3 |
Values that significantly differ from corresponding controls (Igfr,Insrfl/fl) (two-way ANOVA):
P < 0.05.
P < 0.01.
P < 0.001.
Body growth and composition of HiGH mice
Body weights of HiGH mice did not differ at 3 wk of age (Supplemental Fig. 3A). However, as HiGH mice aged, body weights increased above controls in all diet groups and remained approximately 2–3 g greater up to 20 wk of age (Fig. 2 and Supplemental Fig. 3B). Consistent with the anabolic action of GH/IGF-I, there was an overall increase in lean mass, within sex (P < 0.001 in both cases). Of note, independent of genotype, HF feeding significantly increased lean mass (P < 0.001 in males and females), where the combined of effects of elevated GH and HF feeding were synergistic (P < 0.001 in males and females). The increase in lean mass in HiGH mice could be explained, in part, by an increase in skeletal muscle mass, which was not specifically evaluated but was evident by gross morphology (see Supplemental Fig. 3H). In addition, HiGH mice displayed sex- and diet-dependent increases in the absolute and relative weights of the heart, kidney, and spleen (Supplemental Fig. 4). Finally, extracellular water weight was enhanced in HiGH mice, irrespective of diet (Supplemental Fig. 5). However, these increases were parallel to the increase in lean mass, where the water/lean ratio did not differ between genotype within diet [0.14–0.16 extra cellular water (g)/lean mass (g)].
Fig. 2.

Body composition of LF- and HF-fed, IgfIr,InsrrGHpCre (HiGH) and IgfIr,Insrfl/fl (control) male (upper panels) and female (lower panels) mice as assessed by whole-body NMR from 5 to 20 wk of age. A, Absolute total body weight, lean mass, and fat mass in grams. B, Lean mass/total body weight × 100 (% lean mass), fat mass/total body weight × 100 (% fat mass), and fat mass/lean mass. Values are shown as mean ± sem (n = 8–14 mice/group).
Although absolute fat mass increased in response to HF diet, unexpectedly, absolute fat mass of HiGH mice did not differ from controls, within diet (Fig. 2A). However, when lean and fat mass were expressed as percent of whole body weight, a significant increase in relative lean mass and decrease in relative fat mass was observed, but only in male HF-fed HiGH mice, compared with HF-fed control mice, at 16 wk or older (P ≤ 0.05 and P ≤ 0.01, respectively). These differences were reflected by a significant decrease in fat/lean ratio (Fig. 2B). In addition, postmortem analysis of fat depot weights (sc, urogenital, retroperitoneal) revealed a modest sex- and diet-dependent reduction in HiGH mice, compared with controls, where the differences in sc fat depots reached significance in HF-fed male and female mice and in CHOW female mice, when adjusted by body weight (Supplemental Fig. 6).
Surprisingly, the absolute liver weights in LF-fed HiGH male and female mice did not differ from controls. However, when adjusted by body weight, there was a small but significant increase in males, but not females (Fig. 3). In contrast, absolute and relative liver weights of male and female HF-fed HiGH mice were greater than controls (Fig. 3). This increase in HF-fed HiGH mice was associated with an increase in hepatic TG content (Fig. 3), where these changes cannot be readily explained by an increase in substrate availability, because plasma TG and NEFA levels were found to be similar to, or lower than, control values (Table 1). It should be noted that the liver phenotype of CHOW-fed HiGH mice showed an intermediate phenotype, with a modest increase in liver weight (Supplemental Fig. 3F), but the histological appearance of hepatocytes and number of nuclei per square millimeter were similar to that of controls (Supplemental Fig. 3, G and I).
Fig. 3.

Liver phenotype of LF- and HF-fed IgfIr,InsrrGHpCre (HiGH) and IgfIr,Insrfl/fl (control) male (upper panels) and female (lower panels) mice (20 wk of age). Values are shown as mean ± sem (n = 8–14 mice/group). Asterisks (*, P < 0.05; ***, P < 0.001) indicate values that significantly differ from controls. BW, Body weight.
Nutrient use and energy expenditure (EE) of HiGH mice
The use of indirect calorimetry to evaluate whole-body metabolic function revealed that the overall respiratory quotient (RQ) of HiGH male mice was reduced compared with controls (Fig. 4A). This difference was more evident during the postabsorptive state (light period). The reduction in RQ was not associated with significant changes in VO2, VCO2, EE, mean activity levels, or food intake (Fig. 4, B–F, and Supplemental Fig. 7). Interestingly, HiGH mice drank more water than controls, which may be related to changes in body composition (see Fig. 2 below) or due to changes in cardiovascular/renal function.
Fig. 4.

Indirect calorimetry, activity, and food/water intake in LF-fed IgfIr,InsrrGHpCre (HiGH) and IgfIr,Insrfl/fl (control) male mice (10 wk of age). A, Forty-eight-hour profiles of respiratory quotient [RQ = VCO2 (ml/min · kg-lean0.75)/VO2 (ml/min · kg-lean0.75)]. Data were pooled from two separate runs using different mice in each run (n = 10 mice/group). Day and night averages of VO2 (B), VCO2 (C), and EE (kcal/d · kg-lean0.75) (D) activity (horizontal beam breaks) (E) and ad libitum food (F) and water intake (G). Values are shown as mean ± sem (n = 10 mice/group). Asterisks (*, P < 0.05) indicate values that significantly differ from controls.
Glucose homeostasis of HiGH mice
When male and female HiGH mice were fed a LF diet, glucose clearance (assessed by ip GTT), was significantly improved. Specifically, glucose levels at 30 min after glucose load were reduced in HiGH mice compared with LF-fed controls (Fig. 5A), which was associated with a decrease in the area under the curve (Supplemental Table 3), reaching significance in males but not females. The improved glucose clearance in LF-fed HiGH mice was paradoxically associated with a modest decrease in insulin sensitivity (assessed by ip ITT), where glucose levels were significantly higher only in female HiGH mice, 60 min after insulin challenge (Fig. 5A). Improved glucose clearance observed in LF-fed male and female HiGH mice, in the face of low/normal insulin sensitivity, could be explained in part by elevated basal and glucose-stimulated insulin levels (Fig. 5B).
Fig. 5.

Glucose homeostasis regulation in LF- and HF-fed IgfIr,InsrrGHpCre (HiGH) and IgfIr,Insrfl/fl (control) male (upper panels) and female (lower panels) mice. A, GTT (2 g/kg ip, left panels) performed after overnight fasting at 16 wk of age. ITT (1 U/kg ip, right panels) performed in mice fed ad libitum at 18 wk of age. B, Basal and GSIS (left panels) was assessed in t0- and t30-min samples from GTT. HOMA-IR index (right panels) was calculated using glucose and insulin levels measured in t0 samples collected during GTT, using the following formula: [fasting blood glucose (mg/dl) × fasting insulin (μU/ml)/22.5]. Values are shown as mean ± sem (n = 8–14 mice/group). P values indicate the overall effect of genotype. Asterisks (*, P < 0.05; **, P < 0.01) indicate values that differ between genotype, within time, and the letter (a) indicates values that differ between time, within genotype.
In contrast to the improved glucose clearance observed in LF-fed HiGH mice, the response to GTT did not differ between HF-fed HiGH and control mice (Fig. 5A), despite the fact that basal and glucose-stimulated insulin levels remained elevated (Fig. 5B). This could be explained in part by a pronounced decline in insulin sensitivity in HF-fed HiGH mice, where glucose levels were significantly elevated, compared with HF-fed, controls at 30 min (males only) and 60 min (males and females) after insulin challenge (Fig. 5A). In male HiGH mice, this was associated with a significant increase in the area under the curve (Supplemental Table 3). A reduction in insulin sensitivity in HF-fed, HiGH mice is supported by elevated HOMA-IR scores (Fig. 5B). In addition, glucose and 3-hydroxybutyrate (major ketone body in the blood) levels were elevated in HF-fed, HiGH male mice. However, these endpoints did not differ between genotypes in HF-fed females or LF-fed males and females (Table 1). It should be noted that CHOW-fed HiGH mice showed an intermediate phenotype, compared with LF- and HF-fed mice, where the response to GTT was improved, insulin sensitivity was modestly reduced, but HOMA-IR were significantly greater in male, but not female, HiGH mice, relative to controls (Supplemental Fig. 3, D–F, and Supplemental Table 3).
Circulating GH, IGF-I, and IGFBPs in LF- and HF-fed HiGH mice
Consistent with elevated GH levels, circulating IGF-I levels were elevated in LF- and HF-fed HiGH mice, relative to diet-matched controls, where these differences were more pronounced in females (Table 1). In addition, HF feeding had an overall stimulatory effect on IGF-I (P = 0.081 and P ≤ 0.01 in males and females, respectively) (Table 1), where IGF-I levels in HF-fed HiGH females were significantly greater than that observed in LF-fed HiGH and HF-fed control female mice (Table 1). Because circulating IGFBPs can modulate the half-life and biologic effects IGF-I, and the synthesis of IGFBPs has been shown to be regulated by both GH and insulin (38, 39), we examined the impact of GH status and diet on plasma IGFBPs levels, and mean values are shown in Fig. 6. The impact of GH status and diet on plasma IGFBPs levels was assessed within gender by two-way ANOVA, and P values are provided in Fig. 6. Overall, HF feeding suppressed IGFBP1 and stimulated IGFBP3 levels (Fig. 6). In contrast, elevated GH had an overall stimulatory effect on IGFBP1, IGFBP3, IGFBP5, and IGFBP7 levels (Fig. 6).
Fig. 6.
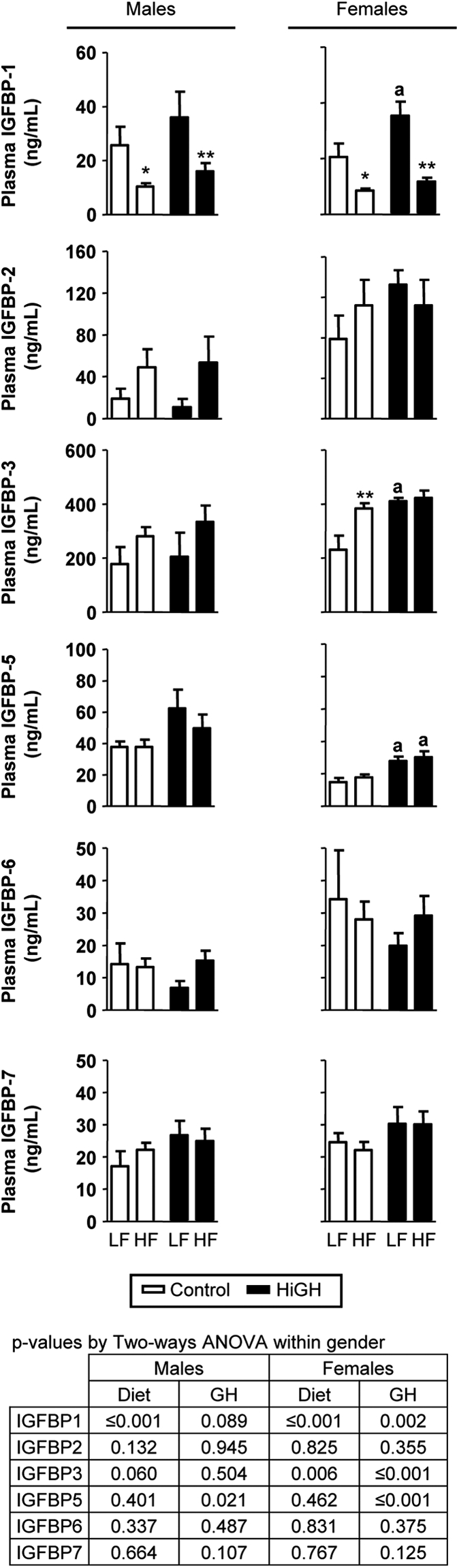
Circulating levels of IGFBPs in LF- and HF-fed IgfIr,InsrrGHpCre (HiGH) and IgfIr,Insrfl/fl (control) male and female mice (20 wk of age). IGFBPs (1–3 and 5–7) was analyzed using a Mouse IGFBPs Magnetic Bead Panel (Millipore). Values are shown as mean ± sem (n = 6–12 mice/group). Values were analyzed by two-way ANOVA within sex, and P values for the overall effect of genotype and diet are provided in the lower panel. Asterisks (*, P < 0.05; **, P < 0.01) indicate values that significantly differ between diet, within genotype, and the letter (a) indicates values that significantly differ (P < 0.05) between diets, within genotype, as assessed by Newman-Keuls post hoc comparisons.
Discussion
The HiGH mouse provides a unique model to study the metabolic impact of elevated endogenous GH and IGF-I. In contrast to previous mouse models of GH excess, where pharmacologic/pathophysiologic levels of GH (>50-fold normal) and IGF-I (2-fold) are associated with almost a doubling of body weight (14, 16, 19, 22, 23), HiGH mice show more modest elevations in GH (2- to 3-fold), IGF-I (1.2- to 1.4-fold), and body weight (2–3 g more than controls). The increase in body weight of HiGH mice is attributed to an increase in absolute lean mass in all diet groups, consistent with the well-known anabolic actions of GH/IGF-I (3–5). It should be noted that HF feeding had an independent stimulatory effect on lean mass that was greater than the impact of elevated GH/IGF-I alone. Increased lean mass in HF-fed mice has also been reported by others (40) and is consistent with the fact the prepubertal obese children have an increased growth velocity. The exact mechanism for this accelerated growth has not been identified but could include increased nutrient availability, direct anabolic actions of insulin, as well as insulin-mediated increase in IGF-I bioavailability via a reduction in hepatic IGFBP1 (41). Indeed, similar to that observed in obese humans, total IGF-I levels were elevated in HF-fed mice, and this was associated with a specific reduction in circulating IGFBP1 levels, consistent with a previous report demonstrating HF feeding decreases IGFBP1 in mice (42). Interestingly, the combined effects of HF diet and elevated GH were additive, where total IGF-I levels were greater in HF-fed, HiGH mice compared with HF-fed controls, which was more pronounced in females. Elevated total IGF-I in HF-fed HiGH mice may be due to both GH-mediated hepatic IGF-I production (data not shown), as well as an increase in the more abundant and long-lasting IGFBP, IGFBP3, which would serve to increase the half-life of circulating IGF-I (38, 39, 43).
Against the widely accepted view that GH prevents fat accumulation, absolute fat mass of HiGH mice did not differ from controls, irrespective of diet up to 20 wk of age. Despite the similarity in total fat mass between HiGH mice and controls, we did observe modest reductions in postmortem fat depot weights, with the most pronounced impact on sc fat, a depot previously reported to be more sensitive to changes in GH input (6, 21). These results suggest that elevated endogenous GH levels in the HiGH model can reduce fat accumulation in a depot-specific fashion. The modest impact of elevated GH on fat accumulation observed in HiGH mice may be due collectively to developmental timing of elevated GH levels, changes in body composition coupled with differential tissue use of FA as an energy source, as well as the balance of GH, IGF-I, and insulin in adults. Specifically, it has been previously shown that GH promotes the proliferation and differentiation of preadipocytes into adipocytes (44, 45), and therefore, early exposure to high levels of GH could establish a larger pool of adipocyte precursor cells. This appears to be the case in the MT-GH mouse models, where absolute fat mass is greater than controls early in life but is associated with smaller adipocyte size (46). With advancing age, absolute fat mass stabilizes in MT-GH mice, whereas wild-type mice continue to accumulate fat stores (23, 46). However, in both MT-GH transgenic (22) and HiGH mice, circulating NEFA levels are modestly suppressed or do not differ from controls under fed or fasted conditions. These results could suggest that the lipolytic action of GH is minimal with chronic exposure, perhaps due to the counterregulatory effects of elevated insulin, as well as elevated IGF-I levels, which in addition to its “insulin-like” activities has been shown to augment the effects of insulin (47). Alternatively, low/normal circulating NEFA in the face of elevated GH could be accounted for by an increase in FA use by the heart and skeletal muscle (1, 2), where it has been previously reported that hearts of MT-GH mice show increased FA use compared with those of wild-type controls (24). This may indeed be the case for HiGH mice that maintain low RQ values during the light period, indicating they are preferentially oxidizing lipids as an energy source during the postabsorptive state. Of note, changes in RQ were not reflected in changes in activity or food intake. In addition, there were no significant alterations in VO2 or EE. At first, these observations seem at odds (i.e. increase in lipid oxidation, without dramatic differences in absolute fat mass and EE). However, recent work by Hoehn et al. (48) demonstrates that FA oxidation and degree of adiposity are not necessarily coupled, and when EE is unchanged, the glucose not used for oxidative metabolism can be recycled and used as substrate for lipogenesis. Although these observations are intriguing, further work is required to determine the exact mechanism by which GH alters substrate use in the HiGH model.
In GH transgenic models (20, 22, 23) and in a GH pituitary tumor model (11, 12), liver weights are almost doubled that of controls, where this increase is attributed to hepatocyte hypertrophy and proliferation (11, 12). In contrast, in HiGH mice fed a LF or CHOW diet, absolute liver weights were similar to controls, and in CHOW-fed HiGH mice, the histological appearance of hepatocytes showed no signs of hypertrophy or an increase in the number of multinucleated cells. Therefore, in the HiGH model, the metabolic changes due to GH-mediated regulation of hepatocyte function are not likely confounded by pathophysiologic changes in hepatocyte size and number. In this context, it is interesting to note that liver weights of HF-fed HiGH mice were greater than those of HF-fed controls, which was associated with an increase in TG content. It is unlikely that the changes in hepatic TG are due to an increase in influx of lipids into the liver, because plasma TG and NEFA levels of HiGH mice were not greater than controls. This suggests that the increase in hepatic lipid content of HiGH mice may be due to the direct actions of GH on hepatic lipid production, where a positive association of GH and hepatic lipid content has been observed by others (10, 34, 49, 50), including our recent observation that hepatic TG content is reduced in a mouse model of adult onset-isolated GHD (AOiGHD), fed a HF diet (34). Curiously, hepatic lipid content is also increased in mice with global or hepatic-specific inactivation of GHR or GHR signaling (51–54). However, because GH is critical for normal hepatocyte proliferation during development (52), and liver of developmental models of GHD or GH resistance (dwarfs) are disproportionately reduced in size relative to changes in body weight (7, 55), the increase in lipid accumulation could be due in part to a mismatched influx of substrate relative to hepatic mass. We also cannot ignore the fact that HiGH mice have elevated insulin levels, where insulin can directly stimulate hepatic lipogenesis, even in the context of diet-induced obesity and insulin resistance (56). Although further studies are required to tease apart the complex interrelationship between GH and insulin on hepatic lipid metabolism, our results suggest that when calories are in excess, elevated endogenous GH levels can promote hepatic lipid accumulation, which would contribute to the development of diabetes, as observed in HF-fed HiGH male mice (see below for further discussion).
When caloric intake is moderate (LF- or CHOW-fed HiGH mice), elevated GH improves glucose clearance, and this occurs in the face of normal/low insulin sensitivity. Improved glucose clearance was also recently reported for GH transgenic mouse models and was attributed, in part, to the increase in lean mass (25). However, in the current study, an increase in lean mass cannot entirely account for improved glucose clearance, because the amount of glucose injected per gram of lean mass did not differ between HiGH and control mice. Alternatively, improved glucose clearance may be directly related to the increased GSIS observed in HiGH mice, where GH has been shown to play a direct stimulatory role in augmenting β-cell function in vitro (57, 58) and in vivo (59). It has also been reported that treatment with IGF-I improves whole-body glucose uptake and glucose tolerance (60), in part through increasing insulin sensitivity (61). Therefore, the elevation in circulating IGF-I levels observed in HiGH mice may work in concert with insulin to maintain glucose homeostasis. In contrast, elevations in GH, IGF-I, and insulin could not protect male HiGH mice against the deleterious effects of diet-induced obesity and actually exacerbated insulin resistance, which was associated with hepatic lipid accumulation, hyperglycemia, and ketosis. It is interesting to note that an opposite phenotype of glucose regulation was observed in our model of AOiGHD (34), where these mice have lower circulating IGF-I and are more insulin sensitive than controls, irrespective of diet. However, when AOiGHD mice are fed a HF diet, insulin and IGF-I levels remain low, and they become glucose intolerant. However, female HiGH mice were somewhat protected from the deleterious effects of HF feeding. Although they did become more insulin resistant (as indicated by an elevated HOMA-IR score) compared with controls, circulating glucose or ketones levels did not differ. It is well documented in humans and animal models that females exhibit less severe obesity-related metabolic disorders, compared with males, which may be related to estrogen-mediated enhancement of adipocyte insulin sensitivity and/or β-cell insulin production (62–64).
In summary, the HiGH mouse model provides a unique opportunity to study the metabolic impact of selectively raising endogenous GH levels within the physiologic range and represents a useful tool to study the role that endogenous circulating GH levels play in regulating health and disease. These initial studies clearly demonstrate that the metabolic phenotype of HiGH mice is diet dependent and unique from that observed in other mouse models of GH excess, as summarized in Table 2. We demonstrate that elevated endogenous GH promotes lean mass and whole-body lipid oxidation but has minimal effects on adiposity, even in response to diet-induced obesity. When caloric intake is moderate, elevated GH improves glucose clearance, which may be explained in part by enhanced IGF-I and insulin output. However, when caloric intake is in excess, elevated GH promotes hepatic lipid accumulation, insulin resistance, hyperglycemia, and ketosis.
Table 2.
Comparison of metabolic endpoints: GH transgenic vs. HiGH mice

Supplementary Material
Acknowledgments
This work was supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development Merit Award, National Institutes of Health Grants R21AG031465 and R01DK088133 (to R.D.K.), Fundacion Caja Madrid, Postdoctoral Grant (M.D.G.), Ministerio de Ciencia e Innovación Grants RYC-2007-00186 and BFU2008-01136/BFI and Ministerio de Educación Grant JC2008-00220 (to R.M.L.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AOiGHD
- adult onset-isolated GHD
- CHOW
- standard rodent chow diet
- EE
- energy expenditure
- GHD
- GH deficient
- GSIS
- glucose-stimulated insulin secretion
- GTT
- glucose tolerance test
- HF
- high fat
- HOMA-IR
- homeostatic model assessment of insulin resistance
- IGFBP
- IGF-I binding protein
- IgfIr
- IGF-I receptor
- Insr
- insulin receptor
- ITT
- insulin tolerance test
- LF
- low fat
- NEFA
- nonesterified fatty acid
- rGHpCre
- rat GH promoter driven Cre recombinase
- RQ
- respiratory quotient
- TG
- triglyceride.
References
- 1. Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D. 2010. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm IGF Res 20:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moller N, Vendelbo MH, Kampmann U, Christensen B, Madsen M, Norrelund H, Jorgensen JO. 2009. Growth hormone and protein metabolism. Clin Nutr 28:597–603 [DOI] [PubMed] [Google Scholar]
- 3. Møller N, Jørgensen JO. 2009. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 30:152–177 [DOI] [PubMed] [Google Scholar]
- 4. Jørgensen JO, Rubeck KZ, Nielsen TS, Clasen BF, Vendelboe M, Hafstrøm TK, Madsen M, Lund S. 2010. Effects of GH in human muscle and fat. Pediatr Nephrol 25:705–709 [DOI] [PubMed] [Google Scholar]
- 5. Birzniece V, Nelson AE, Ho KKY. 2011. Growth hormone and physical perfromance. Trends Endocrinol Metab 22:171–178 [DOI] [PubMed] [Google Scholar]
- 6. Berryman DE, List EO, Kohn DT, Coschigano KT, Seeley RJ, Kopchick JJ. 2006. Effect of growth hormone on susceptibility to diet-induced obesity. Endocrinology 147:2801–2808 [DOI] [PubMed] [Google Scholar]
- 7. Alba M, Fintini D, Salvatori R. 2005. Effects of recombinant mouse growth hormone treatment on growth and body composition in GHRH knock out mice. Growth Horm IGF Res 15:275–282 [DOI] [PubMed] [Google Scholar]
- 8. Bengtsson B-A, Brummer R-J, Eden S, Bosaeus I, Lindstedt G. 1989. Body composition in acromegaly: the effect of treatment. Clin Endocrinol 31:481–490 [DOI] [PubMed] [Google Scholar]
- 9. Quabbe HJ, Plöckinger U. 1996. Metabolic aspects of acromegaly and its treatment. Metabolism 45:61–62 [DOI] [PubMed] [Google Scholar]
- 10. Szendroedi J, Zwettler E, Schmid AI, Chmelik M, Pacini G, Kacerovsky G, Smekal G, Nowotny P, Wagner O, Schnack C, Schernthaner G, Klaushofer K, Roden M. 2008. Reduced basal ATP synthetic flux of skeletal muscle in patients with previous acromegaly. PLoS ONE 3:e3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lloyd RV, Jin L, Chang A, Kulig E, Camper SA, Ross BD, Downs TR, Frohman LA. 1992. Morphologic effects of hGRH gene expression on the pituitary, liver, and pancreas of MT-hGRH transgenic mice. An in situ hybridization analysis. Am J Pathol 141:895–906 [PMC free article] [PubMed] [Google Scholar]
- 12. Luque RM, Soares BS, Peng XD, Krishnan S, Cordoba-Chacon J, Frohman LA, Kineman RD. 2009. Use of the metallothionein promoter-guman growth hormone-releasing hormone (GHRH) mouse to identify regulatory pathways that suppress pituitary somatotrope hyperplasia and adenoma formation due to GHRH-receptor hyperactivation. Endocrinology 150:3177–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yakar S, Kim H, Zhao H, Toyoshima Y, Pennisi P, Gavrilova O, Leroith D. 2005. The growth hormone-insulin like growth factor axis revisited: lessons form IGF-I and IGF-I receptor gene targeting. Pediatr Nephrol 20:251–254 [DOI] [PubMed] [Google Scholar]
- 14. Palmiter RD, Brinster RL, Hammer RE, Trumbauer ME, Rosenfeld MG, Birnberg NC, Evans RM. 1982. Dramatic growth of mice that develop from eggs microinjected with metallothionein-growth hormone fusion genes. Nature 300:611–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palmiter RD, Chen HY, Brinster RL. 1982. Differential regulation of metallothionein-thymidine kinase fusion genes in transgenic mice and their offspring. Cell 29:701–710 [DOI] [PubMed] [Google Scholar]
- 16. Palmiter RD, Norstedt G, Gelinas RE, Hammer RE, Brinster RL. 1983. Metallothionein-human GH fusion genes stimulate growth of mice. Science 222:809–814 [DOI] [PubMed] [Google Scholar]
- 17. Cecim M, Kerr J, Bartke A. 1995. Effects of bovine growth-hormone (bGH) transgene expression or bGH treatment on reproductive functions in female mice. Biol Reprod 52:1144–1148 [DOI] [PubMed] [Google Scholar]
- 18. Eisen EJ, Peterson CB, Parker IJ, Murray JD. 1998. Effects of zinc ion concentration on growth, fat content and reproduction in oMT1a-oGH transgenic mice. Growth Dev Aging 62:173–186 [PubMed] [Google Scholar]
- 19. Olsson B, Bohlooly-Y M, Brusehed O, Isaksson OG, Ahrén B, Olofsson SO, Oscarsson J, Törnell J. 2003. Bovine growth hormone-transgenic mice have major alterations in hepatic expression of metabolic genes. Am J Physiol Endocrinol Metab 285:E504–E511 [DOI] [PubMed] [Google Scholar]
- 20. Oberbauer AM, Stiglich C, Murray JD, Keen CL, Fong DL, Smith LB, Cushwa S. 2004. Dissociation of body growth and adipose deposition effects of growth hormone in oMt1a-oGH transgenic mice. Growth Dev Aging 68:33–45 [PubMed] [Google Scholar]
- 21. Berryman DE, List EO, Coschigano KT, Behar K, Kim JK, Kopchick JJ. 2004. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res 14:309–318 [DOI] [PubMed] [Google Scholar]
- 22. Olsson B, Bohlooly-Y M, Fitzgerald SM, Frick F, Ljungberg A, Ahrén B, Törnell J, Bergström G, Oscarsson J. 2005. Bovine growth hormone transgenic mice are resistant to diet-induced obesity but develop hyperphagia, dyslipidemia, and diabetes on a high-fat diet. Endocrinology 146:920–930 [DOI] [PubMed] [Google Scholar]
- 23. Palmer AJ, Chung MY, List EO, Walker J, Okada S, Kopchick JJ, Berryman DE. 2009. Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology 150:1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bogazzi F, Raggi F, Ultimieri F, Russo D, D'Alessio A, Manariti A, Brogioni S, Manetti L, Martino E. 2009. Regulation of cardiac fatty acids metabolism in transgenic mice overexpressing bovine GH. J Endocrinol 201:419–427 [DOI] [PubMed] [Google Scholar]
- 25. Boparai RK, Arum O, Khardori R, Bartke A. 2010. Glucose homeostasis and insulin sensitivity in growth hormone-transgenic mice: a cross-sectional analysis. Biol Chem 391:1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cordido F, Isidro ML, Nemiña R, Sangiao-Alvarellos S. 2009. Ghrelin and growth hormone secretagogues, physiological and pharmacological aspect. Curr Drug Discov Technol 6:34–42 [DOI] [PubMed] [Google Scholar]
- 27. Ceda GP, Dall'Aglio E, Morganti S, Denti L, Maggio M, Lauretani F, Andrea A, Ceresini G, Cattabiani C, Valenti G. 2010. Update on new therapeutic options for the somatopause. Acta Biomed 81:67–72 [PubMed] [Google Scholar]
- 28. Romero CJ, Ng Y, Luque RM, Kineman RD, Koch L, Bruning JC, Radovick S. 2010. Targeted deletion of somatotroph insulin-like growth factor-I signaling in a cell-specific knockout mouse model. Mol Endocrinol 24:1077–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luque RM, Gahete MD, Cordoba-Chacon J, Childs GV, Kineman RD. 2011. Does the pituitary somatotrope play a primary role in regulating GH output in metabolic extremes? Ann NY Acad Sci 1220:82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luque RM, Amargo G, Ishii S, Lobe C, Franks R, Kiyokawa H, Kineman RD. 2007. Reporter expression, induced by a GH promoter-driven Cre recombinase (rGHp-Cre) transgene, questions the developmental relationship between somatotropes and lactotropes in the adult mouse pituitary gland. Endocrinology 148:1946–1953 [DOI] [PubMed] [Google Scholar]
- 31. Klöting N, Koch L, Wunderlich T, Kern M, Ruschke K, Krone W, Brüning JC, Blüher M. 2008. Autocrine IGF-1 action in adipocytes controls systemic IGF-1 concentrations and growth. Diabetes 57:2074–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brüning JC, Michael MD, Winnay JN, Hayashi T, Hörsch D, Accili D, Goodyear LJ, Kahn CR. 1998. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2:559–569 [DOI] [PubMed] [Google Scholar]
- 33. Lee S, Muniyappa R, Yan X, Chen H, Yue LQ, Hong EG, Kim JK, Quon MJ. 2008. Comparison between surrogate indexes of insulin senstivity and resistance and hyperinsuliniemic euglycemic clamp estimates in mice. Am J Physiol Endocrinol Metab 294:E261–E270 [DOI] [PubMed] [Google Scholar]
- 34. Luque RM, Lin Q, Córdoba-Chacón J, Subbaiah PV, Buch T, Waisman A, Vankelecom H, Kineman RD. 2011. Metabolic impact of adult-onset, isolated, growth hormone deficiency (AOiGHD) due to destruction of pituitary somatotropes. PLoS ONE 6:e15767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kaiyala KJ, Schwartz MW. 2011. Toward a more complete (and less controversial) understanding of energy expenditure and its role in obesity pahtogenesis. Diabetes 60:17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morton NM, Densmore V, Wamil M, Ramage L, Nichol K, Bünger L, Seckl JR, Kenyon CJ. 2005. A polygenic model of the metabolic syndrome with reduced circulating and intra-adipose glucocorticoid action. Diabetes 54:3371–3378 [DOI] [PubMed] [Google Scholar]
- 37. Xu J, Bekaert AJ, Dupont J, Rouve S, Annesi-Maesano I, De Magalhaes Filho CD, Kappeler L, Holzenberger M. 2011. Exploring endocrine GH pattern in mice using rank plot analysis and random blood samples. J Endocrinol 208:119–129 [DOI] [PubMed] [Google Scholar]
- 38. Baxter RC. 1993. Circulating binding-proteins for the insulin-like growth-factors. Trends Endocrinol Metab 4:91–96 [DOI] [PubMed] [Google Scholar]
- 39. Jones JI, Clemmons DR. 1995. Insulin-like growth-factors and their binding-proteins - biological actions. Endocr Rev 16:3–34 [DOI] [PubMed] [Google Scholar]
- 40. Brown JL, Spicer MT, Spicer LJ. 2002. Effect of high-fat diet on body composition and hormone responses to glucose tolerance tests. Endocrine 19:327–332 [DOI] [PubMed] [Google Scholar]
- 41. Brismar K, Fernqvist-Forbes E, Wahren J, Hall K. 1994. Effect of insulin on the hepatic production of insulin-like growth factor-binding protein-1 (Igfbp-1), Igfbp-3, and Igf-I in insulin-dependent diabetes. J Clin Endocrinol Metab 79:872–878 [DOI] [PubMed] [Google Scholar]
- 42. Bielohuby M, Sawitzky M, Stoehr BJ, Stock P, Menhofer D, Ebensing S, Bjerre M, Frystyk J, Binder G, Strasburger C, Wu Z, Christ B, Hoeflich A, Bidlingmaier M. 2011. Lack of dietary carbohydrates induces hepatic growth hormone (GH) resistance in rats. Endocrinology 152:1948–1960 [DOI] [PubMed] [Google Scholar]
- 43. Lehtihet M, Efendic S, Brismar K. 2008. Postprandial paradoxical IGFBP-1 response in obese patients with Type 2 diabetes. Clin Science 115:167–174 [DOI] [PubMed] [Google Scholar]
- 44. Louveau I, Gondret F. 2004. Regulation of development and metabolism of adipose tissue by growth hormone and the insulin-like growth factor system. Domest Anim Endocrinol 27:241–255 [DOI] [PubMed] [Google Scholar]
- 45. Nam SY, Marcus C. 2000. Growth hormone and adipocyte function in obesity. Horm Res 53:87–97 [DOI] [PubMed] [Google Scholar]
- 46. Berryman DE, List EO, Sackmann-Sala L, Lubbers E, Munn R, Kopchick JJ. 2011. Growth hormone and adipose tissue: beyond the adipocyte. Growth Horm IGF Res 21:113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clemmons DR. 2006. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Curr Opin Pharmacol 6:620–625 [DOI] [PubMed] [Google Scholar]
- 48. Hoehn KL, Turner N, Swarbrick MM, Wilks D, Preston E, Phua Y, Joshi H, Furler SM, Larance M, Hegarty BD, Leslie SJ, Pickford R, Hoy AJ, Kraegen EW, James DE, Cooney GJ. 2010. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab 11:70–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lindén D, Sjöberg A, Asp L, Carlsson L, Oscarsson J. 2000. Direct effects of growth hormone on production and secretion of apolipoprotein B from rat hepatocytes. Am J Physiol Endocrinol Metab 279:E1335–E1346 [DOI] [PubMed] [Google Scholar]
- 50. Haluzik M, Yakar S, Gavrilova O, Setser J, Boisclair Y, LeRoith D. 2003. Insulin resistance in the liver-specific IGF-I gene-deleted mouse is abrogated by deletion of the acid-labile subunit of the IGF-binding protein-3 complex. Diabetes 52:2483–2489 [DOI] [PubMed] [Google Scholar]
- 51. Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, Kopchick JJ, Le Roith D, Trucco M, Sperling MA. 2009. Liver-specific deletion of the growth hormone receptor reveals essential role of GH signaling in hepatic lipid metabolism. J Biol Chem 284:19937–19944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cui Y, Hosui A, Sun R, Shen K, Gavrilova O, Chen W, Cam MC, Gao B, Robinson GW, Hennighausen L. 2007. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 46:504–513 [DOI] [PubMed] [Google Scholar]
- 53. Barclay JL, Nelson CN, Ishikawa M, Murray LA, Kerr LM, McPhee TR, Powell EE, Waters MJ. 2011. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology 152:181–192 [DOI] [PubMed] [Google Scholar]
- 54. Sos BC, Harris C, Nordstrom SM, Tran JL, Balázs M, Caplazi P, Febbraio M, Applegate MA, Wagner KU, Weiss EJ. 2011. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J Clin Invest 121:1412–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. List EO, Sackmann-Sala L, Berryman DE, Funk K, Kelder B, Gosney ES, Okada S, Ding J, Cruz-Topete D, Kopchick JJ. 2011. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR−/−) mouse. Endocr Rev 32:356–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li S, Brown MS, Goldstein JL. 2010. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stumulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA 107:3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Svensson C. 1996. Effects of growth hormone in vitro on the glucose metabolism of fetal rat islet β-cells. J Mol Endocrinol 17:131–138 [DOI] [PubMed] [Google Scholar]
- 58. Dalgaard LT, Thams P, Gaarn LW, Jensen J, Lee YC, Nielsen JH. 2011. Suppression of FAT/CD36 mRNA by human growth hormone in pancreatic β-cells. Biochem Biophys Res Commun 410(2):345–350 [DOI] [PubMed] [Google Scholar]
- 59. Wu Y, Liu C, Sun H, Vijayakumar A, Giglou PR, Qiao R, Oppenheimer J, Yakar S, LeRoith D. 2011. Growth hormone receptor regulates β cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J Clin Invest 121:2422–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rao MN, Mulligan K, Tai V, Wen MJ, Dyachenko A, Weinberg M, Li X, Lang T, Grunfeld C, Schwarz JM, Schambelan M. 2010. Effects of insulin-like growth factor (IGF)-I/IGF-binding protein-3 treatment on glucose metabolism and fat distribution in human immunodeficiency virus-infected patients with abdominal obesity and insulin resistance. J Clin Endocrinol Metab 95:4361–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Clemmons DR. 2004. Role of insulin-like growth factor in maintaining normal glucose homeostasis. Horm Metab Res 62:77–82 [DOI] [PubMed] [Google Scholar]
- 62. Macotela Y, Boucher J, Tran TT, Kahn CR. 2009. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes 58:803–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mauvais-Jarvis F. 2011. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol Metab 22:24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Medrikova D, Jilkova ZM, Bardova K, Janovska P, Rossmeisl M, Kopecky J. 3 May 2011. Sex differences during the course of diet-induced obesity in mice: adipose tissue expandability and glycemic control. Int J Obes 10.1038/ijo.2011.87 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.