

Abstract
The projected prevalence of Cushing's syndrome (CS) inclusive of subclinical cases in the adult population ranges from 0.2–2% and it may no longer be considered as an orphan disease (2–3 cases/million/year). The recognition of CS by physicians is important for early diagnosis and treatment. Late-night salivary cortisol, dexamethasone suppressiontesti, or 24-h urine free cortisol are good screening tests. Positively screened cases need stepwise evaluation by an endocrinologist. This paper discusses the importance of screening for CS and suggests a stepwise diagnostic approach to a case of suspected hypercortisolism.
Keywords: Cushing's syndrome, dexamethasone suppression tests, hypercortisolism, salivary cortisol, urinary free cortisol
INTRODUCTION
Cushing's syndrome (CS) is a conglomeration of signs and symptoms resulting from sustained pathological hypercortisolism. On the one hand, diagnosis of CS is often missed initially owing to its rarity and overlapping characteristics with common disorders like metabolic syndrome. It is generally recognized later in its full-blown state. Classical cases typically have a history of symptoms for 1–2 years before the confirmation of diagnosis. Timely diagnosis and treatment of CS is important to decrease morbidity and mortality. To increase awareness about CS amongst patients and physicians alike, United States has declared April 8, 2007 as the National CS Awareness Day. In 2008, The Endocrine Society published clinical practice guidelines for diagnosis and treatment of CS.[1] In this clinical review, a stepwise approach for diagnosis of hypercortisolism is discussed so as to minimize both missing and misdiagnosing CS
Undoubtedly, diagnosis of CS is often missed and delayed for the common metabolic syndrome variants. But the opposite is also true where over diagnosis of this condition can mislead us. In this clinical review, a stepwise approach for diagnosis of hypercortisolism is discussed so as to minimize both missing and misdiagnosing CS.
Step 1: Clinical suspicion and screening for hypercortisolism
There is no rule more invariable than that we are paid for our suspicions by finding what we suspect.
Henry David Thoreau
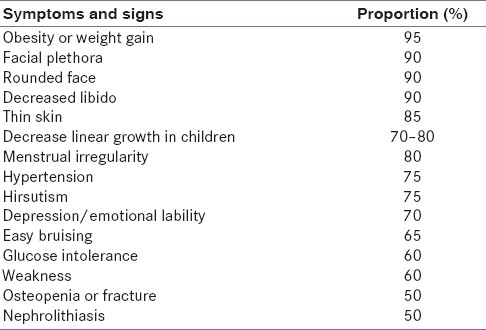
The normal hypothalamic-pituitary-adrenal (HPA) axis exhibits a circadian rhythm. Clinical manifestations of hypercortisolism are the result of an altered rhythm (quality) and excess cortisol load (quantum). Hypercortisolism has a broad spectrum of manifestations.[2] Overt CS is easily recognizable, but it is difficult to delineate subclinical CS from the patients attending obesity, diabetes, and hypertension outpatient clinics. Findings like visceral obesity, glucose intolerance, hypertension, and menstrual irregularities though common in CS are nondiscriminatory. Specific manifestations of hypercortisolism are those affecting skin (livid striae and ecchymoses), muscle (proximal myopathy) and bone (short stature in an obese child, vertebral compression fractures).[1] These catabolic manifestations are subtle in the early course of the disease and manifest with long standing and/or intense hypercortisolism. Hypertrichosis over the forehead is a useful clinical clue. Table 1 summarizes clinical features of CS.
Table 1.
Clinical features of Cushing's syndrome
Clinical intuition backed with an analytical approach is required to establish or exclude a diagnosis of subclinical CS.[3] The Endocrine Society guidelines recommend screening under the following circumstances:
patients with multiple, progressive, and discriminatory findings suggestive of CS
cases with unusual features like hypertension or osteoporosis at a young age
adrenal incidentalomas
children with a decreasing height percentile and increasing weight
Subclinical CS is seen in 5–30% cases of adrenal incidentalomas, which in turn are found in 4–7% of the adult population. Extrapolating this data, the prevalence of subclinical CS in adults would be 0.2–2.0%.[4] CS is one of the differential diagnosis of polycystic ovarian syndrome, which is actually a diagnosis of exclusion.[5] Screening every diabetic patient for CS is not recommended even though the prevalence of occult CS ranges from 1–10% in various series.[6] This is because screening in this group is often associated with very high false positivity. In a study of 171 obese diabetic patients, 31 had unsuppressed overnight dexamethasone suppression cortisol. On follow-up testing, only three of them had elevated 24-h urinary free cortisol, two of whom were shown to have alcoholic pseudo-CS. Finally, there was only one true CS out of 31 patients with unsuppressed overnight dexamethasone suppression cortisol.[7] In such situations where clinical discrimination is poor, the pretest probability of CS in a diabetic individual equals the prevalence of this disorder in a diabetic cohort. With such a poor pretest probabilityeven with the use of specific tests having good likelihood ratio, the posttest probability falls below 20%, which does not make us wiser.[3] Based on this statistical knowledge, routine screening in patients with individual components of metabolic syndrome is not advised at present.
Step 2: Establishing endogenous hypercortisolism
When you have mastered numbers, you will in fact no longer be reading numbers, any more than you read words when reading books. You will be reading meanings.
W. E. B. Du Bois
Before proceeding to the hormonal tests, history of exogenous glucocorticoid intake is imperative to rule out exogenous CS.[8] In India, cases of exogenous CS are commonly encountered, secondary to the ingestion of powdered/injectable medications for pains and aches dispensed indiscriminately. Some features such as increased intraocular pressure, cataracts, benign intracranial hypertension, aseptic necrosis of femoral head, osteoporosis, and pancreatitis are commoner in iatrogenic than in endogenous CS. The diagnosis is obvious, though can be confirmed by drug analysis and suppressed basal serum cortisol and plasma adrenocorticotropic hormone (ACTH).
History of depression, severe obesity, or chronic alcoholism may suggest pseudo-CS, i.e., reversible HPAaxis hyperactivity. Usually such cases are clinically mild, may show suppressed cortisol with dexamethasone suppression test, and reverse to normal with treatment of an associated condition. The insulin tolerance test or loperamide test are rarely indicated.[9]
The test available for the quantitative estimation of cortisol load is 24-h urinary free cortisol. To assess the circadian rhythm and quality of the HPA axis, midnight serum cortisol and late night salivary cortisol tests are useful. The supressibility of the HPA axis is judged by the overnight dexamethasone suppression test (ODS), standard 2-day low-dose dexamethasone suppression
test (LDDS), and LDDS–corticotrophin-releasing hormone (CRH) stimulation test. The methodology of these tests is briefly mentioned in Table 2.[1,10–13] initial evaluation requires the determination of either UFC, late night salivary cortisol, ODS, or LDDS. In patients with a high index of clinical suspicion and an equivocal, discordant or negative initial test result, the subsequent evaluation of either midnight serum cortisol level or LDDS–CRH stimulation test is required. A schematic diagram for diagnosing a patient suspected of having CS is shown in Figure 1. The rationale behind such an approach is that initial tests have a high degree of sensitivity to rule out CS to a large extent. In case of an initial positive result, repeating the test and carrying out other tests increases the overall specificity thereby decreasing the chances of misdiagnosis.[14]
Table 2.
Tests for diagnosis of endogenous hypercortisolism
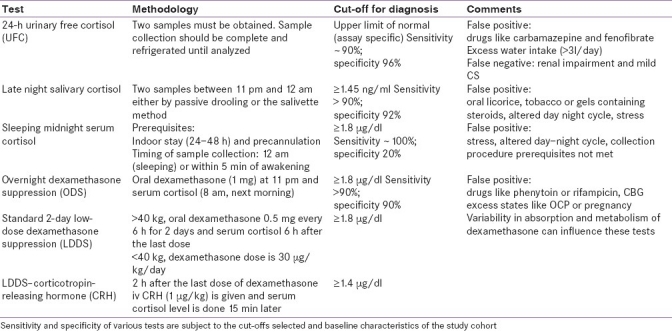
Figure 1.
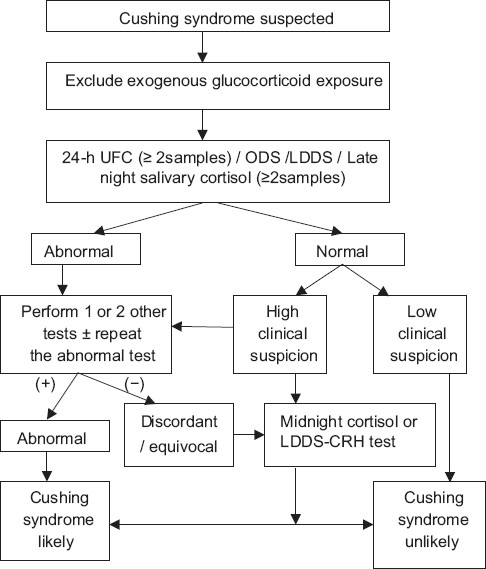
Algorithm for evaluating patients suspected of having CS
In a patient with classical features of CS all tests perform well. The job of an endocrinologist is to delineate subclinical CS from the cohort of metabolic syndrome cases. The specificities of various tests to exclude CS in a cohort of an obese population are reported to be 96% for UFC, 90% for
ODST and 92% for salivary cortisol.[15] The test results seem good; however, in actual sense, for an uncommon disorder like CS even a specificity of up to 99% may not be discriminatory. The following example elucidates this concept. In a cohort of 1000 obese people, consider that there is one case of CS. Even when a test with 99% specificity is applied, there would be 11 persons who would test positive (10 false positive + 1 true positive) for CS. Moreover, it is not easy to delineate the CS case from false positive ones even for the experts. So when the pretest probability is low, in order to increase the likelihood ratio, the endocrinologist must read the same test at higher cut-offs and analyze the results of multiple tests together.[3,14] In case of doubt, a close follow-up of the case with a wait-and-watch policy is best.
Step 3: Determining the source of endogenous hypercortisolism
If you have never missed the diagnosis of ACTH dependent Cushing syndrome, and you have never been fooled attempting to establish its cause, you should refer your patients with suspected hypercortisolism to somebody who has.
James Findling
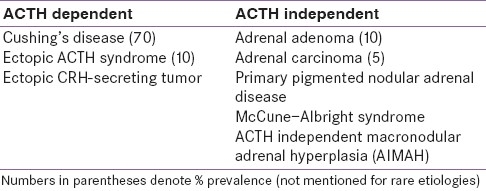
After excluding the exogenous CS, CD is the most common cause of endogenous hypercortisolism. Other causes are mentioned in Table 3.
Table 3.
Various causes of hypercortisolism
Adrenocorticotropic hormone dependency versus independency
Basal plasma ACTH with a two-site immunometric assay is a useful parameter for the diagnosis of adrenal CS. Patients with suppressed morning plasma ACTH (< 5 pg/ml) need adrenal imaging. Normal or elevated ACTH (>15 pg/ml) is suggestive of ACTH-dependent CS. Plasma ACTH values between 5 and 15 pg/ml are equivocal.[9] In our series of CS patients, midnight plasma ACTH value of >7.5 pg/ml was suggestive of CD.[16] Even with an autonomous cortisol secreting adrenal adenoma, plasma ACTH levels may not be completely suppressed. Differentiation may not be easy as unilateral or bilateral adrenal enlargement is frequent in patients with an ACTH hypersecretion. Baseline CRH stimulation test can help to delineate the source in equivocal cases. Plasma ACTH peak levels of <30 pg/ml after corticotrophin releasing hormone CRH stimulation test excludes the presence of an ACTH-dependent hypercortisolism.[17]
Adrenocorticotropic hormone-dependent cases: CD versus ectopic CS
A clinical setting provides important clues towards differential diagnosis. Females with, moderate hypercortisolism, mildly elevated plasma ACTH and normokalemia point towards CD. In contrast, males presenting with severe hypercortisolism, markedly elevated plasma ACTH and hypokalemia are more likely to have an occult ectopic ACTH-secreting tumor.[14]
In case of ACTH-dependent endogenous hypercortisolism, the pretest probability for pituitary origin is >80% overall and >90% in women[14] Hence magnetic resonance imaging (MRI) of the pituitary region is the next step. There is emerging data that the spoiled gradient-recalled acquisition technique is superior to the conventional dynamic contrast spin echo.[18] The reported rate of visualization of pituitary microadenomas on MRI ranges from 50% to 70%. If MRI results are negative or equivocal, further tests are required.
High-dose dexamethasone suppression test (HDDS), CRH stimulation test, and vasopressin stimulation test have been used in the past to differentiate CD from ectopic CS. None of the tests reached specificity of >90%, implying that the information generated through these tests is no better than the empirical pretest probability of it being of pituitary origin. There is hardly any difference between the results of HDDS in patients with CD and those with the ectopic ACTH syndrome. The continued use of this test is not justified.[14] The only test which performs well is CRH-stimulated bilateral inferior petrosal sinus sampling test (IPSS).[19,20] A pituitary: peripheral ACTH gradient of > 3 is suggestive of pituitary origin (specificity 97%). False negative results on IPSS have been attributed to anatomical problems like anomalous venous drainage where as false positive IPSS results have been described with rare case reports of an inactive phase of cyclical ectopic CS and CRH secreting ectopic CS. Lateralization of the tumor with IPSS is possible only in 60–70% of patients, with the reported complication rate being <1% with an experienced vascular intervention radiologist. This test is invasive and the CRH injection has to be imported to India. In clinical practice, IPSS is reserved for patients with normal imaging of the pituitary, neck, chest, and abdomen and hence prior to IPSS, a computed tomography (CT) scan of the neck, chest, and abdomen is performed to rule out an obvious ectopic tumor. If the results of IPSS are negative, then extensive imaging (MRI, CT scan and nuclear scans) are indicated to locate an ectopic source (neuroendocrine tumors like carcinoid of thymus, bronchiole or pancreas, pheochromocytoma, medullary thyroid carcinoma).[21,22] At times, the source of ectopic CS remains occult and may reveal itself on follow-up.
Adrenocorticotropic hormone independent CS: Different etiologies
A CT scan of the adrenal gland can phenotype the adrenal lesion as benign (adenoma) and malignant (carcinomas) depending on the lipid content and contrast wash-out characters. Primary pigmented nodular adrenal disease (PPNAD)and McCune–Albright syndrome are unique causes of childhood CS. The classical paradoxical rise in cortisol after LDDS test is a feature of PPNAD.[23] ACTH independent macronodular adrenal hyperplasia (AIMAH) is a rare familial disorder, in which adrenal steroidogenesis is driven by non-ACTH peptides like vasopressin, glucose-dependent intestinal peptides, catecholamines, luteinizing hormone, etc. A specific test protocol has been described to recognize the stimulating peptide.[24] It is interesting to note that the identification of aberrant adrenal hormone receptors in AIMAH provides opportunities for new specific pharmacological therapies as alternatives to adrenalectomy.
CONCLUSION
Conducting a battery of tests for a clinically florid CS case to prove endogenous hypercortisolism is easy. Real difficulty arises while dealing with the subtle cases, where even the experienced endocrinologists have a tough time interpreting the test results and may resort to the wait-and-watch policy. The basic dictum which still serves as a golden rule is not to proceed further until there is convincing evidence of biochemical hypercortisolism.
While localizing the source, suppressed levels of plasma ACTH and positive adrenal imaging pave an easy path. In case of ACTH-dependent CS, an MRI scan report of clearly defined pituitary adenoma is rewarding. Equivocal or negative findings on an MRI scan mandates CRH-stimulated IPSS. Anyone who has encountered a good number of cases would have regretted his or her decision (at least once), by reliance on the pretest probability of it being pituitary without an IPSS. Localizing an ectopic tumor source is a herculean task.
Finally, efforts to fine-tune our approach to the more difficult aspects of CS must not eclipse with efforts to educate and encourage guided screening for patients t who might benefit from evaluation.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing's syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526–40. doi: 10.1210/jc.2008-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah NS, George J, Acharya SV, Lila AR, Sarathi V, Bandgar TR, et al. Cushing's disease in children and adolescents: Twenty years’ experience from a tertiary care center in India. Endocr Pract. 2011;17:369–76. doi: 10.4158/EP10143.OR. [DOI] [PubMed] [Google Scholar]
- 3.Aron DC. Cushing's syndrome: Why is diagnosis so difficult? Rev Endocr Metab Disord. 2010;11:105–16. doi: 10.1007/s11154-010-9127-3. [DOI] [PubMed] [Google Scholar]
- 4.Chiodini I. Diagnosis and treatment of subclinical hypercortisolism. J Clin Endocrinol Metab. 2011;96:1223–36. doi: 10.1210/jc.2010-2722. [DOI] [PubMed] [Google Scholar]
- 5.Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: The complete task force report. Fertil Steril. 2009;91:456–88. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 6.Tabarin A, Perez P. Pros and cons of screening for occult Cushing syndrome. Nat Rev Endocrinol. 2011 doi: 10.1038/nrendo.2011.51. [In press] [DOI] [PubMed] [Google Scholar]
- 7.Newsome S, Chen K, Hoang J, Wilson JD, Potter JM, Hickman PE. Cushing's syndrome in a clinic population with diabetes. Intern Med J. 2008;38:178–82. doi: 10.1111/j.1445-5994.2007.01434.x. [DOI] [PubMed] [Google Scholar]
- 8.Carroll TB, Findling JW. The diagnosis of Cushing's syndrome. Rev Endocr Metab Disord. 2010;11:147–53. doi: 10.1007/s11154-010-9143-3. [DOI] [PubMed] [Google Scholar]
- 9.Findling JW, Raff H. Cushing's syndrome: Important issues in diagnosis and management. J Clin Endocrinol Metab. 2006;91:3746–53. doi: 10.1210/jc.2006-0997. [DOI] [PubMed] [Google Scholar]
- 10.Newell-Price J, Trainer P, Perry L, Wass J, Grossman A, Besser M. A single sleeping midnight cortisol has 100% sensitivity for the diagnosis of Cushing's syndrome. Clin Endocrinol (Oxf) 1995;43:545–50. doi: 10.1111/j.1365-2265.1995.tb02918.x. [DOI] [PubMed] [Google Scholar]
- 11.Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing's syndrome. Lancet. 2006;367:1605–17. doi: 10.1016/S0140-6736(06)68699-6. [DOI] [PubMed] [Google Scholar]
- 12.Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B. Evidence for the low dose dexamethasone suppression test to screen for Cushing's syndrome–recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34:222–9. doi: 10.1177/000456329703400302. [DOI] [PubMed] [Google Scholar]
- 13.Jeyaraman K, Ammini AC, Nandita G, Dwivedi SN. Late-night salivary cortisol in normal subjects and in patients with Cushing's syndrome. Postgrad Med J. 2010;86:399–404. doi: 10.1136/pgmj.2009.090787. [DOI] [PubMed] [Google Scholar]
- 14.Elamin MB, Murad MH, Mullan R, Erickson D, Harris K, Nadeem S. Accuracy of diagnostic tests for Cushing's syndrome a systematic review and metaanalyses. J Clin Endocrinol Metab. 2008;93:1553–62. doi: 10.1210/jc.2008-0139. [DOI] [PubMed] [Google Scholar]
- 15.Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK. Specificity of screening tests for Cushing's syndrome in an overweight and obese population. J Clin Endocrinol Metab. 2009;94:3857–64. doi: 10.1210/jc.2008-2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.John M, Lila AR, Bandgar T, Menon PS, Shah NS. Diagnostic efficacy of midnight cortisol and midnight ACTH in the diagnosis and localization of Cushing's syndrome. Pituitary. 2010;13:48–53. doi: 10.1007/s11102-009-0197-8. [DOI] [PubMed] [Google Scholar]
- 17.Raff H, Findling JW. A physiologic approach to diagnosis of the Cushing syndrome. Ann Intern Med. 2003;138:980–91. doi: 10.7326/0003-4819-138-12-200306170-00010. [DOI] [PubMed] [Google Scholar]
- 18.Patronas N, Bulakbasi N, Stratakis CA, Lafferty A, Oldfield EH, Doppman J. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003;88:1565–9. doi: 10.1210/jc.2002-021438. [DOI] [PubMed] [Google Scholar]
- 19.Findling JW, Kehoe ME, Raff H. Identification of patients with cushing's disease with negative pituitary adrenacorticotropin gradients during inferior petrosal sinus sampling: Prolactin as an index of pituitary venous effluent. J Clin Endocrinol Metab. 2004;89:6005–9. doi: 10.1210/jc.2004-1378. [DOI] [PubMed] [Google Scholar]
- 20.Kalgikar AM, Chandratreya SA, Goel A, Shrivastava M, Limaye US, Karvat A, et al. Inferior petrosal sinus sampling in the diagnostic evaluation of Cushing's syndrome: K.E.M. experience. J Assoc Physicians India. 2005;53:685–8. [PubMed] [Google Scholar]
- 21.Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing's syndrome due to ectopic corticotropin secretion: Twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab. 2005;90:4955–62. doi: 10.1210/jc.2004-2527. [DOI] [PubMed] [Google Scholar]
- 22.Bhansali A, Walia R, Rana SS, Dutta P, Radotra BD, Khandelwal N. Ectopic Cushing's syndrome: Experience from a tertiary care centre. Indian J Med Res. 2009;129:33–41. [PubMed] [Google Scholar]
- 23.Ganesh HK, George J, Vimal MV, Bandgar T, Menon PS, Shah NS. An unusual variant of Cushing syndrome. Endocr Pract. 2008;14:717–20. doi: 10.4158/EP.14.6.717. [DOI] [PubMed] [Google Scholar]
- 24.Lacroix A. ACTH independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:245–59. doi: 10.1016/j.beem.2008.10.011. [DOI] [PubMed] [Google Scholar]