

Abstract
Background
Several Retinoic Acid Receptors (RAR) agonists have therapeutic activity against a variety of cancer types; however, unacceptable toxicity profiles have hindered the development of drugs. RAR agonists presenting novel structural and chemical features could therefore open new avenues for the discovery of leads against breast, lung and prostate cancer or leukemia.
Results
We have analysed the induced fit of the active site residues upon binding of a known ligand. The derived binding site models were used to dock over 150,000 molecules in silico (or virtually) to the structure of the receptor with the Internal Coordinates Mechanics (ICM) program. Thirty ligand candidates were tested in vitro.
Conclusions
Two novel agonists resulting from the predicted receptor model were active at 50 nM. One of them displays novel structural features which may translate into the development of new ligands for cancer therapy.
Background
The retinoic acid receptors (RAR-α, -β, and -γ) are transcription factors regulating a variety of endocrine metabolic pathways. Unlike anti-estrogens, such as tamoxifen or raloxifene, ligands targeted against the RAR isoforms can present anticancer activity against both estrogen receptor positive and negative breast tumor cells [1]. As a result, such molecules could constitute a novel generation of drugs against breast cancer. For reasons not yet clear, both agonists and antagonists of RAR can present anti-tumor activity against breast, prostate, lung cancer or leukemia [1,2,3,4,5,6,7]. The development of both types of ligands could therefore have important biomedical implications. We have recently demonstrated that antagonists could be discovered rationally, based on a model of the antagonist-bound conformation of the receptor [8]. Our goal here is to discover innovative molecular structures with RAR agonist activity.
Several retinoid and non-retinoid ligands have been described, which activate one or a combination of RAR isoforms. Some of them, such as the natural hormone all-trans retinoic acid (all-trans RA) (Fig. 1a), have been tested clinically, and display unacceptable side effects, such as skin dryness, cheilitis, hypertriglyceridemia and conjunctivitis [9,10]. However, the compounds tested so far belong to limited series of related structures. An increasing amount of data suggests that the RAR-β isoform, which is under the transcriptional control of RAR-α, is involved in suppressing cell growth and tumorigenicity [11,12,13,14,15,16]. Innovative molecules with RAR-α and RAR-β agonist activity could therefore present more favorable toxicity profile than pan-agonists.
Figure 1.

Chemical structure of the RAR ligands discussed in this work. A: All-trans retinoic acid. B: 4- [(5,6,7,8-tetrahydro- 5,5,8,8- tetramethyl-2- naphthalenyl) carboxamido] benzoic acid (Am580). C: 2- (4- carboxyphenyl)- 4- (3-trifluoromethylphenyl) thiazole (Agonist 1). D: 4- (3-(3,5- di-tert-butyl- 4-hydroxyphenyl)-3- oxopropenyl) benzoic acid (Agonist 2) E: 3,5-di-tert-butylchalcone 4'-carboxylic acid (Ch55)
We applied a flexible virtual screening algorithm (Molsoft ICM, virtual library screening module [17]) which rapidly docks hundreds of thousands of flexible compound structures into the ligand binding pocket of RAR, and discovered two novel RAR-β selective agonists. One of these ligands displays original structural and chemical characteristics, which could be used in the development of novel compounds for cancer prevention and therapy.
Results and discussion
We first built a model of the RAR-α agonist binding pocket from the crystal structure of the RAR-γ ligand binding domain (RAR-γ LBD) / all-trans RA complex [18]. All but three amino acids in the vicinity of the ligand are conserved between the two isoforms. These three non-identical residues -A234, M272, and A397- were changed to the RAR-α isoform -S234, I272 and V397- and the energy of the system was minimized (see "Materials and Methods").
In order to address the accuracy of our model of the RAR-α binding pocket, we docked Am580, an RAR-α specific agonist [19], into the receptor (the chemical structure of Am580 is shown Fig. 1b). A rapid docking procedure with flexible ligand and a grid representation of the receptor was followed by an extensive Monte Carlo energy minimization with both ligand and receptor side chains flexible (see "Materials and Methods" for details). The ligand superimposed well with the natural hormone all-trans RA (Fig. 2a). Interestingly, Am580 does not seem to fit in the receptor binding site: the ketone oxygen of the ligand sticks out of the binding pocket, due to too close proximity of residue 234 (Fig. 2b). However, in the complex with RAR-α, this ketone oxygen shares an hydrogen atom with the hydroxyl group of serine 234, and forms a stabilizing hydrogen bond, while a steric clash occurs in the other two RAR isoforms, where residue 234 is an alanine. Consequently, our model provides a rational for Am580 isoform specificity, suggesting that it is relevant and could be used as a template for the discovery of novel RAR-α agonist structures.
Figure 2.
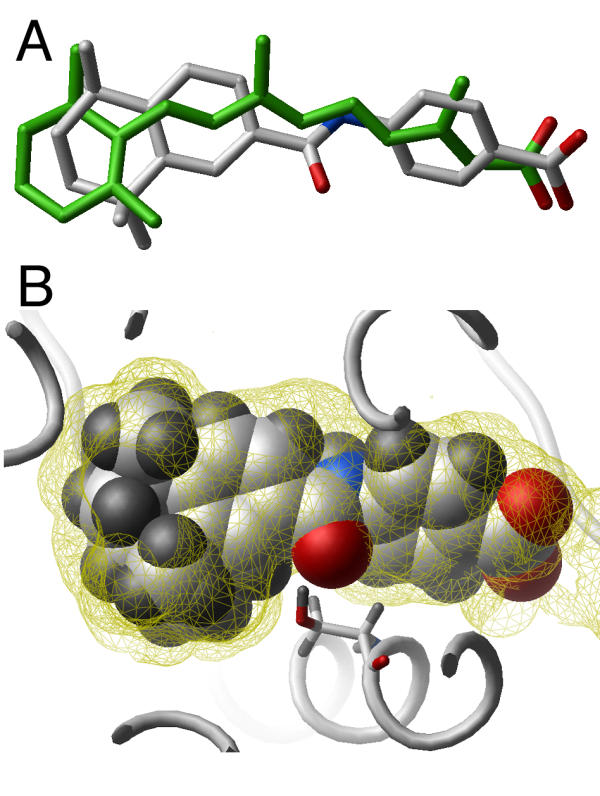
Docking of a known RAR-α specific agonist. The RAR-α selective agonist Am580 was docked into the modeled ligand binding pocket of RAR-α . A: The complexed ligand (white sticks) superimposes with the crystal structure of bound all-trans RA (green). Hydrogens are not shown for clarity. B: Am580 (CPK display) fits tightly into the receptor's pocket (yellow wire), but for a ketone oxygen, which shares an hydrogen with Ser234 of the receptor (displayed as stick). The receptor in the vicinity of the ligand is shown as a white ribbon. Carbons, hydrogens, oxygens and nitrogen are colored white, gray, red and blue respectively. (Image generated with Molsoft ICM)
A high throughput virtual screening was carried out on the Available Chemicals Directory (MDL Information Systems, San Leandro, CA), a compound structure database of over 150,000 molecules. Each compound was automatically docked into a grid representation of RAR-α, as previously described for Am580, and assigned a score according to the quality of the fit [8, 17, 20]. The 5364 ligand candidates which scored better (i.e. lower) than -32 kcal/mol were preselected for a more refined energy minimization procedure, with flexible receptor side chains, and the energy of complexation was predicted as previously described [21] (see "Materials and Methods"). After careful visual examination of the 300 candidates displaying the lowest predicted binding energy, 30 molecules were selected and purchased to be experimentally tested in vitro.
HeLa cells were separately transfected with either of the three wild type hRAR isoforms, and a ΔMTV-IR-CAT reporter gene [22, 23]. The cells were incubated with each ligand at concentrations from 50 nM to 20 μM to stimulate CAT activity. Possible toxicity of the compounds was deduced from the amount of cellular protein extract after 2 days of incubation. The percentage of conversion induced by 1 μM all-trans RA (RARs) or 1 μM 9-cis RA (RXR-β) was used as a positive control to determine the maximum induction. Fig. 3 shows all-trans RA induced conversion of RAR-α as a function of acid concentration. RAR-β could induce 20% of the maximum CAT activity when activated by 50 nM agonist 1 (chemical structure shown Fig. 1c), while RAR-γ was only 12% active and RAR-α not active at all under the same conditions (Fig. 4). At 200 nM of agonist 1, RAR-β was 50% active, RAR-γ 25% and RAR-α 5% active. Similarly, RAR-β could induce 22% of the maximum CAT activity when activated by 50 nM agonist 2 (chemical structure shown Fig. 1d). RAR-α and RAR-γ were 10% and 14% active at the same concentration of agonist, respectively. At 200 nM of agonist 2, RAR-β displayed 48% of its maximal activity, RAR-α 40% and RAR-γ 17%. At 20 μM, agonist 1 induced full activation of RAR-β and RAR-γ, and 80% activation of RAR-α, showing that this compound is a full agonist. Agonist 2 was toxic for the cells at 20 μM, as shown by protein content of cell culture dishes, which is why little transcriptional activity was observed at this concentration. However, little or no toxicity was observed at 8 μM, a concentration at which agonist 2 induced about 50% maximal activity of RAR-α and RAR-β, and 35% maximal activity of RAR-γ. Agonist 1 was not toxic at 20 μM. Finally, RXR-β was activated only weakly by both agonists 1 and 2 at 2 μM, but was significantly activated by agonist 1 at 20 μM (Fig. 4). Comparison of Figs. 3 and 4 also shows that at 200 nM of agonists 1 and 2, RAR-β exhibits the same activity as the positive control RAR-α induced by all-trans RA at the same concentration.
Figure 3.
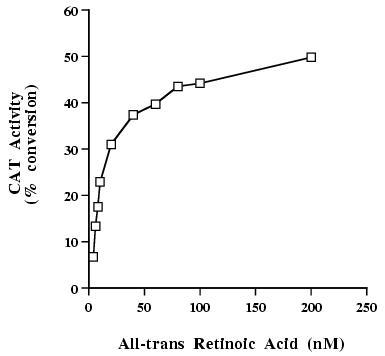
All-trans Retinoic Acid Mediates Dose-Dependent Transcriptional Activation of the IR-CAT Reporter by RAR-α.
Figure 4.
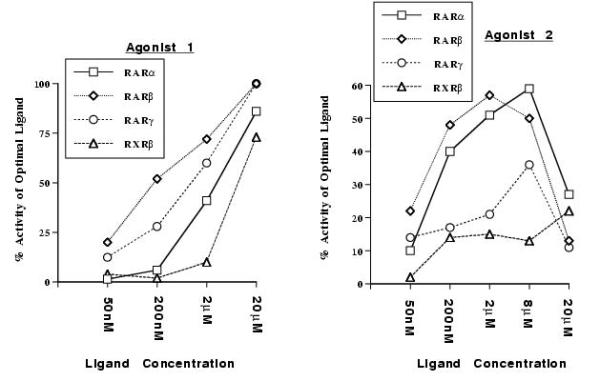
In vitro activity of the novel RAR agonists. HeLa cells were separately transfected with a vector expressing each isoform of the full-length receptor and a ΔMTV-IR-CAT reporter gene. The CAT activity induced by each ligand at concentrations from 50 nM to 20 μM was measured. The activity induced by the natural hormone all-trans RA at 1 μM was used as the reference for maximal induction for RARs, and 1 μM 9-cis RA was used for RXR-β. The activity plotted is the observed percentage of this maximal induction.
The predicted structure of the complex generated by the virtual screening approach, and further optimized with flexible receptor side-chains details clearly the interactions between the receptor and the ligands (Fig. 5). Agonist 1 and 2 both have a carboxylate group which superimposes with the carboxylate of all-trans RA, and makes stabilizing hydrogen bonds with Arg 274 and 278, and the backbone nitrogen of Ser 289 (Fig. 5). All other receptor/ligand interactions are hydrophobic. As a result, the size and flexibility of the ligand as well as the shape complementarity with the receptor are critical for affinity and specificity.
Figure 5.

Structure of the novel RAR agonists. Agonists 1 and 2 are shown (respectively A and B). Left: chemical structure of the compounds. Right: Representation of the compounds docked into the binding pocket of RAR (important residues are displayed as sticks: R274, R278, S289), and superimposed with the crystal structure of all-trans RA (green). The receptor is represented as a white ribbon. Hydrogens are not displayed for clarity. Color coding: carbons, oxygens, nitrogens, sulfurs, fluorides and hydrogens are colored white, red, blue, yellow, magenta and gray respectively.
The structure of agonist 2 is almost similar to that of Ch55 (Fig. 1e), another RAR-β selective agonist [24] with 0.44, 0.04 and 1.7 nM Kd for RAR-α, RAR-β and RAR-γ respectively [4]. However, agonist 2 has an additional hydroxyl group on the aromatic ring. The observation that Ch55 displays some potency against the growth of human non-small cell lung carcinoma cells [4] as well as anti-angiogenic activity [25] suggests that agonist 2 is a good lead candidate for cancer therapy. Since this compound was discovered through a single virtual library screening, this is a very encouraging result which also constitutes a validation of our method.
The structure of agonist 1 is entirely novel; it is the first RAR ligand described so far with either a thiazole ring or a trifluoro group. It presents a very limited level of flexibility and fits tightly into the receptor's binding pocket. This compound illustrates the benefits of the in silico screening procedure, a rational approach which is based solely on the structure of the receptor, and is not biased towards existing ligands. Indeed, structure-based drug design usually derives novel compound structures from ligands previously described. Our approach enabled the discovery ab initio of RAR agonists displaying some activity at 50 nM with a 7% success rate.
The specificity profile of the two RAR ligands described here are similar to that of Ch55, a compound with ten times higher affinity for RAR-β than RAR-α, and which can block the growth of certain cancer types [4]. It will be interesting to test these molecules for anti-proliferative activity, both in vitro and in vivo. The observation that agonist 1 does not belong to any of the series of RAR ligands described so far makes this compound particularly interesting for further development.
While a model of RAR-α was used to conduct the virtual screening, agonists 1 and 2 have an EC-50 of 200 nM for RAR-β, and an EC-50 of 4 and 2 μM respectively for RAR-α. This observation emphasizes the primary goal of structure-based screening procedures, which is to significantly accelerate the initial steps of lead identification projects by automatically discriminating between binders and non-binders, rather than ranking ligands according to their affinity for the receptor. The discovery of 2 RAR-α agonists out of 30 molecules tested is a good illustration of this approach.
Conclusions
This report details the rapid discovery of RAR agonists with novel structural features, thanks to a powerful virtual ligand screening approach, and a research strategy where considerations on existing ligands are avoided. One of the molecules presented here constitute a good framework for the development of a novel series of RAR ligands very different from all structures described so far. Such ligands could present more favorable specificity and toxicity profiles, and have important applications in cancer therapy.
Materials and Methods
Modeling of RAR-alpha ligand binding pocket
The crystal structure of RAR-γ-LBD complexed to all-trans RA was used as a template [18] and the three residues in the vicinity of the ligand which are not conserved between the two isoforms were modified accordingly: A234, M272, and A397 were changed to S234, I272 and V397 respectively. The rotation variables of the side chains within 3.5 Å of the modified residues were unfixed and the energy of the system was minimized in the internal coordinate space, according to the ICM method [26, 29].
Docking of AM580 into RAR-alpha
The flexible ligand was docked into a combination of five potential map representations of the RAR-α ligand binding pocket, which account for two Van der Waals boundaries, hydrophobicity, electrostatics and hydrogen bonding profiles [20,27]. This rapid docking procedure was followed by a more refined energy minimization of the complex, with a full atom representation of the receptor, and flexible receptor side chains, according to the ICM stochastic global optimization algorithm [26, 27] as implemented in the Molsoft ICM 2.7 program [17].
Virtual screening of the compound structure database
The ICM program [17] was used to perform both receptor modeling and virtual ligand screening. The procedure followed was similar than previously described [8]: each flexible ligand of the Available Chemicals Directory (MDL Information Systems, San Leandro) was docked automatically into the combination of potential maps described above, and assigned a score according to its fit with the receptor. The scoring function included continuum as well as discreet electrostatics, hydrophobicity and entropy parameters [20]. The screening of the database of over 150,000 ligands took less than a month on 10 "194 MHZ IP25" processors. Using the same computing power, the 5364 compounds which scored better (i.e. lower) than -32 were preselected for a second automatic round of selection: they were all docked in two days into a full atom representation of the receptor, with flexible receptor side chains, according to a global energy optimization in internal coordinates [17, 26,27,28]. The structure generated by the initial library screening was used as a starting point for this second, more refined, docking procedure. The binding energy of the compounds was then evaluated with a boundary elements implementation of solvation electrostatics [21] and the 300 compounds showing the lowest binding energy were selected for further examination. After careful visual inspection for shape complementarity, hydrogen bonding network, compound flexibility, and potential Van der Waals clashes, 30 molecules out of 300 were selected and purchased to be experimentally tested in vitro. We should stress out here that the Van der Waals term is too noisy to be added to the binding energy function, as discussed in details elsewhere [21]. As a consequence, some compounds with a low predicted binding energy were not retained, because they seemed likely to clash sterically with the receptor.
Biological activity of the ligand candidates
HeLa cells were transfected by calcium phosphate precipitation using 1 μg of plasmid expressing the full length receptor isoform and 1 μg of a ΔMTV-IR-CAT reporter gene, as previously described [22, 23]. Ligands were dissolved in DMSO at 20 mM final concentration. Cell cultures were supplemented with indicated ligands immediately after addition of the calcium phosphate/DNA precipitate. Media and ligands were replaced after 24 h and cells were harvested and assayed for CAT activity 24 h later: acetylated and unreacted [14C] chloramphenicol was excised from a thin layer chromatography plate and quantitated in a liquid scintillation counter. The amount of cellular protein extract after two days of incubation was measured to determine the compounds toxicity. Agonist 1 was purchased from Bionet Research (catalog number 1G-433S). Agonist 2 was purchased from Sigma-Aldrich (Sigma Aldrich library of rare chemicals. Catalog number S08503-1).
Acknowledgments
Acknowledgments
We would like to thank Maxim Totrov for his help and support throughout the project, Claudio Cavasotto for helping with the manuscript, and the Department of Defense (Grants DAMD179818133, DAMD179919318 and DAMO179919323) for supporting the project.
This work was also supported by NIH grant DK16636 and NYS Empire Award C015710 to HHS.
Contributor Information
Matthieu Schapira, Email: schapira@saturn.med.nyu.edu.
Bruce M Raaka, Email: bruce.raaka@med.nyu.edu.
Herbert H Samuels, Email: herbert.samuels@med.nyu.edu.
Ruben Abagyan, Email: abagyan@scripps.edu.
References
- Fitzgerald P, Teng M, Chandraratna RA, Heyman RA, Allegretto EA. Retinoic acid receptor alpha expression correlates with retinoid-induced growth inhibition of human breast cancer cells regardless of estrogen receptor status. Cancer Res. 1997;57:2642–2645. [PubMed] [Google Scholar]
- Fanjul AN, Piedrafita FJ, Al-Shamma H, Pfahl M. Apoptosis induction and potent antiestrogen receptor-negative breast cancer activity in vivo by a retinoid antagonist. Cancer Res. 1998;58:4607–4610. [PubMed] [Google Scholar]
- Lu XP, Fanjul A, Picard N, Shroot B, Pfahl MA. Selective retinoid with high activity against an androgen-resistant prostate cancer cell type. Int J Cancer. 1999;80:272–278. doi: 10.1002/(SICI)1097-0215(19990118)80:2<272::AID-IJC17>3.3.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57:4931–4939. [PubMed] [Google Scholar]
- Sun SY, Yue P, Wu GS, El-Deiry WS, Shroot B, Hong WK, Lotan R. Implication of p53 in growth arrest and apoptosis induced by the synthetic retinoid CD437 in human lung cancer cells. Cancer Res. 1999;59:2829–2833. [PubMed] [Google Scholar]
- Mologni L, Ponzanelli I, Bresciani F, Sardiello G, Bergamaschi D, Gianni M, Reichert U, Rambaldi A, Terao M, Garattini E. The novel synthetic retinoid 6-[3-adamantyl-4-hydroxyphenyl]-2-naphthalene carboxylic acid (CD437) causes apoptosis in acute promyelocytic leukemia cells through rapid activation of caspases. Blood. 1999;93:1045–1061. [PubMed] [Google Scholar]
- Chandaratna RA. Future trends: a new generation of retinoids. J Am Acad Dermatol. 1998;39(4 Pt 2):S149–S152. doi: 10.1016/s0190-9622(98)70313-5. [DOI] [PubMed] [Google Scholar]
- Schapira M, Raaka B, Samuels HH, Abagyan R. Rational discovery of nuclear receptor antagonists. Proc Natl Acad Sci U S A. 2000;97:1008–1013. doi: 10.1073/pnas.97.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman SM, Benner SE, Hong WK. Retinoid chemoprevention studies in upper aerodigestive tract and lung carcinogenesis. Cancer Res. 1994;54:2025s–2028s. [PubMed] [Google Scholar]
- Costa A, De Palo G, Decensi A, Formelli F, Chiesa F, Nava M, Camerini T, Marubini E, Veronesi U. Retinoids in cancer chemoprevention clinical trials with the synthetic analogue fenretinide. Ann N Y Acad Sci. 1995;768:148–162. doi: 10.1111/j.1749-6632.1995.tb12118.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lee MO, Wang HG, Li Y, Hashimoto Y, Klaus M, Reed JC, Zhang X. Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol Cell Biol. 1996;16:1138–1149. doi: 10.1128/mcb.16.3.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter M, Berger J, Daxenbichler G, Muller-Holzner E, Widschwendter A, Mayr A, Marth C, Zeimet AG. Loss of retinoic acid receptor beta expression in breast cancer and morphologically normal adjacent tissue but not in the normal breast tissue distant from the cancer. Cancer Res. 1997;57:4158–4161. [PubMed] [Google Scholar]
- Li Y, Dawson MI, Agadir A, Lee MO, Long L, Hobbs PD, Zhang XK. Regulation of RAR-β expression by RAR- and RXR-selective retinoids in human lung cancer cell lines: effect on growth inhibition and apoptosis induction. Int J Cancer. 1998;75:88–95. doi: 10.1002/(SICI)1097-0215(19980105)75:1<88::AID-IJC14>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Xu XC, Liu X, Tahara E, Lippman SM, Lotan R. Expression and up-regulation of retinoic acid receptor-beta is associated with retinoid sensitivity and colony formation in esophageal cancer cell lines. Cancer Res. 1999;59:2477–2483. [PubMed] [Google Scholar]
- Lin B, Chen G, Xiao D, Kolluri SK, Cao X, Su H, Zhang XK. Orphan receptor COUP-TF is required for induction of retinoic acid receptor beta growth inhibition and apoptosis by retinoic acid in cancer cells. Mol Cell Biol. 2000;20:957–970. doi: 10.1128/MCB.20.3.957-970.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY, Wan H, Yue P, Hong WK, Lotan R. Evidence that retinoic acid receptor beta induction by retinoids is important for tumor cell growth inhibition. J Biol Chem. 2000;275:17149–17153. doi: 10.1074/jbc.M000527200. [DOI] [PubMed] [Google Scholar]
- Molsoft LLC. ICM 2.7 Program Manual. 1998.
- Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Crystal structure of the RAR-γ ligand-binding domain bound to all-trans retinoic acid. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- Delescluse C, Cavey MT, Martin B, Bernard BA, Reichert U, Maignan J, Darmon M, Shroot B. Selective high affinity retinoic acid receptor alpha or beta-gamma ligands. Mol Pharmacol. 1991;40:556–562. [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Derivation of sensitive discrimination potential for virtual ligand screening. Proceedings of the 3rd annual international conference on computational molecular biology (RECOMB 99) Lyon France, ACM Press. 1999. pp. 312–317.
- Schapira M, Totrov M, Abagyan R. Prediction of the binding energy for small molecules peptides and proteins. J Mol Recognit. 1999;12:177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.3.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Qi JS, Desai-Yajnik V, Greene ME, Raaka BM, Samuels HH. The ligand-binding domains of the thyroid hormone/retinoid receptor gene subfamily function in vivo to mediate heterodimerization gene silencing and transactivation. Mol Cell Biol. 1995;15:1817–1825. doi: 10.1128/mcb.15.3.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Desai-Yajnik V, Lo E, Schapira M, Abagyan R, Samuels HH. NRIF3 is a novel coactivator mediating functional specificity of nuclear hormone receptors. Mol Cell Biol. 1999;19:7191–7202. doi: 10.1128/mcb.19.10.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiso Y, Kitagawa K, Nishino H, Iwashima A, Shudo K. Suppression of c-mos expression in teratocarcinoma cells with a new type of inducer of differentiation 35-di-tert-butylchalcone 4'-carboxylic acid. Exp Cell Res. 1987;173:262–266. doi: 10.1016/0014-4827(87)90351-x. [DOI] [PubMed] [Google Scholar]
- Oikawa T, Hirotani K, Nakamura O, Shudo K, Hiragun A, Iwaguchi T. A highly potent antiangiogenic activity of retinoids. Cancer Lett. 1989;48:157–162. doi: 10.1016/0304-3835(89)90054-2. [DOI] [PubMed] [Google Scholar]
- Abagyan RA. Protein structure prediction by global energy optimization Computer Simulation of Biomolecular Systems. Theoretical and Experimental Applications Volume 3 (Edited by van Gunsteren WF et al.) Leiden, ESCOM Science Publishers BV. 1997.
- Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins. 1997;Suppl 1:215–220. doi: 10.1002/(SICI)1097-0134(1997)1+<215::AID-PROT29>3.3.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Abagyan RA, Totrov MM, Kuznetsov DN. ICM - a new method for protein modelling and design applications to docking and structure prediction from the distorted native conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]