

Abstract
Amyloid beta (Aβ), the putative causative agent in Alzheimer disease, is known to affect glutamate receptor trafficking. Previous studies have shown that Aβ downregulates the surface expression of N-methyl D-aspartate type glutamate receptors (NMDARs) by the activation of STriatal-Enriched protein tyrosine Phosphatase 61 (STEP61). More recent findings confirm that STEP61 plays an important role in Aβ-induced NMDAR endocytosis. STEP levels are elevated in human AD prefrontal cortex and in the cortex of several AD mouse models. The increase in STEP61 levels and activity contribute to the removal of GluN1/GluN2B receptor complexes from the neuronal surface membranes. The elevation of STEP61 is due to disruption in the normal degradation of STEP61 by the ubiquitin proteasome system. Here, we briefly discuss additional studies in support of our hypothesis that STEP61 contributes to aspects of the pathophysiology in Alzheimer's disease. Exogenous application of Aβ-enriched conditioned medium (7PA2-CM) to wild-type cortical cultures results in a loss of GluN1/GluN2B subunits from neuronal membranes. Abeta-mediated NMDAR internalization does not occur in STEP knock-out cultures, but is rescued by the addition of active TAT-STEP to the cultures prior to Aβ treatment.
Key words: Alzheimer disease, amyloid beta, NMDA receptor, protein tyrosine phosphatases, STEP, synaptic plasticity
In Alzheimer disease (AD), the abnormal accumulation of soluble Aβ peptides has a profound impact on cognitive function.1 Aβ peptides disrupt synaptic plasticity, a molecular mechanism involved in learning and memory.2,3 N-methyl D-aspartate type glutamate receptors (NMDAR) play an important role in the development of synaptic strengthening. Aβ downregulates the surface expression of NMDARs by activation of STriatal-Enriched protein tyrosine Phosphatase 61 (STEP61).4 STEP61 is a brain-specific phosphatase that opposes the development of synaptic strengthening.5 STEP61 is present in postsynaptic terminals, immunoprecipitates with the NMDAR complex and decreases NMDA channel function.6,7 The reduced channel function is mediated, at least in part, by an increased internalization of the NMDAR complex, as STEP dephosphorylates the GluN2B subunit at a regulatory tyrosine (tyr1472) leading to NMDAR endocytosis. Knocking down STEP with interfering RNA increases NMDAR trafficking to synaptic membranes.4,8 A previous study suggested that Aβ leads to the activation of STEP through a calcineurin-mediated pathway, which subsequently increased internalization of surface NMDAR.4 A recent study has demonstrated that STEP is also regulated by the ubiquitin proteasome system, and an Aβ-mediated disruption of the proteasome leads to increased STEP61 levels in human Alzheimer's disease (AD) brains and AD mouse models.9 Taken together, these studies suggest that an increase in the activity of STEP61 contributes to the cognitive deficits in AD by increasing the internalization of NMDAR from synaptic membrane surfaces.
STEP Activity Regulates the Expression of GluN1/GluN2B Subunits on Neuronal Surface
To clarify the role of STEP in GluN1/GluN2B endocytosis, we overexpressed STEP protein in wild-type (WT) cortical cultures by transducing WT TAT-STEP protein and examined surface levels of GluN1/GluN2B using biotinylation.4,10 Incubation of WT TAT-STEP protein (2 µM; 1 hr) resulted in a significant decrease in surface GluN1 (76.9 ± 1.9%) and GluN2B (75.0 ± 5.7%) compared to the control (Fig. 1A). Incubation of a catalytically inactive STEP protein C300S (TAT-STEP C-S) failed to decrease surface levels of GluN1 (108.0 ± 6.9%) and GluN2B (103.8 ± 3.9%) (Fig. 1A). This result suggests that active STEP protein is required for internalization of GluN1 and GluN2B subunits from the neuronal surface membranes.
Figure 1.

STEP activity regulates GluN1/GluN2B trafficking. (A) Surface biotinylation of wild-type cortical cultures after treatment with WT TAT-STEP OR an inactive variant TAT-STEP C-S and probed for surface and total GluN1 and GluN2B with respective antibodies. Biotinylated receptors were normalized to total receptor level and then to loading control to determine percent change. Quantitation shows a significant decrease in surface GluN1 and GluN2B subunits in WT TAT-STEP treated, compared to TAT-STEP C-S or untreated control (*p < 0.05; one-way ANOVA with post hoc tukey's test; n = 3). (B) Surface biotinylation of wild-type and STEP KO cortical cultures after treatment with CHO (control) or 7PA2-CM (Aβ-enriched) medium in the presence or absence of WT TAT-STEP protein and probed for surface and total GluN1 and GluN2B with respective antibodies. Quantitation show a significant decrease in surface GluN1 and GluN2B subunits in wild-type cultures upon 7PA2-CM or 7PA2-CM + WT TAT-STEP treatment compared to CHO treated, whereas the effect of 7PA2-CM was absent in STEP KO cultures and was rescued in the presence of WT TAT-STEP protein (*p < 0.05, **p < 0.01; one-way ANOVA with post hoc Tukey's test; n = 4).
Aβ Treatment Fails to Induce NMDAR Endocytosisin STEP KO Cultures
To study the role of STEP in Aβ-mediated NMDAR endocytosis, we took advantage of STEP KO cultures.11 We examined Aβ-mediated NMDAR endocytosis in STEP KO cultures using two complementary techniques: surface biotinylation and immunofluoresence as described previously.4,9 We used 7PA2-conditioned medium (7PA2-CM) as a source of Aβ. These cell secrete soluble amyloid-beta oligomers into the medium that were concentrated for use in these experiments.12,13 Cortical cultures derived from WT and STEP KO (18 DIV) were treated with 7PA2 (Aβ-enriched) or CHO (control) medium and GluN1 and GluN2B levels were determined by surface biotinylation. The surface GluN1 and GluN2B levels were significantly decreased in WT cultures after 7PA2-CM (Aβ-enriched) treatment compared to control (GluN1: 75.2 ± 1.8%; GluN2B: 64.8 ± 3.7%) (Fig. 1B). As previously shown, there was no change in the surface GluN2A levels upon 7PA2-CM treatment9 (data not shown). As expected, STEP KO cultures showed increase in basal levels of GluN1 and GluN2B compared to WT (GluN1: 138.9 ± 3.5%; GluN2B: 130.0 ± 4.4%) and 7PA2 treatment failed to significantly decrease the surface GluN1 and GluN2B receptors (GluN1: 129.9 ± 3.7%; GluN2B: 129.6 ± 10.4%) (Fig. 1B). Preincubation with WT TAT-STEP protein during 7PA2-CM treatment rescued GluN1/GLUN2B internalization (GluN1: 89.2 ± 3.3%; GluN2B: 63.7 ± 5.6%) in STEP KO cultures compared to CHO controls (Fig. 1B).
Next, we examined the Aβ-mediated NMDAR endocytosis in WT and STEP KO cortical cultures using immunofluorescence to measure the colocalization of GluN1 with the presynaptic marker synapsin I.4 Treatment of neuronal cultures from WT mice with 7PA2-CM significantly decreased surface GluN1 staining (4.78 ± 0.24 puncta per 10 µm, p < 0.001) as compared to untreated WT (8.69 U ± 0.47 puncta per 10 µm) cultures (Fig. 2A and B). Untreated STEP KO cultures had significantly higher numbers of GluN1 puncta (15.33 ± 0.83 puncta per 10 µm, p < 0.001) compared to WT cultures (Fig. 2A and C). Aβ-mediated endocytosis was abolished in STEP KO cultures (16.31 U± 0.78 puncta per 10 µm, p > 0.99 as compared to KO control) (Fig. 2C and D).
Figure 2.
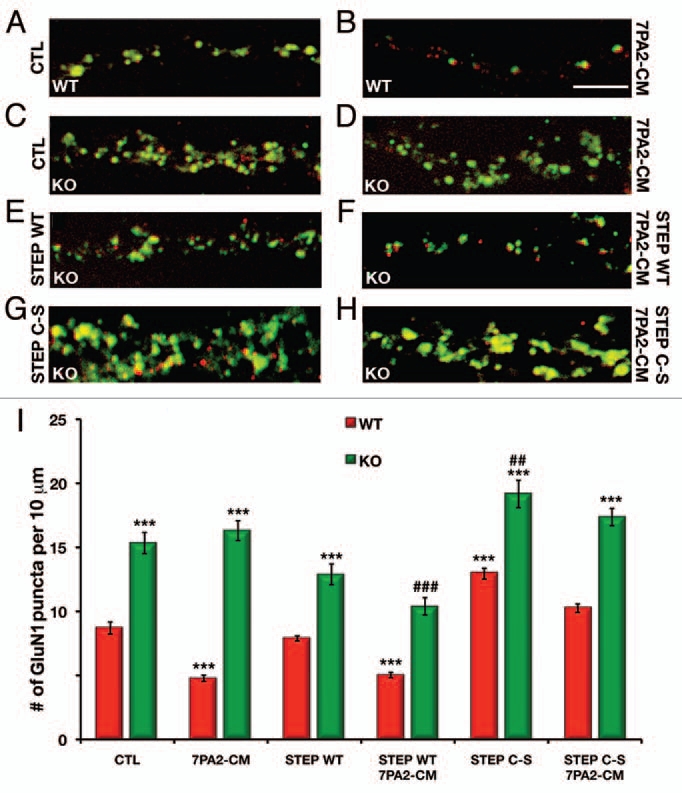
STEP KO cultures show no decrease in GluN1 surface levels after 7PA2-CM (Aβ-enriched) treatment. (A–D) representative images from STEP WT and KO cortical cultures either untreated or treated with 7PA2-CM (Aβ-enriched) showing Glun1 surface staining (green) and the pre-synaptic marker synapsin I (red). (E and F) STEP KO cortical cultures pretreated with WT TAT-STEP alone or WT TAT-STEP followed by 7PA2-CM. (G and H) STEP KO cortical cultures pretreated with WT TAT-STEP C-S (inactive) alone or TAT-STEP C-S followed by 7PA2-CM. (I) Histogram showing the quantification of the surface GluN1 staining that colocalized with the presynaptic marker, synapsin I. STEP KO cultures showed significantly higher levels of surface GluN1 compared to WT controls. Treatment of WT cultures with 7PA2-CM resulted in a significant loss of GluN1; however, this was abolished in the STEP KO cultures. Restoration of STEP WT protein, but not inactive STEP C-S, rescued the 7PA2-CM-mediated endocytosis of GluN1 in the KO cultures (***p < 0.001 as compared to WT control; ###p < 0.001 or ##p < 0.01 as compared to KO control, two-way ANOVA, n = 4 independent experiments, scale bar: 5 µm).
We examined whether we could rescue Aβ-induced GluN1 endocytosis by restoring STEP protein to the KO cultures. The WT cortical cultures treated with WT TAT-STEP were similar to untreated WT cultures either in the presence or absence of 7PA2-CM (Fig. 2I). Treatment of KO cortical cultures with WT TAT-STEP reduced GluN1 surface expression (12.89 ± 0.81 puncta per 10 µm, p > 0.20 as compared to KO control, Fig. 2E) and resulted in significant Aβ-induced GluN1 internalization (10.39 ± U0.67 puncta per 10 µm, p < 0.001) as compared to both KO cultures and KO cultures treated with 7PA2-CM (Fig. 2F). The surface GluN1 levels in KO cultures treated with WT TAT-STEP followed by 7PA2-CM were not significantly different from WT cultures (p > 0.74), suggesting that restoration of WT STEP protein in KO cultures rescues Aβ-induced GluN1 endocytosis. In contrast, treatment of KO cortical cultures with inactive TAT-STEP C-S fusion protein resulted in a significant increase in surface expression of GluN1 receptors (19.17 ± 1.07 puncta per 10 µm, p < 0.002 as compared to KO control, Fig. 2G) and the inactive STEP (C-S) fusion protein did not rescue Aβ-induced endocytosis of GluN1 subunits (17.36 U± 0.68 puncta per 10 µm, p > 0.99) (Fig. 2H). The WT cultures treated with TAT-STEP C-S showed a significant increase in GluN1 surface expression and partially blocked the Aβ mediated GluN1 internalization (Fig. 2I).
In summary, these studies indicate that active STEP protein is required for Aβ-induced internalization of NMDARs. It also suggests a new therapeutic strategy by which inhibiting STEP activity could lead to the development of a new family of therapeutic agents in Alzheimer disease.
Acknowledgements
We thank laboratory members for helpful discussions and critical reading of the manuscript. The work was funded by The American Health Assistance Foundation, the Institute for the Study of Aging and NIH grants MH01527 and MH52711 to P.J.L.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/12910
References
- 1.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, et al. Beta-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 5.Braithwaite SP, Paul S, Nairn AC, Lombroso PJ. Synaptic plasticity: one STEP at a time. Trends Neurosci. 2006;29:452–458. doi: 10.1016/j.tins.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oyama T, Goto S, Nishi T, Sato K, Yamada K, Yoshikawa M, et al. Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: a light and electron microscopic study with a complementary DNA-generated polyclonal antibody. Neuroscience. 1995;69:869–880. doi: 10.1016/0306-4522(95)00278-q. [DOI] [PubMed] [Google Scholar]
- 7.Pelkey KA, Askalan R, Paul S, Kalia LV, Nguyen TH, Pitcher GM, et al. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 8.Braithwaite SP, Adkisson M, Leung J, Nava A, Masterson B, Urfer R, et al. Regulation of NMDA receptor trafficking and function by striatal-enriched tyrosine phosphatase (STEP) Eur J Neurosci. 2006;23:2847–2856. doi: 10.1111/j.1460-9568.2006.04837.x. [DOI] [PubMed] [Google Scholar]
- 9.Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, et al. Aβ-mediated NMDA receptor endocytosis in Alzheimer's disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010;30:5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tashev R, Moura PJ, Venkitaramani DV, Prosperetti C, Centonze D, Paul S, et al. A substrate trapping mutant form of striatal-enriched protein tyrosine phosphatase prevents amphetamine-induced stereotypies and long-term potentiation in the striatum. Biol Psychiatry. 2009;65:637–645. doi: 10.1016/j.biopsych.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkitaramani DV, Paul S, Zhang Y, Kurup P, Ding L, Tressler L, et al. Knockout of striatal enriched protein tyrosine phosphatase in mice results in increased ERK1/2 phosphorylation. Synapse. 2009;63:69–81. doi: 10.1002/syn.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 13.Poling A, Morgan-Paisley K, Panos JJ, Kim EM, O'Hare E, Cleary JP, et al. Oligomers of the amyloidbeta protein disrupt working memory: confirmation with two behavioral procedures. Behav Brain Res. 2008;193:230–234. doi: 10.1016/j.bbr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]