

Abstract
In contrast to other chromatographic methods for purifying proteins (e.g. gel filtration, affinity, and ion exchange), hydrophobic interaction chromatography (HIC) commonly requires experimental determination (referred to as screening or "scouting") in order to select the most suitable chromatographic medium for purifying a given protein 1. The method presented here describes an automated approach to scouting for an optimal HIC media to be used in protein purification.
HIC separates proteins and other biomolecules from a crude lysate based on differences in hydrophobicity. Similar to affinity chromatography (AC) and ion exchange chromatography (IEX), HIC is capable of concentrating the protein of interest as it progresses through the chromatographic process. Proteins best suited for purification by HIC include those with hydrophobic surface regions and able to withstand exposure to salt concentrations in excess of 2 M ammonium sulfate ((NH4)2SO4). HIC is often chosen as a purification method for proteins lacking an affinity tag, and thus unsuitable for AC, and when IEX fails to provide adequate purification. Hydrophobic moieties on the protein surface temporarily bind to a nonpolar ligand coupled to an inert, immobile matrix. The interaction between protein and ligand are highly dependent on the salt concentration of the buffer flowing through the chromatography column, with high ionic concentrations strengthening the protein-ligand interaction and making the protein immobile (i.e. bound inside the column) 2. As salt concentrations decrease, the protein-ligand interaction dissipates, the protein again becomes mobile and elutes from the column. Several HIC media are commercially available in pre-packed columns, each containing one of several hydrophobic ligands (e.g. S-butyl, butyl, octyl, and phenyl) cross-linked at varying densities to agarose beads of a specific diameter 3. Automated column scouting allows for an efficient approach for determining which HIC media should be employed for future, more exhaustive optimization experiments and protein purification runs 4.
The specific protein being purified here is recombinant green fluorescent protein (GFP); however, the approach may be adapted for purifying other proteins with one or more hydrophobic surface regions. GFP serves as a useful model protein, due to its stability, unique light absorbance peak at 397 nm, and fluorescence when exposed to UV light 5. Bacterial lysate containing wild type GFP was prepared in a high-salt buffer, loaded into a Bio-Rad DuoFlow medium pressure liquid chromatography system, and adsorbed to HiTrap HIC columns containing different HIC media. The protein was eluted from the columns and analyzed by in-line and post-run detection methods. Buffer blending, dynamic sample loop injection, sequential column selection, multi-wavelength analysis, and split fraction eluate collection increased the functionality of the system and reproducibility of the experimental approach.
Protocol
1. Buffer and Sample Preparation
Technical note: All buffers should be refrigerated and each step of this protocol should be performed on ice or at 4 °C, unless otherwise noted. The low temperature is essential to prevent degradation of the protein to be purified and to ensure optimum operating conditions. HIC media may produce different separation results if operated at room temperature, and the chromatography system should be operated in a 4 °C cold room or cold box.
Prepare 500 ml of the following buffers: 200 mM NaH2PO4 (Buffer A1), 200 mM Na2HPO4, (Buffer A2), and 4.8 M (NH4)2SO4 (Buffer B2). Deionized water (Buffer B1) will also be used. Degas the buffer to ensure optimal chromatographic operating conditions. The system maximizer will blend together Buffers A1 and A2 to generate a low-salt phosphate elution buffer, and it will blend Buffers B1 and B2 to generate the high-salt ammonium sulfate start buffer (2.4 M (NH4)2SO4).
Prepare 20 ml of sample to be loaded by mixing 10 ml of cell lysate containing the protein to be purified with 10 ml of Buffer B2. The final ammonium sulfate concentration of the cell lysate sample solution should be approximately 2.4 M (NH4)2SO4, making it roughly equivalent to the start buffer. Cell lysate can be prepared by resuspending the bacterial cell pellet in TE buffer (10 mM Tris 1 mM EDTA, pH 8.0) followed by sonication or the addition of 1 mg/ml lysozyme and centrifugation at 3,700 × g for 10 minutes.
Filter the lysate-buffer mixture through a low protein binding 0.45 μm syringe filter to remove particulate matter from the solution. Keep the sample on ice until it is ready to be loaded into the system.
2. Physical Setup and Plumbing of DuoFlow Chromatography System
Ensure all device power and communication connections are made. Devices should be visible in the Biologic DuoFlow chromatography software manual control window. A summary of devices are listed in Table 1. An illustration of the system and plumbing diagram is in Figure 2.
Attach electrical connections for the valves to the pump workstation and system maximizer ports such that the port numbering follows a logical sequence. Label each valve with its respective port number for future reference. The pump workstation and system maximizer serve as the main communication connections between the computer/controller and chromatography system. Valves will be automatically recognized and displayed in the software when connected to either device.
Technical note: While operating the chromatography system in manual mode, valves are controlled independently. Take care to verify that the desired flow path is configured, particularly when switching valves, before initiating buffer flow.
Submerge Teflon tubing into Buffers A1, A2, B1, and B2. Ensure adequate volumes of buffer are available for entire protocol and that tubing will continue to remain submerged. Preparing 500 ml of Buffers A1, A2, and B2 and 1 L of Buffer B1 will be adequate.
Plumb sample loading valve and dynamic sample loop as illustrated in Figure 2 and per manufacturer's instructions. Connect the sample loop to the sample valve inlet (Position 3) using the smallest length of tubing possible. This will minimize sample dilution during the sample load process.
Minimize the tubing length between the sample, sample loading pump, and sample loading valve inlet (Position 2). This will decrease the amount of sample necessary to load the sample loop.
Prepare 8 identically sized pairs of 1/16" OD PEEK tubing, with each pair joined together by a 1/4-28 female-to-1/4-28 female union. Connect the pairs to Positions 1-8 of the two column selection valves. The 7 unions connected in-line to column selection valve Positions 2-8 will later be replaced by the chromatography columns to be tested. The union connected inline with Position 1 will be maintained as a chromatography column bypass.
Technical note: Ensure each pair of tubing is connected to the same position on both column selection valves.
Turn on both the 4-wavelength UV/Vis Detector (QuadTec) lamp and fixed-wavelength 280 nm UV Detector lamp. The wavelengths to be measured by the QuadTec for this protocol include 214 nm, 280 nm, and 397 nm. Measuring 280 nm with both the fixed-wavelength UV Detector and QuadTec provides system redundancy and ensures data validity.
Confirm all detectors are calibrated per the manufacturer's instructions. The backpressure regulator is necessary to reduce air bubble accumulation in detectors and should be installed down-stream of the detectors but up-stream of the pH monitor.
The system presented here includes two fraction collectors, which provides substantial convenience for post-run eluate analysis. Attach the fraction collectors, stream splitter valve, and splitter controller as illustrated in Figure 2. The fraction collector connected to Position 1 of the splitter valve will collect 900 μl eluate fractions in 12 ml culture tubes, while the fraction collector connected to Position 2 will collect 100 μl eluate fractions in 96 well microtiter plates.
The Position 2 fraction collector, splitter valve, and splitter controller are operated offline of the chromatography workstation. The splitter controller is contained within a Bio-Rad Econo Gradient Pump; however, the pump component of the device is not used. A "% Split" setting option will be displayed on the Econo Gradient Pump when the splitter valve is connected to it.
Set % Split to 10. The Position 1 fraction collector will collect 90% of the eluate (900 μl/fraction), and the Position 2 fraction collector will collect 10% (100 μl/fraction). The fraction collectors will advance in synchrony allowing for fraction numbers of both collectors to correspond to each other.
Replace the standard fraction collector drop head on the Position 2 fraction collector with a microplate drop head. The microplate drop head has a smaller aperture, which permits collection of 50% smaller drop sizes (i.e. 25 μl instead of the standard 50 μl) and is particularly beneficial when collecting ≤ 500 μl fractions.
Using the onscreen control panel, set the Position 2 fraction collector to collect fractions in 96-well plates and advance at a rate of 1 sample/minute. The splitter controller and Position 2 fraction collector will be started manually immediately following the start of the first chromatography run.
Flush the Position 2 fraction collector and microplate drop head with Buffer B1.
3. Priming System Lines, Programming Run Method, and Equilibrating HIC Columns
With system maximizer inlet lines submerged into degassed Buffers A1, A2, B1, and B2, prime the workstation pumps, rinse sample loop, and flush the chromatography system per manufacturer's instructions. Complete priming step with all 4 maximizer system inlets (A1, A2, B1, and B2) in order to remove remaining air bubbles.
Preparing the system requires manual operation of the system controller via the BioLogic software. Confirm correct positioning of sample load valve and column selection valves prior to starting buffer flow.
Prepare to start a manual 1 ml/minute flow of elution buffer (0 %B) through the system, as described in Steps 3.4-3.5.
In the Setup window of the BioLogic program software, select "Use Buffer Blending" and "Phosphate Buffer with Ammonium Sulfate". Confirm Buffers A1, A2, B1, and B2 listed in the Setup window correspond to those prepared.
Manually set the flow rate to 1 ml/minute of elution buffer (0 %B). Observe backpressure, conductivity, UV tracings, and pH remain consistent and within normal operating limits. Abnormalities indicate air or column blockage that should be addressed prior to proceeding.
Flush all 8 column selection lines with 5 ml elution buffer. Do this by first stopping the flow, setting both column selection valves to Position 2, and resuming the 1 ml/minute flow, 0 %B. Repeat for column selection lines in Positions 3-8.
Connect 1 ml HiTrap HIC columns to Positions 2-8 of column selection valves. Complete steps 3.8-3.13 with one column at a time. Take note of which HIC column is placed in which selection valve position. A summary of the HIC column media characteristics is listed in Table 2.
The physical connection of GE Healthcare HiTrap columns to the Bio-Rad DuoFlow system requires a series of fittings utilizing 1/4-28, M6, and Luer fittings. Remove the union connecting the Position 2 column selection valves. Align the Bio-Rad and GE Healthcare fittings as illustrated in Figure 3.
Attach upstream column fittings to the DuoFlow. Avoid trapping air bubbles in the column or tubing by firmly attaching the upstream column fittings to the upstream selection valve fitting. Run elution buffer through the fittings until all air is expelled and a large drop of buffer is present. Temporarily stop buffer flow.
Remove the stopper connected to the column inlet and place a large drop of low-salt buffer (0 %B) on the top of the column. Attach the column to the upstream fittings. Having both the column inlet and inlet fitting overflowing with drops of buffer ensures a connection free of air bubbles.
If the column is being used for the first time, snap off the column outlet end. Attach the outlet to the downstream column fittings and selection valve tubing. Verify all fittings are tightly fastened.
Set the backpressure limit to 40 psi. Wash the column with 5 column volumes (5 column volumes = 5 ml) of elution buffer (0 %B) at a flow rate of 1 ml/minute. Continue column wash, if necessary, until pH, UV tracings and backpressure have stabilized.
Wash column with 10 column volumes (10 ml) start buffer (100 %B) at a flow rate of 1 ml/minute. Continue column wash, if necessary, until system parameters listed in the preceding step as well as conductivity have stabilized.
The HIC column prepared above is now ready for sample loading. Once all columns to be tested are prepared, manually switch the column selection valves to Position 1 (union) and stop buffer flow.
4. Sample Loading
Submerge sample inlet tubing into 20 ml sample to begin the process of loading sample into the dynamic sample loop. From the manual setting window of the BioLogic software, switch sample loop load/wash valve to Position 1 (Sample) and the sample loading valve to Purge.
Initiate action of the sample loop loading pump, drawing up sample into the pump tubing at a flow rate of 1 ml/minute until immediately after sample reaches the sample loading valve. This removes buffer and/or air from the sample load line.
Stop the flow of the loading pump and switch the sample loading valve from Purge to Load. Restart the sample loop loading pump at a flow rate of 1 ml/minute until 10 ml of sample has been drawn into the dynamic sample loop.
Sufficient sample is now loaded into the dynamic sample loop for up to 10 sequential HIC column scouting runs.
5. Programming the Software Method and Running the Column Scouting Protocol
From the Setup window on the BioLogic software, select the system devices marked with an asterisk in Table 1.
Select 6 HIC columns to assay. Program a linear descending salt gradient and 6-column scouting method, as illustrated in Table 3. The method program is a modification of the BioLogic software HIC column setup and has been optimized for the current application.
A 6-column scouting method is recommended due to fraction collector limitations. All 7 columns can be scouted if the program method is adjusted to collect larger fraction volumes. Do not add the scouting step until the program is otherwise complete, as selecting the scouting feature prevents subsequent editing.
Begin the scouting run. Manually commence start buffer (100 %B) flow, 1 ml/minute. Press start on the splitter controller to commence 90%-10% eluate stream-splitting between the 12 ml culture tube fraction collector (Position 1) and the 96-well plate fraction collector (Position 2), respectively. Technical note: Recall the 96-well plate fraction collector is operated offline from the BioLogic software.
Run the program method. Immediately after starting the program method run, press start on the onscreen control panel of the offline 96-well plate fraction collector. If the two fraction collectors are not 100% synchronous, manually advance the offline 96-well plate fraction collector as the other fraction collector advances to the second fraction collection tube.
Observe the real time display to confirm anticipated run parameters. Backpressure, conductivity, and %B (Figure 4) are run parameters that can be monitored to ensure column-to-column reproducibility of run conditions. Valve positioning, pH, and sample loading should also be monitored. UV tracings can be compared to verify similar flow-through elution peaks.
Utilize in-line and post-run eluate analysis techniques to identify the elution profile and purification of the protein of interest (i.e. "target protein").
For samples containing GFP, observe the elution in-line using the protein's unique UV absorbance at 397 nm. Compare the GFP-specific 397 nm absorbance tracing to the general protein 280 nm absorbance tracing to estimate relative separation and purification using the different HIC media being scouted (Figure 5).
Post-run analysis of GFP and other target proteins can be accomplished through multiple methods. Aliquots of 5-50 μl from the 96-well plate fractions can be transferred to a fresh plate and assayed for specific protein content by ELISA or western blot, and total protein content of each fraction can be analyzed by using conventional SDS-PAGE or an Experion microfluidic electrophoresis system. GFP fluorescence can be detected in 12 ml culture tubes using UV light (Figure 6).
Identification of most promising HIC media can be determined by comparison of GFP elution relative to other proteins contained in the sample. Purification with the most promising HIC media can then be optimized according to other parameters, including type and concentration of start buffer and pH.
Technical note: At the conclusion of the experiment, place all inlet lines into Buffer B1 (degassed H2O) and thoroughly rinse the pumps, valves, and all tubing with water. Follow manufacturers' instructions for cleaning and long-term storage of chromatography system and HIC columns.
6. Representative Results:
Representative HIC salt gradient, conductivity, and column pressure of the scouting runs are presented in Figure 4. The change in salt concentration (blue line), as measured by the percentage of buffer drawn from high-salt buffer lines is typical of HIC methodology. As the salt concentration decreases, proteins bound to the column elute. Conductivity (red line), which corresponds to observed salt concentration, is measured in-line immediately following the QuadTec and UV detectors. The off-set between salt gradient and conductivity tracings indicates the time required for buffer to travel from the buffer inlet, through the system, and to the conductivity monitor. Throughout the sample run, the system pressure (grey lines) and pH remain relatively constant.
Figure 5 shows chromatograms of sequential HIC column scouting runs. The in-line detection of total protein (A280, blue line) and GFP (A397, green line) is accomplished by measuring the absorbance of light at 280 nm and 397 nm, respectively. It is possible to approximate the relative GFP abundance and separation for each scouting run by comparing the two lines. GFP bound to the tested HIC columns with different degrees of affinity and its elution profile varied. Selection of a preferred HIC column for future purifications is based on identifying the column that produces the sharpest GFP elution peak and greatest separation from other proteins.
In Figure 6, culture tubes for Fractions 10, 12, 14, 16, and 18 of the Phenyl FF (high sub) scouting run were visualized under ambient room (fluorescent) light and ultraviolet (UV) light. Tubes were viewed both face-forward (left panels) and top-down (right panels) in order to observe the characteristic GFP emission spectrum. GFP (green) is clearly detected in Fraction 14 in both UV images. These post-run data correspond nicely with the in-line detection of GFP by measuring eluate absorbance at 397 nm (A397, green line), which also identifies Fraction 14 as containing the major peak of eluted GFP. The diffuse blue in the left UV panel is light emitted from the UV lamp.
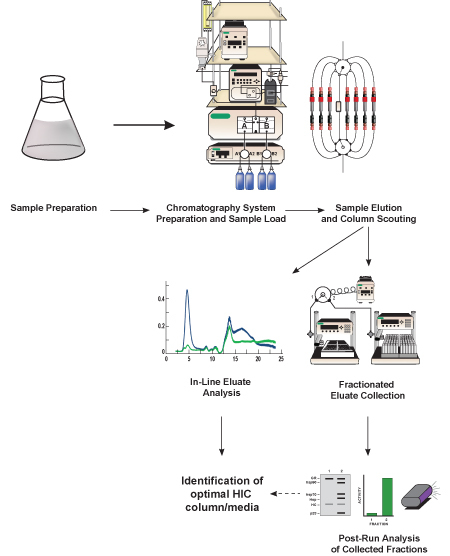 Figure 1. Schematic representation of this protocol. Bacterial lysate containing protein of interest (GFP, target protein) is prepared, diluted into a high-salt buffer equivalent to the chromatography start buffer, and filtered. Once the liquid chromatography system is prepared, sample is loaded and the target protein is separated from other proteins contained in the lysate using a hydrophobic chromatography medium contained in a pre-packed HIC column. The separation method is repeated several times using different chromatography media (referred to as column scouting) in order to determine which media provides the best GFP separation. The eluted proteins (eluate) is analyzed in-line using detectors included in the chromatography system and it is collected into smaller fractions for subsequent (post-run) analysis. Based on the in-line and post-run analysis of GFP activity and separation, an optimum HIC column and chromatographic medium are identified.
Figure 1. Schematic representation of this protocol. Bacterial lysate containing protein of interest (GFP, target protein) is prepared, diluted into a high-salt buffer equivalent to the chromatography start buffer, and filtered. Once the liquid chromatography system is prepared, sample is loaded and the target protein is separated from other proteins contained in the lysate using a hydrophobic chromatography medium contained in a pre-packed HIC column. The separation method is repeated several times using different chromatography media (referred to as column scouting) in order to determine which media provides the best GFP separation. The eluted proteins (eluate) is analyzed in-line using detectors included in the chromatography system and it is collected into smaller fractions for subsequent (post-run) analysis. Based on the in-line and post-run analysis of GFP activity and separation, an optimum HIC column and chromatographic medium are identified.
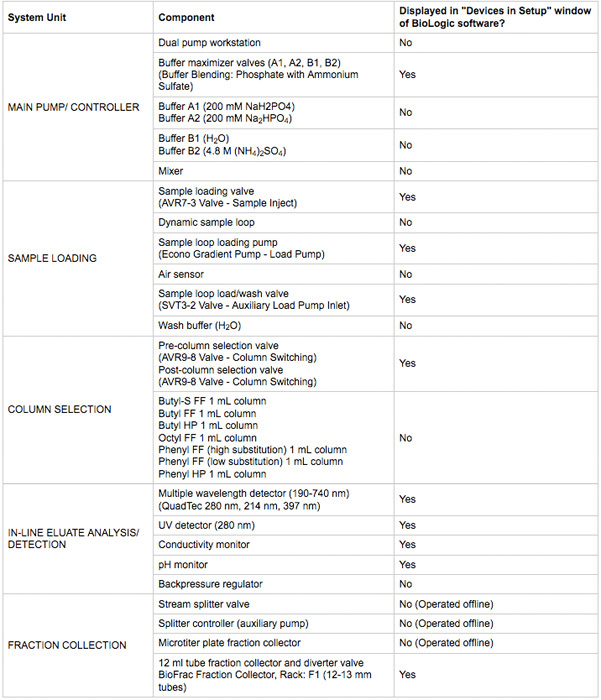 Table 1. Key chromatography system components used in this protocol.
Table 1. Key chromatography system components used in this protocol.
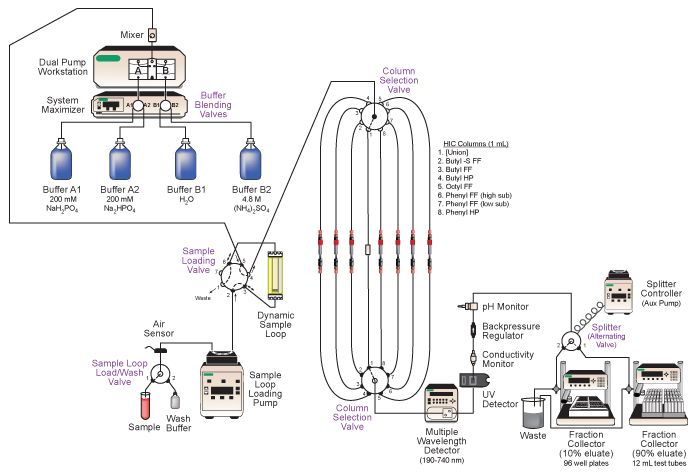 Figure 2. Diagram of Bio-Rad DuoFlow medium-pressure liquid chromatography system employed. Key features of the system include automated buffer blending, loading of sample for sequential column runs, automated selection of different chromatography columns, in-line analysis of the eluate, and tandem collection of the fractionated eluate. See text and Table 1 for details.
Figure 2. Diagram of Bio-Rad DuoFlow medium-pressure liquid chromatography system employed. Key features of the system include automated buffer blending, loading of sample for sequential column runs, automated selection of different chromatography columns, in-line analysis of the eluate, and tandem collection of the fractionated eluate. See text and Table 1 for details.
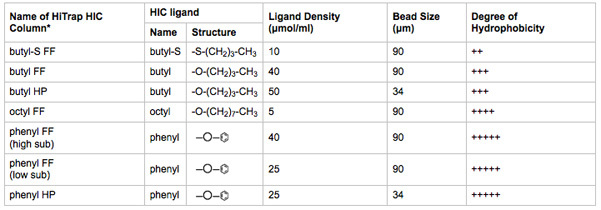 Table 2. Biophysical characteristics of HIC media tested. The name of the HiTrap HIC column is indicative of the ligand, ligand density, and bead size.
Table 2. Biophysical characteristics of HIC media tested. The name of the HiTrap HIC column is indicative of the ligand, ligand density, and bead size.
*FF = fast flow; HP = high performance. Larger bead size increases ligand binding capacity and flow rate. Smaller bead size increases chromatographic resolution. Information derived from literature provided by the manufacturer.
 Figure 3. Connection of GE Healthcare HiTrap HIC column to Bio-Rad DuoFlow system.To the upstream 1/4-28 Delrin nut and ferrule (A1), a male Luer-to-female 1/4-28 fitting (B1) and male 1/16"-to-female Luer fitting (C) are connected. The fittings are attach to the HiTrap column after being flushed with buffer and having all bubbles removed. The column outlet is connected to a female 1/16"-to-male M6 fitting (D), male Luer-to female M6 fitting (E), and female Luer-to-female 1/4-28 fitting (B2). This entire assembly is connected to the downstream 1/4-28 Delrin nut and ferrule (A2). The upper panel shows the fittings separated and the lower panel shows the fittings assembled.
Figure 3. Connection of GE Healthcare HiTrap HIC column to Bio-Rad DuoFlow system.To the upstream 1/4-28 Delrin nut and ferrule (A1), a male Luer-to-female 1/4-28 fitting (B1) and male 1/16"-to-female Luer fitting (C) are connected. The fittings are attach to the HiTrap column after being flushed with buffer and having all bubbles removed. The column outlet is connected to a female 1/16"-to-male M6 fitting (D), male Luer-to female M6 fitting (E), and female Luer-to-female 1/4-28 fitting (B2). This entire assembly is connected to the downstream 1/4-28 Delrin nut and ferrule (A2). The upper panel shows the fittings separated and the lower panel shows the fittings assembled.
| Step # | Volume (mL) | Description | Parameters | |
| 1 | 0.00 | Collect 1.0 mL fractions during entire run | ||
| 2 | 0.00 | Switch Columns | HIC Column 1 (Position 2) | |
| 3 | 0.00 | Isocratic Flow | pH: 6.80 100% B | Volume: 2.00 mL Flow: 1.00 mL/min |
| 4 | 2.00 | Zero Baseline | QuadTec | |
| 5 | 2.00 | Zero Baseline | UV Detector | |
| 6 | 2.00 | Inject Sample | Sample Load Dynamic Loop | Auto Inject Valve Volume: 0.50 mL Flow: 1.00 mL/min |
| 7 | 3.00 | Isocratic Flow | pH: 6.80 100%B | Volume: 5.00 mL Flow: 1.00 mL/min |
| 8 | 8.00 | Linear Gradient | pH: 6.80 100%B -> 0%B | Volume: 10.00 mL Flow: 1.00 mL/min |
| 9 | 18.00 | Isocratic Flow | pH: 6.80 0%B | Volume: 3.00 mL Flow: 1.00 mL/min |
| 10 | 21.00 | Isocratic Flow | pH: 6.80 100%B | Volume: 5.00 mL Flow: 1.00 mL/min |
| 11 | 26.00 | Switch Columns | Union (Position 1) | |
| End | 26.00 | End of Protocol | Automatically Repeats with 5 additional HIC columns (Positions 3-7) | |
| Scout type | Number of Runs: 6 | Number of Steps Scouted: 1 |
Table 3. BioLogic software protocol for HIC scouting method employed.
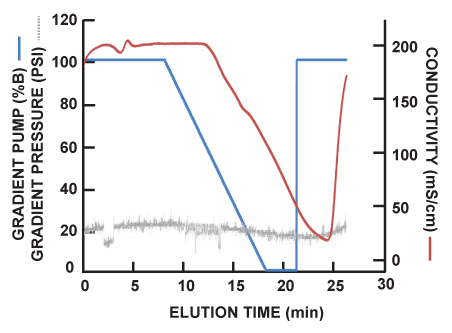 Figure 4. Representative HIC salt gradient, conductivity, and column pressure. As the salt concentration (blue line) decreases, conductivity (red line) does as well. The off-set between salt gradient and conductivity tracings indicates time required for buffer to travel from the buffer inlet to the conductivity monitor. System pressure (grey lines) remains relatively constant for the duration of the scouting run.
Figure 4. Representative HIC salt gradient, conductivity, and column pressure. As the salt concentration (blue line) decreases, conductivity (red line) does as well. The off-set between salt gradient and conductivity tracings indicates time required for buffer to travel from the buffer inlet to the conductivity monitor. System pressure (grey lines) remains relatively constant for the duration of the scouting run.
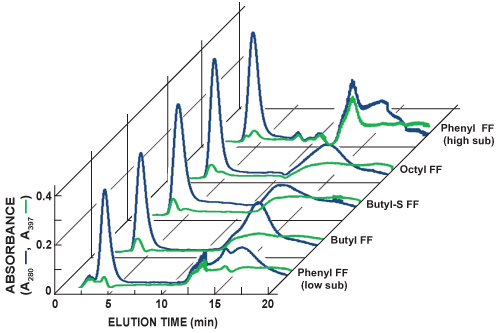 Figure 5. Compiled chromatograms of sequential HiTrap HIC column scouting runs. The in-line detection of total protein (A280, blue line) and GFP (A397, green line) is accomplished by measuring the absorbance of light at 280 nm and 397 nm, respectively. In this series of experiments, the sharpest GFP elution peak was observed with the Phenyl FF (high sub) column. The Phenyl FF (high sub) column also appeared to provide the greatest separation between GFP and other proteins. Additional 1 ml HiTrap HIC columns tested included Phenyl fast flow low substitution (Phenyl FF (low sub)), butyl fast flow (butyl FF), butyl-S fast flow (butyl-S FF), and octyl fast flow (octyl FF).
Figure 5. Compiled chromatograms of sequential HiTrap HIC column scouting runs. The in-line detection of total protein (A280, blue line) and GFP (A397, green line) is accomplished by measuring the absorbance of light at 280 nm and 397 nm, respectively. In this series of experiments, the sharpest GFP elution peak was observed with the Phenyl FF (high sub) column. The Phenyl FF (high sub) column also appeared to provide the greatest separation between GFP and other proteins. Additional 1 ml HiTrap HIC columns tested included Phenyl fast flow low substitution (Phenyl FF (low sub)), butyl fast flow (butyl FF), butyl-S fast flow (butyl-S FF), and octyl fast flow (octyl FF).
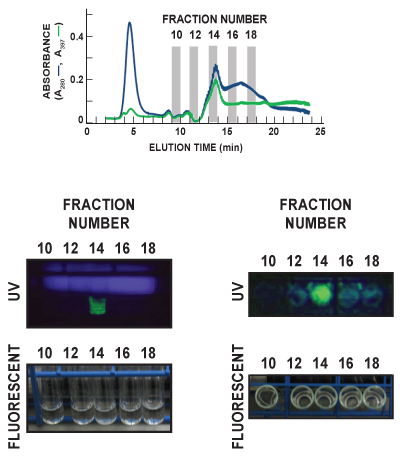 Figure 6. Representative post-run visualization of GFP in sample eluate. Eluate fractions were collected at a rate of 1/minute, and each was analyzed for GFP content. In this representative figure, culture tubes for Fractions 10, 12, 14, 16, and 18 of the Phenyl FF (high sub) scouting run were visualized under ambient room (fluorescent) light and ultraviolet (UV) light. Tubes were viewed both face-forward (left panels) and top-down (right panels). The diffuse blue in the left UV panel is light emitted from the UV lamp. GFP (green) is clearly detected in Fraction 14 in both UV images. The upper panel indicates the total protein (A280, blue line) and GFP (A397, green line) chromatogram tracings of the 5 fractions being visualized.
Figure 6. Representative post-run visualization of GFP in sample eluate. Eluate fractions were collected at a rate of 1/minute, and each was analyzed for GFP content. In this representative figure, culture tubes for Fractions 10, 12, 14, 16, and 18 of the Phenyl FF (high sub) scouting run were visualized under ambient room (fluorescent) light and ultraviolet (UV) light. Tubes were viewed both face-forward (left panels) and top-down (right panels). The diffuse blue in the left UV panel is light emitted from the UV lamp. GFP (green) is clearly detected in Fraction 14 in both UV images. The upper panel indicates the total protein (A280, blue line) and GFP (A397, green line) chromatogram tracings of the 5 fractions being visualized.
Discussion
Liquid chromatography techniques have proven invaluable for preparing highly purified proteins necessary for conducting immunological 6, biochemical 7, and structural 8 studies. HIC purification methods most often require empirical determination of a preferred medium, and ligand structure, ligand density, and matrix bead properties all have been shown to impact chromatographic results 2, 3. Automated column scouting is an efficient approach for selecting a HIC medium for subsequent optimization and protein purification 4. The automated column scouting method presented can be readily adapted to various HIC protein purification strategies. Alterations in salt concentration, choice of salt, salt gradient, and pH may further improve purification conditions, and the effects of varying these essential parameters have been previously reviewed 13,14. HIC eluate containing a partially purified target protein can be further purified using a complementary chromatographic technique, such as ion exchange chromatography (IEX) or gel filtration/size exclusion chromatography 9,10.
Because of its unique light absorbance and emission characteristics, the elution profile of GFP can be identified using in-line and post-run approaches. To that end, this protocol can further be adapted for purification of recombinant GFP-fusion proteins, in which an unrelated target protein is “tagged” with GFP. GFP-tagged target proteins can be detected with UV light 11 and purified using various chromatographic approaches, including the HIC purification strategy described above. GFP purification has also become a pedagogical staple in biochemistry labs for the teaching of modern protein science techniques 12.
While basic HIC protein purification can be conducted using a substantially less robust chromatography system, the instrumentation presented here has a number of distinct advantages to aid in obtaining favorable results. Notable benefits of utilizing a highly automated system include improved run-to-run condition reproducibility, time-savings, and decreased opportunities for air to be introduced into the system 10. System-controlled buffer blending, which is facilitated by the system maximizer, allows for increased consistency in buffer preparation and experimental reproducibility. Drawing enough sample load into the dynamic sample loop for all scouting runs further ensures uniformity of the sample being added to each column and allows for sequential runs without interruption or manual reloading. Controlled sample injection from the sample loop onto the column decreases variability that may occur with manual sample loading. A pair of column selection valves allow for consecutive sample runs, each using a different HIC column, without having to replumb the system. Performing multi-wavelength analysis is particularly beneficial when assaying a protein with a unique spectrophotometric profile. In addition to GFP, cytochromes, flavoproteins, and other heme-containing proteins may benefit from this technique. Conductivity and pH monitoring devices allow for verification of real-time experimental conditions. Split fraction eluate collection allows for improved fraction handling and enables easy transfer for post-run analysis methods that require a minimal sample volume (e.g. ELISA, SDS-PAGE, western blot, and Experion microfluidic electrophoresis). While operating the second fraction collector offline requires manual synchronization, it allows for maximum flexibility in fraction collector selection. The most significant drawbacks to utilizing such a robust chromatography system for HIC column scouting and protein purification include the initial time and budgetary expenditures associated with instrument acquisition and operator training.
The protocol presented here utilizes a Bio-Rad DuoFlow chromatography system; however, equally robust instrumentation from other manufacturers, such as the ÄKTA Avant from GE Healthcare, may also be utilized and are capable of producing equivalent results. Even comparable chromatography system have unique characteristics (e.g. method programming, component nomenclature, operator preference, and scalability limitations) that should be considered before initiating a purification procedure or instrument acquisition.
Disclosures
Select chromatography reagents and supplemental instrumentation were provided by Bio-Rad.
Acknowledgments
This work was funded by National Institutes of Health grant GM086822 and National Science Foundation major research instrumentation grant DBI-0960313. The authors would like to thank Drs. Jon Miyake & Donna Hardy (Bio-Rad) and Jennifer Loertscher (Seattle University) for their technical expertise. Select chromatography reagents and supplemental instrumentation were generously provided by Bio-Rad.
References
- Cummins PM, O'Connor BF. Hydrophobic interaction chromatography. Methods Mol. Biol. 2011;681:431–437. doi: 10.1007/978-1-60761-913-0_24. [DOI] [PubMed] [Google Scholar]
- Nagrath D, Xia F, Cramer SM. Characterization and modeling of nonlinear hydrophobic interaction chromatographic systems. J. Chromatogr. A. 2011;1218:1219–1226. doi: 10.1016/j.chroma.2010.12.111. [DOI] [PubMed] [Google Scholar]
- To BC, Lenhoff AM. Hydrophobic interaction chromatography of proteins. IV. Protein adsorption capacity and transport in preparative mode. J. Chromatogr. A. 2011;1218:427–440. doi: 10.1016/j.chroma.2010.11.051. [DOI] [PubMed] [Google Scholar]
- Huddleston JG, Wang R, Lyddiatt A. On the use of mild hydrophobic interaction chromatography for "method scouting" protein purification strategies in aqueous two-phase systems: a study using model proteins. Biotechnol Bioeng. 1994;44:626–635. doi: 10.1002/bit.260440509. [DOI] [PubMed] [Google Scholar]
- Arun KH, Kaul CL, Ramarao P. Green fluorescent proteins in receptor research: an emerging tool for drug discovery. J. Pharmacol. Toxicol. Methods. 2005;51:1–23. doi: 10.1016/j.vascn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Terrizzi SC, Banh C, Brossay L.A Protocol for the Production of KLRG1 Tetramer. J. Vis. Exp. 2010;(35):e1701–e1701. doi: 10.3791/1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PJM, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem. 2005;280:33792–33799. doi: 10.1074/jbc.M506997200. [DOI] [PubMed] [Google Scholar]
- Zeytuni N, Zarivach R. Purification of the M. magneticum strain AMB-1 Magnetosome Associated Protein MamAΔ41. J. Vis. Exp. 2010;(37):10–3791. doi: 10.3791/1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Li X, Bai G, Ma G, Su Z. An automatic system for multidimensional integrated protein chromatography. J. Chromatogr. A. 2010;1217:6898–6904. doi: 10.1016/j.chroma.2010.08.067. [DOI] [PubMed] [Google Scholar]
- Camper DV, Viola RE. Fully automated protein purification. Anal. Biochem. 2009;393:176–181. doi: 10.1016/j.ab.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammon J, Palanivelu DV, Chen J, Patel C, Minor DL. A green fluorescent protein screen for identification of well-expressed membrane proteins from a cohort of extremophilic organisms. Protein Sci. 2009;18:121–133. doi: 10.1002/pro.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. Using green and red fluorescent proteins to teach protein expression, purification, and crystallization. Biochem. Mol. Biol. Educ. 2008;36:43–54. doi: 10.1002/bmb.117. [DOI] [PubMed] [Google Scholar]
- Queiroz JA, Tomaz CT, Cabral JM. Hydrophobic interaction chromatography of proteins. J. Biotechnol. 2001;87:143–159. doi: 10.1016/s0168-1656(01)00237-1. [DOI] [PubMed] [Google Scholar]
- McCue JT. Theory and use of hydrophobic interaction chromatography in protein purification applications. Methods Enzymol. 2009;463:405–414. doi: 10.1016/S0076-6879(09)63025-1. [DOI] [PubMed] [Google Scholar]