

SUMMARY
Recognition of nucleic acids as a signature of infection by Toll-like receptors (TLRs) 7 and 9 exposes the host to potential self-recognition and autoimmunity. It has been proposed that intracellular compartmentalization is largely responsible for reliable self versus non-self discrimination by these receptors. We have previously shown that TLR9 and TLR7 require processing prior to activation, which may further reinforce receptor compartmentalization and tolerance to self, yet this possibility remains untested. Here we report that residues within the TLR9 transmembrane (TM) region conferred the requirement for ectodomain proteolysis. TLR9 TM mutants responded to extracellular DNA, and mice expressing such receptors died from systemic inflammation and anemia. This inflammatory disease did not require lymphocytes and appeared to require recognition of self-DNA by dendritic cells. To our knowledge, these results provide the first demonstration that TLR-intrinsic mutations can lead to a break in tolerance.
INTRODUCTION
Toll-like Receptors (TLRs) are receptors of the innate immune system that have evolved to recognize conserved features of microbes. A subset of TLRs (TLR3, TLR7, TLR8, and TLR9) link the recognition of nucleic acids to induction of innate and adaptive immune responses (Medzhitov, 2007; Takeda et al., 2003). Although this strategy enables detection of viral nucleic acid within the degradative environment of intracellular compartments, the cost of this strategy is the potential recognition of self DNA and RNA, which has been implicated in autoimmune diseases such as systemic lupus erythematosus and psoriasis (Lande et al., 2007; Marshak-Rothstein, 2006). Accordingly, regulatory mechanisms must exist that prevent responses to self-derived nucleic acids. One such mechanism appears to be the intracellular localization of TLR9 and TLR7, which sequesters these receptors from extracellular self nucleic acid released from necrotic cells or apoptotic cells that undergo secondary necrosis (Barton and Kagan, 2009; Marshak-Rothstein, 2006). Ligand recognition occurs in endocytic compartments, although the mechanistic details of receptor trafficking and localization remain somewhat enigmatic and, in some cases, controversial (Brinkmann et al., 2007; Ewald et al., 2008; Kim et al., 2008; Latz et al., 2004; Leifer et al., 2006; Leifer et al., 2004).
Nevertheless, mechanisms that facilitate delivery of self nucleic acid to these compartments, such as internalization via surface receptors or association with cationic peptides, can trigger TLR activation (Lande et al., 2007; Leadbetter et al., 2002; Marshak-Rothstein, 2006). It is noteworthy that all examples of TLR-mediated recognition of self nucleic acids, especially in vivo, involve aberrant ligand delivery as opposed to receptor intrinsic mutations. One exception comes from our previous work in which we showed that a chimeric TLR9-TLR4 receptor (TLR9N4C), which traffics to the cell surface, gains the ability to respond to extracellular vertebrate DNA in vitro (Barton et al., 2006). However, the TLR4-based signaling of this receptor prevented us from performing definitive experiments regarding the effect of altered TLR9 localization (Barton et al., 2006). Unlike TLR9 that signals only through the common TLR adaptor MyD88, TLR4 utilizes both MyD88 and TRIF, another TLR adaptor, so the in vitro gain of function we observed with this chimera can not be unequivocally attributed to altered localization. Moreover, this receptor does not result in autoimmune disease when expressed in vivo (Fig. S1A). This lack of disease may be due to the inability of this receptor to signal in plasmacytoid dendritic cells (pDCs) and B cells but could also imply that additional regulatory mechanisms exist that prevent responses to self.
We and others have recently described another regulatory step that may limit TLR activation to endolysosomes; namely, the ectodomain of TLR9 is cleaved prior to receptor activation (Ewald et al., 2008; Park et al., 2008). We have recently demonstrated that TLR7 and TLR3 are similarly regulated, suggesting that ectodomain proteolysis may be a general mechanism to restrict nucleic acid sensing by TLRs to intracellular compartments (Ewald et al., 2011). Receptor processing consists of at least two distinct proteolytic steps mediated by asparagine endopeptidase and cathepsins (Ewald et al., 2011; Ewald et al., 2008; Park et al., 2008; Sepulveda et al., 2009), and treatment of cells with inhibitors of these proteases prevents TLR9, TLR7, and TLR3 activation (Ewald et al., 2011). An attractive possibility is that this requirement for proteolysis by lysosomal proteases prevents mislocalized receptors (e.g., receptors that access the cell surface en route to the endolysosome) from responding to self nucleic acid because they remain unprocessed and thus non-functional (Barton and Kagan, 2009; Ewald et al., 2008). In this way, the requirement for proteolysis may obviate the need for strict receptor compartmentalization; however, the importance of receptor processing for maintaining tolerance to self nucleic acid remains unexplored.
In the work reported here, we describe TLR9 mutants that no longer require ectodomain proteolysis for activation. Expression of these dysregulated receptors in mice results in lethal autoinflammatory disease. We find that this disease is independent of B and T cells yet requires dendritic cells. Thus, dysregulated activation of TLR9 can lead to autoinflammatory disorders in certain contexts. Overall, this work clearly demonstates that mutations in TLR9 can break tolerance to self-nucleic acid. In addition, we provide, to our knowledge, the first evidence that receptor proteolysis has evolved to regulate self versus non-self discrimination by nucleic acid sensing TLRs.
RESULTS
Mutations in the TLR9 transmembrane (TM) domain enable signaling in the absence of ectodomain proteolysis
A direct way to test the role of receptor processing in self versus non-self discrimination would be to express TLR9 truncation mutants already lacking the portion of the ectodomain that is normally removed by cleavage. We constructed a number of different “pre-cleaved” receptors, including a truncated TLR9 mutant recently reported to complement TLR9-deficiency (Park et al., 2008); however, when expressed in HEK293T cells or Tlr9-/- macrophages, these receptors responded very poorly or not at all to CpG oligonucleotides (ODN) (Fig. S1B). Analysis of N-linked sugars indicated that truncated TLR9 mutants were unable to exit the endoplasmic reticulum (ER), presumably due to misfolding of the ectodomains (Fig. S1C). Thus, we concluded that expression of pre-truncated receptors would not allow us to test the physiological relevance of receptor cleavage for maintaining tolerance.
As an alternative approach, we sought to construct mutant TLR9 receptors that bypass the requirement for ectodomain processing prior to activation. Ligand binding studies indicate that proteolysis does not regulate the receptor’s ability to bind ligands; instead, processing of the ectodomain may be required to allow ligand-bound receptors to undergo a conformational change that initiates MyD88 recruitment and signal transduction. Indeed, the initiation of signal transduction by TLR9 homodimers appears to require a membrane proximal shift in the cytosolic domains. These data as well as the presence of atypical amino acids for a membrane spanning region led us to focus on the transmembrane (TM) domain of TLR9 as potentially involved in mediating the requirement for ectodomain processing. Our strategy was to create a chimeric receptor (hereafter called TLR9TM-MUT) in which the TLR9 TM region was replaced with a more conventional, largely hydrophobic membrane spanning sequence (Fig. 1A).
Figure 1. TLR9TM-MUT does not require processing for activation.
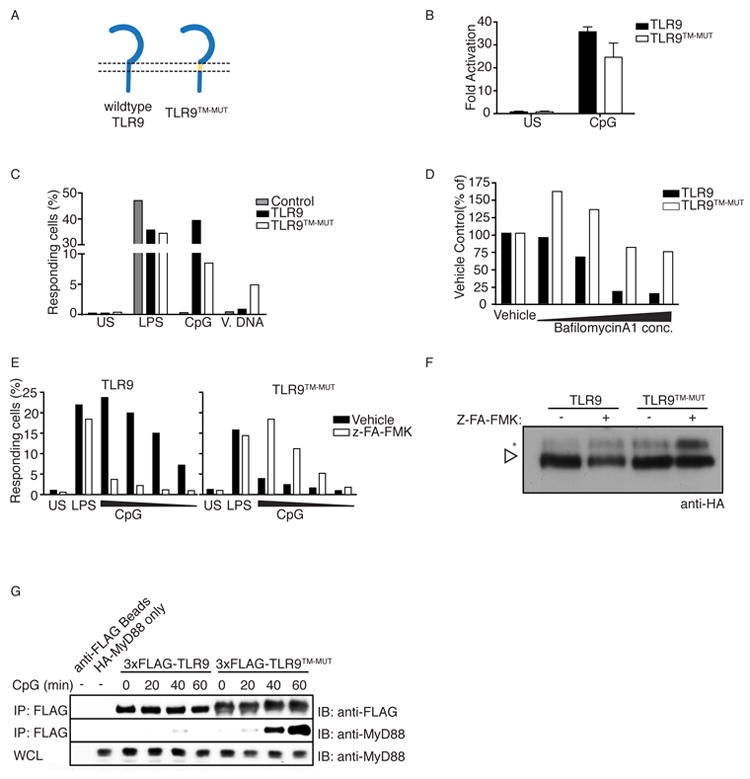
A, Schematic of TLR9 and TLR9TM-MUT. TLR9TM-MUT consists of the ectodomain and cytosolic domain of TLR9 (blue) and the transmembrane domain of TLR3 (yellow). B, TLR9TM-MUT can respond to CpG DNA ligands. HEK293 cells stably expressing a NF-κB luciferase reporter and TLR9 or TLR9TM-MUT were stimulated with CpG ODN (phosphorothioate backbone) as indicated and luciferase activity was measured 16hr later. C, Macrophages expressing TLR9TM-MUT respond to CpG ODN and extracellular genomic DNA. TLR9-deficient macrophages transduced with retroviruses encoding TLR9, TLR9TM-MUT or empty vector (control) were stimulated as indicated and the percentage of GFP-positive cells producing TNF was measured by intracellular staining and flow cytometry. Data shown are representative of two experiments. D and E, Signaling by TLR9TM-MUT does not require proteolysis. TLR9-deficient macrophages expressing TLR9 or TLR9TM-MUT were pretreated with bafilomycinA1 for 2 hours (D) or Z-FA-FMK overnight (E), stimulated with CpG ODN, and stained to measure TNF production, as described in (C). F, Stabilization of shifted form of TLR9TM-MUT in the presence of Z-FA-FMK. Anti-HA immunoblot of TLR9 and TLR9TM-MUT from lysates of macrophage treated with DMSO (vehicle) or Z-FA-FMK. Open triangle indicates full-length TLR9. Asterisk denotes the shifted form of the full-length receptor. Immunoblot shown is representative of two experiments. G, Full-length TLR9TM-MUT can recruit MyD88. TLR9-deficient macrophages expressing N-terminal FLAG-TLR9 or FLAG-TLR9TM-MUT were stimulated with CpG ODN and recruitment of MyD88 was measured by immunoprecipitation (IP) and immunoblot (IB) as indicated. The immunoblot of whole cell lysates (WCL) shows equivalent amounts of MyD88. Data shown are representative of two experiments. Unless stated otherwise, experiments in this figure were performed at least three times with similar results.
To determine the impact of TM substitutions on TLR9 signaling, we tested the ability of cells expressing TLR9TM-MUT to respond to TLR9 ligands. HEK293 cells transduced with TLR9TM-MUT responded to CpG ODN (with the nuclease-resistant phosphorothioate backbone), although the response was weak relative to cells expressing wildtype TLR9 (Fig. 1B). Similar results were observed in TLR9-deficient macrophages and dendritic cells reconstituted with each receptor (Fig. 1C and Fig. S1D). Remarkably, despite the reduced signaling of TLR9TM-MUT to CpG DNA ligands mentioned above, activation of TLR9TM-MUT was no longer blocked by bafilomycinA1 or Z-FA-FMK (Fig. 1D, E), compounds that potently block receptor processing (Ewald et al., 2011; Ewald et al., 2008; Park et al., 2008). BafilomycinA1, in particular, leads to accumulation of a post-ER full-length form of TLR9, the immediate precursor of the cleaved receptor (Ewald et al., 2008). This unprocessed form of TLR9TM-MUT also accumulated with Z-FA-FMK treatment (Fig. 1F, see asterisk). However, in contrast to TLR9, this accumulation of the full-length form of TLR9TM-MUT correlated with increased responsiveness to ligands, further supporting the conclusion that mutations in the TM enable TLR9TM-MUT to signal independently of processing.
To test directly whether TLR9TM-MUT no longer requires proteolytic processing prior to activation we compared the abilities of full-length wildtype TLR9 and full-length TLR9TM-MUT to recruit MyD88 in response to ligand. As expected, in immortalized TLR9-deficient macrophages reconstituted with N-terminal FLAG-tagged TLR9, MyD88 did not associate with the full-length receptor (isolated by FLAG immunoprecipitation) after stimulation with CpG ODN (Fig. 1G). These data agree with our previous published findings that only cleaved TLR9 recruits MyD88 (Ewald et al., 2008). In contrast, full-length TLR9TM-MUT did recruit MyD88 under similar conditions (Fig. 1G). This difference was not due to defective signaling of the wildtype TLR9 expressing cells, as both receptors were capable of inducing tumor necrosis factor (TNF) production (Fig S1E). Thus, mutations in the TM of TLR9TM-MUT enable signaling in the absence of ectodomain processing.
TLR9TM-MUT can access the cell surface and respond to extracellular ligands
We next determined how bypassing the requirement for ectodomain processing impacts self versus non-self discrimination by TLR9. We compared the responses of TLR9 and TLR9TM-MUT to phosphodiester CpG ODN and salmon sperm genomic DNA, ligands to which wildtype TLR9 responds poorly due to degradation of ligands before reaching the TLR9-containing endolysosome. TLR9TM-MUT responded robustly to both ligands while TLR9 did not, suggesting that TLR9TM-MUT had gained the ability to respond to ligands at the cell surface (Fig. 2A). To test this possibility directly, we compared the abilities of cells expressing TLR9 and TLR9TM-MUT to respond to biotin-CpG ODN immobilized by conjugation to streptavidin-coated plates. While cells expressing wildtype TLR9 were not activated, TLR9TM-MUT expressing cells responded robustly (Fig. 2B). These data indicate that bypassing the cleavage requirement enables TLR9TM-MUT to access and respond to ligands at the cell surface.
Figure 2. TLR9TM-MUT can access the cell surface and responds to extracellular ligands.
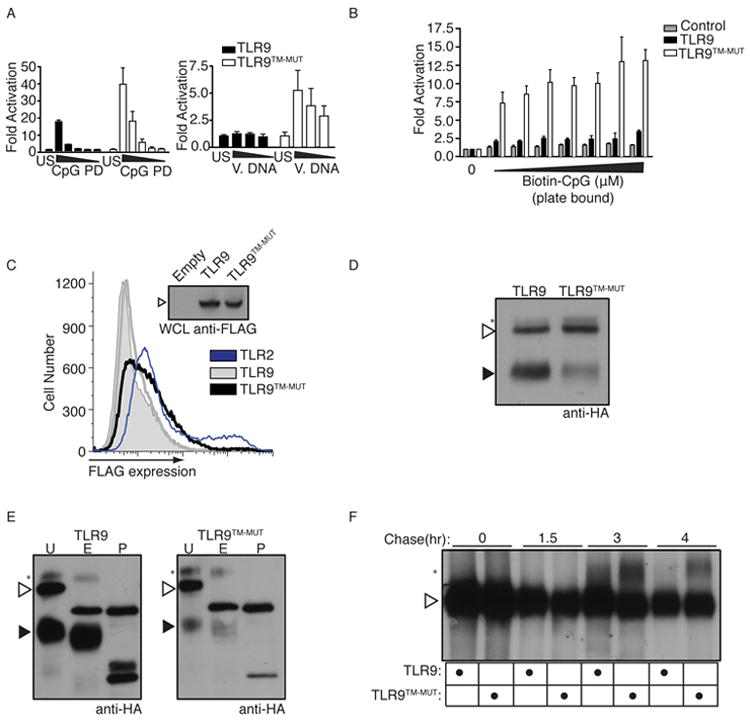
A, TLR9TM-MUT is more responsive to extracellular DNA ligands. Luciferase assays were performed as described in (B) except stimulations were with either phosphodiester (PD) backbone CpG ODN or salmon sperm DNA (V. DNA). Data shown are representative of two experiments. B, TLR9TM-MUT can respond to immobilized CpG ODN at the cell surface. Cells described in (Fig.1B) were plated on streptavidin coated plates to which biotinylated-CpG ODN had been previously conjugated. Luciferase activity was measured in lysates after 6h. C, Full-length TLR9TM-MUT is expressed at the cell surface. Flow cytometry histograms of anti-FLAG staining of HEK293T cells stably expressing N-terminal FLAG-TLR9 (grey), FLAG-TLR9TM-MUT (black), FLAG-TLR2 (blue), or empty vector control (shaded). Anti-FLAG immunoblot of whole cell lysates (WCL) of the indicated cells (inset). See also Figure S2A. D, The ectodomain of TLR9TM-MUT is processed inefficiently. Anti-HA immunoblot of lysates from TLR9-deficient macrophages expressing C-terminally HA-tagged TLR9 or TLR9TM-MUT. Open triangle indicates full-length TLR9. Closed triangle indicates processed TLR9. Asterisk denotes the shifted form of the full-length receptor. E, TLR9TM-MUT traffics through the Golgi apparatus. Immunoprecipitated TLR9 or TLR9TM-MUT from macrophage lysates were treated with Endoglycosidase H (E), PNGase F (P) or left untreated (U). F, TLR9 and TLRTM-MUT exit the ER with similar efficiency and kinetics. Pulse-chase analysis of macrophages expressing TLR9 or TLR9TM-MUT. TLR9 or TLR9TM-MUT were harvested at the indicated chase times, immunoprecipitated and visualized by SDS-PAGE. Data presented are representative of at least three independent experiments.
Whether TLR9 trafficking includes a plasma membrane step is not well understood. Most studies have not detected the receptor at the cell surface, although it remains possible that the receptor localizes to the plasma membrane in certain cell types or transiently during an intermediate trafficking step (Latz et al., 2004; Lee et al., 2006). To examine these possibilities for TLR9 and TLR9TM-MUT, we measured surface expression of N-terminal FLAG-tagged receptors by flow cytometry. Use of an N-terminal tag allows us to assess how much, if any, of the unprocessed receptors are at the cell surface. TLR9TM-MUT was clearly detectable on the cell surface at amounts higher than TLR9 albeit less than TLR2 (Fig. 2C). This difference in localization was not due to differences in overall expression, as anti-FLAG immunoblot revealed similar amounts of each protein, and the expression of a bicistronic human CD2 reporter was similar (Fig. 2C, inset, and Fig. S2A). Consistent with the slightly elevated cell surface expression of TLR9TM-MUT, the cleaved form of TLR9TM-MUT was reduced relative to wild-type TLR9, suggesting that TLR9TM-MUT does not efficiently traffic to the endolysosome (Fig. 2D, filled arrowheads). This reduction in processing was not due to inefficient ER exit, as the shifted form of the full-length receptor (representing the pool of TLR9 that has exited the ER and trafficked through the Golgi) was detectable in cells expressing TLR9TM-MUT (see asterisk in Fig. 2D). As expected, this shifted form of the protein was resistant to endoglycosidase H (endoH) treatment (Fig. 2E and (Ewald et al., 2008)). Pulse-chase analysis confirmed that the post-ER full-length forms of TLR9 and TLR9TM-MUT appeared with similar kinetics (asterisk in Fig. 2F). However, the shifted form of TLR9TM-MUT was more stable than the shifted form of TLR9 (Fig. 2F, 3h versus 4h), which agrees with the slightly higher amounts of TLR9TM-MUT at the cell surface. Finally, as expected, TLR9TM-MUT signaling still required the ER-resident protein, Unc93b1 (Fig. S2B), and expression of TLR9TM-MUT did not perturb the balance between Unc93b1 and other nucleic acid sensing TLRs, as TLR7 and TLR3 responses were largely unaltered in TLR9TM-MUT expressing DCs and macrophages (Fig. S1D and S2C).
Expression of TLR9TM-MUT in vivo causes lethal autoinflammatory disease
Because the TM mutations enable TLR9TM-MUT to signal independently of ectodomain proteolysis, we used TLR9TM-MUT to test the relevance of this regulatory mechanism in vivo. We transduced TLR9-deficient hematopoietic stem cells (HSCs) with retroviruses encoding TLR9, TLR9TM-MUT, or empty vector (control) and transferred these HSCs into lethally irradiated C57BL/6 recipients (Fig. 3A). HSCs were transduced with similar efficiencies and expressed similar amounts of GFP driven from a bicistronic vector (Fig. S3A). Strikingly, all mice receiving TLR9TM-MUT HSCs died within 4 weeks of transfer while mice receiving control or TLR9 HSCs remained healthy and viable (Fig. 3B).
Figure 3. Expression of TLR9TM-MUT in vivo leads to fatal inflammation.
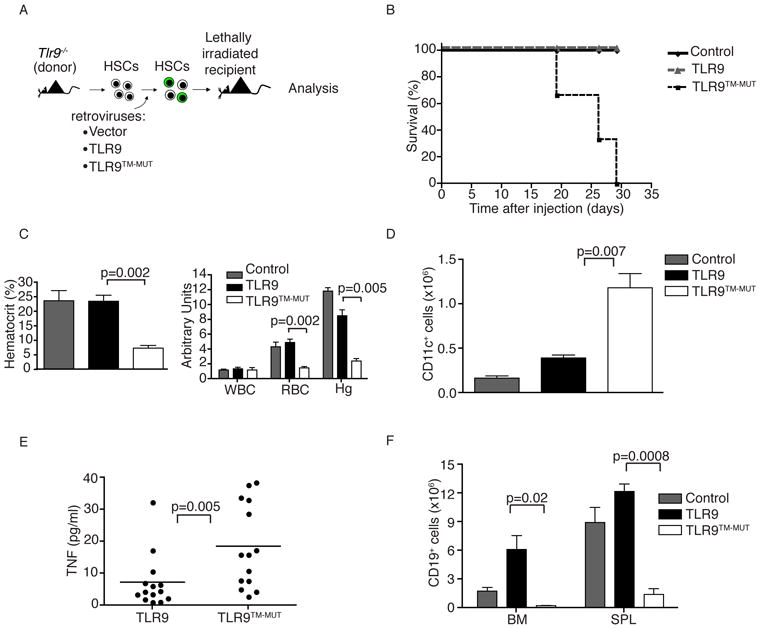
A, Schematic of the approach used to express TLR9 or TLR9TM-MUT in mice. Hematopoietic stem cells (HSC) from TLR9-deficient mice were transduced with retroviruses encoding TLR9, TLR9TM-MUT, or vector control followed by transfer into lethally irradiated C57BL/6 recipient mice. B, Mice expressing TLR9TM-MUT die from inflammatory disease. Survival plot of radiation chimeric mice receiving HSCs expressing either TLR9, TLR9TM-MUT, or empty vector (control). C, TLR9TM-MUT expressing mice develop anemia. Mean values of hematocrit, red blood cells (RBC), and hemoglobin (Hg) in the blood of mice at day 19 post-HSC transfer. D, Expression of TLR9TM-MUT in mice causes an expansion of DCs. Graph showing the total number of GFP-positive, CD11c+ cells in the bone marrow at day 19 after injection of HSCs. E, TLR9TM-MUT expression leads to inflammatory cytokine induction. TNF was measured in sera from the indicated mice at day 19-29 post-HSC transfer. Each point represents an individual mouse. Horizontal line represents the mean. F, Expression of TLR9TM-MUT in mice leads to block in B cell development. The total number of CD19+ cells in the bone marrow and spleens of the indicated mice, as measured by flow cytometry, is shown. Unless stated otherwise, experiments in this figure were performed at least three times with similar results. See also Figure S3.
TLR9TM-MUT expressing mice were profoundly anemic, as measured by hematocrit, erythrocyte and hemoglobin counts (Fig. 3C). The overall cellularity of bone marrow in TLR9TM-MUT expressing mice was reduced relative to TLR9-expressing mice, whereas the cellularity of the spleen was similar between all groups (Fig. S3C, D). Analysis of the cellular composition of bone marrow and spleens from TLR9TM-MUT expressing mice revealed an expansion of CD11c+ cells relative to mice receiving TLR9 and control transduced HSCs (Fig. 3D and Fig. S3B, E). Moreover, TLR9TM-MUT-expressing mice had elevated amounts of a number of inflammatory cytokines, including TNF, Rantes, and interleukin-18 (IL-18), consistent with TLR9TM-MUT activation in DCs (Fig. 3E and Fig. S3H). Surprisingly, the bone marrow and spleens of TLR9TM-MUT mice had drastically reduced numbers of CD19+ B cells, indicating a block in B cell development at an early stage (Fig. 3F). B cell development was blocked for both transduced and non-transduced cells, supporting the conclusion that this phenotype was not due to cell autonomous defects in HSCs (Fig. S3G). No abnormalities in the blood by complete blood count (CBC) or in the cellular composition of the spleen and bone marrow were observed in mice receiving control or TLR9 expressing HSCs (Fig. 3C-F).
TLR9TM-MUT induced autoinflammatory disease is independent of B and T cells but dependent on CD11c+ dendritic cells
The expansion of DCs and lack of B cells suggested that disease in TLR9TM-MUT mice was driven by DC activation and did not require autoantibody production. To test this possibility directly, we generated radiation chimeras using retrovirally transduced HSCs from Rag1-deficient mice. Mice receiving wildtype or Rag1-deficient HSCs expressing TLR9TM-MUT developed similarly severe disease, indicating that lymphocytes were not required for TLR9TM-MUT induced disease (Fig. 4A, B). Therefore, we considered the possibility that expression of TLR9TM-MUT on DCs enabled these cells to recognize DNA released from apoptotic cells undergoing secondary necrosis, as might be expected in irradiated mice. Consistent with this hypothesis, we detected increased DNA in the sera of irradiated mice (Fig. S4C). Moreover, expansion of DCs was only observed in GFP-positive cells, indicating that this aspect of the phenotype was cell autonomous and required TLR9TM-MUT (Fig. S3E, F). These DCs expressed high amounts of MHC class II, CD11b, and CD11c but were negative for sialic acid Ig-binding lectin-H (SiglecH), which suggests they were conventional DCs (Fig. 4C).
Figure 4. TLR9TM-MUT mediated inflammatory disease is driven by CD11c+ dendritic cells.
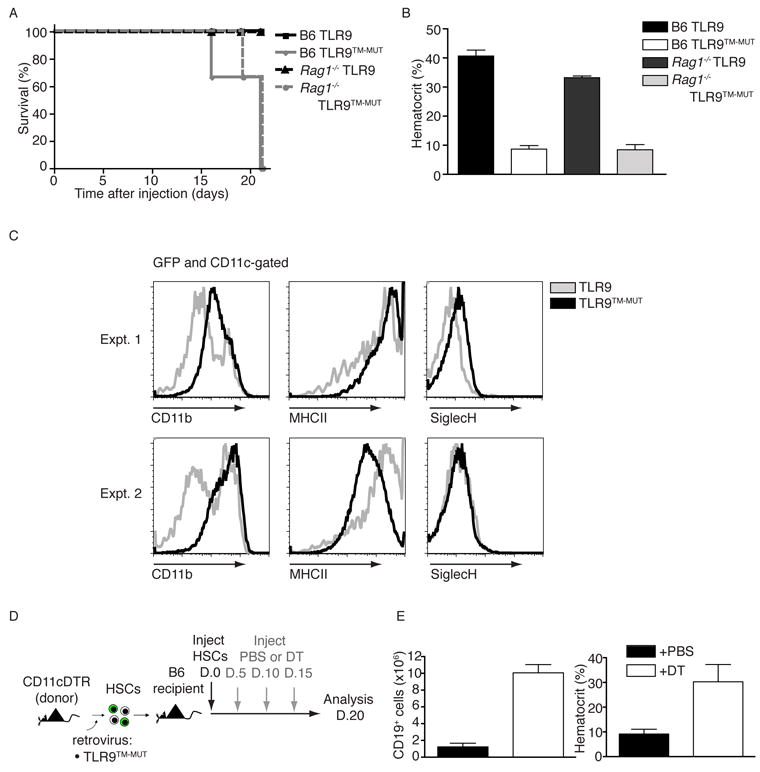
A and B, Disease caused by TLR9TM-MUT does not require B or T cells. Experiments were performed as in (3B-D), except Rag1-deficient HSCs were used to generate chimeric mice. A, Survival plot of chimeric mice receiving transduced HSCs of the indicated genotypes. B, Blood analyses at day 19 post-HSC transfer are shown. The data shown are representative of two independent experiments. C, Surface phenotype of CD11c+ cells in TLR9TM-MUT radiation chimeras. Bone marrow cells from radiation chimeras expressing the indicated receptors were analyzed for cell surface marker expression by flow cytometry (d20 post-transfer). The histograms show expression of the CD11b, MHC class II, and SiglecH on GFP-gated CD11c+ cells. Representative histograms are shown for individual mice in two separate experiments containing four mice. D and E, CD11c+ cells are required for inflammatory disease in TLR9TM-MUT mice. D, Schematic of experimental setup for depletion of CD11c+ cells in radiation chimeras expressing TLR9TM-MUT. Mice were given either PBS and Diphtheria Toxin (DT) every five days after introduction of TLR9TM-MUT transduced HSCs and analyzed twenty days after HSC injection. E, Block in B cell development and anemia seen in TLR9TM-MUT radiation chimeras is rescued by depletion of CD11c+ cells. The total number of CD19+ cells in the spleens of the indicated mice as well as mean hematocrit are shown for PBS or Diphtheria toxin (DT)-treated mice. Data are representative of two experiments with four mice per group. See also Figure S4A and B.
To test more directly the role of DCs in TLR9TM-MUT-mediated disease we transduced HSCs from mice expressing the diphtheria toxin receptor driven by the CD11c promoter (CD11c-DTR mice) and transferred these HSCs into irradiated recipients. Injecting these radiation chimeras with diphtheria toxin (DT) depleted CD11c-positive cells and prevented anemia and B cell depletion, while mice receiving saline still developed disease typical of TLR9TM-MUT expressing mice (Fig. 4D, E, Fig. S4A). As expected, the rescue mediated by DT was only apparent when HSCs expressing CD11c-DTR were used, ruling out any non-specific effects of DT treatment (Fig. S4B). Collectively, these data indicate that DCs in TLR9TM-MUT expressing mice are necessary for the anemia and B cell depletion; although, we cannot formally rule out that another CD11c-positive population of cells is depleted by DT treatment and contributes to disease. Importantly, these results further support that the observed inflammatory disease is not simply due to cell autonomous defects in the HSCs associated with ectopic TLR9TM-MUT expression since depletion of a specific cell type rescues these mice from disease.
Autoinflammatory disease in TLR9TM-MUT is partially rescued in the absence of TNF but remains intact interferon-α receptor (IFNAR) deficient mice
Systemic inflammation may account for the anemia and B cell deficiency observed in TLR9TM-MUT mice, as inflammatory mediators such as TNF and type I interferon (IFN) have been shown to negatively impact B cell development and erythropoiesis (Lin et al., 1998; Ueda et al., 2005; Ueda et al., 2004; Yamada et al., 1991; Zoumbos et al., 1985). Indeed, when we generated radiation chimeras using Tnfa-/-Lta-/-Ltb-/- HSCs, the mice expressing TLR9TM-MUT showed prolonged survival, reduced anemia, and less severe B cell depletion (Fig. 5A-C and data not shown). Using mice lacking the type I IFN receptor (IFNAR) or IFNAR-deficient HSCs, however, did not ameliorate the disease (Fig 5D-F, Fig. S5A-D), which was consistent with our observation that the expanded DCs did not express the plasmacytoid DC marker SiglecH (Fig. 4C), and we could not detect elevated amounts of type I interferon in the serum of these mice (data not shown).
Figure 5. Lack of TNF and lymphotoxin αβ but not IFNAR partially rescues inflammatory disease in TLR9TM-MUT expressing mice.
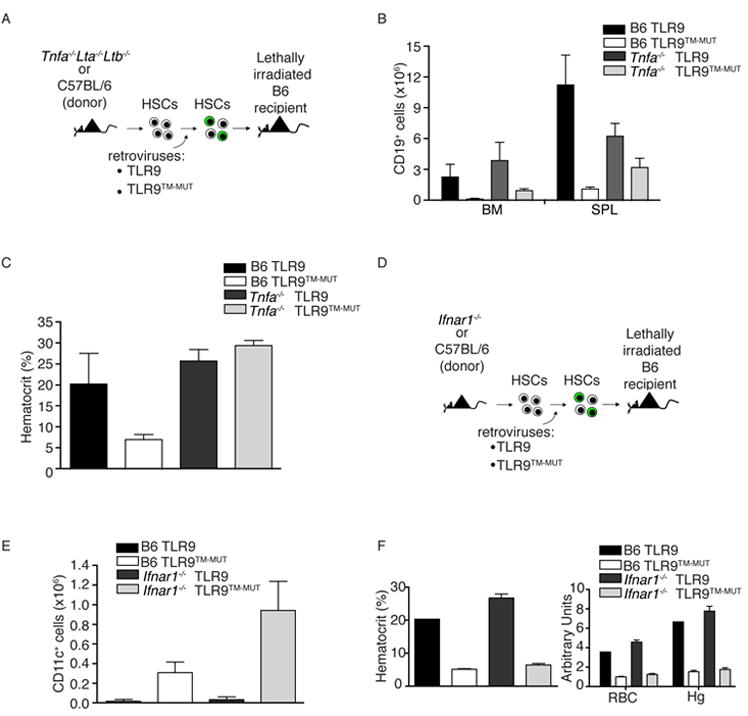
A-C, Autoinflammatory disease mediated by TLR9TM-MUT is partially ameliorated in the absence of tnf and lymphotoxinαß. A, Schematic of bone marrow chimera experiment to address role of TNF in TLR9TM-MUT mediated disease. B, Partial recovery of B cell development in TLR9TM-MUT expressing radiation chimeras generated with Tnfa-/-Lta-/-Ltb-/- HSCs. The number of CD19+ B cells in the bone marrow (BM) and spleen (SPL) of TLR9 and TLR9TM-MUT expressing radiation chimeras in C57BL/6 or Tnfa-/-Lta-/-Ltb-/- cells (d20 post-transfer) is shown. C, Tnfa-/-Lta-/-Ltb-/- TLR9TM-MUT expressing mice do not develop anemia at early time points. Mean values of hematocrit in the blood of mice at day 20 post-HSC transfer. Data shown are representative of two experiments. D-F, Loss of Ifnar1 does not rescue TLR9TM-MUT mediated disease in vivo. D, Schematic of bone marrow chimera experiment to address role of type I interferon in TLR9TM-MUT mediated disease. Initial experiment utilized IFNAR-deficient or C57BL/6 donors. E, Expression of TLR9TM-MUT using IFNAR-deficient HSCs in mice causes an expansion of dendritic cells similar to wild type. Graph showing the average number of GFP-gated CD11c+ cells in the bone marrow and spleen at day 25 after injection of HSCs. F, TLR9TM-MUT expression in IFNAR-deficient HSCs expressing mice develop severe anemia. Mean values of hematocrit, red blood cells (RBC), and hemoglobin (Hg) in the blood at day 25 post-HSC transfer. See also Figure S5.
While these data suggested that bypassing the requirement for ectodomain proteolysis leads to a break in tolerance, an alternative interpretation is that TLR9TM-MUT has altered signaling properties or engages pathways not normally accessible to wildtype TLR9. We tested this possibility by determining the role of known TLR9 signaling components in TLR9TM-MUT-based disease. As expected for a receptor with unaltered downstream signaling, the disease was entirely MyD88-dependent and did not require TRIF (Fig. S5E-G). These data, along with our in vitro studies (Fig S1D-E) argue that the TLR9TM-MUT signaling pathways are unaltered relative to wildtype TLR9, other than the ability to access extracellular ligands at the cell surface and signal independently of cleavage.
Altering 5 amino acids within the TLR9 transmembrane region is sufficient to cause autoinflammatory disease
The striking phenotype associated with expression of the TLR9TM-MUT receptor suggests that elements of the TLR9 TM impose the requirement for ectodomain proteolysis. Loss of this regulation can result in systemic inflammatory disease. To define more precisely which residues are required to maintain TLR9 tolerance, we made additional mutants (TLR9TM-MUT2, TLR9TM-MUT3, and TLR9TM-MUT4) in which fewer residues within the TLR9 TM domain were altered (Fig. 6A). Analysis of the processing of each receptor indicated that only one of the mutants, TLR9TM-MUT4, recapitulated the altered trafficking and proteolysis we had observed with TLR9TM-MUT (Fig. 6B). Moreover, TLR9TM-MUT4 responded, albeit more weakly than TLR9TM-MUT, to plate-bound CpG ODN and was insensitive to protease inhibitors, while TLR9TM-MUT2 and TLR9TM-MUT3 behaved like wildtype TLR9 in these assays (Fig. 6C, D). Consistent with these functional data, the amounts of TLR9TM-MUT4 on the cell surface were below our detection limit (data not shown). Altogether these data suggest that TLR9TM-MUT4 has a weaker gain-of-function phenotype than TLR9TM-MUT. Nevertheless, mice receiving HSCs expressing TLR9TM-MUT4 succumbed to inflammatory disease characterized by severe anemia much like TLR9TM-MUT expressing mice, while TLR9TM-MUT2 and TLR9TM-MUT3 expressing mice were unaffected (Fig. 6E). These results indicate that replacement of only five amino acids (Leu, Ser, Trp, Asp, and Cys) within the TLR9 TM is sufficient to render the receptor self reactive, although we cannot formally rule out that these residues are positioned just outside the TM region rather than within the membrane. Regardless, this region of the protein clearly plays a key regulatory role in vivo.
Figure 6. TLR9 TM mutants identify residues critical in preventing self-reactivity.
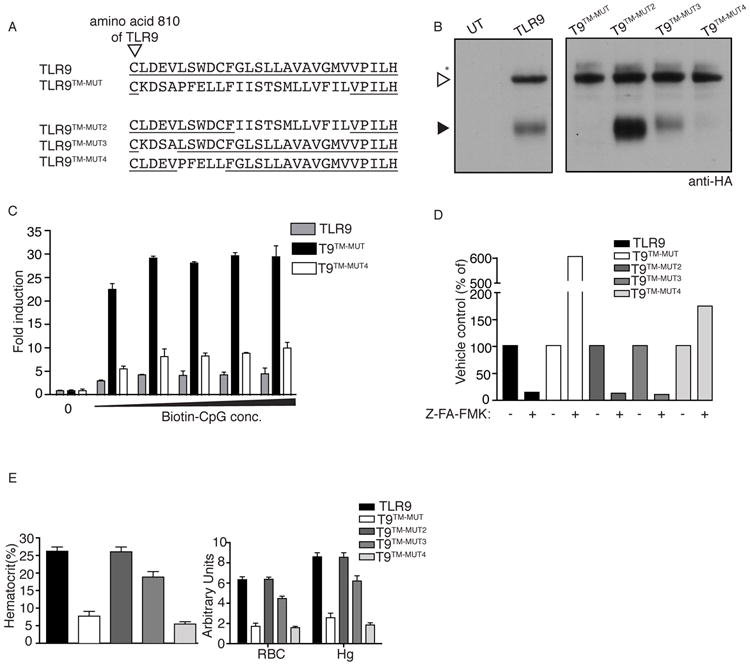
A, Schematic showing an alignment of TM regions of TLR9 mutants. Underline indicates residues from TLR9. Arrowhead indicates the last conserved cysteine of the ectodomain. B, TLR9TM-MUT4 has altered trafficking to the endolysosome. Anti-HA immunoblots of lysates from TLR9-deficient macrophages expressing the indicated TLR9 TM mutants, as described in (A). C, TLR9TM-MUT4 can respond to ligands at the cell surface. HEK293 cells stably expressing TLR9 or the indicated TLR9 mutants were plated on streptavidin-coated plates to which biotinylated CpG ODN had been previously conjugated. Luciferase activity was measured in lysates after 6hr. D, TLR9TM-MUT4 activation does not require proteolysis. TLR9-deficient macrophages expressing the indicated TLR9 TM mutants were stimulated with CpG ODN after treatment with DMSO (vehicle) or Z-FA-FMK. TNF production was measured by flow cytometry. The data are normalized for each TLR9 TM mutant by dividing by the percentage of responding cells in vehicle treated control samples. Results shown for B-D are representative of at least three experiments. E, TLR9TM-MUT4 induces inflammatory disease in vivo. Mean values of hematocrit, red blood cells (RBC), and hemoglobin (Hg) in the blood of mice at day 19 post-HSC transfer. Data are representative of two independent experiments.
DISCUSSION
The results presented here demonstratate that mutations within a TLR can result in a break of tolerance. There are many examples of autoimmune or autoinflammatory diseases resulting from perturbations in cell-extrinsic pathways that ultimately lead to excessive generation or altered delivery of self-ligands for innate immune receptors (Hanayama et al., 2004; Mevorach et al., 1998; Napirei et al., 2000; Yasutomo et al., 2001; Yoshida et al., 2005). One key piece of evidence indicating that TLR9 can contribute to the break in tolerance in autoimmune disease has been work demonstrating TLR9’s ability to synergize with autoreactive B cell antigen receptors bound to antibody-chromatin immune complexes and lead to potent activation of these cells (Leadbetter et al., 2002). This work has suggested that TLR9 signaling in response to self-DNA can lead to the production of potentially pathological self-reactive antibodies, likely due to delivery of ligands to intracellular compartments. What distinguishes our work from these previous studies is the finding that receptor-intrinsic mutations can alter TLR activation and break tolerance to self. The biophysical changes associated with signal transduction remain poorly described for TLR family members, but our work suggests that residues in the TM domain play a key regulatory role for TLR9 and impose the requirement for ectodomain processing that is necessary to avoid self recognition.
Use of radiation chimeras in our studies demonstrated that bypassing the requirement for TLR9 ectodomain processing enabled cells to respond to self-DNA released from dying cells. Activation of these cells resulted in production of inflammatory cytokines that fed back on hematopoiesis. The features of inflammatory disease that we describe here are similar to those observed in other models of dysregulated innate immune activation (Deane et al., 2007; O’Connell et al., 2008; Yoshida et al., 2004). Most relevant to our work is the recent observation that repeated CpG ODN injection in mice leads to inflammatory disease that is independent of B and T cells and results in anemia (Behrens et al., 2011). Additionally, transgenic mice overexpressing TLR7 develop a lupus-like disease characterized by DC activation and anemia within two to ten months (Deane et al., 2007). Importantly, though, the disease in TLR9TM-MUT expressing animals is much more acute and cannot simply be attributed to overexpression, as TLR9 expressing mice in our system are healthy in the time frame analyzed. Anemia is also observed in DNAse II-deficient mice, which produce type I IFN due to undigested DNA expelled during erythropoiesis (Yoshida et al., 2004). The disease induced by TLR9TM-MUT was not rescued in IFNAR-deficient mice but was partially ameliorated when HSCs lacking the genes encoding TNF, lymphotoxin α, and lymphotoxin β were used, suggesting that a combination of inflammatory cytokines may be responsible for the hematopoietic defects in these mice. The resulting anemia we observed in TLR9TM-MUT expressing mice was accompanied by reduced white blood cells, red blood cells and platelet counts which is similar to aplastic anemia in humans, a form of anemia which is largely idiopathic and correlates with increases in serum TNF.
One unexpected aspect of the disease was the independence of disease progression on B and T cells. However, as mentioned earlier, recent in vivo studies with repeated CpG ODN injection leads to an inflammatory disease that did not require lymphocytes (Behrens et al., 2011). Disease progression is also RAG-independent in the autoinflammatory arthritis observed in DNaseII-IFNAR doubly-deficient mice (Kawane et al., 2010). In our model, this independence may be due to the severity of the phenotype resulting from the abundance of self-nucleic acid that is present after irradiation and the presence of a dysregulated mutant of TLR9 that can respond to those ligands. Although the observed disease is not lupus-like, it clearly affirms the gain of function to self nucleic acid we initially observed in vitro; the resulting loss of self-tolerance in vivo demonstrates the importance of the necessity for linking receptor function with proteolysis within intracellular compartments.
Perhaps the most surprising aspect of our study was that the TM region of TLR9 conferred the regulatory requirement for ectodomain proteolysis. Precisely how these amino acids mediate this regulation remains unclear. Experiments using split green fluorescent protein (GFP) molecules fused to the TLR9 ectodomain and TM indicate that receptor activation leads to increased proximity of TM domains within the TLR9 dimer (Latz et al., 2007). Previous work from our group has shown that both cleaved and uncleaved TLR9 can bind CpG ODN, yet adapter recruitment and receptor activation is only a property of cleaved receptors (Ewald et al., 2008). Thus, amino acids within the TLR9 TM domain may prevent the formation of this active conformation prior to processing of the ectodomain. Therefore, we postulate that TLR9TM-MUT has gained the ability to undergo ligand-induced conformational changes in the absence of cleavage due to alteration of these residues in the TM. Notably, the disrupted TM region did not result in constitutive activation of TLR9TM-MUT since signaling did not occur in the absence of ligand. Structural studies of TLR9 may provide clarity on whether the ectodomain conformation after ligand binding is indeed altered upon cleavage of receptor.
Although the requirement for processing is common to all nucleic acid sensing TLRs, it is becoming increasingly clear that these TLRs are each regulated through distinct mechanisms. For example, as mentioned previously, TLR7 overexpression is sufficient to break tolerance in vivo (Deane et al., 2007). However, the lack of disease in wild-type TLR9 expressing radiation chimeras suggests that the disease in mice expressing TLR9TM-MUT is due to a different form of dysregulation and not simply overexpression. Additionally, in the MRL/lpr model of lupus, TLR9-deficient animals have exacerbated disease while TLR7-deficient mice are partially protected (Christensen et al., 2006). This surprising result suggests that, despite similar expression profiles and signaling by both receptors, there are important differences in the regulation of TLR7 and TLR9 that influence responses to self-ligands that remain poorly understood. Whether the proteolysis of TLR7 and TLR3 is regulated through residues within the TM of these receptors is not yet clear, and it is possible that there are other regions of these proteins that regulate processing for each receptor. We attempted to make a comparable “TLR7TM-MUT”, but we were unable to generate a functional receptor. There is little similarity between the TM regions of these receptors which may suggest that regulation of proteolysis occurs through distinct mechanisms for each of the nucleic acid sensing TLRs.
It is also possible that the TLR9 TM mediates interaction with an as yet unidentified regulatory protein that influences signaling and trafficking of the receptor. It is notable that this region of TLR9 contains an aspartic acid (D818), which is quite unusual in TM domains. Charged residues within TM domains can mediate association with other proteins, or in this case may govern the interactions between the TM domains within the TLR9 dimer (Latz et al., 2007). Our strategy did not directly address the importance of D818; however, a TLR9 mutant with only D818 changed to alanine was inhibited by protease inhibitors similarly to wildtype TLR9, indicating that this amino acid does not, by itself, dictate the requirement for ectodomain proteolysis (data not shown). Therefore, it seems likely that multiple residues cooperate to confer this regulation or that the phenotypes of TLR9TM-MUT and TLR9TM-MUT4 are dependent on the precise amino acids that we substituted into these receptors.
Regarding the likelihood that mutations in the TM region of TLR9 could predispose individuals to autoimmune or autoinflammatory disorders, the severe phenotype observed in TLR9TM-MUT expressing mice suggests that similar alleles in humans may be fatal as congenital mutations. For this reason, genome-wide association studies may be unlikely to identify such mutations, unless certain substitutions result in less severe, non-fatal alleles. In our system, we were unable to identify any substitutions that resulted in such alleles, although this possibility certainly remains possible. It would seem more likely, though, that TM mutations could play a role in somatic cells, resulting in rare cells with self-reactive TLRs. These cells may facilitate the initial break in tolerance responsible for certain autoimmune disorders.
EXPERIMENTAL PROCEDURES
Reagents
All chemicals and reagents, unless noted otherwise, were purchased from Fisher Scientific. CpG ODN (TCCATGACGTTCCTGACGTT, all phosphorothioate linkages) and 5’-biotinylated CpG ODN of the same sequence were from Integrated DNA Technologies. LPS, salmon sperm DNA (endotoxin free), and R848 were purchased from Invivogen. PolyI:C was purchased from Sigma. GM-CSF was purchased from Peprotech. The following antibodies were from eBioscience unless otherwise stated: anti-HA (clone 3F10, Roche), anti-FLAG (M2 and M5, Sigma), polyclonal anti-MyD88 (Enzo), PE-Cy7–conjugated anti-CD11c (N418), PE-Cy5-conjugated anti-B220 (RA3-6B2), PE–conjugated anti-TNF (MP6-XT22), PE-Cy7–conjugated CD19 (1D3), PE-Cy5-conjugated anti-CD2 (RPA-2.10, BDBiosciences), APC-Alexa750-conjugated anti-CD11b (M1/70), APC-conjugated anti-MHC II (M5), Alexa647-conjugated anti-SiglecH (ebio440c), and Alexa-647 goat anti-mouse IgG (Invitrogen). TNF was quantified in cell culture supernatants with the CBA mouse inflammation kit (BD Biosciences).
DNA cloning
TLR9TM-MUT was constructed by ‘PCR sewing’ of cDNA corresponding to amino acids 1–810 and 834-1032 of mouse TLR9 and amino acids 698-720 of mouse TLR3. cDNA encoding precleaved TLR9 (corresponding to amino acids 471-1032) was kindly provided by H. Ploegh (MIT). All TLR-encoding cDNAs were cloned into the mouse stem cell virus–based retroviral vector MSCV2.2, provided by M. Schlissel (UC Berkeley). TLR9-HA and TLR9TM-MUT-HA were constructed by the addition of a hemagglutinin epitope (YPYDVPDYA) to the C-terminal end of each receptor in MSCV2.2. Unc93b1 and the non-functional H412R mutant were cloned into MSCV2.2. Flag TLR9 and Flag TLR9TM-MUT were constructed in pCMV-Flag-8 (Sigma) and were then transferred into MigR2 (a MSCV2.2-based vector in which GFP has been replaced by human CD2).
Cell lines, plasmids and tissue culture
HEK293 and HEK 293T cells were from American Type Culture Collection. Cell lines were cultured in DMEM supplemented with 10% (vol/vol) FCS, L-glutamine, penicillin-streptomycin, sodium pyruvate and HEPES, pH 7.2 (Invitrogen). Macrophages and DCs were cultured in M-CSF and GM-CSF, respectively, containing RPMI-1640 medium supplemented with 10% (vol/vol) FCS, L-glutamine, penicillin-streptomycin, sodium pyruvate and HEPES, pH 7.2 as previously described (Barbalat et al., 2009). Unless otherwise noted, stable lines were generated by transducing cells with MSCV2.2 retroviruses encoding the target cDNA. Immortalized macrophages were generated as previously described(Blasi et al., 1985). Briefly, bone marrow-derived macrophages were cultured in supernatant containing M-CSF as well as virus encoding both v-raf and v-myc. After 8 days, macrophages were removed from M-CSF containing media and cultured in RPMI-1640 media with added supplements as described above.
Mice
C57BL/6, Rag1-/-, and Tnfa-/-Ltα-/-Ltβ-/- mice were purchased from Jackson Laboratories. Tlr9-/-, Ticam1-/- referred to as TRIF in the text), and Myd88-/-Ticam1-/- mice (provided by S. Akira) were backcrossed at least seven generations onto the C57BL/6 background. Ifnar1-/- mice on the C57BL/6 background were provided by D. Portnoy (UC Berkeley). CD11c-DTR transgenic mice (Jung et al., 2002) on the C57BL/6 background were kindly provided by the Cancer Research Lab at UC Berkeley. Unc93b13d/3d mice (generated by B. Beutler) were obtained from the MMRRC at UC Davis. All mice were housed in the animal facilities at the University of California, Berkeley according to guidelines of the Institutional Animal Care and Use Committee.
Retroviral transduction
Transduction of HSCs was performed by first enriching for HSCs by isolating bone marrow from donor mice that had been injected intraperitoneally 4d earlier with 5mg of 5-fluorouracil. Cells were cultured in stem cell media (DMEM supplemented with 15% FCS, 10mM sodium pyruvate, 2mM L-glutamine, 100U/ml of penicillin, 100g/ml of streptomycin, 100ng/ml of stem cell factor, 10ng/ml of IL-6 and 10ng/ml of IL-3, cytokines from R&D systems). 48h later, these cells were transduced with retroviral supernatant (supplemented with stem cell factor, IL-6, IL-3 and polybrene) on two successive days. Virus was produced with the ØNX-E packaging line (provided by G. Nolan, Stanford University). After the second transduction, cells were washed three times with PBS before injection into recipient mice. For retroviral transduction of immortalized macrophages, VSV-G–psuedotyped retroviral supernatant was made with GP2-293 packaging cells as previously described (Ewald et al., 2008).
Bone Marrow chimera mice
At least 1 wk before irradiation and BM transfer, mice were placed on trimethoprim-sulfamethoxazole (via drinking water). Recipient mice received lethal total body irradiation (1000 rad) from a 137Cs source. Twenty-four hours later they were reconstituted with syngeneic HSCs that had been cultured and transduced as specified in the retroviral transduction section. After reconstitution, animals were monitored until they displayed severe morbidity and/or loss of 10% of their starting body weight. For DC depletion experiments, HSCs derived from CD11c-DTR mice (Jung et al., 2002) were used to generate chimeras as described above followed by intravenous injection with 200ng of diphtheria toxin (Sigma) or saline as indicated in the schematic in Figure 4D. At the time of sacrifice, bone marrow and spleens were harvested, processed into single-cell suspensions for counting, and analyzed as previously described (Barbalat et al., 2009). Experimental groups typically consisted of three to five mice. Mice were analyzed individually.
Blood collection and analysis
Blood was collected by retroorbital bleeding or cardiac puncture. After collection, blood was divided into aliquots for preparation of sera and for haematological analysis. Serum was prepared using serum separator tubes (BD Biosciences) according to manufacturer’s instructions. Erythrocyte, leukocyte, and hematocrit counts were assayed by the UCSF Comprehensive Cancer Center Mouse Pathology Core facility, using a HEMAVET Multispecies Hematology Analyzer. Cytokines in sera samples were analyzed using SearchLight custom multiplex protein array (Pierce Biotechnology, Inc.). Serum DNA concentrations were measured using Quant-iT dsDNA Assay kit (Invitrogen).
Immunoprecipitation and western blot analyses
Cells were lysed as previously described (Ewald et al., 2008). Briefly, cells were lysed in TNT buffer (20 mM Tris pH 8.0, 200 mM NaCl, 1% Triton X-100) or Digitonin lysis buffer (50 mM Tris pH 7.4, 150mM NaCl, 5mM EDTA pH 8.0, 1% Digitonin) supplemented with Complete protease inhibitor cocktail (Roche). Lysates were cleared of insoluble material by centrifugation. For immunoprecipitations, lysates were incubated with anti-HA matrix (Roche) or anti-FLAG matrix (Sigma) and precipitated proteins were boiled in SDS buffer, separated by SDS–PAGE, and probed by anti-HA, anti-FLAG or anti-MyD88 immunoblot.
Luciferase assays
Cells were stimulated with ligands and lysed with Passive Lysis buffer (Promega) after 16h. Luciferase activity was measured on a LMaxII-384 luminometer (Molecular Devices). For plate-bound CpG assays, strepavidin coated plates (Pierce) were blocked overnight with 0.1% BSA in PBS followed by incubation with biotin-CpG ODN (2h at room temperature). After extensive washing, plates were seeded with HEK293 cells stably expressing a NF-κB luciferase reporter and the TLR of interest. In parallel, cells were seeded onto TC plates for stimulation with soluble CpG ODN. Cells were lysed after 6hr and luciferase activity was measured as described above. All assays were performed in triplicate.
Pulse–chase analysis
Cells were starved for 1 h in cysteine and methionine-free media, then pulsed with 0.25 mCi 35S-cysteine and methionine (Perkin-Elmer). After a 45-min pulse, cells were washed and cultured in 5 ml chase media with 10,000-fold molar excess of L-cysteine, L-methionine or harvested as the zero time point. Time points were harvested as follows: cells were washed twice in 2 ml PBS and lysed in 1 ml RIPA plus protease inhibitor cocktail. Labeled HA-tagged proteins were immunoprecipitated using anti-HA-matrix and visualized using SDS-PAGE.
Flow cytometry
For surface marker analysis, cells were pre-incubated with anti-CD16-CD32 (2.4G2; Monoclonal Antibody Core, University of California, San Francisco) followed by staining with the indicated antibodies. To measure TNF production, brefeldinA was added 30 min after stimulation, cells were collected after an additional 4h, and cells were stained for intracellular cytokines with a Fixation & Permeabilization kit according to manufacturer’s instructions (eBioscience). For FLAG-TLR surface expression, HEK293T cells stably expressing FLAG-TLR9, FLAG-TLR9TM-MUT, or FLAG-TLR2 were stained with anti-FLAG (M5) antibody followed by Alexa-647 goat anti-mouse IgG secondary antibody. All data were collected on LSR II (Becton Dickinson) or FC-500 (Beckman Coulter) flow cytometers and were analyzed with FloJo software (TreeStar).
Statistical analysis
Student’s paired t-test was used for statistical comparison. P-values represent comparison of TLR9TM-MUT with wild type TLR9. Prism software was used for all analyses (Graphpad Software).
Supplementary Material
Acknowledgments
We thank R. Vance and the Barton and Vance labs for helpful discussions. Supported by grants from the NIH (AI072429) and the Lupus Research Institute to G.M.B. M.L.M. was supported by a Ruth L. Kirschstein NRSA Predoctoral Fellowship (F31AI083012).
Footnotes
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, Kambayashi T, Koretzky GA. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 2011;121:2264–2277. doi: 10.1172/JCI43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Mathieson BJ, Varesio L, Cleveland JL, Borchert PA, Rapp UR. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318:667–670. doi: 10.1038/318667a0. [DOI] [PubMed] [Google Scholar]
- Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim Y-M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. The Journal of Cell Biology. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J Exp Med. 2011;208:643–651. doi: 10.1084/jem.20100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi G-P, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (New York, NY) 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G-I, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawane K, Tanaka H, Kitahara Y, Shimaoka S, Nagata S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc Natl Acad Sci U S A. 2010;107:19432–19437. doi: 10.1073/pnas.1010603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–238. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, Cao W, Wang Y-H, Su B, Nestle FO, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- Lee J, Mo J-H, Katakura K, Alkalay I, Rucker AN, Liu Y-T, Lee H-K, Shen C, Cojocaru G, Shenouda S, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- Leifer CA, Brooks JC, Hoelzer K, Lopez JL, Kennedy MN, Mazzoni A, Segal DM. Cytoplasmic targeting motifs control localization of toll-like receptor 9. J Biol Chem. 2006 doi: 10.1074/jbc.M607511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173:1179–1183. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Dong C, Cooper MD. Impairment of T and B cell development by treatment with a type I interferon. J Exp Med. 1998;187:79–87. doi: 10.1084/jem.187.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25:177–181. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- O’Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J, Paquette RL, Baltimore D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–594. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B, Brinkmann MM, Spooner E, Lee CC, Kim Y-M, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda FE, Maschalidi S, Colisson R, Heslop L, Ghirelli C, Sakka E, Lennon-Duménil A-M, Amigorena S, Cabanie L, Manoury B. Critical Role for Asparagine Endopeptidase in Endocytic Toll-like Receptor Signaling in Dendritic Cells. Immunity. 2009;31:737–748. doi: 10.1016/j.immuni.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;201:1771–1780. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med. 2004;199:47–58. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada G, Ogawa M, Akagi K, Miyamoto H, Nakano N, Itoh S, Miyazaki J, Nishikawa S, Yamamura K, Taniguchi T. Specific depletion of the B-cell population induced by aberrant expression of human interferon regulatory factor 1 gene in transgenic mice. Proc Natl Acad Sci U S A. 1991;88:532–536. doi: 10.1073/pnas.88.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28:313–314. doi: 10.1038/91070. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nature immunology. 2004 doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- Zoumbos NC, Gascon P, Djeu JY, Young NS. Interferon is a mediator of hematopoietic suppression in aplastic anemia in vitro and possibly in vivo. Proc Natl Acad Sci U S A. 1985;82:188–192. doi: 10.1073/pnas.82.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.