

Abstract
This study tested the hypothesis that a novel mitochondria-targeted SS-31 peptide attenuates the burn injury-induced apoptosis and endoplasmic reticulum (ER) stress and improves insulin sensitivity in the skeletal muscle. Following 30% total body surface area burn or sham-burn, mice were injected daily with SS-31 peptide (5 mg/kg body weight) and the rectus abdominis muscles collected on post-burn days 1, 3, and 7. The tissues were subjected to various biochemical and immunohistochemical analyses. Treatment with SS-31 peptide prevented burn-induced increases in the caspase 3 activity (p < 0.05) and apoptosis (p < 0.01) on post-burn day 7. The SS-31 peptide treatment also prevented the increase in the expression levels of phosphatase and tensin homolog (PTEN) on post-burn days 3 and 7. Burn injury-induced increases in the levels of two ER stress markers, binding immunoglobulin protein (BiP) and protein disulfide isomerase (PDI), were significantly decreased by the SS-31 peptide treatments on post-burn day 7 and on day 3 for BiP as well (p < 0.05). The effects of SS-31 appear to be, in part, due to its ability to reduce oxidative stress in burned mice, evidenced by reduced expression of oxidized proteins that were clearly evident on post-burn day 7. Our results demonstrate a possible therapeutic potential of SS-31 peptide to ameliorate the adverse effects of burn injury in skeletal muscle.
Keywords: SS-31 peptide, burn injury, reactive oxygen species (ROS), insulin resistance
INTRODUCTION
Patients with major burn injury develop the classical signs of insulin resistance, including hyperglycemia or glucose intolerance, increased protein catabolism, and impaired insulin-stimulated glucose uptake in muscle (1, 2). Moreover, the lack of anabolic effects of insulin during critical illness leads to loss of muscle mass with time and decreased tension generating capacity. The muscle weakness leads to decreased mobilization, hypoventilation, dependence on respirators, difficulties in weaning off respirators, and prolonged rehabilitation and hospitalization, and even death (3). The molecular mechanisms of burn injury-induced insulin resistance in the skeletal muscle remain poorly understood and efforts to establish treatments for patients with such debilitating muscle wasting have met with little success thus far.
Numerous reports have implicated increased oxidative stress with mitochondrial dysfunction (4, 5) and endoplasmic reticulum (ER) stress (6, 7) as contributing factors causing insulin resistance in various diabetic animal models. Consistent with these findings, previous studies demonstrated that burn injury induces insulin resistance as well as mitochondrial dysfunction in the rodent skeletal muscle (8–12). In fact, the lack of anabolic (insulin) signaling has been shown to cause apoptotic cell death in muscle (10–12). Evidence for ER stress and production of reactive oxygen species (ROS) have been observed in the liver following burn injury (13–15). Since mitochondria and ER are well-known sites of ROS production (6, 16), we hypothesized that burn injury induces oxidative stress by increasing the local production of ROS, particularly in the muscle.
Oxidative stress has been shown to activate stress/inflammatory signaling pathways including ER stress and C-Jun-N-terminal kinase (JNK) pathway (17, 18), which subsequently disrupt normal insulin signaling by inhibiting the activation of the downstream effectors such as AKT/PKB and increasing a tumor suppressor phosphatase and tension homolog (PTEN) expression (19). SS (Szeto-Schiller) peptides are small cell-permeable cellular organelles-targeted antioxidants that possess intrinsic ability to scavenge ROS. By reducing mitochondrial and possibly endoplasmic ROS, these peptides inhibit mitochondrial permeability transition and cytochrome c release, thus preventing oxidant-induced cell death (20). Treatment with SS peptides has been shown to promote improvements in oxidative stress-mediated pathophysiological conditions including ischemic-perfusion and myocardial infarction in in vivo animal models (20, 21).
Herein, we test the hypothesis that burn injury-induced apoptosis, ER stress, and altered insulin signaling in the skeletal muscle of mice could be ameliorated by treatment with SS-31 peptide (H-D-Arg-Dimethyl Tyr-Lys-Phe-NH2).
RESEARCH DESIGN AND METHODS
Animal Treatments
All protocols for animal use and euthanasia were approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital in accordance with National Institutes of Health guidelines. Six to eight week old male C57BL/6J mice were subjected to approximately 30% total body surface area burn injury by immersing the back of the trunk for 8 s and the abdomen for 6 s in 80°C under general anesthesia using pentobarbital sodium (60 mg/kg body weight, i.p.). This procedure, based on previous experience and histology results (data not shown), has been shown to cause direct heat damage only to the skin and not to deeper tissues (22). All animals received a single dose of butorphanol (2 mg/kg i.p.) after the induction of anesthesia for post operative analgesia. Immediately following the burn injury, the mice were subjected to a daily injection of SS-31 peptide at 5 mg/kg body weight dose (Burn SS-31 group) or saline of same volume (Burn saline group). SS-31 peptides (obtained from Dr. Szeto) were prepared by dissolving in saline solution at 1μg/μl and the animals were injected appropriate volume based on the animals’ body weight determined every morning prior to injection. We chose the 5 mg/kg body weight SS-31 dose based on a previous study that showed maximum protective effects without any side-effects (23). A weight- and age-matched sham-burn group of animals, subjected to immersion in lukewarm (~37°C) water under anesthesia but otherwise treated exactly as burns served as controls (Sham saline and Sham SS-31). All animals were fluid resuscitated with i.p. injection of 2 ml of 0.9% NaCl, applied with 1% silver sulfadiazine cream to the treated surface, and kept warm with a heat blanket until full recovery from anesthesia. This burn injury model has been used widely in burn research and previously demonstrated to not only reduce protein synthesis but also increase proteolytic pathways and stimulate insulin resistance, similar to the catabolic response seen in patients with burn injury (24, 25).
Although the hyperinsulinemic-euglycaemic clamp technique is considered to be the “gold-standard” for measuring insulin sensitivity, due to complications encountered with performing this technique on burn injured mice we performed a surrogate method of glucose tolerance test instead. Briefly, the mice were fasted overnight, beginning at 6 pm on day 2 of burn or sham burn injury until 9 am the following day and then injected with glucose (2 g/kg i.p.). Blood glucose levels were determined by sampling from tail vein at 0 (before glucose injection), 15, 30, 60, and 90 min after glucose injection using a Precision Xtra glucometer (Abbot, Alameda, CA). Burn injury dramatically alters hemoglobin and hematocrit concentrations, which can affect the glucose concentration measurements. The manufacturer is unable to confirm if this instrument takes that into account. Twelve mice per treatment group were used for this study unless otherwise indicated.
On days 1, 3, and 7 following burn- or sham-burn injury, the rectus abdominis tissues were collected for various biochemical studies including apoptosis and expression of markers for insulin signaling (phosphorylated Akt, phophatase and tension homolog, PTEN) and ER stress (binding immunoglobin protein, BiP and protein disulphide isomerase, PDI). We chose these days based on previous studies (10, 15) that demonstrated the effects of burn injury on apoptosis and ER stress change during days following burn injury with maximal effects observed 3–7 days after the burn injury. The tissues were collected 24 hours after the last injection of SS-31 peptide or saline solution. In order to examine the treatment effects on insulin signaling, the animals were fasted beginning at 6 pm the evening before and continued until the next morning when human insulin (10 Unit of Humulin R/kg body weight; Eli Lilly, Indianapolis, IN) was injected via tail vein after approximately 15–16 hours of fasting. Ten minutes after insulin injection the animals were euthanized by cervical dislocation and the rectus abdominis muscle was immediately collected. The tissues were snap-frozen in liquid nitrogen and stored until subsequent studies. Eight mice per treatment group for each of the three post-burn days 1, 3, and 7 were used for biochemical studies unless otherwise indicated.
Western Blot Analysis
The snap-frozen samples were pulverized and homogenized using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY) in ice-cold lysis buffer (1:10, wt/vol) containing 20 mM Tris·HCl (pH 7.4), 1%Triton-X 100, 50 mM NaCl, 250 mM sucrose, 50 mM NaF, 5 mM sodiumpyrophosphate, 2 mM dithiothreitol (DTT), 4 mg/l leupeptin, 50 mg/l trypsin inhibitor, 0.1 mM benzamidine, and 0.5 mM PMSF, followed by centrifuging at 14,000 g for 30 min at 4°C. The supernatants were collected and protein concentrations were determined via the Bradford assay. Proteins samples (40 μg)were resolved by SDS-PAGE, transferred to 0.2 μm nitrocellulose membrane (BioRad Inc., Hercules, CA) and blots were probed with the respective antibodies. The primary antibodies for phosphorylated (Serine 473) Akt, PTEN, PDI, and BiP were from Cell Signaling Technology (Beverly, MA). On detection of pAkt, PTEN, PDI and BiP, membranes were stripped using Restore Western Blot buffer (Thermo Scientific, Rockford, IL) and reprobed with anti-total Akt (Cell Signaling Technology, Beverly, MA) or with Anti-GAPDH antibody (Abcam, Cambridge, MA) to normalize protein loading during Western blot assay. Antibody-bound proteins were visualized on film using chemiluminescence detection reagents (Millipore, Billerica, MA) and protein bands were scanned and quantitated using NIH ImageJ software.
Terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine 5-triphosphate nick end-labeling (TUNEL) assay for detection of apoptosis
Apoptosis in muscle was examined at 1, 3 and 7 days after burn. Following euthanasia by cervical dislocation, the rectus abdominis muscle tissues were obtained and fixed in 10% formalin overnight at room temperature for TUNEL staining. A commercial kit (ApopTag In Situ Apoptosis Detection, Millipore, Billerica, MA), which links digoxigenin-nucleotide to DNA by TdT, was used. Five μm-thickness sections were deparaffined with xylene, rehydrated with a descending series of ethanol, incubated with proteinase K, immersed in 3% aqueous hydrogen peroxide, and then pretreated with equilibration buffer. DNA was labeled at the 3′-end by incubating sections with a mixture of digoxigenin deoxynucleotide triphosphate, unlabeled deoxynucleotide triphosphate, and TdT enzyme at 37°C for 1 h. Slides were washed with phosphate buffered saline (PBS) and incubated with antidigoxigenin antibody conjugated to peroxidase at room temperature for 30 min. Slides were washed again in PBS, incubated with 3, 3-diaminobenzidine peroxidase substrate, counterstained with methyl green, mounted, and sealed.
Cell Death Assay
Quantitative determination of internucleosomal DNA fragmentation was performed using Cell Death Detection ELISAPLUS kit (Roche Applied Biosciences) similar to those described previously (26, 27) at 1, 3 and 7 days after burn or sham burn. Briefly, 100 μg of lysates were incubated with anti-histone biotin and anti-DNA POD in 96-well plate for 2 hrs, capturing mononucleosome- and oligonucleosome-associated histone-DNA complexes to the bottom of the plate well. The complexes were then visualized by chromagen development with a peroxidase substrate. The absorbance at 405 nm and 490 nm (reference wavelength) were determined with a microplate reader(Molecular Devices Corp., Sunnyvale, CA).
Caspase 3 Activity Assay
Detection of caspase activity was carried out using The EnzCheck Caspase-3 Assay kit#1 (Invitrogen, Carlsbad, CA). Protein lysates (250 μg/well) were mixed with the substrate and reaction was carried out for 2 hr at 37°C and fluorescence levels were detected at ex/em: 355/460 nm using a fluorescence microplate reader (Molecular Devices Corp., Sunnyvale, CA).
Statistical Analysis
Comparisons among more than two groups were assessed with one-way ANOVA followed by Newman-Keuls’ Multiple Comparison Test. A P value <0.05 was considered statistically significant.
RESULTS
SS-31 treatment prevents burn-induced apoptosis in the skeletal muscle
TUNEL staining showed a marked increase in the number of apoptotic cells in the rectus abdominis muscle on post-burn day 7. The treatments with SS-31 peptides appeared to prevent cells from undergoing apoptosis as evidenced by the reduced number of apoptotic nuclei in the muscle of Burn SS-31 mice compared to that of Burn mice (Fig. 1A).
Fig. 1.



Effects of burn injury and SS-31 peptide on apoptosis in skeletal muscle. Representative TUNEL-labeled photomicrographs of rectus abdominis muscle collected on postburn day 7 are shown. Compared with sham saline control, burn injury dramatically increased the number of apoptotic cells, but daily injections of SS-31 peptide attenuated the increased in the number of cells undergoing apoptosis. The peptides by itself had no effects on apoptosis (sham SS-31 panel). Arrows indicate representative TUNEL-positive cells. The images are of the same magnification and captured at 400× magnifications. Cell death (B) and caspase activity (C) assays showed that burn injury increased apoptotic DNA fragmentation and caspase activity significantly on postburn day 7, but the treatment with SS-31 peptide reduced each of these values to a level that was not statistically different from that of time-matched sham control group (*P< 0.05); n= 8 for cell death data and n = 7 for caspase activity data.
To quantitatively evaluate the induction of apoptosis, we measured the proportion of cytoplasmic internucleosomal DNA fragmentation as well as the activity of caspase 3 in the lysates from rectus abdominis muscle. Consistent with the TUNEL results, burn injury caused a significant increase, compared to sham control group, in the level of apoptotic DNA fragmentation (Fig. 1B) and the activity of caspase-3 assessed by increases in fluorescence (Fig. 1C) on post-burn day 7. Treatment with SS-31 peptide attenuated these increases induced by burn injury (Fig. 1B & 1C). On post-burn day 1 and 3, however, the apoptotic cell death, caspase-3 activity and SS-31 peptide changes did not reach statistical significance. It should be noted, however, that on days 1 and 3 post-burn apoptosis was not significantly increased and therefore the beneficial effects of SS-31 peptide did not became manifest on these days.
Taken together, these results provide evidence that burn injury increases apoptosis by post-burn day 7 and this increase was prevented by treatment with SS-31 peptide.
Evidence that SS-31 treatment may ameliorate the deleterious effects of burn on insulin signaling function in the skeletal muscle
Glucose tolerance test, conducted on post-burn day 3, demonstrated that burn injury impaired glucose clearance from the circulation but SS-31 peptide treatment resulted in significantly improved glucose tolerance (Fig. 2A). SS-31 peptide seemed to have beneficial effects on Sham Saline glucose levels too, although this did not reach statistical significance. To examine the effects of SS-31 peptide on burn injury-induced insulin signaling proteins, we evaluated levels of phosphorylated form of Akt/PKB (S473) and protein expression of PTEN using Western blot analysis. Burn injury caused significant reduction in insulin-stimulated phosphorylation of Akt (Fig. 2B) on post-burn days 3 and 7 while increasing PTEN expression levels (Fig. 2C) on all three days examined. Either decreased phosphorylation of Akt or increased expression of PTEN can attenuate downstream signaling of insulin. A preliminary study demonstrated that, when insulin was not injected, Akt phosphorylation levels were undetectable and did not differ between sham and burn injured animals (data not shown). The proportion of phosphorylated form of Akt in burned mice treated with SS-31 peptides tended to be greater than that of burned mice on all three days, but none of these reached statistical significance (Fig. 2B). However, the treatment with SS-31 peptide did attenuate the increased expression of PTEN (p < 0.05) observed in burned animals on days 3 and 7 of burn injury (Fig. 2C).
Fig. 2.



Effects of SS-31 peptide on glucose tolerance and insulin signaling in burned mice. Glucose tolerance test (A, upper panel) showed that burn injury resulted in an upward shift of glucose tolerance curve that was prevented by the SS-31 peptide treatment. Area-under-curve (AUC) analysis of glucose concentration-time curves (the lower panel) showed that burn caused a significant increase in the AUC, which was reversed with the SS-31 peptide treatments (P< 0.001). The number at the base of each column indicates the number of animals used. Compared with time-matched sham control group, burn injury induced the reduction in the levels of phosphorylated form of Akt (S473) on days 3 and 7 (B) and increased PTEN expression (C) in the rectus abdominis on all days examined. There was general trend of increased phosphorylated form of Akt following the SS-31 peptide treatments, but no statistical significance was reached on any of the days examined. However, the SS-31 peptide treatment did attenuate the burn-induced increase in the PTEN expression levels on postburn days 3 and 7. The phosphorylated form of Akt and PTEN bands were normalized to total Akt and GAPDH, respectively. The results in B and C are presented as percentage of control group on the same day; n = 8 for both pAkt and PTEN data; *P< 0.05, **P< 0.01. Representative Western blots for phosphorylated Akt and PTEN are shown in the upper panel of B and C, respectively.
SS-31 treatment ameliorates burn-induced ER stress in the skeletal muscle
To test the hypothesis that burn injury induces ER stress in muscle, we analyzed protein expression levels of BiP (also known as glucose regulated protein 78, GRP78) and PDI proteins, two well-known ER stress markers. As shown in Figures 3A and 3B, burn injury dramatically increased these proteins levels on post-burn days 1, 3 and 7. The treatments with SS-31 peptide attenuated burn-induced increases in BiP expression levels significantly on post-burn days 3 and 7 (Fig. 3A) while their effects on PDI were limited to post-burn day 7 only (Fig. 3B).
Fig. 3.


Effects of SS-31 peptide on ER stress markers in skeletal muscle of burned mice. Burn injury induced dramatic increases in the expression of BiP (A) and PDI (B) compared with control group on postburn days 1, 3, and 7. Treatment of SS-31 peptide significantly attenuated burn injury induced increase in the expression of BiP on days 3 and 7, whereas the treatment effects on the PDI expression levels were seen only on day 7 of burn injury. The results in A and B are presented as percentage of control on the same day; n = 7 for BiP data and n = 8 for PDI data; *P< 0.05, **P< 0.01. Representative Western blots for BiP and PDI are shown in the upper panel of A and B, respectively.
SS-31 peptide renders its effect by attenuating oxidative stress
To examine involvement of oxidative stress, OxyBlot™ assay, which is one of the commonly used methods for detecting oxidative damage to proteins, was performed. Carbonyl groups of oxidized proteins in muscle were derivatized to a 2, 4-dinitrophenylhydrazone (DNP) moiety and subsequently detected using anti-DNP antibodies. As shown in Figure 4, burn injury appeared to increase the intensity of protein bands detected clearly on post-burn day 7 and the beneficial effects of SS-31 peptide on reducing the oxidized proteins were clearly evident on the same day (Fig. 4).
Fig. 4.
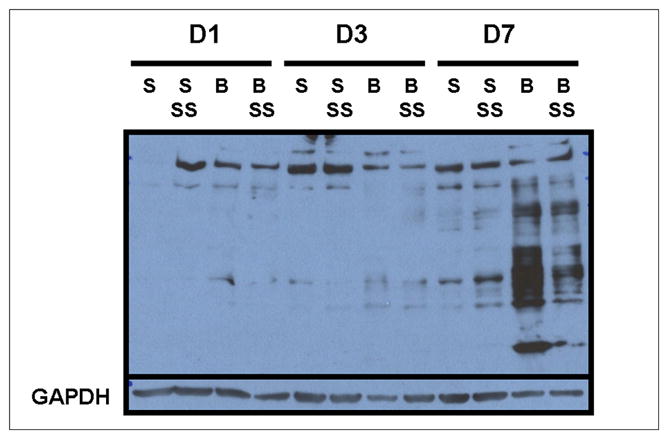
The representative OxyBlot showing the effects of burn injury and SS-31 peptide on oxidized proteins in skeletal muscle (n = 3). Burn injury appeared to increase the amount of oxidized proteins most clearly on postburn day 7 as evaluated by OxyBlot assay, but the increases seem to be prevented by the SS-31 peptide treatments. The lower panel shows GAPDH protein bands to demonstrate equal loading of proteins on each lane.
DISCUSSION
This study shows that the treatment with novel antioxidant SS-31 peptide ameliorated the burn-induced apoptosis, the increased expression of PTEN and the markers of ER stress, BiP and PDI on post-burn day 7 in rectus abdominis muscle. The effect of SS-31 peptide on PTEN and BiP expression were also observed on post-burn day 3. Moreover, we provide evidence that burn injury induces increased oxidative stress in muscle on post-burn day 7, which may be directly or indirectly related to the changes in insulin signaling proteins. Mitochondria and endoplasmic reticulum are known sites of ROS production (6, 16). Previous reports (8, 10, 12, 13, 15, 28) also show these organelles to have dysfunction in the liver associated with burn injury. It is likely that the burn injury-induced increase in the production of ROS leads to ER stress and programmed cell death that were observed in the current study.
Muscle wasting stemming from burns, sepsis, and immobilization share a common feature in that they all display symptoms of decreased anabolic signaling via insulin (insulin resistance) and apoptotic cell death in muscle (1, 2, 29, 30). The mechanisms for decreased anabolic signaling and apoptotic cell death may be different for each. Therefore, understanding the mechanism(s) by which burn injury induces these changes likely will render potential treatment for not only patients of burn injury but also possibly those suffering from sepsis- and immobilization-induced muscle wasting. The involvement of mitochondrial dysfunction as one of several underlying causes of muscle wasting in burns has been previously reported by our laboratory (10, 12) and others (8, 31). There seems to be connection between mitochondrial dysfunction and changes in insulin signaling proteins. However, ER stress as another cellular mechanism responsible for burn-induced altered insulin signaling has only begun to receive scientific attention and is limited to the hepatic tissues to date (13, 15, 32). Herein, we report for the first time that ER stress also occurs in the skeletal muscle of burn injured mice as evidenced by PDI and BiP expression. Interestingly, unlike the previous studies (13, 15, 32) our attempt to detect other known ER stress markers such as p-protein kinase R-like endoplasmic reticulum kinase (p-PERK) and inositol-requiring enzyme 1α (IRE-1α) generated inconclusive results (data not shown). It may be due to the differences in the ways burn injury treatments were carried out or the fact that different tissues (the liver vs. skeletal muscle) behave differently.
The SS peptides, so called because they were designed by Hazel H. Sezto and Peter W. Schiler, are small cell-permeable peptides of less than ten amino acid residues that specifically target to inner mitochondrial membrane and possess mitoprotective properties (20). There have been a series of SS peptides synthesized and characterized, but for our study, we decided to use SS-31 peptide (H-D-Arg-Dimethyl Tyr-Lys-Phe-NH2) for its well-documented efficacy (20). The SS-31 peptide exerts its mitoprotective effects by scavenging ROS and reducing its production within mitochondria, and maintaining mitochondrial membrane potential.(20). Consistent with their well-known antioxidative effects, this study provides evidence that the SS-31 peptides reduced expression of oxidized proteins and contribute to the overall beneficial effects of SS-31 peptides in preventing apoptosis and improving ER stress brought on by burn injury. Interestingly, the beneficial effects of the SS-31 peptide treatment on the variables that we examined were observed on days 3–7 after burn injury was induced. Previous studies demonstrated that apoptosis in skeletal muscle peaks around 3 – 7 days after burn injury (10), consistent with hypercatabolic state observed during this period. Thus, peak adverse effects of burn injury are known to manifest several days after the burn injury itself (2, 30) and therefore the beneficial effects of SS-31 peptide became manifest at this time. However, the utility of SS-31 peptide to treat burn-induced muscle wasting and muscle weakness needs further studies.
Recent studies provide evidence for a direct link between insulin signaling pathway and ER stress (33, 34). Chemical induction of ER stress has been shown to dephosphorylate Akt and cause subsequent cell death (34) whereas an inhibition of Akt phosphorylation sensitized chondrocytes to ER stress (33). Interestingly, both studies report that only a PI3K inhibitor, but not a MEK1 inhibitor, is capable of inducing ER stress (34), suggesting a complex nature of cross-talk between ER stress and insulin signaling pathway. Our observation of increased expression of PTEN following burns is consistent with the decreased activation of PI3-K, evidenced as decreased phosphorylation of Akt.
In the current study, we chose to examine the rectus abdominis muscle because it represents the local site of burn injury where the effects of burn injury would be expected to be maximal. Therefore, we investigated the effects of treatment with SS-31 peptide in the most severely affected muscle after burn injury. It is important to note that this model causes no direct damage to muscle (22) and no evidence for that was present in rectus muscles examined. Nonetheless, previous studies demonstrated that burn injury causes apoptosis at sites distant from burn (10, 35) and it would be interesting to examine if SS-31 peptides can also prevent/reverse burn-induced adverse effects on apoptosis and ER stress at distant sites such as the tibialis anterior or gastrocnemius muscle in the hindlimb.
In summary, we demonstrate that burn injury induces apoptosis and ER stress in the rectus abdominis and these abnormalities likely contribute to metabolic and functional derangements that occur following burn injury. Moreover, we provide evidence that novel antioxidant SS-31 peptides ameliorated burn-induced apoptosis, ER stress and elevated PTEN expression on day 7 of burned mice, and its effects may be mediated by reducing oxidative stress. Our data point to a novel therapeutic approach to treat burn injury-induced insulin resistance and muscle wasting.
Acknowledgments
We would like to thank Dr. H. Szeto for providing us with SS-31 peptides and advice regarding the peptide use.
References
- 1.Cree MG, Wolfe RR. Postburn trauma insulin resistance and fat metabolism. Am J Physiol Endocrinol Metab. 2008;294(1):E1–9. doi: 10.1152/ajpendo.00562.2007. [DOI] [PubMed] [Google Scholar]
- 2.Demling RH, Seigne P. Metabolic management of patients with severe burns. World J Surg. 2000;24(6):673–680. doi: 10.1007/s002689910109. [DOI] [PubMed] [Google Scholar]
- 3.Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol. 2005;37(10):1948–1961. doi: 10.1016/j.biocel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 5.Yokota T, Kinugawa S, Hirabayashi K, Matsushima S, Inoue N, Ohta Y, Hamaguchi S, Sobirin MA, Ono T, Suga T, et al. Oxidative stress in skeletal muscle impairs mitochondrial respiration and limits exercise capacity in type 2 diabetic mice. Am J Physiol Heart Circ Physiol. 2009;297(3):H1069–1077. doi: 10.1152/ajpheart.00267.2009. [DOI] [PubMed] [Google Scholar]
- 6.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 7.Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond ) 2008;32(Suppl 7):S52–54. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Padfield KE, Astrakas LG, Zhang Q, Gopalan S, Dai G, Mindrinos MN, Tompkins RG, Rahme LG, Tzika AA. Burn injury causes mitochondrial dysfunction in skeletal muscle. Proc Natl Acad Sci U S A. 2005;102(15):5368–5373. doi: 10.1073/pnas.0501211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugita H, Kaneki M, Sugita M, Yasukawa T, Yasuhara S, Martyn JA. Burn injury impairs insulin-stimulated Akt/PKB activation in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288(3):E585–591. doi: 10.1152/ajpendo.00321.2004. [DOI] [PubMed] [Google Scholar]
- 10.Yasuhara S, Perez ME, Kanakubo E, Yasuhara Y, Shin YS, Kaneki M, Fujita T, Martyn JA. Skeletal muscle apoptosis after burns is associated with activation of proapoptotic signals. Am J Physiol Endocrinol Metab. 2000;279(5):E1114–1121. doi: 10.1152/ajpendo.2000.279.5.E1114. [DOI] [PubMed] [Google Scholar]
- 11.Astrakas LG, Goljer I, Yasuhara S, Padfield KE, Zhang Q, Gopalan S, Mindrinos MN, Dai G, Yu YM, Martyn JA, et al. Proton NMR spectroscopy shows lipids accumulate in skeletal muscle in response to burn trauma-induced apoptosis. FASEB J. 2005;19(11):1431–1440. doi: 10.1096/fj.04-2005com. [DOI] [PubMed] [Google Scholar]
- 12.Yasuhara S, Kaneki M, Sugita H, Sugita M, Asai A, Sahani N, Chon JY, Tompkins RG, Martyn JA. Adipocyte apoptosis after burn injury is associated with altered fat metabolism. J Burn Care Res. 2006;27(3):367–376. doi: 10.1097/01.BCR.0000216777.94365.47. [DOI] [PubMed] [Google Scholar]
- 13.Jeschke MG, Gauglitz GG, Song J, Kulp GA, Finnerty CC, Cox RA, Barral JM, Herndon DN, Boehning D. Calcium and ER stress mediate hepatic apoptosis after burn injury. J Cell Mol Med. 2009;13(8B):1857–1865. doi: 10.1111/j.1582-4934.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parihar A, Parihar MS, Milner S, Bhat S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns. 2008;34(1):6–17. doi: 10.1016/j.burns.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Song J, Finnerty CC, Herndon DN, Boehning D, Jeschke MG. Severe burn-induced endoplasmic reticulum stress and hepatic damage in mice. Mol Med. 2009;15(9–10):316–320. doi: 10.2119/molmed.2009.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu Y, Hendershot LM. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid Redox Signal. 2009;11(9):2317–2331. doi: 10.1089/ars.2009.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vinayagamoorthi R, Bobby Z, Sridhar MG. Antioxidants preserve redox balance and inhibit c-Jun-N-terminal kinase pathway while improving insulin signaling in fat-fed rats: evidence for the role of oxidative stress on IRS-1 serine phosphorylation and insulin resistance. J Endocrinol. 2008;197(2):287–296. doi: 10.1677/JOE-08-0061. [DOI] [PubMed] [Google Scholar]
- 19.Kim JS, Saengsirisuwan V, Sloniger JA, Teachey MK, Henriksen EJ. Oxidant stress and skeletal muscle glucose transport: roles of insulin signaling and p38 MAPK. Free radical biology & medicine. 2006;41(5):818–824. doi: 10.1016/j.freeradbiomed.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 20.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10(3):601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 21.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18(3):215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 22.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8(6):1049–1051. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, Calingasan NY, Szeto HH, Beal MF. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98(4):1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- 24.Fang CH, Li BG, Tiao G, Wang JJ, Fischer JE, Hasselgren PO. The molecular regulation of protein breakdown following burn injury is different in fast- and slow-twitch skeletal muscle. Int J Mol Med. 1998;1(1):163–169. doi: 10.3892/ijmm.1.1.163. [DOI] [PubMed] [Google Scholar]
- 25.Ikezu T, Okamoto T, Yonezawa K, Tompkins RG, Martyn JA. Analysis of thermal injury-induced insulin resistance in rodents. Implication of postreceptor mechanisms. J Biol Chem. 1997;272(40):25289–25295. doi: 10.1074/jbc.272.40.25289. [DOI] [PubMed] [Google Scholar]
- 26.Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, Gunaseelan S, Sinko PJ, Stein S, Farmanfarmaian A, Minko T. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci U S A. 2005;102(36):12962–12967. doi: 10.1073/pnas.0504274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pakunlu RI, Wang Y, Tsao W, Pozharov V, Cook TJ, Minko T. Enhancement of the efficacy of chemotherapy for lung cancer by simultaneous suppression of multidrug resistance and antiapoptotic cellular defense: novel multicomponent delivery system. Cancer research. 2004;64(17):6214–6224. doi: 10.1158/0008-5472.CAN-04-0001. [DOI] [PubMed] [Google Scholar]
- 28.Cree MG, Fram RY, Herndon DN, Qian T, Angel C, Green JM, Mlcak R, Aarsland A, Wolfe RR. Human mitochondrial oxidative capacity is acutely impaired after burn trauma. Am J Surg. 2008;196(2):234–239. doi: 10.1016/j.amjsurg.2007.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biolo G, Fleming RY, Maggi SP, Nguyen TT, Herndon DN, Wolfe RR. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. The Journal of clinical endocrinology and metabolism. 2002;87(7):3378–3384. doi: 10.1210/jcem.87.7.8699. [DOI] [PubMed] [Google Scholar]
- 30.Herndon DN, Tompkins RG. Support of the metabolic response to burn injury. Lancet. 2004;363(9424):1895–1902. doi: 10.1016/S0140-6736(04)16360-5. [DOI] [PubMed] [Google Scholar]
- 31.Tzika AA, Mintzopoulos D, Mindrinos M, Zhang J, Rahme LG, Tompkins RG. Microarray analysis suggests that burn injury results in mitochondrial dysfunction in human skeletal muscle. Int J Mol Med. 2009;24(3):387–392. doi: 10.3892/ijmm_00000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauglitz GG, Halder S, Boehning DF, Kulp GA, Herndon DN, Barral JM, Jeschke MG. Post-Burn Hepatic Insulin Resistance Is Associated with Er Stress. Shock. 2010;33(3):299–305. doi: 10.1097/SHK.0b013e3181b2f439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price J, Zaidi AK, Bohensky J, Srinivas V, Shapiro IM, Ali H. Akt-1 mediates survival of chondrocytes from endoplasmic reticulum-induced stress. Journal of cellular physiology. 2010;222(3):502–508. doi: 10.1002/jcp.22001. [DOI] [PubMed] [Google Scholar]
- 34.Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochemical and biophysical research communications. 2006;340(1):286–290. doi: 10.1016/j.bbrc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 35.Lightfoot E, Jr, Horton JW, Maass DL, White DJ, McFarland RD, Lipsky PE. Major burn trauma in rats promotes cardiac and gastrointestinal apoptosis. Shock. 1999;11(1):29–34. doi: 10.1097/00024382-199901000-00004. [DOI] [PubMed] [Google Scholar]