

Abstract
Ionizing radiation (IR) triggers many signaling pathways primarily originating from either damaged DNA or non-nuclear sources such as growth factor receptors. Thus, to study the DNA damage-induced signaling component alone by irradiation would be a challenge. To generate DNA double-strand breaks (DSBs) and minimize non-nuclear signaling, human cancer cells having bromodeoxyuridine (BrdU)—substituted DNA were treated with the photosensitizer Hoechst 33258 followed by long wavelength UV (UV-A) treatment (BrdU photolysis). BrdU photolysis resulted in well-controlled, dose-dependent generation of DSBs equivalent to radiation doses between 0.2–20 Gy, as determined by pulsed-field gel electrophoresis and accompanied by dose-dependent ATM (ser-1981), H2AX (ser-139), Chk2 (thr-68) and p53 (ser15) phosphorylation. Interestingly, low levels (≤2 Gy equivalents) of BrdU photolysis stimulated ERK phosphorylation whereas higher (>2 Gy eq.) resulted in ERK dephosphorylation. ERK phosphorylation was ATM-dependent whereas dephosphorylation was ATM-independent. The ATM-dependent increase in ERK phosphorylation was also seen when DSBs were generated by transfection of cells with an EcoRI expression plasmid or by electroporation of EcoRI enzyme. Furthermore, AKT was critical for transmitting the DSB signal to ERK. Altogether, our results show that low levels of DSBs trigger ATM- and AKT-dependent ERK pro-survival signaling and increased cell proliferation whereas higher levels result in ERK dephosphorylation consistent with a dose-dependent switch from pro-survival to anti-survival signaling.
Key words: bromodeoxyuridine, DNA repair, MAP kinase, p53, KU-55933, U87 glioma cells
Introduction
DNA double-strand breaks (DSBs) occur in response to various DNA damaging agents such as ionizing radiation (IR), radiomimetic drugs and during normal DNA replication. The ability to repair DNA damage with the highest fidelity is a fundamental part of cell survival.1,2 The cell responds to DNA damage by triggering a number of signaling pathways known as the DNA damage response (DDR). Activation of the DDR triggers cell cycle checkpoints that stall cell cycle progression to allow for DNA repair or when the damage is too severe initiate apoptosis and cell death.3–5
The ATM (ataxia telangiectasia mutated) protein is the principal regulator of the DDR in response to IR. ATM phosphorylates more than 700 proteins involved in cell cycle control, DNA repair, apoptosis and modulation of chromatin structure, including p53, Brca1, Chk2, 53BP1, SMC-1 and histone H2AX.6 ATM-dependent H2AX phosphorylation is one of the earliest signs of DNA damage and is necessary for efficient DSB repair.7–9 ATM is not limited to regulating the response to DSBs but also plays an important role in the response to oxidative stress and for modulating cell growth through growth factor receptors.10–13
Our previous work with human glioma cells has shown that radiation is able to induce ERK signaling important for ATM-dependent foci formation.14 Furthermore, activation of the epidermal growth factor receptor (EGFR) by epidermal growth factor, transforming growth factor-α and radiation lead to the stimulation of prosurvival signaling through the ERK and the phosphoinositide 3-kinase (PI3K)/AKT pathways that contribute to cellular processes that regulate cell survival and apoptosis (review in ref. 15). In that vein, we recently showed that EGFR-ERK and AKT signaling positively affects DSB repair in an ATM-dependent manner.16 Another recent study showed that EGFR and hyperactivated PI3K-AKT signaling promotes DNA-PKcs activation and DSB repair.17
The DDR is very complex as radiation not only elicits DNA damage signaling but additionally triggers independent non-nuclear signaling that emanates from activated growth factor receptors and inactivated cytoplasmic protein phosphatases. Collectively, these signals influence cell cycle regulation, DNA repair and apoptosis.15,18–20 Thus, the effect of DNA damage alone on cell fate would be difficult to study in irradiated cells. In the present study we used BrdU photolysis,21,22 expression of ectopic EcoRI, and electroporation of EcoRI enzyme to generate DSBs and minimize non-nuclear effects. We then examined the signaling responses resulting from these DSBs. We show here that ATM regulates ERK phosphorylation and we report on a bi-phasic ERK response that could play an important role in determining cellular fate in response to DNA damage and the balance between cell survival and death. Indeed, we show that low levels of DSBs stimulate cell proliferation. Furthermore, we demonstrate that AKT is positioned downstream from ATM and is important for transmitting the DDR to the ERK pathway.
Results
Radiation induces dose-dependent ERK phosphorylation in human glioma cells.
In general, MAPK signaling pathways are activated by extra-cellular or cytoplasmic events that result in the translocation of activated MAPKs to the nucleus. While radiation directly activates growth factor receptors in the plasma membrane additional signals flow in the opposite direction from damaged DNA to the cytoplasm (inside-out signaling).15 To examine the nature of ERK1/2 (ERK) phosphorylation in response to DNA damage and establish a reference for further investigation, we first carried out an IR dose and time course experiment with human U87 glioma cells. We observed a dose-dependent maximum of approximately 3-fold above basal levels that peaked at 5 min and declined over time, but remained elevated for at least 1 h post-irradiation (Fig. 1). These results are in agreement with previous reports that demonstrated rapid ERK signaling in a variety of irradiated human tumor cell lines (reviewed in ref. 18).
Figure 1.

Time course and IR dose response of ERK phosphorylation. (A) U87 cells were exposed to 0 Gy (lanes 1, 5, 9 and 13), 2 Gy (lanes 2, 6, 10 and 14), 5 Gy (lanes 3, 7, 11 and 15) and 20 Gy (lanes 4, 8, 12 and 16). Cells were collected at 5 min (lanes 1–4), 15 min (lanes 5–8), 30 min (9–12) and 60 min (13–16) and analyzed for ERK phosphorylation by western blotting. ERK denotes equal protein loading. The figure shows representative western blots of triplicate repeats. (B) Densitometry analysis of (A) presented in graph form. Data points, ERK phosphorylation levels plotted against radiation dose. Error bars, SEM; n = 3. Fold (x) denotes changes in p-ERK levels compared to control (0 Gy) normalized to total ERK protein levels. *p < 0.05.
BrdU photolysis generates DSBs with high efficiency.
The introduction of DSBs by non-radiological methods might provide a more suitable approach for studying the significance of signals that result specifically from DNA damage while minimizing non-nuclear influence. Previously, BrdU photolysis was shown to generate DSBs at high yields,21,22 suitable for studying DNA repair.8 Thus, we treated human glioma cells with BrdU for two days followed by the addition of the photosensitizer Hoechst 33258 and UV-A irradiation, which we refer to as BrdU photolysis. The UV-A dose was used to control the extent of DSBs.
To establish that we indeed generated DSBs after BrdU photolysis, we first carried out pulsed-field gel electrophoresis (PFGE) of DNA isolated from treated glioma cells (Fig. 2A). DSBs reached maximum levels 1 h after UV-A exposure and then began to decline to undetectable levels by 24 h (Fig. 2B). PFGE was able to detect DSBs resulting from IR in the range of 10–40 Gy, producing a linear relationship between dose and DSBs (Fig. 2C). We were able to detect DSBs with UV-A doses as low as 500 J/m2 (Fig. 2D), corresponding to ∼5 Gy equivalents (5 Gy eq.) derived from the extrapolation of IR-induced DSBs and assuming linear IR and UV-A dose responses. We found that the level of DSBs resulting from BrdU photolysis treatment at a dose of 150 J/m2 was equivalent to ∼2 Gy, while the level of DSBs resulting from a UV-A dose of 1500 J/m2 was equivalent to ∼20 Gy (Fig. 2C and D). Thus, the extent of BrdU photolysis was defined as low (≤2 Gy eq.) or high (>2 Gy eq.).
Figure 2.

Double-strand breaks detected by PFGE in response to BrdU photolysis and IR. (A) Autoradiogram showing the extent of DNA released after BrdU photolysis and IR. U87 cells were exposed to a UV-A dose of 1,500 J/m2; without BrdU (Lanes 1–6), with BrdU (lanes 7–12) and collected at 0.5, 1, 3, 6 and 24 h or U87 cells were exposed to 10, 20 and 40 Gy of IR (lanes 13–15) and collected immediately. (B and C) Densitometic analysis of (A) presented in graph form. (B) Data points, DNA released from lanes 1–12 plotted over time. (C) Data points, DNA released from lanes 13–15 plotted against IR dose. (D) UV-A dose response of DSBs from BrdU photolysis at 1 h. Data points, fraction of DNA released plotted against the indicated UV-A doses. The intercept of the dotted lines indicates the minimum UV-A dose resulting in DSBs detected by PFGE. Using the graphs in (C) and (D) and assuming a linear relationship between DSBs and IR and UV-A dose, respectively, the Gy equivalent doses for 150, 500 and 1,500 J/m2 of UV-A corresponds to 2, 5, 6 and 18–20 Gy, respectively.
In contrast to the results obtained with IR, which instantaneously generates maximal levels of DSBs, BrdU photolysis produced peak levels of DSBs at 1 h (Fig. 2B), suggesting that BrdU photolysis generates DSBs with slower kinetics. The half-life of the DSBs resulting from BrdU photolysis was 8–10 h, which is considerably slower than the <1 h half-life of IR-induced DSBs when p-(ser-139) H2AX (γ-H2AX) was measured,23 suggesting that BrdU photolysis generates more complex DSBs that require more time for repair. Our findings are in agreement with those of Limoli and Ward.21,22
BrdU photolysis triggers a typical DDR response.
To demonstrate that BrdU photolysis triggered a DDR, we carried out dose and time course experiments examining p53, Chk2, H2AX and ATM phosphorylation using western blot analyses. BrdU photolysis increased p53 and Chk2 phosphorylation as early as 30 min after UV-A exposure and remained maximally elevated between 1 and 6 h and then declined between 6 and 24 h (Sup. Fig. 1). Such increases did not occur if any of the three treatments, BrdU, Hoechst dye or UV-A was omitted (Sup. Fig. 1A, compare lanes 1–4 and 6–8, and data not shown). We observed a saturation of p53 (serine-15) phosphorylation at a UV-A dose as low as 125 J/m2 (1.5 Gy eq.) which remained unchanged up to 20 Gy eq. On the other hand, γ-H2AX levels linearly increased with escalating doses of UV-A (Sup. Fig. 2). We also observed ATM (serine-1981) phosphorylation with a UV-A dose as low as 50 J/m2 or 0.7 Gy eq. (Sup. Fig. 3). Similar results were seen with MCF-7 cells (ATM+ p53+) (Sup. Fig. 4), suggesting that the response to BrdU photolysis is general in nature and not cell line specific. Altogether, except for differences in the kinetics of DNA damage accumulation and repair BrdU photolysis resulted in a DDR reminiscent of that triggered by IR.
BrdU photolysis at low doses increases ERK phosphorylation whereas high doses result in dephosphorylation.
To determine the effects of DSBs produced by BrdU photolysis on pro-survival signaling, we first focused on the ERK pathway. We observed ∼2-fold increases in ERK phosphorylation when cells were exposed to BrdU photolysis with low UV-A doses (≤150 J/m2) corresponding to ≤2 Gy eq (Fig. 3A). However, at the intermediate (5 Gy eq.) and higher doses (≥20 Gy eq.), phosphorylation decreased below basal levels without any reduction in overall ERK protein levels, reflecting an apparent bi-phasic effect on ERK phosphorylation (Fig. 3A). Dephosphorylation was more pronounced at 6 h than at 3 h. Omitting either BrdU, Hoechst dye or UV-A did not increase ERK phosphorylation (data not shown), suggesting that all three components are required for triggering ERK signaling. A time course experiment revealed that increased p-ERK levels occurred as early as 1 h, peaked at 3 h (1.9-fold) and declined by 6 h (Fig. 3B). A UV-A dose response at 3 h showed that maximum p-ERK levels (3.3-fold) occurred after a dose of 50 J/m2 (∼0.7 Gy eq.) (Fig. 3C). At doses >500 J/m2 (>5 Gy eq.), p-ERK levels were reduced below basal level.
Figure 3.

Bi-phasic effects on ERK phosphorylation in response to BrdU photolysis. (A) U87 cells were treated with BrdU, Hoechst dye and increasing doses of UV-A [0 J/m2 (lanes 1 and 9); 15 J/m2 (lanes 2 and 10); 50 J/m2 (lanes 3 and 11); 150 J/m2 (lanes 4 and 12); 500 J/m2 (lanes 5 and 13); 1,500 J/m2 (lanes 6 and 14); 3,000 J/m2 (lanes 7 and 15) and 6,000 J/m2 (lanes 8 and 16)]. Cells were collected at 3 and 6 h and analyzed by western blotting. ERK and β-actin denote equal loading. (B) Time course response of ERK phosphorylation. U87 cells were labeled with BrdU, treated with Hoechst dye and irradiated with UV-A at 150 J/m2 (+) or not (−). Cells were collected at 1, 3 and 6 h after UV-A treatment for western blotting and quantification by densitometric analysis. (C) UV-A dose response. U87 cells were labeled with BrdU, treated with Hoechst dye and irradiated with UV-A at 15, 50, 150, 500 and 1,500 J/m2 or left untreated (0). Cells were collected after 3 h for western blot analyses and subsequent densitometric determination. Data points, ERK phosphorylation levels. Error bars, SEM; n = 3. Fold (x) denotes changes in p-ERK levels compared to control (no UV-A) normalized to total ERK protein levels. *p < 0.05.
To show that the bi-phasic p-ERK response induced by BrdU photolysis was not cell line specific, we examined this response in HEK293 cells (ATM+ p53+). A dose response experiment with UV-A doses ranging between 15 and 1,500 J/m2 was carried out. We observed a maximum increase in ERK phosphorylation of 2.6-fold at 50 J/m2, (0.7 Gy eq.), and again at higher UV-A doses the level of ERK phosphorylation decreased below basal level (Fig. 4). Furthermore, phosphorylation of ATM and p53 increased with UV-A dose. These results are similar to the ones obtained with U87 and MCF-7 cells suggesting that not only are the p53 responses superimposable, but the ERK response is also almost identical, strengthening the conclusion that the bi-phasic effect on ERK phosphorylation is general in nature. In addition, the ERK phosphorylation response is not likely p53-dependent since we observed a saturation of p53 phosphorylation at very low UV-A doses at which p-ERK levels were elevated whereas at higher doses ERK phosphorylation was abolished but p53 phosphorylation was not (Sup. Fig. 5).
Figure 4.
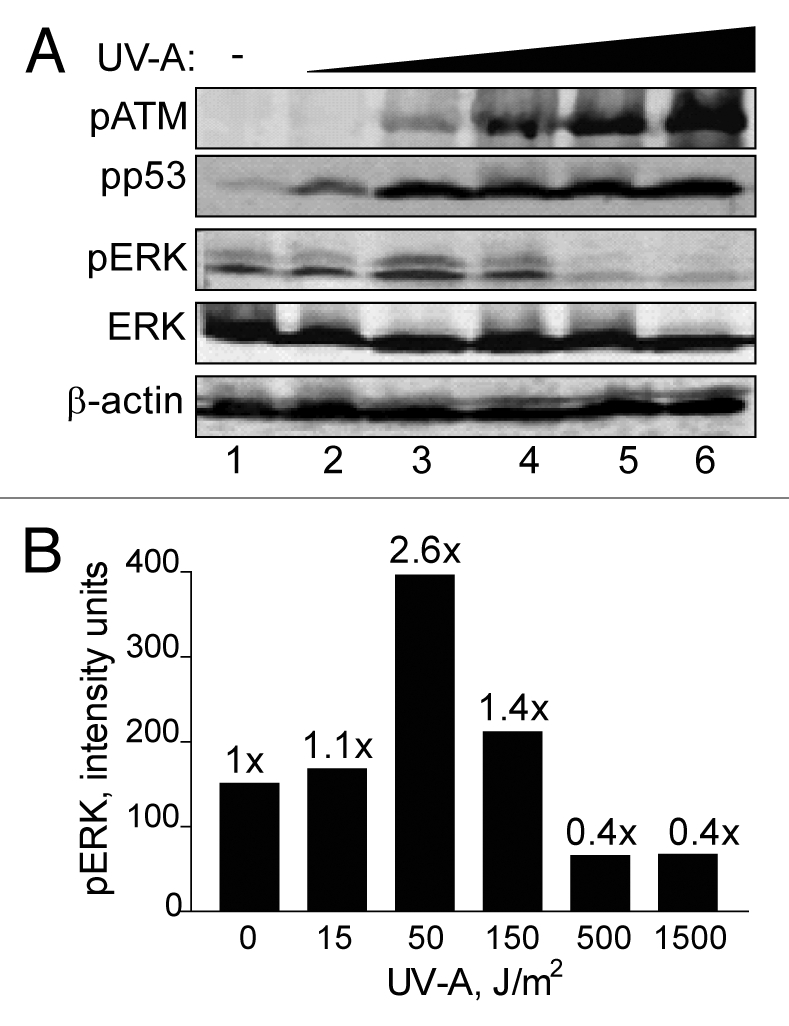
Brdu photolysis dose response of ERK phosphorylation in HEK293 cells. (A) HEK293 cells were treated by BrdU photolysis with increasing doses of UV-A; 15 J/m2 (lane 2), 50 J/m2 (lane 3), 150 J/m2 (lane 4), 500 J/m2 (lane 5), 1,500 J/m2 (lane 6) or untreated (lane 1). Cells were collected after 3 h for western blot analyses. ERK and β-actin denote equal loading. (B) Densitometic analysis of (A) presented in graph form. Data points, ERK phosphorylation levels plotted against UV-A dose. Fold (x) denotes changes in p-ERK levels compared to control (no UV-A) normalized to total ERK levels.
ERK phosphorylation in response to BrdU photolysis is ATM-dependent.
To further investigate ATM's role in ERK phosphorylation, we examined these responses in SV40-immortalized human AT5BIVA (ATM−) and normal GM637 (ATM+) fibroblasts. We found that ERK phosphorylation increased in GM637 cells but not in AT5BIVA cells in response to low levels of BrdU photolysis (Fig. 5), suggesting that ATM is important for ERK phosphorylation in response to DSBs. On the other hand, at higher levels ERK was similarly dephosphorylated in both GM637 and AT5BIVA cells. However, ERK dephosphorylation occurred in AT5BIVA cells at the considerably lower UV-A dose of 150 J/m2 (2 Gy eq.), a dose which increased p-ERK levels in GM637 cells (Fig. 5B). Thus, increased phosphorylation of ERK at lower levels of BrdU photolysis was dependent on ATM whereas ERK-dephosphorylation was ATM-independent but DSB dose-dependent. These results suggest that ERK pro-survival or growth stimulatory signals are generated by DSBs at lower levels whereas at higher levels they are reduced or abrogated altogether.
Figure 5.

ERK phosphorylation in response to BrdU photolysis is impaired in A-T cells. (A) AT5BIVA (lanes 1–4) and GM637 (lanes 5–8) cells were labeled with BrdU, treated with Hoechst dye, and irradiated with UV-A [no UV-A (lanes 1 and 5); 150 J/m2 (lanes 2 and 6); 500 J/m2 (lanes 3 and 7); and 1,500 J/m2 (lanes 4 and 8)]. Cells were collected 1 h after irradiation and processed for western blot analyses. ERK denotes equal loading. (B) Densitometic analysis of (A) presented in graph form. Data points, ERK phosphorylation levels plotted against UV-A dose. Fold (x) denotes changes in p-ERK levels compared to control (no UV-A) normalized to total ERK levels.
To further support the finding that ATM is involved in causing the increase of ERK phosphorylation in response to low levels of BrdU photolysis, we examined ERK phosphorylation with or without the presence of a specific ATM inhibitor, KU-55933.14,24 In the absence of KU-55933, we noted a ∼2-fold increase in ERK phosphorylation after a dose of 2 Gy eq. However, in the presence of KU-55933 this increase was greatly diminished and ERK phosphorylation levels remained close to basal levels (Fig. 6). This result supports the finding with A-T cells and suggests that increased ERK phosphorylation in response to DSBs induced by BrdU photolysis is dependent on ATM signaling.
Figure 6.

ATM inhibitor KU-55933 inhibits ERK phosphorylation in response to BrdU photolysis. (A) U87 cells labeled with BrdU, either left untreated lane (lane1–4) or treated with KU-55933 at 10 µM (lane 5–8) for 30 min before treatment with Hoechst dye and UV-A irradiation (Lanes 1 and 5, no UV-A; 2 and 6, 15 J/m2; 3 and 7, 50 J/m2; and 4 and 8, 150 J/m2). Cells from each set were collected 3 h after irradiation and processed for western blotting. ERK denotes equal loading. The figure shows representative western blots of triplicate repeats. (B) Densitometic analysis of (A) presented in graph form. Data points, ERK phosphorylation levels plotted against UV-A dose, error bars. SEM; n = 3. Fold (x) denotes changes in p-ERK levels compared to control (no UV-A) normalized to total ERK levels. *p < 0.05.
BrdU photolysis increases ERK activation via MEK.
MEK1 and MEK2 are positioned immediately upstream of ERK.19 To determine if the DSB signal that activates ERK is transmitted through MEK1/2 (MEK), U87 cells were treated with the MEK inhibitor U0126 prior to low levels of BrdU photolysis. In the absence of inhibitor a 1.9-fold increase in ERK phosphorylation was observed, and in the presence complete inhibition occurred (Sup. Fig. 6). Combined, this result suggests that ATM signals in response to low levels of DSBs generated by BrdU photolysis, which in turn activates ERK via MEK.
ATM signals to MEK-ERK via AKT in response to DSBs.
Recently, ATM was implicated as an upstream mediator of radiation-induced AKT (ser-473) phosphorylation/activation.25 AKT is considered to be a pro-survival kinase that prevents stress-induced JNK signaling and apoptosis and promotes growth.15,26 To better define a role for AKT in DSB signaling through the ERK pathway, we used an experimental system based on genetically manipulating HEK239 cells. We transfected HEK293 cells with a plasmid expressing the restriction endonuclease EcoRI to induce DSBs enzymatically which is expected to digest genomic DNA and trigger the DDR as it does in yeast.27 Similar to the results seen with BrdU photolysis, expression of EcoRI resulted in a 2.4-fold increase in ERK phosphorylation as well as a similar increase in H2AX phosphorylation (Fig. 7A), suggesting that ATM was activated. ERK phosphorylation increased in a time and dose-dependent manner likely due to the expression of EcoRI and the generation of DSBs (Fig. 7B). Importantly, blocking ATM kinase activity with KU-55933 reduced ERK phosphorylation to below background levels (Fig. 7C), supporting the result seen with BrdU photolysis and KU-55933 (see Fig. 6). Phosphorylation of AKT at ser-473 also increased (1.9-fold) in cells expressing EcoRI, and this increase was also blocked with KU-55933 (Fig. 7D), suggesting that ATM signals to MEK-ERK via AKT.
Figure 7.

EcoRI-induced DSBs increase H2AX, ERK and AKT phosphorylation in an ATM-dependent manner. (A) HEK293 cells were transfected with 5 µg of either pcDNA3 or pcDNA3-NLS-HA-EcoRI and cells collected for western blotting 72 h post transfection. Fold (x) denotes changes in p-ERK levels compared to control (no EcoRI) normalized to total ERK levels. (B) HEK293 cells transfected with increasing quantities of pcDNA3-NLS-HA-EcoRI (0.1–5.0 µg) and pcDNA3 to a total of 5 µg. Cells were collected after 24, 48 and 72 h for western blotting and subsequent densitometric analysis. (C and D) HEK293 cells were transfected with pcDNA3-NLS-HA-EcoRI (5 µg) and treated or not with KU-55933 at 10 µM and collected after 72 h for western blotting and densitometric analysis. Data points, phosphorylation levels as a function of plasmid DNA quantity, error bars. SEM; n = 3. Fold (x) denotes changes in phosphorylation levels compared to control (pcDNA3, no EcoRI) normalized to ERK levels. *p < 0.05.
To further pinpoint the role of AKT in DSB signaling through the ERK pathway, we electroporated EcoRI restriction enzyme into HEK293 cells and determined the effect on ERK phosphorylation. We were able to recapitulate the DSB dose-dependency seen with BrdU photolysis and show that only very low levels of EcoRI were able to increase ERK phosphorylation whereas higher levels did not, but, in fact, reduced them (Fig. 8A and B). On the other hand, γ-H2AX levels, indicative of DSBs, showed a linear increase with the highest units of EcoRI producing the most significant increase (Fig. 8A and C and Sup. Fig. 7).
Figure 8.

EcoRI-induced DSBs stimulate ERK phosphorylation similar to BrdU photolysis. (A) HEK293 cells were electroporated with either electroporation buffer only (lane 1–4) or increasing doses of EcoRI enzyme as indicated. Cells were collected at 2, 10, 30 and 60 min for western blotting. ERK denotes equal loading. (B and C) Densitometic analysis of (A) presented in graph form. Data points, phosphorylation levels plotted against time normalized to ERK.
To confirm a regulatory role for AKT in ERK signaling, we established HEK293 cells harboring pcDNA3 (empty) or dominant-negative (K179M) AKT1 expression plasmids. A radiation dose response of these two cell populations showed that the expression of Myc-tagged DN-AKT1 reduced radiation-induced ERK phosphorylation levels (Fig. 9A). Interestingly, when DN-AKT was expressed γ-H2AX levels increased. EcoRI at very low levels (2.5 units) was then electroporated into the two cell populations and the effect on ERK phosphorylation determined. We found that cells expressing DN-AKT completely blocked ERK phosphorylation whereas control cells produced a 2-fold increase suggesting an important role of AKT in directing DSB signaling to the ERK pathway (Fig. 9B). All combined, electroporation of EcoRI showed similar if not identical ERK phosphorylation responses as those from BrdU photolysis, i.e., low levels of DSBs increased phosphorylation whereas higher levels decreased it. Whereas ERK phosphorylation was bi-phasic and AKT-dependent, γ-H2AX levels increased linearly with the amount of EcoRI electroporated. Thus, pro-survival signaling through ERK in response to low levels of DSBs is modulated by AKT.
Figure 9.

Dominant-negative AKT reduces ERK phosphorylation in response to IR and EcoRI-induced DSBs. HEK293 cells stably expressing pcDNA3 or DN-AKT-Myc plasmids were: (A) exposed to 0, 5 or 10 Gy and collected after 10 min for western blotting and subsequent densitometric analysis. ERK denotes equal loading. Myc indicates expression of DN-AKT. Data points, phosphorylation levels plotted against radiation dose normalized to ERK or (B) electroporated with either buffer alone or buffer with EcoRI enzyme (2.5 units) and cells collected after 5 min for western blotting and subsequent densitometric analysis. Data points, ERK phosphorylation levels plotted against buffer alone or EcoRI normalized to ERK. Error bars, SEM; n = 3. Fold (x) denotes changes in p-ERK levels compared to control (pcDNA3, no EcoRI) normalized to ERK. *p < 0.05.
Low levels of DSBs increase human glioma cell proliferation.
To determine the impact of low levels of DNA damage on cell survival, we examined the effects of electroporating increasing doses of EcoRI on U87 proliferative capacity. U87 (p53+) cells are known to respond to repeated low doses of radiation by senescing instead of undergoing apoptosis and cell death.28 Thus, we also monitored senescence by staining for β-galactosidase. Indeed, we show that low levels of EcoRI resulted in significant 1.1 to 1.4-fold increases in proliferation over a 4 day period (Fig. 10A). Whereas there was a clear dose-dependent increase in H2AX phosphorylation indicative of escalating extent of DSBs (Fig. 10B), it is apparent that these cells are not as amenable to electroporation as the HEK293 cells (see Fig. 8 and Sup. Fig. 7). We were unable to induce significant cell killing even with EcoRI as high as 1,500 units (data not shown). Nevertheless, at the relative low effective EcoRI doses used (compare γ-H2AX signals in Figs. 8 and 10B), we also did not see significant increases in senescence as determined by β-galactosidase staining (Fig. 10C). On the other hand, 3 days of serum starvation clearly induced a large extent of senescence/autophagy. Altogether, these results show that low levels of DNA damage inflicted by EcoRI increases cell proliferation in a dose-dependent manner in line with prosurvival responses elicited via ERK and AKT signaling.
Figure 10.

Low levels of DSBs increase cell proliferation. (A) Human U87 cells were electroporated with buffer alone, 2.5 U or 200 U of EcoRI enzyme as described in the legend to Figure 8. Four days after electroporation cells were collected from triplicate dishes, stained with Trypan Blue and cell viability determined by flow cytometry as described.40 (B) Electroporated U87 cells from (A) were cultured for 1 or 6 h and then collected and processed for western blotting with GAPDH serving as loading control. (C) Electroporated U87 cells from (A) were stained for β-galactosidase (top fields). Starved or unstarved U87 cells were stained for β-galactosidase after 72 h (bottom fields). Data points, survival fraction as a function of electroporated EcoRI. Error bars, SEM; n = 3. Fold (x) denotes changes in live cell levels compared to control (buffer, no EcoRI). *p < 0.05; ***p < 0.0005.
Discussion
The primary objective of this study was to shed light on the role of ATM-dependent “inside-out” signaling in response to DSBs and the effect on ERK and AKT pro-survival signaling.15 We have previously shown that EGFR signaling via ERK is important for repair foci formation and ATM-mediated activation of AKT phosphorylation in response to IR.14,16 To further delineate the nuclear signals generated by DSBs while minimizing the influence of non-nuclear signaling, we utilized BrdU photolysis to generate DSBs and for studying the ensuing signaling events.21,22
We found that although the kinetics of formation and removal of DSBs caused by BrdU photolysis were different from those seen after IR, still many of the DNA damage responses were similar. BrdU photolysis was strictly dependent on all three components, i.e., BrdU incorporation into DNA, Hoechts dye and UV-A, for triggering of the DDR. We were able to show that ATM autophosphorylation occurred at UV-A doses 1,000-fold lower than those used previously for studying ATM activation with UV-A alone.12 We extensively demonstrated that BrdU photolysis increased ATM-dependent phosphorylation of various proteins involved in the classical DDR signaling network,29–31 suggesting that BrdU photolysis can be used to study DSB signaling and at the same time minimize non-nuclear signaling. Taken together, even though other types of DNA damage, including single-strand breaks and DNA-protein cross-links, were reported to occur by BrdU photolysis,22 our results show a strong dependence on ATM that firmly support the idea that it is the DSBs that trigger these responses and not other types of DNA damage.
We documented a strict linear relationship between IR dose and ERK phosphorylation in human glioma cells. Because of the relatively high basal ERK phosphorylation levels in cancer cells compared to normal cells it is very difficult to discern increases in ERK phosphorylation at doses <2 Gy (unpublished observations). However, this is possible in primary human diploid cells.32 On the other hand, ERK responses generated by either BrdU photolysis or electroporated EcoRI enzyme were bi-phasic in nature with increased ERK phosphorylation seen at low levels of DSBs that decreased and even dropped below basal levels at high.
The bi-phasic ERK response seen with BrdU photolysis was not detected after treatment with IR as the induction of DSBs is considered to be rapid and removal occurs in a matter of minutes to hours. In contrast, BrdU photolysis produced maximum DSBs at 1 h and the half-life of removal was extended to 8 h. The difference in time needed to reach peak ERK phosphorylation in response to IR and BrdU photolysis, respectively, could perhaps be explained by differences in the kinetics of generating and removing (DNA repair) the DSBs. Thus, the prolonged half-life of the more severe BrdU DNA lesions might sustain the DDR so that it can be detected by western blotting. However, we were also able to document the same bi-phasic ERK reponse by EcoRI electroporation. Thus, it appears as if it is not the complexity and the sustained nature of the DNA damage that is critical since EcoRI cuts are considered relatively benign and quickly repaired. Rather, we favor the idea that IR is able to prevent or counteract ERK dephosphorylation occurring at high levels of DSBs. This is an important observation since this would mean that clinically relevant doses of radiation could be counter-productive and, in fact, might stimulate cancer growth.
The fact that the increase in BrdU photolysis-induced ERK phosphorylation was accompanied by ATM phosphorylation that was abolished with KU-55933 suggests an important role for ATM in ERK signaling in agreement with our previous study in reference 14. Moreover, the absence of such increase in ERK phosphorylation in A-T cells also suggests that ERK phosphorylation is dependent on ATM. These results are in agreement with a previous report demonstrating that ERK activation in response to the DNA damaging agent etoposide was abolished in ATM− fibroblasts.33 When human glioma M059J (DNA-PKcs−) and M059K (DNA-PKcs+) cells were examined for the ability to trigger a DDR in response to BrdU photolysis we found them both competent and indistinguishable from U87 cells, suggesting that DNA-PKcs is not important for ERK signaling in response to DSBs (data not shown). Also in agreement with this report, we found that the increased ERK phosphorylation by low levels of BrdU photolysis appears to be p53-independent. The M059K and M059J cells express mutant p53,34 whereas U87 cells are p53 wild-type. Thus, neither DNA-PKcs nor p53 seem critical for increasing ERK phosphorylation by low levels of BrdU photolysis.
When we examined the effect of a MEK inhibitor on ERK activation in response to BrdU photolysis, we found that the inhibitor abrogated ERK phosphorylation, suggesting that the signal that activates ERK is transmitted via MEK. Taken together, our results suggest that BrdU photolysis separately activates ERK and p53 signaling pathways in ATM-dependent manners. However, these pathways might still function cooperatively in controlling cell cycle checkpoints, DNA repair and apoptosis depending on the extent of the DNA damage. A similar dose-dependent, biphasic response to DNA damage was recently reported to occur when cells switch from a senescent to a quiescent state as a result of p53′s ability to suppress mTOR signaling at high levels of DNA damage.35,36 However, whether this response is related to our bi-phasic ERK response is currently not known. The increase in ERK and AKT signaling resulted in increased cell proliferation after electroporation with EcoRI at low doses unable to induce senescence.
Our data did not indicate a direct activation of the MEK-ERK pathway by ATM, but rather suggest the involvement of an unknown intermediate. On the other hand, ERK dephosphorylation in response to high levels of DSBs could occur through the alleviation of a negative regulatory loop, for example by (a) member(s) of the dual-specificity MAPK phosphatase family.37 We found here that dephosphorylation of ERK occurs in both ATM+ and ATM− cells suggesting that ATM does not regulate ERK dephosphorylation under these conditions. In a recent study it was reported that ATM negatively regulates radiation-induced ERK signaling via MKP1 dephosphorylation of ERK,38 a finding which is in agreement with our BrdU photolysis results at doses >2 Gy eq. except in our study this occurred independently of ATM. The reason for this difference is presently unclear but could be related to how the cells were propagated and maintained in the two studies.
AKT phosphorylation at ser-473 is critical for full AKT kinase activation.39 Using the ATM kinase-specific inhibitor KU-55933, we showed that AKT phosphorylation was blocked in response to DSBs generated by EcoRI expression thus supporting earlier findings demonstrating that ATM is critical for regulating insulin and IR-induced AKT activation.25,40 In our study, we now also show that AKT regulates ERK signaling in response to low levels of DSBs. We were able to recapitulate the BrdU photolysis effects with the use of an EcoRI expression plasmid and by direct electroporation of the enzyme to generate ‘pure’ DSB signaling. The two latter experimental approaches provided further evidence that ATM and AKT control ERK phosphorylation in response to low levels of DSBs. Importantly, we were able to demonstrate a significant and dose-dependent increase in cell numbers with escalating doses of EcoRI suggesting that at the low levels of DSBs able to induce ERK and AKT signaling results in proliferative responses.
Collectively our data show that cross-talk between the AKT and ERK pathways and survival signaling occur during the DDR triggered by low levels of DSBs without any influence from non-nuclear sources. How AKT transmits a pro-survival signal to ERK is presently not known except that the signal seems to go through MEK. RAS and RAF have both been associated with AKT-ERK cross-over signaling,15 thus making it possible that the DDR is shunted from AKT to ERK. Alternatively, rather than being controlled by (a) kinase(s), (a) protein phosphatase(s) could regulate the DSB-induced prosurvival signaling. For example, we recently reported on the possibility that a phosphatase might negatively control the phosphorylation of AKT at ser-473 in an ATM-dependent manner.40 Protein phosphatase 2A has strong links with ATM, AKT and ERK signaling and could possibly control a multitude of these DSB-induced responses.41–44
In summary, our observations suggest that the “inside-out” signaling events resulting from low levels of DSBs are mediated by ATM and signal through AKT to the MEK-ERK pathway. This is in line with pro-survival roles for ATM, AKT and ERK in regulating DSB repair at low levels of DNA damage, thus preserving cellular homeostasis and promoting cell proliferation and survival.16,17 At higher levels of DSBs, however, ERK signaling is blunted by ERK dephosphorylation in an ATM-independent fashion expected to stop growth, trigger senescence, and even result in cell death or apoptosis. Our study has uncovered important AKT-ERK signaling mechanisms stemming from DNA damage that are separate from radiation-induced growth factor receptor-mediated signaling previously reported by our group. “Inside-out” and “outside-in” signaling are both expected to occur during standard radiotherapy. With proper molecular targeting using small molecule inhibitor(s), growth-promoting signaling could perhaps be reduced while cell killing is increased by inhibiting the DDR.
Materials and Methods
Cell culture, chemicals and irradiation.
Cells were obtained from ATCC (Manassas, VA) and the Coriell Cell Repository (Camden, NJ). U87 (p53+), M059K (DNA-PKcs+) and M059J (DNA-PKcs−) human glioma cells were cultured at 37°C and 5% CO2 in α-MEM medium (Invitrogen, Carlsbad, CA). AT5BIVA (ATM−) and GM637 (ATM+) SV40-transformed human fibroblasts, MCF-7 human breast carcinoma cells and HEK293 human embryonic kidney cells were cultured in DMEM medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (Irvine Scientific, Santa Ana, CA) and penicillin/streptomycin as described in reference 45. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA) unless otherwise indicated. KU-55933 (2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one) (KuDOS Pharmaceuticals, Cambridge, United Kingdom) was dissolved in DMSO, stored at −20°C and used at a final concentration of 10 µM added to cell culture medium such that the final DMSO concentration was ≤0.1%.24 The MEK1/2 inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis (2-aminophenylthio) butadiene] (EMD Biosciences, San Diego, CA) was dissolved in DMSO and stored at −20°C. U0126 was used at a final concentration of 5 µM.46,47 The inhibitors were added 30 min prior to the addition of the DNA intercalator/photosensitizer Hoechst 33258 dye and UV-A irradiation or transfection and remained in the medium throughout the experiment. Cell irradiations were performed using an MDS Nordion Gammacell 40 (ON, Canada) research irradiator with a Cs-137 source delivering a dose of 1.05 Gy/min.
Plasmids.
The EcoRI expression plasmid pcDNA3-NLS-HA-EcoRI was generated by assemblying DNA from PCR of pJR1152,27 and Ad-I-SceI-NG,48 and cloned into pcDNA3 (Invitrogen, Carlsbad, CA). The EcoRI plasmid was propagated in EcoRI methylase expressing bacteria to prevent digestion of DNA by EcoRI endonuclease and the DNA sequence verified by DNA sequencing prior to use. pcDNA-NLS-HA-EcoRI has a nuclear localization signal (NLS) fused to the HA epitope at the N-terminus of EcoRI. The pcDNA3-DN-AKT1 plasmid was constructed from pUSEamp-AKT1 (K179M) expressing Myc-tagged, dominant-negative mouse AKT1 (K179M) from Upstate (Lake Placid, NY) and pcDNA3.
BrdU photolysis and pulsed-field gel electrophoresis (PFGE).
Sub-confluent cells were treated with 5-bromo-2′-deoxyuridine (BrdU)/thymidine at final concentrations of 0.3 and 2.4 µM, respectively and cultured for 48 h,21 serum starved for 12 h by washing cells three times with serum-free media and maintained in serum free medium throughout the experiment. Under these conditions >95% of the cells were positive for BrdU incorporation by using anti-BrdU-fluorescence-based flow cytometry and in situ immunofluorescence microscopy. To generate DSBs, Hoechst 33258 (Invitrogen, Carlsbad, CA) was added to the cell culture medium to a final concentration of 10 µM 5 min prior to UV-A (365 nm) irradiation.21 After irradiation, the cells were collected at the indicated times, centrifuged and cell pellets stored at −80°C until use. All procedures were performed in dim light. Cells were processed and analyzed by PFGE essentially as described in reference 49, but with the following modifications—cell pellets were resuspended in PBS at 4 × 106 cells/ml and mixed 1:1 with 1% InCert agarose (Cambrex, East Rutherford, NJ). Plugs were cast, allowed to solidify and digested in 0.5 mM EDTA, 1% w/v Sarkosyl with 1 mg/ml of proteinase K overnight at 37°C. The digested plugs were washed several times in 10 mM Tris-HCl, 1 mM EDTA (pH 7.5) and then stored at 4°C in 0.5 mM EDTA (pH 8.0). Electrophoresis was performed on a CHEF-DR II PFGE unit (Bio-Rad, Hercules, CA), operating at 40 V with a switch-time of 75 s for 16 h using a 0.5% agarose gel in 0.5x TBE. Following DNA separation, Southern transfer of the DNA onto a GeneScreen Plus nylon membrane (PerkinElmer, Boston, MA) was performed over a 48 h period using capillary transfer in 0.5 M NaOH/2 M NaCl. The membrane was hybridized to random-primed human genomic DNA labeled with 32P-dCTP, the signal detected using a Bio-Rad Molecular Imager FX and the extent of DNA released quantified using Quantity-One software (BioRad, Hercules, CA). The relative levels of DSBs generated by BrdU photolysis were calculated using the formula: DNAreleased/(DNAplug + DNAreleased) and compared to DSBs generated by IR so that Gy-equivalents could be calculated. Background levels of DNA released were subtracted prior to calculations.
Electroporation of EcoRI.
1 × 106 HEK293 cells were electroporated with EcoRI (New England Biolabs, Ipswich, MA) using the Amaxa Nucleofector Kit and the Nucleofector II Device (Lonza, Koln, Germany; www.lonza.com/research). Briefly, cells were resuspended in Nucleofector solution buffer with (2.5–200 U) or without EcoRI followed by electroporation using program A023 (HEK293) or X001 (U87). The cells were gently transfered into microfuge tubes, incubated at 37°C for the indicated times, and then collected for western blot analyses. Survival experiments were carried out by Trypan Blue-flow cytometry as described in reference 40. Senescent U87 cells were detected by staining for β-galactosidase using low-pH buffer as described in reference 28.
Antibodies and western blotting.
Cell lysates were separated by SDS-PAGE and transferred onto PVDF membranes for western blot analyses as described in reference 14. Antibodies against p-(ser-15) p53, p-(thr-68) Chk2, p-(ser-473) AKT, p-(ser-1981) ATM and myc-epitope were from Cell Signaling, Inc., (Beverly, MA). p-(ser-139) H2AX antibody was from Trevigen (Gaithersburg, MD). β-actin, ERK1/2 (ERK), GAPDH and p-(thr-185/tyr-187) ERK1/2 (p-ERK) antibodies were from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA). Primary antibodies were detected using antibodies conjugated to HRP followed by detection with the western Lighting system (PerkinElmer, Boston, MA) and exposure to X-ray film, or using infrared tagged secondary antibodies (Molecular Probes, Eugene, OR) and detection with the Odyssey Infrared Imaging system (LI-COR BioSciences, Lincoln, NE). The bands were quantified using Quantity-One software (Bio-Rad, Hercules, CA) and the Odyssey software.
Statistics.
Experimental data was analyzed for significance by unpaired Student's t test and expressed as mean ± SEM and p-values ≤0.05 were considered significant.
Acknowledgements
Work was supported in part by NIH P01CA72955, 1R01NS064593, R21ES016636 (K.V.); R01CA40615 (L.F.P.); and T32CA085159 and American Brain Tumor Association (S.E.G.).
Author contributions
A.K., R.N.M., B.R.A., S.E.G., S.M.D., and E.R. carried out experiments. L.F.P. provided technical expertise and comments on the manuscript. A.K. and K.V. conceived the experiments and wrote the manuscript. All authors read and approved the manuscript.
Supplementary Material
References
- 1.Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res. 2002;511:145–178. doi: 10.1016/s1383-5742(02)00009-1. [DOI] [PubMed] [Google Scholar]
- 2.Karran P. DNA double strand break repair in mammalian cells. Curr Opin Genet Dev. 2000;10:144–150. doi: 10.1016/s0959-437x(00)00069-1. [DOI] [PubMed] [Google Scholar]
- 3.Jackson SP. Detecting, signalling and repairing DNA double-strand breaks. Biochem Soc Trans. 2001;29:655–661. doi: 10.1042/0300-5127:0290655. [DOI] [PubMed] [Google Scholar]
- 4.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 5.Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22:5792–5812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- 6.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 7.Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 9.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peretz S, Jensen R, Baserga R, Glazer PM. ATMdependent expression of the insulin-like growth factor-I receptor in a pathway regulating radiation response. Proc Natl Acad Sci USA. 2001;98:1676–1681. doi: 10.1073/pnas.041416598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiloh Y. ATM (ataxia telangiectasia mutated): expanding roles in the DNA damage response and cellular homeostasis. Biochem Soc Trans. 2001;29:661–666. doi: 10.1042/0300-5127:0290661. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Ma WY, Kaji A, Bode AM, Dong Z. Requirement of ATM in UVA-induced signaling and apoptosis. J Biol Chem. 2002;277:3124–3131. doi: 10.1074/jbc.M110245200. [DOI] [PubMed] [Google Scholar]
- 13.Adamson AW, Kim WJ, Shangary S, Baskaran R, Brown KD. ATM is activated in response to N-methyl-N'-nitro-N-nitrosoguanidine-induced DNA alkylation. J Biol Chem. 2002;277:38222–38229. doi: 10.1074/jbc.M204409200. [DOI] [PubMed] [Google Scholar]
- 14.Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair and the DNA damage response. Cancer Res. 2007;67:1046–1053. doi: 10.1158/0008-5472.CAN-06-2371. [DOI] [PubMed] [Google Scholar]
- 15.Valerie K, Yacoub A, Hagan MP, Curiel DT, Fisher PB, Grant S, et al. Radiation-induced cell signaling: inside-out and outside-in. Mol Cancer Ther. 2007;6:789–801. doi: 10.1158/1535-7163.MCT-06-0596. [DOI] [PubMed] [Google Scholar]
- 16.Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, Valerie K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol Ther. 2009;8:730–738. doi: 10.4161/cbt.8.8.7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69:4252–4259. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dent P, Yacoub A, Contessa J, Caron R, Amorino G, Valerie K, et al. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat Res. 2003;159:283–300. doi: 10.1667/0033-7587(2003)159[0283:sariao]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 19.Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt-Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Signal transduction and cellular radiation responses. Radiat Res. 2000;153:245–257. doi: 10.1667/0033-7587(2000)153[0245:stacrr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 21.Limoli CL, Ward JF. Response of bromodeoxyuridine-substituted Chinese hamster cells to UVA light exposure in the presence of Hoechst dye #33258: survival and DNA repair studies. Radiat Res. 1994;138:312–319. [PubMed] [Google Scholar]
- 22.Limoli CL, Ward JF. A new method for introducing double-strand breaks into cellular DNA. Radiat Res. 1993;134:160–169. [PubMed] [Google Scholar]
- 23.Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 25.Viniegra JG, Martinez N, Modirassari P, Losa JH, Parada Cobo C, Lobo VJ, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem. 2005;280:4029–4036. doi: 10.1074/jbc.M410344200. [DOI] [PubMed] [Google Scholar]
- 26.Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnes G, Rine J. Regulated expression of endonuclease EcoRI in Saccharomyces cerevisiae: nuclear entry and biological consequences. Proc Natl Acad Sci USA. 1985;82:1354–1358. doi: 10.1073/pnas.82.5.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J Neurosurg. 2006;105:111–118. doi: 10.3171/jns.2006.105.1.111. [DOI] [PubMed] [Google Scholar]
- 29.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 30.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 31.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki K, Kodama S, Watanabe M. Extremely low-dose ionizing radiation causes activation of mitogen-activated protein kinase pathway and enhances proliferation of normal human diploid cells. Cancer Res. 2001;61:5396–5401. [PubMed] [Google Scholar]
- 33.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, et al. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 34.Anderson CW, Allalunis-Turner MJ. Human Tp53 from the malignant glioma-derived cell lines M059J and M059K has a cancer-associated mutation in exon 8. Radiat Res. 2000;154:473–476. doi: 10.1667/0033-7587(2000)154[0473:htftmg]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 35.Leontieva OV, Gudkov AV, Blagosklonny MV. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010;9:4323–4327. doi: 10.4161/cc.9.21.13584. [DOI] [PubMed] [Google Scholar]
- 36.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010;2:344–352. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–4615. doi: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- 38.Nyati MK, Feng FY, Maheshwari D, Varambally S, Zielske SP, Ahsan A, et al. Ataxia telangiectasia mutated downregulates phospho-extracellular signal-regulated kinase 1/2 via activation of MKP-1 in response to radiation. Cancer Res. 2006;66:11554–11559. doi: 10.1158/0008-5472.CAN-06-1935. [DOI] [PubMed] [Google Scholar]
- 39.Meier R, Alessi DR, Cron P, Andjelkovic M, Hemmings BA. Mitogenic activation, phosphorylation and nuclear translocation of protein kinase Bbeta. J Biol Chem. 1997;272:30491–30497. doi: 10.1074/jbc.272.48.30491. [DOI] [PubMed] [Google Scholar]
- 40.Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling and inhibits migration and invasion. Mol Cancer Ther. 2009;8:2894–2902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, et al. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–4461. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 43.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 44.Adams DG, Coffee RL, Jr, Zhang H, Pelech S, Strack S, Wadzinski BE. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J Biol Chem. 2005;280:42644–42654. doi: 10.1074/jbc.M502464200. [DOI] [PubMed] [Google Scholar]
- 45.Rosenberg E, Hawkins W, Holmes M, Amir C, Schmidt-Ullrich RK, Lin PS, et al. Radiosensitization of human glioma cells in vitro and in vivo with acyclovir and mutant HSV-TK75 expressed from adenovirus. Int J Radiat Oncol Biol Phys. 2002;52:831–836. doi: 10.1016/s0360-3016(01)02754-7. [DOI] [PubMed] [Google Scholar]
- 46.Scherle PA, Jones EA, Favata MF, Daulerio AJ, Covington MB, Nurnberg SA, et al. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J Immunol. 1998;161:5681–5686. [PubMed] [Google Scholar]
- 47.DeSilva DR, Jones EA, Favata MF, Jaffee BD, Magolda RL, Trzaskos JM, et al. Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J Immunol. 1998;160:4175–4181. [PubMed] [Google Scholar]
- 48.Golding SE, Rosenberg E, Khalil A, McEwen A, Holmes M, Neill S, et al. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J Biol Chem. 2004;279:15402–15410. doi: 10.1074/jbc.M314191200. [DOI] [PubMed] [Google Scholar]
- 49.Macann AM, Britten RA, Poppema S, Pearcey R, Rosenberg E, Allalunis-Turner MJ, et al. DNA double-strand break rejoining in human follicular lymphoma and glioblastoma tumor cells. Oncol Rep. 2000;7:299–303. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.