

Abstract
The broadly prescribed anti-tumor drug cisplatin coordinates to DNA, altering the activity of cellular proteins whose functions rely upon sensing DNA structure. Cisplatin is also known to coordinate to RNA, but the effects of RNA-Pt adducts on the large number of proteins that process the transcriptome are currently unknown. In an effort to address how platination of an RNA alters the function of RNA processing enzymes, we have determined the influence of [Pt(NH3)2]2+-RNA adducts on the activities of 3’ → 5’ and 5’→3’ phosphodiesterases, a purine-specific endoribonuclease, and a reverse transcriptase. Single Pt(II) adducts on RNA oligonucleotides of form (5’-U6-XY-U5-3’: XY=GG, GA, AG, GU) are found to block exonucleolytic digestion. Similar disruption of endonucleolytic cleavage is observed, except for the platinated XY= GA RNA where RNase U2 uniquely tolerates platinum modification. Platinum adducts formed with a more complex RNA prevent reverse transcription, providing evidence that platination is capable of interfering with RNA’s role in relaying sequence information. The observed disruptions in enzymatic activity point to the possibility that cellular RNA processing may be similarly affected, which could contribute to the cell-wide effects of platinum anti-tumor drugs. Additionally, we show that thiourea reverses cisplatin-RNA adducts, providing a chemical tool for use in future studies regarding cisplatin targeting of cellular RNAs.
Keywords: Cisplatin, RNA, MALDI-MS, exonuclease, RNase, reverse transcription, platinum removal
INTRODUCTION
Cisplatin, cis-diamminedichloroplatinum(II), is broadly employed in the treatment of many cancers and is frequently used as a model for the development of nucleic acid targeted anti-tumor complexes. Years of extensive study have provided evidence that cisplatin’s activity stems from drug binding to neighboring purine nucleotides in sequences of genomic DNA, inducing structural distortions.1–3 Cellular proteins that recognize these distortions start a cascade of events, ultimately resulting in apoptosis.4–6 Despite RNA’s chemical similarity to DNA and a growing appreciation of the cellular resources devoted to producing, maintaining, and regulating the transcriptome,7–10 relatively little is known about how cisplatin affects biological processes involving RNA.
Previous reports have described the disruption by cisplatin of translation11 and splicing12 in cell extracts. These results are complemented by in vitro studies describing RNA oligonucleotide targeting by cisplatin13–18 and by several Pt-drug conjugates.19,20 In addition to these studies, our lab has recently reported that cisplatin is capable of crosslinking structurally complex RNAs.21 Elmroth and coworkers18 and our laboratory21 have reported that in vitro, RNA binding is kinetically preferred over drug coordination to DNA. Taken together these initial findings raise important questions regarding which types of cellular RNAs may be targeted by cisplatin, and whether cellular RNA function is significantly impaired as a result.
In RNA life cycles, a number of enzymes carry out the chemical reactions required to produce, regulate, and recycle the transcriptome.7,8,10,22 Because RNA-protein interactions are sensitive to RNA structure, we hypothesized that platinum binding to RNA would disrupt the function of enzymes that process RNA. Here we have determined how RNA processing by 5’→3’ and 3’→5’ exonucleases, a purine-specific endoribonuclease, and a reverse transcriptase are affected by cisplatin-RNA adducts in vitro. Each of these enzymes is inhibited at the site of a Pt(II)-RNA adduct, but with slightly different products that depend on the adduct and the enzyme. This enzymatic inhibition likely results from a combination of nucleobase modification and more general structural distortions caused by platinum binding. Additionally, we have examined the reaction of platinated RNAs with thiourea and report that cisplatin-RNA adducts are reversed by prolonged incubation, providing an experimental means to address future biological queries requiring platinum removal from RNA.
RESULTS AND DISCUSSION
Inhibition of directional exonucleases by Pt(II)-RNA adducts
Because RNA damage and mis-processing are increasingly implicated in disease mechanisms,23–25 it is of interest to investigate the mechanisms by which platination of RNA may be capable of disrupting the dynamic function of the transcriptome. Exonucleases, specifically directional phosphodiesterases, have been previously shown to stop at metallated DNA sequences.26 Exonucleolytic cleavage by both the 3’→5’ phosphodiesterase from Croatalus adamanteus (VPD, Phosphodiesterase I) and the 5’→3’ phosphodiesterase from bovine spleen (SPD, Phosphodiesterase II) is blocked by binding of Pt and Rh complexes to DNA nucleobases.27–33 The RNA transcriptome requires the function of many exonucleases that act in RNA maturation and degradation processes and facilitate post-transcriptional control of gene expression.22,34 Examples of important, directional cellular RNA exonucleases include Xrn1 (5’→3’) and components of the exosome complex (3’→5’).35 mRNA turnover by these enzymes is highly dependent on RNA stability elements such as 5’-m7-G caps, 3’-poly(A) tails,35 and stable secondary structures.36 RNA platination may also inhibit or alter exonuclease activity, affecting RNA processing pathways.
VPD and SPD exonucleases have been used previously to locate transplatin crosslinks in RNA duplexes,37 and to identify the binding locations of platinum complexes to 4-mer GA3 RNAs.38 In these experiments, exonuclease digestion proceeds up to nucleotides involved in platinum binding, producing RNA fragments that retain bound metals. We systematically tested whether similar enzymatic inhibition resulted from cisplatin binding to single-stranded 13-mer RNAs containing isolated purine sequences that are known to be targeted by cisplatin. In these experiments, reaction of 5’-(U)6-XY-(U)5 (XY = GG, AG, GA, or GU) RNAs with an aquated form of cisplatin produced a dominant product containing a single [Pt(NH3)2] fragment when analyzed by MALDI-MS (Figures 1a and b).21 Peaks from platinated RNAs display masses corresponding to the parent RNA plus 227 amu, depicting two less H’s than would be expected from attachment of a Pt(NH3)2 fragment (229 amu). This is due to the proton transfers necessary to offset binding of Pt2+ and still produce singly charged molecular ions.39 Platinated RNAs were subsequently digested using VPD and SPD exonucleases, after which MALDI-MS was used to characterize the products of each reaction. The results of these experiments are summarized in Table 1. Spectra corresponding to each entry are included as Supplementary Information, Figures S3-S6.
Figure 1. Platination and VPD Exonuclease Digestion of RNAs.

MALDI-MS spectra of a) 5’-(U)6-AG-(U)5-3’ RNA b) platinated 5’-(U)6-AG-(U)5-3’ RNA c) platinated 5’-(U)6-AG-(U)5-3’ RNA digested by VPD (3’→5’) exonuclease d) platinated 5’-(U)6-GU-(U)5-3’ RNA digested by VPD (3’→5’) exonuclease.
Table 1.
Products from Enzymatic Processing of Platinated 13-mer RNAs

|
VPD: 3’→5’ Venom Phosphodiesterase.
SPD: 5’→3’ Spleen Phosphodiesterase.
U2: RNase U2 , cleaves 3’ to A or G.
“Pt” indicates [Pt(NH3)2].
In all cases VPD (3’→5’) digestion of platinated RNAs containing two neighboring internal purines extends up to, but not beyond the 3’ purine (Figure 1c). These results correspond well with previous experiments using DNA, where metallation is presumed to alter DNA structure and disrupt enzyme recognition, protecting an oligonucleotide from further digestion.40,41 VPD digestion of the platinated RNA strand containing a single G reproducibly gives [UUUUUUG + Pt(NH3)2 +H]+ as the major detected product, as expected for monofunctional adducts forming at the single G. Interestingly, major secondary peaks representing [UUUUUUGU + Pt(NH3)2 + H]+ and [UUUUUUGUU + Pt(NH3)2 + H]+ RNA fragments are also detected (Figure 1d). The differences between these products and those obtained from digestion of the platinated XY= GA, AG, and GG RNAs presumably reflects differential processing of mono- versus difunctional platinum adducts. A detailed examination of MALDI-MS spectra obtained from the platinated 5’-(U)6-GU-(U)5 RNA, however, does not show masses for an additional Cl, H2O, or OH platinum ligand as expected to be present in a monofunctional adduct (Figure S1). Although we suspect that this ligand may be lost during ionization processes,42,43 it is also possible that the fourth Pt(II) coordination site is occupied by an RNA ligand, perhaps by a neighboring U nucleobase. In biologically relevant pH ranges, the potential N3 amine ligands in uridine (and thymidine) nucleobases are not typically considered to be targets for cisplatin because they are protonated at pH values below 10.44 Platination of thymine and uracil have, however, both been observed.45,46 In addition, characterization of a platinated d(TpG) DNA dinucleotide by NMR spectroscopy found evidence for solvent-dependent Pt(II) coordination to either the N3 of a neighboring pyrimidine base in aqueous solution,47 or alternatively, interaction with non-bridging oxygens in the phosphodiester backbone in less polar solvents,48 The multiple products resulting from nuclease digestion of the platinated 5’-(U)6-GU-(U)5 RNA may reflect a combination of these types of Pt(II) interactions.
Digestion of the same singly platinated RNAs using SPD (5’→3’) also shows that this exonuclease is inhibited at platinated RNA sequences. For RNAs containing two internal purines, SPD digestion stops at the 5’U that precedes each predicted platinum adduct (Figure 2a). These results, and similar observations by Chottard and coworkers,34 contrast with what is typically observed for cisplatin diadducts occurring with DNA, where SPD cleavage proceeds all the way up to a platinum binding site.40,41 In addition, it is again found that the platinum adducts formed with the 5’-(U)6-GU-(U)5 RNA disrupt exonuclease activity differently than platinum adducts formed with the XY= GA, AG, and GG RNAs. SPD digestion of the platinated XY= GU RNA, produces [GUUUUU + Pt + H]+ and [UGUUUUUU + Pt + H]+ RNA fragments (Figure 2b). As with VPD digestion, the presence of multiple digestion products may reflect different types of platinum coordination occurring at this site. One model explaining the variances observed in how both VPD and SPD process different types of platinum adducts is that each type of adduct uniquely alters RNA structure. Platinum coordination to a single nucleobase is expected to cause less distortion in the phosphodiester backbone of an RNA than that resulting from platinum chelation. While cisplatin broadly disrupts exonuclease processing, these results show that the precise outcome of this inhibition relies on both the type of platinum adduct formed with an RNA and on how a given protein recognizes specific aspects of RNA structure.
Figure 2. SPD Digestion of Platinated RNAs.

MALDI-MS spectra of a) SPD (5’→3’) digestion of platinated 5’-(U)6-AG-(U)5-3’ RNA b) SPD (5’→3’) digestion of platinated 5’-(U)6-GU-(U)5-3’ RNA.
Cellular 5’→3’ and 3’→5’ exonucleases often act concertedly to degrade RNAs and have synergistic responsibilities in RNA processing.35 For this reason a single platinum adduct occurring on an RNA may eventually be approached by nucleases from each direction. Prolonged incubation of the platinated RNAs used in this study with both 3’→5’ and 5’→3’ exonucleases results in low molecular weight products of only a few nucleotides in length (data not shown). In some cases, these products may represent the end of a drug-damaged RNA’s cellular lifecycle.
Influence of Pt(II)-RNA adducts on processing by RNase U2
In addition to exonucleases, a number of sequence- and structure-specific endoribonucleases (RNases) are used to trim and modify RNAs in a cell. RNases such as RNase P and Drosha (RNase III) are critical in maturation of the transcriptome.49,50 New evidence showing that certain RNases are active in cellular stress response51 suggests that if platination is capable of disrupting RNase function, cells may then become handicapped in their ability to respond to platinum damage.
RNases T1 and U2 specifically recognize purine ribonucleotides and catalyze cleavage of the 3’ phosphodiester bond attached to a G or an A.52,53 Monofunctional [Pt(NH3)3Cl]+ coordination to the N7 atom of purine nucleobases has been shown to disrupt molecular recognition by RNase T1 and RNase U2.54 Cleavage patterns produced by partial RNase T1 digestion have been used in several studies in order to infer platination sites and monitor broad structural changes in RNA resulting from platination.16–18 Our findings using processive exonucleases led us to investigate whether endonucleolytic cleavage was similarly dependent on the type of platinum adduct formed or dependent upon additional factors such as RNA sequence. To test this, platinated 13-mer RNAs were reacted with RNase U2 (under conditions that cleave both A and G), and digestion products were again characterized by MALDI-MS. For platinated sequences containing XY=GG, AG, and GU, RNase U2 cleavage is not observed, being blocked at the purine nucleotides predicted to be involved in platinum binding (Figure 3a and b). Surprisingly, however, digestion of the platinated 5’-(U)6-GA-(U)5-3’ RNA shows a reaction product corresponding to [UUUUUUGA + Pt(NH3)2 + H],+ depicting RNase cleavage 3’ to an adenosine predicted to be involved in a cisplatin diadduct (Figure 3c and d). In DNA, closure of monofunctional 5’-d(AG*)-3’ cisplatin adducts is preferred over closure of 5’-d(G*A)-3’ adducts,55 raising the possibility that cisplatin does not readily form a diadduct with the GA-containing RNA. However, data obtained from VPD (3’→5’) digestion of the same platinated 5’-(U)6-GA-(U)5-3’ RNA (Figure S2) shows no digestion beyond the 3’ adenosine, supporting its involvement in a cisplatin diadduct. These cumulative data suggest that the platinated G*A* RNA adduct is uniquely recognized and processed by RNase U2, whereas other purine-Pt(II) adducts are not. The basis for this selectivity may arise from the RNase’s known A>G preference, allowing the endonuclease in some cases to partially recognize the 3’ platinated A despite platinum modification. Together, the inhibition of both 5’→3’ and 3’→5’ exonucleases and an endoribonuclease show that cisplatin-RNA adducts disrupt a representative of each of the 3 major classes of intracellular RNA-degrading enzymes22 and suggests that platination may disrupt RNA processing reactions in drug-treated cells.
Figure 3. U2 Digestion of Platinated RNAs.

MALDI-MS spectra obtained following a) RNase U2 digestion of 5’-(U)6-GU-(U)5-3’ RNA b) RNase U2 digestion of platinated 5’-(U)6-GU-(U)5-3’ RNA c) RNase U2 digestion of 5’-(U)6-GA-(U)5-3’ RNA. d) RNase U2 digestion of platinated 5’-(U)6-GA-(U)5-3’ RNA.
Pt(II)-RNA adducts block cDNA synthesis by reverse transcription
Accessing the information stored within an RNA transcript typically relies on successful recognition of an RNA sequence through the formation of Watson-Crick base pairs. In addition to processes such as si- and miRNA genetic regulation that rely on sequence complementarity in trans, cellular machines such as the ribosome and reverse transcriptases (RTs) move over long sequences of RNA where even relatively infrequent platination could potentially stall or otherwise disrupt function. Correspondingly, reverse transcription is commonly used to study both natural (e.g. hypermodified nucleobases such as wybutosine) and unnatural (e.g. 2’-OH acylation used in SHAPE chemistry) RNA modifications,56,57 making it a technique amenable to monitor platination of RNA. Recently Rijal, and Chow have reported that cisplatin binding to exposed purines in the E. coli ribosome may be studied by RT-based primer extension of RNA isolated from drug-treated cells.15 We extended this methodology to locate platinum adducts with an RNA termed PEBBD (Primer-Extended BBD) (Figure 4a). We have recently reported that the internal loop contained in the PEBBD sequence forms intramolecular crosslinks when reacted with cisplatin.21 To explore the ability of Pt(II) adducts to affect RT-based primer extension, PEBBD RNA was reacted with increasing amounts of an aquated form of cisplatin, annealed to a 5’ end-labeled DNA primer and reverse transcribed using M-MuLV RT. Primer extension was monitored using sequencing dPAGE. (Figure 4b). cDNA synthesis proceeds 5’→3’ (toward the 5’ end of the RNA), and shows several platinum-concentration dependent disruptions. The first major disruption at A32 occurs immediately 3’ to a G that we identified previously, using chemical footprinting, to be involved in platinum crosslinking across the internal loop of this RNA.21 A second major interruption (A26) is observed immediately prior to a GG sequence adjacent to a capping GAAA tetraloop, indicating platination at this site. Additional RT stops in the GAAA tetraloop may indicate platinum binding to this region of PEBBD. Tetraloops of the general GNRA sequence are highly conserved RNA structural motifs and have been predicted to contain metal-binding sites.58–60 It is interesting to postulate that RT inhibition in this region results from cisplatin targeting of this semi-“native” metal ion-binding site. Information from nucleotides on the 5’ side of the internal loop is obscured by platinum-independent inhibition under these RT conditions, perhaps due to formation of stable secondary structures with remaining regions of the 5’ primer sequence. In conjunction with previous data,15 these data suggest that reverse transcription is broadly applicable to the identification of platinated sites in RNA, and further suggest that platination could disrupt similar enzymatic processing events requiring RNA recognition in vivo.
Figure 4. Reverse Transcription of Platinated PEBBD.

a) Sequence of the BBD region of PEBBD. 5’ and 3’ sequences attached for in vitro PCR amplification of coding DNA, transcription, and reverse transcription are represented by blue lines. RT stops are represented by arrows. b) Reverse transcription of platinated PEBBD analyzed by 15% sequencing dPAGE along with a plot of band intensities quantified using ImageQuant software.
Reversing Pt(II)-RNA adducts with thiourea
The removal of cisplatin adducts from DNA through reaction with sulfur-containing small molecules has been studied pursuant to an understanding of how biomolecular thiol and thioketone nucleophiles might compete for cisplatin binding or remove drug adducts in the cell.61 Additionally, platinum removal is a necessary step in several common experimental methods used to study cisplatin binding to DNA, and S-donors provide a milder alternative to the reaction conditions required for more commonly used cyanide salts.26,62,63 We therefore studied the reaction of thiourea with platinated RNAs, using MALDI-MS to monitor the disappearance of platinated 5’-(U)6-GU-(U)5-3’and 5’-(U)6-GG-(U)5-3’ RNAs. When a 200 μM solution of platinated RNA is reacted with an equal volume of a saturated thiourea solution at 37 ºC, products corresponding to platinated RNAs disappear while products representing un-modified RNAs increase in relative abundance (Figure 5). While MALDI-MS is not strictly a quantitative technique and therefore cannot be used to assess the kinetics of the platinum removal, the spectra indicate the complete disappearance of platinated RNAs at 72 h under these conditions with no detectable degradation of the RNA. This reaction should be amenable to future studies seeking to remove cisplatin from pools of RNA isolated from drug-treated cells, and suggests that sulfur containing biomolecules have the potential to reverse cisplatin-RNA adducts.
Figure 5. Removal of Cisplatin From RNA by Thiourea.
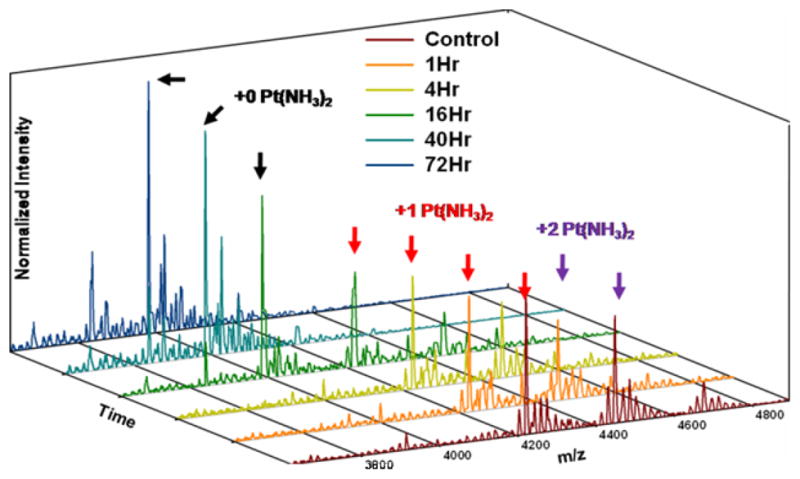
MALDI-MS spectra of the reaction of platinated 5’-(U)6-GG-(U)5-3’ RNA with thiourea taken over the course of 72 h. Similar results are observed with a platinated 5’-(U)6-GU-(U)5-3’ RNA (data not shown).
Summary
The results of these experiments demonstrate that single [Pt(NH3)2] adducts in an RNA oligonucleotide inhibit exonuclease, endonuclease, and reverse-transcription activities in a sequence-specific manner. Combined, these results show that platination of RNA disrupts the function of a number of enzymes that are analogous to those necessary for proper function of the transcriptome. Successful RNA maturation, splicing, regulation, translation, and degradation are all reliant upon the chemical fidelity of RNA transcripts. Platinum modification of individual nucleobases or more general alterations in RNA tertiary structure resulting from treatment with Pt(II) compounds may disrupt these critical processes in vivo. In the complex environment of a cell, additional factors may influence the processing of platinated RNAs, such as recently discovered RNA surveillance mechanisms responsible for handling RNA damaged by oxidative stress.25,64 Taken together, these results drive interest in new studies that seek to deconvolute the effects of RNA platination from other cell-wide responses.
MATERIALS AND METHODS
Nucleic Acid Substrates
The 13-mer RNAs used in this study were ordered from Dharmacon, Inc., deprotected following the manufacturer’s protocol and were reacted without further purification. DNA was purchased from Integrated DNA Technologies. PEBBD RNA was transcribed from a dsDNA template containing primers necessary for PCR amplification and for transcription by T7 RNA polymerase. Following transcription PEBBD RNA was purified using 15–20% dPAGE, eluted then desalted and concentrated using Millipore YM-3 Centricon tubes. Subsequent buffer exchange and desalting was often accomplished with G-25 Sephadex size exclusion resin (GE Healthcare) on laboratory prepared microcentrifuge columns (BioRad). Oligonucleotide concentrations were determined using a Varian Cary 300 Bio UV-visible spectrophotometer.
Cisplatin Aquation
Cisplatin (Sigma-Aldrich) was stored as a 1 mM solution in 10 mM NaCl and stored in the dark at 4 °C. Immediately before use, cisplatin was aquated by reaction with 0.95 equiv. of 12 mM AgNO3 (stored in the dark). The aquation reaction was incubated at 50 °C for 1 h, at which time AgCl was precipitated by centrifugation. The supernatant solution was removed and diluted accordingly. The predominant platinum species formed varies by pH with [Pt(NH3)2Cl(OH2)]+ expected to be the main Pt-complex present at pH 7.0 (by 195Pt NMR, data not shown).
Platination of 13-mer RNAs
A solution of 80 μM RNA (typically 8 nmol) was reacted with 240 μM aquated cisplatin in the presence of 1 mM Mg(NO3)2, 100 mM NaNO3, in 10 mM Na2PO4 buffer (pH 7.0) for 16 h at 37 °C. Reactions were stopped by application to G-25 Sephadex spin-columns to remove unbound platinum, then eluted and dried to completion by SpeedVac, and resuspended in deionized water. At this time aliquots were removed and analyzed by MALDI-MS in order to establish proper platination. MALDI-MS showed these reaction conditions to produce predominantly mono-platinated RNAs that were then used in nuclease-processing studies without further purification.
No significant amount of intermolecular crosslinked products were observed, even between platinated 5’-(U)6-GU-(U)5-3’ RNA strands, and attempts to force such crosslinking were unsuccessful (data not shown). Presumably, even under the moderately strong ionic conditions employed (120 mM Na+, 1 mM Mg2+) electrostatic repulsion between two negatively charged and non-complementary RNA oligonucleotides prevents platinum crosslinking.
3’→5’ Venom Phosphodiesterase (VPD) Digestion of Platinated RNAs
Venom phosphodiesterase from Crotalus adamanteus (Phosphodiesterase I, VPD) was obtained as a lyophilized solid (Sigma-Aldrich). Approximately 5 mg of the white solid (≥0.01 unit/mg) was dissolved in 500 uL of a 50% glycerol solution containing 10 mM Zn(OAc)2 and used as a enzyme stock of ~1 × 10−4 U/ul. Digestion reactions were carried out by addition of ~1 × 10−4 U of VPD stock to 400 pmol of platinated RNA (100 μM in H2O). Reactions were incubated 2 h at 50 °C at which time protein was removed by phenol-chloroform extraction and two washes with 24:1 chloroform:isoamylalcohol. The aqueous layer was retained and dried to completion by SpeedVac. RNA was further purified by C18 ZipTips (Millipore) before MALDI-MS (vida infra).
5’→3’ Spleen Phosphodiesterase (SPD) Digestion of Platinated RNAs
Calf spleen phosphodiesterase (Phosphodiesterase II, SPD) was obtained as a 0.1 U/uL 50% glycerol solution (EMD Chemicals) and used as received. Digestion reactions were carried out by addition of 0.1 U SPD to 400 pmol of platinated RNA (~100 °M in H2O). Reactions were incubated 4hr at 50 °C at which time protein was removed by phenol-chloroform extraction and two washes with 24:1 chloroform:isoamylalcohol. The aqueous layer was retained and dried to completion by SpeedVac.
RNA was further purified by C18 ZipTips before MALDI-MS
RNase U2 Digestion of Platinated RNAs. RNase U2 was obtained as a 1 U/uL, 50% glycerol solution (ThermoScientific) and used as received. Digestion reactions were carried out by addition of 1 U RNase U2 to 400 pmol platinated or un-platinated RNA (~100 μM in H2O). Reactions were incubated 1 h at 50°C and were then purified by C18 ZipTips prior to MALDI-MS analysis.
MALDI-MS
Dried RNA samples were resuspended in deionized water and purified using C18 ZipTips using a procedure modified from the manufacturer’s protocol for RNA.65 ZipTips were washed by aspiration three times with 1:1 MeCN:H2O, and equilibrated by washing three times with 0.1% trifluoroacetic acid (TFA). RNAs were bound to the tip by repeated aspiration of the analyte solution. Bound RNA was washed three times by aspiration with 0.1% TFA, three times with deionized water, and then eluted from the column using two washes of 1:1 MeCN:H2O. The eluent was dried to completion by SpeedVac and resuspended in a matrix consisting of 375 mM 2’,4’,6’-trihydroxyacetophenone (THAP, Sigma-Aldrich), 30 mM diammonium citrate in 3:1 EtOH:H2O, with added NH4+ loaded Dowex cation exchange beads (Aldrich) and applied to the sample plate. MALDI-MS analysis was performed on a Waters QToF Premier mass spectrometer in positive-ion mode using V-mode optics.
5’ End-Labeling
The DNA primer used for reverse transcription of PEBBD was radiolabeled with T4 poly-nucleotide kinase (Fermentas) using γ32P-ATP (Perkin-Elmer). The end-labeled primer was purified by 20% dPAGE, imaged by phosphor screen and excised. Excised gel bands were eluted overnight in deionized water. The resulting eluent was ethanol precipitated and further desalted using G-25 Sephadex spin-columns.
Platination of PEBBD
A solution of 20 μM (200 pmol) PEBBD RNA was annealed and platinated with 0 to 1 equiv activated cisplatin in 1 mM Mg(NO3)2, 10 mM NaNO3, 5 mM 3-morpholinopropanesulfonic acid (MOPS), pH 6.8 for 12 h at 37 °C. Reactions were passed through G-25 Sephadex spin-columns to remove unbound platinum, and the eluent was dried to completion by SpeedVac. Reactions were resuspended in deionized water prior to reverse transcription.
Reverse Transcription of Platinated PEBBD
Platinated PEBBD samples were reverse transcribed using M-MuLV reverse transcriptase (Fermentas) using a procedure based on the manufacturer’s protocol. Briefly, 100 pmol of the appropriate 5’-end-labeled DNA primer was annealed to platinated PEBBD in the manufacturer’s supplied reaction buffer and then incubated at 42 °C for 1 h after addition of the enzyme. The reactions were diluted with formamide loading buffer containing 0.005% (w/v) xylene cyanol and bromophenol blue and analyzed using 15% dPAGE. ImageQuant version 5.0 software (Molecular Dynamics) was used to quantify band intensities. Microsoft Excel 2004 was used to normalize and graph Image Quant generated intensity values.
Sequence referencing lanes were generated with a Sequenase Version 2.0 DNA Sequencing kit (USB Corporation) following the manufacturer’s protocol, using an appropriate DNA template and the same 5’ end-labeled primer used with PEBBD.
Platinum Removal by Thiourea
A solution of platinated 5’-(U)6-GU-(U)5-3’ or 5’-(U)6-GG-(U)5-3’ RNA (200 μM in H2O, 2 uL) was reacted with an equal volume (2 uL) of a saturated thiourea solution. Reactions were incubated at 37 °C for varying amounts of time, purified using C18 ZipTips and analyzed by MALDI-MS.
Supplementary Material
Acknowledgments
We thank Kimberly Bushnell for help in optimizing conditions for the reverse transcription of PEBBD and Luke Ward for helpful discussions. We thank Dr. Steve Golledge and the Center for Advanced Materials Characterization in Oregon for support of MALDI-MS facilities, which were obtained under NSF-CRIF CHE-0639170. Funding from the NIH (GM058096), the Robert A. Welch Foundation, and the University of Oregon is gratefully acknowledged.
Footnotes
Supporting Information Available. MALDI-MS spectra referenced in the text showing Pt(NH3)2 adduct formation and detailed assignment of secondary peaks in the spectra of platinated 5’-(U)6-GU-(U)5-3’ RNA, a MALDI-MS spectrum showing VPD digestion products of platinated 5’-(U)6-GA-(U)5-3’, along with MALDI-MS data for the each entry in Table 1 are included as supplementary information. This information is available free of charge and is accessible on the World Wide Web at http://pubs.acs.org.
References
- 1.Hambley TW. Dalton Trans. 2001:2711–2718. [Google Scholar]
- 2.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 3.Reedijk J. PNAS. 2003;100:3611–3616. doi: 10.1073/pnas.0737293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D, Lippard SJ. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 5.Jung YW, Lippard SJ. Chem Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 6.Fuertes MA, Castilla J, Alonso C, Perez JM. Curr Med Chem. 2003;10:257–266. doi: 10.2174/0929867033368484. [DOI] [PubMed] [Google Scholar]
- 7.Doma MK, Parker R. Cell. 2007;131:660–668. doi: 10.1016/j.cell.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 8.Parker R, Song HW. Nat Struct Mol Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- 9.Bellacosa A, Moss EG. Current Biology. 2003;13:R482–R484. doi: 10.1016/s0960-9822(03)00408-1. [DOI] [PubMed] [Google Scholar]
- 10.Kelly SM, Corbett AH. Traffic. 2009;10:1199–1208. doi: 10.1111/j.1600-0854.2009.00944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg J, Sato P. Molecular Pharmacology. 1988;33:611–616. [PubMed] [Google Scholar]
- 12.Schmittgen TD, Ju JF, Danenberg KD, Danenberg PV. Int J Oncology. 2003;23:785–789. [PubMed] [Google Scholar]
- 13.Hagerlof M, Hedman H, Elmroth SKC. Biochem Biophys ResComm. 2007;361:14–19. doi: 10.1016/j.bbrc.2007.06.131. [DOI] [PubMed] [Google Scholar]
- 14.Hagerlof M, Papsai P, Hedman HK, Jungwirth U, Jenei V, Elmroth SKC. J Biol Inorg Chem. 2008;13:385–399. doi: 10.1007/s00775-007-0327-6. [DOI] [PubMed] [Google Scholar]
- 15.Rijal K, Chow CS. Chem Comm. 2009:107–109. doi: 10.1039/b816633a. [DOI] [PubMed] [Google Scholar]
- 16.Papsai P, Aldag J, Persson T, Elmroth SKC. Dalton Trans. 2006:3515–3517. doi: 10.1039/b603833f. [DOI] [PubMed] [Google Scholar]
- 17.Papsai P, Snygg AS, Aldag J, Elmroth SKC. Dalton Tran. 2008:5225–5234. doi: 10.1039/b719542g. [DOI] [PubMed] [Google Scholar]
- 18.Hagerlof M, Papsai P, Chow CS, Elmroth SKC. J of Biol Inorg Chem. 2006;11:974–990. doi: 10.1007/s00775-006-0157-y. [DOI] [PubMed] [Google Scholar]
- 19.N'Soukpoe-Kossi CN, Descoteaux C, Asselin E, Bariyanga J, Tajmir-Riahi HA, Berube G. DNA and Cell Biology. 2008;27:337–343. doi: 10.1089/dna.2008.0727. [DOI] [PubMed] [Google Scholar]
- 20.Boer J, Blount KF, Luedtke NW, Elson-Schwab L, Tor Y. Angew Chem-Int Edit. 2005;44:927–932. doi: 10.1002/anie.200461182. [DOI] [PubMed] [Google Scholar]
- 21.Hostetter AA, Chapman EG, DeRose VJ. J Am Chem Soc. 2009;131:9250–9257. doi: 10.1021/ja809637e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houseley J, Tollervey D. Cell. 2009;136:763–776. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 23.Mendell JT, Dietz HC. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- 24.Philips AV, Cooper TA. Cell Mol Life Sci. 2000;57:235–249. doi: 10.1007/PL00000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li ZW, Wu JH, DeLeo CJ. IUBMB Life. 2006;58:581–588. doi: 10.1080/15216540600946456. [DOI] [PubMed] [Google Scholar]
- 26.Tullius TD, Lippard SJ. J Am Chem Soc. 1981;103:4620–4622. [Google Scholar]
- 27.Inagaki K, Kasuya K, Kidani Y. Chem Lett. 1983:1345–1348. [Google Scholar]
- 28.Asara JM, Hess JS, Lozada E, Dunbar KR, Allison J. J Am Chem Soc. 2000;122:8–13. [Google Scholar]
- 29.Gonnet F, Kocher F, Blais JC, Bolbach G, Tabet JC, Chottard JC. J Mass Spectrom. 1996;31:802–809. doi: 10.1002/(SICI)1096-9888(199607)31:7<802::AID-JMS358>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 30.Gonnet F, Reeder F, Kozelka J, Chottard JC. Inorg Chem. 1996;35:1653–1658. doi: 10.1021/ic951136e. [DOI] [PubMed] [Google Scholar]
- 31.Chifotides HT, Koomen JM, Kang M, Tichy SE, Dunbar KR, Russell DH. Inorg Chem. 2004;43:6177–6187. doi: 10.1021/ic040040u. [DOI] [PubMed] [Google Scholar]
- 32.Rao L, Bierbach U. J Am Chem Soc. 2007;129:15764. doi: 10.1021/ja077390a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reeder F, Guo ZJ, Murdoch PD, Corazza A, Hambley TW, Berners-Price SJ, Chottard JC, Sadler PJ. Euro J Biochem. 1997;249:370–382. doi: 10.1111/j.1432-1033.1997.00370.x. [DOI] [PubMed] [Google Scholar]
- 34.Danckwardt S, Hentze MW, Kulozik AE. Embo J. 2008;27:482–498. doi: 10.1038/sj.emboj.7601932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garneau NL, Wilusz J, Wilusz CJ. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 36.Poole TL, Stevens A. Biochem Biophys Res Comm. 1997;235:799–805. doi: 10.1006/bbrc.1997.6877. [DOI] [PubMed] [Google Scholar]
- 37.Alguero B, de la Osa JL, Gonzalez C, Pedroso E, Marchan V, Grandas A. Angew Chem-Int Edit. 2006;45:8194–8197. doi: 10.1002/anie.200603128. [DOI] [PubMed] [Google Scholar]
- 38.Bombard S, Kozelka J, Favre A, Chottard JC. Euro J Biochem. 1998;252:25–35. doi: 10.1046/j.1432-1327.1998.2520025.x. [DOI] [PubMed] [Google Scholar]
- 39.Iannitti-Tito P, Weimann A, Wickham G, Sheil MM. Analyst. 2000;125:627–633. doi: 10.1039/a908920i. [DOI] [PubMed] [Google Scholar]
- 40.Fichtinger-Schepman AMJ, Van der Veer JL, Den Hartog JHJ, Lohman PHM, Reedijk J. Biochemistry. 2002;24:707–713. doi: 10.1021/bi00324a025. [DOI] [PubMed] [Google Scholar]
- 41.Inagaki K, Kidani Y. Inorg Chim Acta-Bioinorganic Chemistry. 1985;106:187–191. [Google Scholar]
- 42.Costello CE, Nordhoff E, Hillenkamp F. Int J Mass Spectrom Ion Process. 1994;132:239–249. [Google Scholar]
- 43.Nyakas A, Eymann M, Schurch S. J Am Soc Mass Spectrom. 2009;20:792–804. doi: 10.1016/j.jasms.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 44.Lippert B. Progress in Inorganic Chemistry. 1989;37:1–97. [Google Scholar]
- 45.Lippert B. Coord Chem Rev. 2000;200:487–516. [Google Scholar]
- 46.Campbell MA, Miller PS. Biochemistry. 2008;47:12931–12938. doi: 10.1021/bi801000w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elizondo-Riojas MA, Gonnet F, Chottard JC, Girault JP, Kozelka J. J Biol Inorg Chem. 1998;3:30–43. [Google Scholar]
- 48.Kozelka J, Barre G. Chem Euro J. 1997;3:1405–1409. [Google Scholar]
- 49.Mandel CR, Bai Y, Tong L. Cell Mol Life Sci. 2008;65:1099–1122. doi: 10.1007/s00018-007-7474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lau PW, MacRae IJ. J Cell Mol Med. 2009;13:54–60. doi: 10.1111/j.1582-4934.2008.00520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson DM, Parker R. Cell. 2009;138:215–219. doi: 10.1016/j.cell.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Pace CN, Heinemann U, Hahn U, Saenger W. Angew Chem-Int Edit Engl. 1991;30:343–360. [Google Scholar]
- 53.Noguchi S, Satow Y, Uchida T, Sasaki C, Matsuzaki T. Biochemistry. 1995;34:15583–15591. doi: 10.1021/bi00047a025. [DOI] [PubMed] [Google Scholar]
- 54.Escaffre M, Favre A, Chottard JC, Bombard S. Anal Biochem. 2002;310:42–49. doi: 10.1016/s0003-2697(02)00279-8. [DOI] [PubMed] [Google Scholar]
- 55.Mantri Y, Lippard SJ, Baik MH. J Am Chem Soc. 2007;129:5023–5030. doi: 10.1021/ja067631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motorin Y, Muller S, Behm-Ansmant I, Brantant C. Rna Modification. Vol. 425. Elsevier Academic Press Inc; San Diego: 2007. pp. 21–53. [Google Scholar]
- 57.Mortimer SA, Weeks KM. Nat Protocols. 2009;4:1413–1421. doi: 10.1038/nprot.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mundoma C, Greenbaum NL. J Am Chem Soc. 2002;124:3525–3532. doi: 10.1021/ja012268b. [DOI] [PubMed] [Google Scholar]
- 59.Maderia M, Horton TE, DeRose VJ. Biochemistry. 2000;39:8193–8200. doi: 10.1021/bi000140l. [DOI] [PubMed] [Google Scholar]
- 60.Costa M, Michel F. Embo J. 1997;16:3289–3302. doi: 10.1093/emboj/16.11.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reedijk J. Chem Rev. 1999;99:2499–2510. doi: 10.1021/cr980422f. [DOI] [PubMed] [Google Scholar]
- 62.Ober M, Lippard SJ. J Am Chem Soc. 2007;129:6278–6286. doi: 10.1021/ja0706145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monjardet-Bas V, Bombard S, Chottard JC, Kozelka M. Chem Euro J. 2003;9:4739–4745. doi: 10.1002/chem.200305085. [DOI] [PubMed] [Google Scholar]
- 64.Wurtmann EJ, Wolin SL. Crit Rev Biochem Mol Biol. 2009;44:34–49. doi: 10.1080/10409230802594043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ragas JA, Simmons TA, Limbach PA. Analyst. 2000;125:575–581. doi: 10.1039/a909709k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.