

Abstract

Macrocyclic natural products are a powerful class of leadlike chemical entities. Despite commonly violating Lipinski's “rule of 5”, these compounds often demonstrate superior druglike physicochemical and pharmacokinetic attributes when compared to their acyclic counterparts. However, the elaborate structural architectures of such molecules require rigorous synthetic investigation that complicates analogue development and their application to drug discovery programs. To circumvent these limitations, a conformation-based approach using limited structure–activity relationships and molecular modeling was implemented to design simplified analogues of trienomycin A, in which the corresponding analogues could be prepared in a succinct manner to rapidly identify essential structural components necessary for biological activity. Trienomycin A is a member of the ansamycin family of natural products that possesses potent anticancer activity. These studies revealed a novel trienomycin A analogue, monoenomycin, which manifests potent anticancer activity.
Keywords: Trienomycin A, conformation, anticancer, ansamycin synthesis, structure−activity relationship
The macrocycle is an important structural motif amongst bioactive natural products and imparts favorable physicochemical and pharmacological attributes. Macrocyclic geometry maintains a delicate balance between flexibility and rigidity that commonly affects solubility, permeability, and binding to biological targets. Despite their inherent druglike properties, the complicated architecture exhibited by macrocyclic natural products provides a significant challenge toward their utility in drug development. While the discovery of novel chemical methodologies has led to the de novo synthesis of numerous macrocyclic natural products, approaches toward simplified analogues remain underinvestigated. In this letter, a strategy utilizing molecular modeling combined with a conformation-based approach was used to develop simplified ansamycin analogues that maintain biological activity.
Mycobacteria produce the ansamycin family of natural products, which manifest diverse biological activities. Structural characteristics of this family include a macrocyclic polyketide bridged by an aromatic core. Several ansamycins display significant clinical attributes, including rifampicin, which manifests antimicrobial activity for the treatment tuberculosis,1 and geldanamycin, derivatives of which have entered phase III clinical trials for the treatment of cancer.2 In general, the ansamycin family exhibits a high degree of broad inhibitory activities that include antiviral, antifungal, and immunosuppressant activities.3,4
Trienomycin A (Figure 1, 1) is a member of the ansamycin family that was first isolated from Streptomyces sp. no. 83-16 by Umezawa and co-workers.5 In contrast to other members of the ansamycin family that possess a p-quinone or p-hydroquinone moiety within the aromatic bridge, trienomycin A contains a nonredox active phenol. In addition, trienomycin A displays a biological profile that contrasts the activity manifested by other ansamycins. For example, mycotrienin II, which is structurally identical to trienomycin A with the exception of a p-hydroquinone moiety, is a potent antifungal agent as well as a promising anticancer agent.3,6 In contrast, trienomycin A manifests potent anticancer activity (IC50 of 128 nM against HeLa cervical cancer cells)7 but displays no antifungal activity, nor does it display any significant antimicrobial, antiviral, or immunosuppressant activities.3,5,7−9
Figure 1.

Structures of rifampicin, geldanamycin, trienomycin A, and mycotrienin II.
A previous report indicated that while trienomycin A exhibits potent anticancer and antitumor activities, it was found to be significantly less toxic to nontransformed cells.8 Thus, the unique biological activity manifested by trienomycin A poses this natural product as an attractive lead compound for cancer chemotherapeutic development. Unfortunately, limited structure–activity relationships (SAR) for trienomycin A have been reported, and the mechanism of action remains unknown. Furthermore, significant quantities of the natural product are not available, and only one total synthesis has been reported, which produces the natural product in 31 linear steps.10 Although elegant in nature, this route does not afford a succinct method for evaluation of SAR. Therefore, a generalized method for the production of synthetically useful trienomycin A analogues employing a conformation-based approach was pursued.
Previously reported semisynthetic modifications to the natural product including acetylation of the 13-OH (2), saturation of the triene motif (3), and deletion of the N-cyclohexylcarbonyl d-Ala (NCxDA) side chain (4) resulted in almost complete ablation of anticancer activity (Figure 3).3 However, methylation of the free phenol did produce an analogue equipotent to the natural product (Figure 2, 5). Without apparent SAR trends and the lack of activity for semisynthetic derivatives, no obvious hypothesis for further exploration was available. Therefore, an alternate approach was pursued based on the overall conformation of the macrocyclic compounds and the lowest energy conformations of such analogues.
Figure 3.
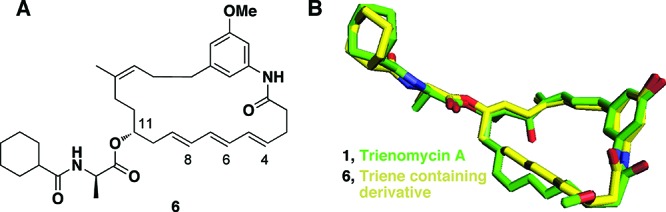
(A) Structure of theoretical trienomycin A analogue lacking the 13-OH, 12-CH3, and 3-OMe functional groups. (B) Overlay of trienomycin A and the theoretical derivative lacking the 13-OH, 12-CH3, and 3-OCH3 functional groups (6). The theoretical analogue exhibited an energy value of 21.527 kcal/mol and a similarity score of 0.7935 with the trienomycin A (1).
Figure 2.

(A) Overlay of energy-minimized trienomycin A and select semisynthetic derivatives using the Ligand Similarity tool (rigid superposition) from the Surflex software suite. (B) Relative energies and similarity score from the Ligand Similarity tool.
The natural product (1) and four semisynthetic derivatives (2–5) were constructed in the lowest energy conformations using SYBYL. The three-dimensional geometries of these compounds were then analyzed using the Surflex Ligand Similarity tool (default parameter settings).11 Rigid superposition maintained low energy conformations and provided informative differences in ligand similarity (Figure 2). Semisynthetic derivatives 2–4 exhibited significant conformational perturbations in macrocycle geometry and placement of NCxDA side chain, but methyl-phenyl ether 5 retained the native geometry. Because these semisynthetic derivatives maintain most of the hydrogen-bonding capabilities of trienomycin A, we hypothesized that orientation of the NCxDA side chain and projection of the phenol are critical to the biological activity manifested by trienomycin A.
The efficiency by which simplified analogues of trienomycin A could overlay with the natural product was evaluated in silico, also via Surflex calculations. Sequential removal of functionality and subsequent comparison of the energy-minimized derivatives to the natural product were analyzed. The 13-OH, 12-CH3, and 3-OCH3 functionalities appeared to exhibit little effect on the overall conformation, which suggested that they could be omitted without significantly affecting biological activity (Figure 3).
The triene motif was evaluated next, with the aim of generating the most synthetically accessible derivatives. Twelve energy-minimized “monoene” derivatives, containing all possible olefin isomers and the methyl ether of the reported derivative, were evaluated based upon their ability to adopt a conformation that allowed both the phenol and the NCxDA side chain to occupy the same conformational space as the natural product. Olefin geometry was critical in these structures and was responsible for orienting both the NCxDA side chain and the phenol into conformations that were similar to the natural product. Most of the monoene derivatives exhibited significant differences in macrocycle geometry as compared to trienomycin A. However, derivatives 7–10 manifested a macrocycle geometry similar to the natural product and produced relatively high similarity scores (Figure 4). Monoene A, which contains trans-olefins at both C8 and C14, occupied the closest three-dimensional orientation to trienomycin A and exhibited the highest similarity score (0.7815).
Figure 4.

(A) Labeled monoene derivative key. (B) Comparison of monoene derivatives and trienomycin A. (C) Relative energies and similarity score from the Ligand Similarity tool.
Because monoene A (7) exhibited the highest similarity score to trienomycin A, it was the target for chemical synthesis. Synthesis of 7 was envisioned retrosynthetically to occur through a Wittig olefination between triphenylphosphonium iodide salt 12 and ketone 13,12 followed by Mitsunobu coupling of the d-Ala side chain 14(13) and subsequent ring-closing metathesis (Figure 5).14 This synthetic route would also provide access to monoene E (11), which exhibited the lowest similarity score and could be used as a negative control to evaluate our conformation-based approach.
Figure 5.

Retrosynthetic analysis of compound 7, monoene A.
Synthesis of Wittig salt 12 commenced with nucleophilic aromatic substitution of methyl 3,5-dinitrobenzoate 15 by lithium methoxide to provide methyl-phenyl ether 16,15 which was then reduced to aldehyde 17 by diisobutylaluminum hydride.16 Aldehyde 17 was subjected to Horner–Wadsworth–Emmons conditions to furnish 18,17 which was subjected to diimide mediated reduction to provide 19.18 The ethyl ester of 19 was reduced to the alcohol using diisobutylaluminum hydride to yield 20, the nitro moiety of which was reduced to the aniline 21 by palladium on carbon under a hydrogen atmosphere. Selective amidation of compound 21 with non-8-enoic acid was accomplished using (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU) to afford amido-alcohol 22.19 Until this point, no chromatographic separation was required, enabling the preparation of large quantities. Amido-alcohol 22 was then iodinated and converted to Wittig salt 12 (Scheme 1).
Scheme 1. Synthesis of Wittig Salt 12.

Reagents and conditions: (a) LiOMe, MeOH, reflux, 97%. (b) DIBAl-H, DCM, −78 °C, 93%. (c) Triethylphosphonoacetate, NaH, THF, 0 °C, 94%. (d) Potassium azodicarboxylate, AcOH, DME, 50 °C, 97%. (e) DIBAl-H, PhMe, 0 °C. (f) 10% Pd/C, H2, EtOAc. (g) Non-8-enoic acid, COMU, DIPEA, DMF, 93% over two steps. (h) PPh3, imidazole, I2, DCM, 83%. (i) PPh3, CAN, 90%.
Synthesis of ketone 13 was similarly straightforward. Oxidation of ketol 23 to the ketoaldehyde and treatment of the dicarbonyl species with 0.5 equiv of Brown's allylborane furnished optically active ketol 24.20 Synthesis of ketone 13 was completed following silyl ether formation (Scheme 2).
Scheme 2. Synthesis of Ketone 13.
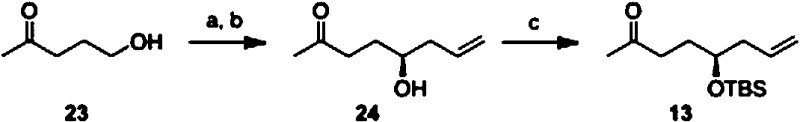
Reagents and conditions: (a) PCC, DCM. (b) Allyl magnesium bromide, (+)-B-methoxydiisopinocampheylborane, Et2O, ee > 95%, 82% (BRSM) over two steps. (c) TBSCl, imidazole, DMF, 79%.
N-Cyclohexylcarbonyl d-alanine acid 14 was constructed in one step by treating d-alanine with cyclohexylcarbonyl chloride in the presence of tribasic potassium phosphate in tetrahydrofuran (Scheme 3).
Scheme 3. Synthesis of N-Cyclohexylcarbonyl d-Alanine Acid 14.
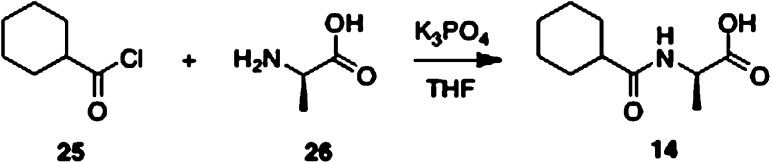
Reagents and conditions: (a) K3PO4, THF, 76%.
With the synthons in hand, Wittig olefination reaction between 12 and 13, followed by silyl deprotection, and Mitsunobu coupling between 25 and 14 led to metathesis precursors 26 (trans) and 27 (cis) in a 1:1 mixture, which were separable by chromatography. Originally, it was desired to obtain the macrocyclic alcohol after ring-closing metathesis, which could subsequently undergo diversification at a later stage. However, the macrocyclic alcohol failed to efficiently undergo esterification via Mitsunobu conditions or direct acylation, due to steric constraints resulting from the macrocyclic conformation. Therefore, the NCxDA side was appended prior to cyclization. Each isomer was subjected to ring-closing metathesis with Grubbs first generation catalyst to yield products 7 and 11 as a 2:1 and 5:1 (trans:cis) mixture of olefin isomers, respectively (Scheme 4). The isomeric mixtures were evaluated for antiproliferative activity against MCF-7 and HeLa cancer cells. As predicted in our conformation-based design approach, compound 7 retained antiproliferative activity (IC50 = 0.47 μM) while compound 11 was inactive.
Scheme 4. Synthesis of Monoene Derivatives of Trienomycin A.

Reagents and conditions: (a) KHMDS, toluene, 78%. (b) TBAF, THF, 97%. (c) Diethyl azodicarboxylate, PPh3, 14, 37% (26), 39% (27). (d) Grubbs first generation catalyst, DCM, 83%.
The anticancer activity exhibited by compound 7 prompted investigations into the amino acid side chain. Specifically, the effects of increasing steric bulk in lieu of the methyl substituent and alterations of the amide to include small alkyl groups were pursued. The various side chains were synthesized from the corresponding amino acids and appropriate acyl chlorides similar to compound 14. Once synthesized, the side chains were coupled with compound 25 using Mitsunobu conditions and subsequently cyclized with Grubbs first generation catalyst (Figure 6, 28–34).
Figure 6.

Structures of monoenomycin analogues 28–34.
These compounds were evaluated for antiproliferative activity against HeLa and MCF-7 cancer cells (Table 1). From the data presented, deviation in steric bulk, with functionalities that exhibit smaller or larger groups than methyl, is not tolerated at the α-position of the d-Ala side chain. Similarly, decreasing steric bulk of the alkyl amide decreases biological activity.
Table 1. Anti-proliferative Effects of Trienomycin A Compound Library against HeLA and MCF-7 Cancer Cell Linesa.
| IC50 (μM) |
||
|---|---|---|
| compound | HeLa | MCF-7 |
| 7 | 0.47 ± 0.04 | 0.53 ± 0.02 |
| 11 | >100 | >100 |
| 28 | 13 ± 0.5 | 12 ± 0.5 |
| 29 | 1.7 ± 0.03 | 1.9 ± 0.05 |
| 30 | 9.1 ± 0.09 | 1.0 ± 0.04 |
| 31 | 4.6 ± 0.09 | 5.1 ± 0.07 |
| 32 | >100 | >100 |
| 33 | 1.6 ± 0.09 | 0.72 ± 0.5 |
| 34 | 2.5 ± 0.9 | 1.7 ± 0.4 |
| geldanamycin | >5 | 0.053 ± 0.001 |
IC50 values based on MTS assay run in triplicate (see the Supporting Information).
In conclusion, molecular modeling was used to predict simplified derivatives of trienomycin A that were energetically predisposed to adopt a conformation similar to the natural product. The analogues manifested potent antiproliferative activities against MCF-7 and HeLa cancer cell lines and provided the first SAR for these natural product analogues. Because trienomycin A is synthetically complex, simplified derivatives provide a method that enables rapid elucidation of SAR and provide a potential tool by which the biological target may be discovered through subsequent studies. Further investigation into the SAR and mechanism of action for monoenomycin is ongoing and will be reported in due course.
We acknowledge NIH/NCI for financial support, CA109265. G.E.L.B. acknowledges the Madison and Lila Self Graduate Fellowship and the American Foundation for Pharmaceutical Education for financial support and Dr. Geraldine Calvet for preliminary studies.
Supporting Information Available
Full experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lalloo U. G.; Ambarram A. New antituberculous drugs in development. Curr. HIV/AIDS Rep. 2010, 7, 143–151. [DOI] [PubMed] [Google Scholar]
- Brandt G. E. L.; Blagg B. S. J. Alternate strategies of Hsp90 modulation for the treatment of cancer and other diseases. Curr. Top. Med. Chem. 2009, 9, 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama S.; Anraku Y.; Mita A.; Yang Z.-B.; Shibata K.; Komiyama K.; Umezawa I.; Omura S. Structure-activity relationship of a novel antitumor ansamycin antibiotic trienomycin A and related compounds. J. Antibiot. 1988, 41, 1223–1230. [DOI] [PubMed] [Google Scholar]
- Grunicke H.; Pushendorf B.; Werchau H. Mechanism of action of distamycin A and other antibiotics with antiviral activity. Rev. Physiol. Biochem. Pharmacol. 1976, 75, 69–96. [DOI] [PubMed] [Google Scholar]
- Funayama S.; Okada K.; Komiyama K.; Umezawa I. Structure of trienomycin A, a novel cytocidal ansamycin antibiotic. J. Antibiot. 1985, 38, 1107–1109. [DOI] [PubMed] [Google Scholar]
- Masse C. E.; Yang M.; Solomon J.; Panek J. S. Total Synthesis of (+)-mycotrienol and (+)-mycotrienin I: Application of asymmetric crotylsilane bond constructions. J. Am. Chem. Soc. 1998, 120, 4123–4134. [Google Scholar]
- Funayama S.; Okada K.; Iwasaki K.; Komiyama K.; Umezawa I. Structures of trienomycins A, B and C, novel cytocidal ansamycin antibiotics. J. Antibiot. 1985, 38, 1677–1683. [DOI] [PubMed] [Google Scholar]
- Komiyama K.; Hirokawa Y.; Yamaguchi K.; Funayama S.; Masuda K.; Anraku Y.; Umezawa I.; Omura S. Antitumor activity of trienomycin A on murine tumors. J. Antibiot. 1987, 40, 1768–1772. [DOI] [PubMed] [Google Scholar]
- Umezawa I.; Funayama S.; Okada K.; Iwasaki K.; Satoh J.; Masuda K.; Komiyama K. Studies on a novel cytocidal antibiotic, trienomycin A taxonomy, fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. 1985, 38, 699–705. [DOI] [PubMed] [Google Scholar]
- Smith A. B. III; Barbosa J.; Wong W.; Wood J. L. Total syntheses of (+)-trienomycins A and F via a unified strategy. J. Am. Chem. Soc. 1996, 118, 8316–8328. [Google Scholar]
- Jain A. N. Ligand-based structural hypotheses for virtual screening. J. Med. Chem. 2004, 47, 947–961. [DOI] [PubMed] [Google Scholar]
- Maryanoff B. E.; Reitz A. B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar]
- Mitsunobu O.; Yamada M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar]
- Vougioukalakis G. C.; Grubbs R. H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010, 110, 1746–1787. [DOI] [PubMed] [Google Scholar]
- Herlt A. J.; Kibby J. J.; Rickards R. W. Synthesis of unlabelled and carboxyl-labelled 3-Amino-5-hydroxybenzoic acid. Aust. J. Chem. 1981, 34, 1319–1324. [Google Scholar]
- Ziegler K.; Martin H.; Krupp F. Metallorganische verbindungen, XXVII aluminiumtrialkyle und dialkyl-aluminiumhydride aus aluminiumisobutyl-verbindungen. Justus Liebigs Ann. Chem. 1960, 629, 14–19. [Google Scholar]
- Wadsworth W. S. Jr.; Emmons W. D. The utility of phosphonate carbanions in olefin synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar]
- Adam W.; Eggelte J. Cyclic peroxides. 57. Prostanoid endoperoxide model compounds: 2,3-Dioxabicyclo[2.2.1]heptane via selective diimide reduction. J. Org. Chem. 1977, 42, 3987–3988. [DOI] [PubMed] [Google Scholar]
- El-Faham A.; Funosas R. S.; Prohens R.; Albericio F. COMU: A safer and more effective replacement for benzotriazole-based uronium coupling reagents. Chem.—Eur. J. 2009, 15, 9404–9416. [DOI] [PubMed] [Google Scholar]
- Jadhav P. K.; Bhat K. S.; Perumal P. T.; Brown H. C. Chiral synthesis via organoboranes. 6. Asymmetric allylboration via chiral allyldialkylboranes. Synthesis of homoallylic alcohols with exceptionally high enantiomeric excess. J. Org. Chem. 1986, 51, 432–439. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.