

Abstract
All eukaryotic and some prokaryotic ClC anion transport proteins have extensive cytoplasmic C-termini containing two cystathionine-β-synthase (CBS) domains. CBS domain secondary structure is highly conserved and consists of two α-helices and three β-strands arranged as β1-α1-β2-β3-α2. ClC CBS domain mutations cause muscle and bone disease and alter ClC gating. However, the precise functional roles of CBS domains and the structural bases by which they regulate ClC function are poorly understood. CLH-3a and CLH-3b are C. elegans ClC anion channel splice variants with strikingly different biophysical properties. Splice variation occurs at cytoplasmic N- and C-termini and includes several amino acids that form α2 of the second CBS domain (CBS2). We demonstrate that interchanging α2 between CLH-3a and CLH-3b interchanges their gating properties. The “R-helix” of ClC proteins forms part of the ion-conducting pore and selectivity filter and is connected to the cytoplasmic C-terminus via a short stretch of cytoplasmic amino acids termed the “R-helix linker”. C-terminus conformation changes could cause R-helix structural rearrangements via this linker. X-ray structures of three ClC protein cytoplasmic C-termini suggest that α2 of CBS2 and the R-helix linker could be closely apposed and may therefore interact. We found that mutating apposing amino acids in α2 and the R-helix linker of CLH-3b was sufficient to give rise to CLH-3a-LIKE gating. We postulate that the R-helix linker interacts with CBS2 α2, and that this putative interaction provides a pathway by which cytoplasmic C-terminus conformational changes induce conformational changes in membrane domains that in turn modulate ClC function.
Key words: ClC channel, chloride channel, homology model
Introduction
Members of the ClC superfamily of anion transport proteins have been identified in plants, yeast, eubacteria, archaebacteria and various invertebrate and vertebrate animals. ClC genes encode anion channels and Cl−/H+ exchangers and are expressed in plasma and intracellular organelle membranes where they perform essential physiological functions including transepithelial Cl− transport, systemic ion homeostasis, regulation of membrane potential and regulation of cytoplasmic and intra-organelle Cl− and H+ levels. Mutations in three of the nine human ClC encoding genes give rise to muscle, bone and kidney diseases.1–4
Extensive electrophysiological and molecular biological studies have shown that ClC proteins are homodimers and that each monomer forms an independently gated protopore that functions as an anion channel or a Cl−/H+exchanger.5 X-ray crystallography studies of bacterial ClCs have confirmed this model and demonstrate that each monomer comprises 18 α-helical domains (designated “A–R”), 17 of which are membrane embedded.6,7 The overall three-dimensional membrane structure of the bacterial ClC proteins is likely conserved throughout the ClC superfamily.6,8–11
All eukaryotic and some bacterial ClC proteins have extensive cytoplasmic C-terminal domains following the R-helix.1–4 These intracellular C-termini contain two cystathionine-β-synthase (CBS) domains.12–15 The CBS domain is a 50–60 amino acid motif found in numerous diverse proteins.16,17 Mutations in CBS domains give rise to several diseases16 and alter ClC gating12,18–23 indicating that they play a critical role in protein structure/function. Structural and biochemical studies have demonstrated that the cytoplasmic C-termini of eukaryotic ClCs interact to form dimers.13–15,24
Six ClC genes, termed clh (Cl− channel homolog)-1-6,25 are present in the C. elegans genome.26–28 C. elegans CLH proteins are representative of the three mammalian ClC subfamilies.27,28 clh-3 encodes two splice variants, CLH-3a and CLH-3b, that exhibit striking differences in sensitivity to depolarizing voltages, activating voltages and extracellular pH and Cl−.18,29
The structural bases by which the cytoplasmic C-terminus controls ClC gating and activity are poorly understood. We have exploited the distinct functional differences of CLH-3a and CLH-3b to address this question. Splice variation of the two channels occurs in their cytoplasmic N- and C-termini. Mutagenesis studies have shown that the gating properties of CLH-3b can be converted to those of CLH-3a by deletion mutations in its unique cytoplasmic C-terminus or in the first CBS domain (CBS1).18
In the present study, we use a combination of homology modeling and mutagenesis to further characterize the role of C-terminus splice variation in regulating CLH-3b gating. Splice variation of CLH-3a and CLH-3b includes the second α-helix (α2) of the second CBS domain (CBS2). Interchanging α2 between the two channels interchanges their gating properties. X-ray structures of ClC-0, ClC-Ka and ClC-5 cytoplasmic C-termini13–15 suggest that α2 lies close to and could therefore interact with a short stretch of cytoplasmic amino acids, termed the R-helix linker, that connect membrane helix R to the intracellular C-terminus. The R-helix forms part of the channel pore and selectivity filter.7 Given the structural role played by the R-helix in ion conduction and its direct connection to a large cytoplasmic C-terminus, Dutzler et al.7 proposed that it could provide a pathway by which conformational changes in intracellular domains regulate channel/transporter function. Consistent with this model, we found that mutating apposing amino acids in α2 and the R-helix linker of CLH-3b was sufficient to give rise to CLH-3a-like gating. We propose that the R-helix linker interacts with CBS2 α2, and that this putative interaction provides a pathway by which cytoplasmic C-terminus conformational changes induce conformational changes in membrane domains that in turn modulate ClC function. Our studies provide novel insights into the role of CBS domains, the R-helix linker and cytoplasmic C-terminus conformational changes in regulating ClC gating properties.
Results
Large deletion mutations in CLH-3b CBS domains give rise to CLH-3a-like gating.
Figure 1 shows the sequence of the predicted intracellular domains of CLH-3a and CLH-3b. The membrane-associated domains of the two channels are identical29 and have been omitted from the figure for clarity. Splice variation occurs in cytoplasmic regions of the channels and includes a 71 amino acid extension of the CLH-3a N-terminus, a 101 amino acid insert located between CBS1 and CBS2 in CLH-3b and a 160 amino acid extension of the CLH-3b C-terminus. In addition, eight of the last nine C-terminal amino acids of CLH-3a are distinct from those in the same region of CLH-3b. Six of these nine amino acids are predicted to be located at the end of CBS2.
Figure 1.

Sequence of predicted intracellular N- and C-termini of CLH-3a and CLH-3b. Membrane-associated domains have identical sequence and are omitted for clarity. Double vertical lines indicate start of membrane-associated sequence, which ends at residues 613 for CLH-3a and 542 for CLH-3b. Splice variations in CLH-3a and CLH-3b are highlighted in yellow and green, respectively. Location of CBS domains were predicted using the domain recognition programs PFAM,30 InterProScan31 and MyHits32 and are outlined with boxes.
Studies using C. elegans deletion mutants18 have demonstrated that disruption of CLH-3b CBS1 domain structure gives rise to channels with CLH-3a functional properties. In addition, deletion of the last 169 amino acids of CLH-3b, which includes part of CBS2 (Fig. 1), yields channels that have gating properties identical to those of CLH-3a. To determine whether disruption of CBS2 structure per se was responsible for this effect, we deleted the last seventeen amino acids (residues 822–838) of CBS2 in CLH-3b. Figure 2 shows the characteristics of wild-type CLH-3a and CLH-3b and the mutant channel CLH-3bΔ822-838. Both CLH-3a and CLH-3b show strong inward rectification and activation at hyperpolarized voltages (Fig. 2A). However, CLH-3a activation requires much stronger hyperpolarization. Mean activation voltages were −54 mV for CLH-3a and −27 mV for CLH-3b (Fig. 2B). Deletion of the last seventeen amino acids of CBS2 in CLH-3b induced a hyperpolarizing shift in activation voltage (Fig. 2A and B). The mean activation voltage of CLH-3bΔ822-838 was ∼-52 mV and was not significantly (p > 0.6) different from that of CLH-3a.
Figure 2.

CLH-3b CBS domain deletion mutations give rise to CLH-3a-like gating. Current-to-voltage relationships, activation voltages and activation by bath acidification are shown in (A–C), respectively. **p < 0.001 and *p < 0.01 compared to wild-type CLH-3b. Effects of depolarized holding potentials are shown in (D–F). (D) Representative current traces showing hyperpolarization induced current activation in cells held at depolarized holding potentials. Cells were held at voltages of −20 to 60 mV for 3 sec before currents were activated by stepping to −120 mV. (E) Current activation by depolarized holding potentials. Peak current amplitudes are normalized to that measured following a holding potential of −20 mV (I−20 mV holding potential). (F) Effect of holding potential on current inactivation. Mean pseudo-steady-state current (Isteady state) was measured over the last 20 msec of the −120 mV test pulse and normalized to peak current (Ipeak) amplitude. Values in (A–C, E and F) are means ± S.E. (n = 5–12). CLH-3bΔ822-838 is a deletion mutant lacking the last 17 amino acids of CBS2.
ClC channels are activated by extracellular acidification.4 Reduction of bath pH to 5.9 activates CLH-3a and CLH-3b ∼3.2-and ∼1.6-fold, respectively. Deletion of amino acids 822–838 significantly (p < 0.001) increased the pH sensitivity of CLH-3b resulting in a ∼4.4-fold activation by reducing bath pH to 5.9 (Fig. 2C).
CLH-3a shows unique sensitivity to depolarized holding voltages termed pre-depolarization induced potentiation.18,19,29 As shown in Figure 2D and E, increasing the degree of holding potential depolarization increases the extent of or “potentiates” hyperpolarization-induced current activation. Currents potentiated by pre-depolarization also undergo slow, partial inactivation (Fig. 2D and F). Wild-type CLH-3b is insensitive to depolarized holding voltages (Fig. 2D–F). However, the deletion mutation in CBS2 induced sensitivity to pre-depolarization that resembled that of CLH-3a (Fig. 2D–F). Taken together, data in Figure 2 and our previous studies18,19 demonstrate that disruption of either CBS1 or CBS2 structure in CLH-3b gives rise to channels with CLH-3a-like biophysical properties.
In contrast to CLH-3b, deletion of the last 11 amino acids of CBS1 or the last 17 amino acids of CBS2 in CLH-3a had no effect on voltage and pH sensitivity (Fig. 3A and B) or the response to depolarized holding potentials (Fig. 3C and D). These data demonstrate that the unique gating properties of CLH-3a are insensitive to disruption of CBS domain conformation.
Figure 3.

CLH-3a gating is unaffected by large deletion mutations in CBS1 or CBS2. (A) Activation voltage. (B) Activation by bath acidification. (C) Current activation by depolarized holding potentials. (D) Effect of holding potential on current inactivation. Data in (C and D) were acquired as described in Figure 2 legend. Values are means ± S.E. (n = 4–12). CLH-3aΔ675-685 and CLH-3aΔ792-808 are deletion mutants lacking the last 11 and last 17 amino acids of CBS1 and CBS2, respectively.
Splice variation of CBS2 determines the gating characteristics of CLH-3a and CLH-3b.
The primary sequence of CBS domains in diverse proteins is variable, but the motif has a highly conserved secondary structure consisting of an N-terminal β-strand (β1) followed by an α-helix (α1), two β-strands (β2 and β3) and an α-helix (α2).16,17 As shown recently by Dutzler and coworkers,13,15 CBS1 and CBS2 domains play an important role in determining the overall homodimeric structure of the cytoplasmic regions of ClC proteins. Large deletions in either CBS1 or CBS2 are expected to dramatically disrupt C-terminus conformation and function. Our deletion mutation studies (Figs. 2 and 3) as well as previous work18,19 demonstrate that overall CBS domain architecture plays a critical role in determining CLH-3b but not CLH-3a functional properties.
As shown in Figure 1, CBS2 domains of CLH-3a and CLH-3b predicted using the domain recognition programs PFAM,30 InterProScan31 and MyHits32 are alternatively spliced at the last six amino acids. This region forms part of α2. The sequences of the six splice variant amino acids in CLH-3a and CLH-3b are VCFLIS and LRLAIE, respectively.
We postulated that splice variation of α2 may be a critical determinant of channel biophysical properties. To test this hypothesis, we interchanged the six alternatively spliced amino acids between the channels. CLH-3b + VCFLIS exhibited a hyperpolarized activation voltage (Fig. 4A), increased pH sensitivity (Fig. 4B) and sensitivity to depolarized holding potentials (Fig. 4C and D). Replacement of the last six amino acids in CBS2 of CLH-3a with the analogous amino acids in CLH-3b (CLH-3a + LRLAIE) had little effect on activation voltage or pH sensitivity (data not shown).
Figure 4.

Interchanging alternatively spliced amino acids in CBS2 interchanges channel functional properties. (A) Activation voltage. **p < 0.0001 compared to wild-type CLH3b (B) Activation by bath acidification. *p < 0.05 compared to wild-type CLH-3b (C) Current activation by depolarized holding potentials. (D) Effect of holding potential on current inactivation. Data in (C and D) were acquired as described in Figure 2 legend. Values are means ± S.E. (n = 5–11). CLH-3b + VCFLIS and CLH-3a + LRLAIEYLQ are chimeras in which alternatively spliced amino acids in CBS2 have been interchanged between the two channels.
The response of CLH-3a + LRLAIE to pre-depolarization was variable between cells. Of the 15 cells that were patch-clamped from two separate transfections, we found that two cells showed a strong response to pre-depolarization resembling that of wild-type CLH-3a, seven cells showed no response and six cells exhibited a weak response (data not shown). This variability suggested to us that additional spliced amino acids may contribute to channel gating properties.
To examine CBS2 structure in greater detail than that predicted by domain recognition programs, we used Rosetta software33,34 to develop 20,000 candidate structures of amino acids 803–811 (i.e., VCFLISRKK) for CLH-3a and 833–841 (i.e., LRLAIEYLQ) for CLH-3b. Figure 5 is a histogram showing the number of amino acids in this region that adopt a helical conformation and the number of Rosetta models that predict this conformation. For CLH-3a and CLH-3b, the median number of amino acids that are predicted to adopt a helical conformation are five and seven, respectively. This difference between the splice variants is highly significant (p < 0.0001) and indicates that α2 of CLH-3b is predicted to be longer than that of CLH-3a and encompasses amino acids immediately C-terminal to the CBS2 domain predicted by domain recognition programs.
Figure 5.
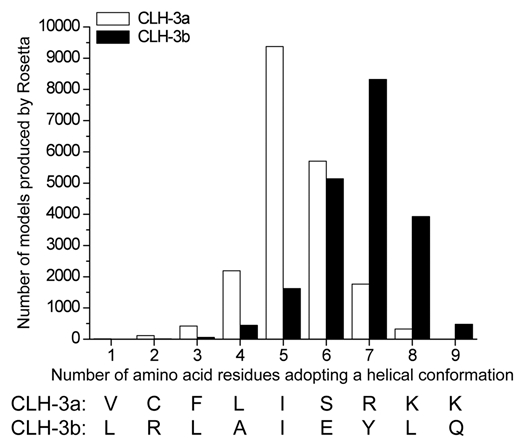
Rosetta modeling33,34 of amino acids comprising CBS2 in CLH-3a and CLH-3b. Sequence of the nine amino acids modeled are shown at the bottom of the graph and includes the predicted last six amino acids of CBS2. The graph shows the number of amino acids adopting a helical conformation and the number of Rosetta models that predicted the structure. The median number of helical amino acids in CLH-3a and CLH-3b are five and seven and are predicted by ∼9,300 and ∼8,300 Rosetta models, respectively. The difference between CLH-3a and CLH-3b in the number of helical residues is highly significant (p < 0.0001).
Given the possible differences in α2 length and the variable response of CLH-3a + LRLAIE to depolarizing voltages, we interchanged the last nine amino acids of CLH-3a with the analogous amino acids of CLH-3b. The activation voltage of CLH-3a + LRLAIEYLQ remained hyperpolarized and pH sensitivity was similar to that of wild-type CLH-3a (Fig. 4A and B). However, in contrast to CLH-3a + LRLAIE, none of the currents recorded from CLH-3a + LRLAIEYLQ expressing cells (10 cells in six separate transfections) showed sensitivity to depolarized holding potentials (Fig. 4C and D).
To determine whether the last three amino acids of CLH-3a (i.e., RKK) independently regulate channel properties, we exchanged them with the analogous amino acids of CLH-3b (i.e., YLQ). Replacing RKK with YLQ (i.e., CLH-3a + YLQ) had no significant effect on pH sensitivity or activation voltage (p > 0.7) or sensitivity to depolarized voltages (data not shown). Similarly, CLH-3b + RKK showed reduced pH sensitivity similar to that of wild-type CLH-3b and was insensitive to pre-depolarization (data not shown). However, the activation voltage for CLH-3b + RKK was significantly (p < 0.0001) hyperpolarized to a mean ± S.E. value of −51 ± 1 mV (n = 13), which is similar to that of wild-type CLH-3a (see Fig. 2B).
The R-helix linker plays a critical role in determining CLH-3a and CLH-3b gating properties.
Data in Figure 4 demonstrate that splice variation of α2 of CBS2 plays a major role in determining the gating characteristics of CLH-3a and CLH-3b. Differences in the primary and secondary structure of α2 could alter local interactions within the channel that give rise to unique gating properties. To explore this possibility, we generated homology models of the cytoplasmic C-termini of CLH-3a and CLH-3b based on the 1.6 angstrom crystal structure of ClC-Ka (Fig. 6).13 The model of the cytoplasmic C-terminal domain was manually docked to the membrane-associated region of CLC-ec1 (Protein Data Bank accession CODE 1OTS) to provide context for the model (Fig. 6B). The cytoplasmic C-termini of both channels were modeled as homodimers as has been shown experimentally for ClC-0, ClC-5, ClC-Ka and ClC-Kb.13–15,24 Each pair of CBS domains within a monomer interact to form a dimer and the monomer-monomer interface is located between the two CBS2 domains13 (Fig. 6B).
Figure 6.

Structural features of the CLH-3b cytoplasmic C-terminus. (A) Amino acid alignment of ClC-Ka and CLH-3b used for developing the CLH-3b C-terminus homology model. Secondary structure assignments and CBS domain locations are shown for ClC-Ka and are based on its crystal structure.13 ClC-Ka CBS domains are denoted β1-α1-β2-β3-α2. Amino acid numbering is based on CLH-3b sequence. (B) Ribbon diagram of CLH-3b C-terminus homology model (amino acids 549–615 and 783–841). To provide context for the C-terminus structure, it is shown manually docked to the CLC-ec1 membrane-associated region (gray and magenta). Protein Data Bank accession code 1OTS.6 The membrane-associated R-helix domain is shown in magenta and the cytoplasmic R-helix linker is shown in black. Each monomer of the cytoplasmic C-terminus dimer contains one CBS1 (red) and one CBS2 (green) domain. The six amino acid splice variation of CBS2 is shown in blue. The region (amino acids 616–782) between CBS1 and CBS2 is not modeled. Also not modeled are the first nine amino acids of the R-helix linker. (C) Sequence of the R-helix and R-helix linker in CLH-3b. Asterisks denote amino acids in the R-helix linker that were mutated (see Fig. 7). (D) Ribbon diagrams showing the relationship of mutated amino acids in CBS2 α2 (shown in blue and green) to F559 (shown in gray) in the R-helix linker and CBS2 α1 (shown in green).
A stretch of 20 cytoplasmic amino acids termed the R-helix linker connects the C-terminus of the R-helix of CLH-3a and CLH-3b to the N-terminus of CBS1 (Fig. 6C). The R-helix forms part of the channel pore and selectivity filter. The conformation of the R-helix and thus channel properties may be modulated by cytoplasmic domains.7 Crystal structures of ClC-0, ClC-Ka and ClC-5 cytoplasmic C-termini13–15 indicate that part of the cytoplasmic R-helix linker lies close to α2 of CBS2 (Fig. 6B). This close apposition suggests that the linker could interact with CBS2 and that this putative interaction may in turn play a role in mediating the differences in CLH-3a and CLH-3b gating induced by α2 splice variation.
Examination of the homology models indicated that F559 in the R-helix linker of CLH-3b lies very close to A836 in α2 of CBS2 (Fig. 6D). The orientation of the homologous phenylalanine in CLH-3a, F630, could be altered due to the presence of a bulkier leucine residue, L806, at the corresponding position in α2. To test whether F559 plays a role in regulating channel properties, we mutated it to aspartate (F559D). As shown in Figure 7A–D, this mutation gave rise to CLH-3a-like voltage and pH sensitivity and sensitivity to depolarized holding potentials. In contrast, mutation of F630 to aspartate in CLH-3a had no effect on channel properties (data not shown).
Figure 7.

A single point mutation in the R-helix linker gives rise to CLH-3a-like gating. (A) Activation voltage. (B) Activation by bath acidification. (C) Current activation by depolarized holding potentials. (D) Effect of holding potential on current inactivation. Data in (C and D) were acquired as described in Figure 2 legend. Values are means ± S.E. (n = 5–12). *p < 0.0001 compared to wild-type CLH-3b. H552, M554, E557, F559 and M560 are amino acids located in the R-helix linker of CLH-3b (see Fig. 6C).
To determine whether the channel gating was uniquely sensitive to mutation of F559, we also performed charge addition or charge swapping mutations to the neighboring amino acids H552, M554, E557 or M560 (see Fig. 6C). None of these amino acids were predicted to have strong interactions with α2. Consistent with this prediction, mutation of M554 or E557 to lysine or H552 or M560 to aspartate had no effect on CLH-3b gating (Fig. 7A–D).
As we have shown previously,18 CLH-3a and CLH-3b mutants in which the extracellular glutamate residue that forms the fast gate is mutated to cysteine show 7–8-fold differences in reactivity to the sulfhydryl modifying reagent MTSET. Deletion mutations in the CLH-3b C-terminus give rise to MTSET reactivity very similar to that of CLH-3a suggesting that cytoplasmic domains modulate the conformation of channel extracellular regions.18 To determine whether the F559D mutation alters the conformation of CLH-3b extracellular domains, we mutated the fast gate glutamate residue to cysteine (i.e., E167C). Consistent with our previous observations,18 this mutation gave rise to constitutively active channels that are no longer sensitive to voltage and extracellular protons (data not shown).
Exposure of the E167C mutant to 1 mM MTSET inhibited whole cell current amplitude as we have shown previously.18 The mean ± S.E. time constant for inhibition was 283 ± 34 sec (n = 6). Mutation of F559 to aspartate reduced the inhibition time constant nearly 7-fold to a mean ± S.E. value of 41 ± 6.6 sec (n = 5). The striking difference in rates of MTSET inhibition for E167C and the E167C/F559D double mutant is remarkably similar to the difference observed between CLH-3a and CLH-3b.18 These data together with the results shown in Figure 7 demonstrate that mutation of a single amino acid in the R-helix linker, F559, to aspartate converts CLH-3b gating properties and extracellular cysteine reactivity to those resembling CLH-3a.
As shown in Figure 6D, the homology model predicts that F559 lies close to A836 in α2 suggesting that the two amino acids could interact. To test this possibility, we mutated A836 to aspartate. The A836D mutant gave rise to a small hyperpolarizing shift in activation voltage and potentiation behavior that was considerably less than that observed with wild-type CLH-3a (data not shown). We therefore mutated A836 to a larger, positively charged lysine. The A836K mutant exhibited voltage and pH sensitivity nearly identical to those of CLH-3a (Fig. 8A–D).
Figure 8.

Mutation of amino acids in α2 of CBS2 predicted to interact with F559 in the R-helix linker or α1 amino acids give rise to CLH-3a-like gating. (A) Activation voltage. (B) Activation by bath acidification. (C) Current activation by depolarized holding potentials. (D) Effect of holding potential on current inactivation. Data in (C and D) were acquired as described in Figure 2 legend. Values are means ± S.E. (n = 5–8). *p < 0.01 and **p < 0.0001 compared to wild-type CLH-3b. L833, L835, A836 and I837 are amino acids located in α2 of CBS2 of CLH-3b.
We also mutated amino acids immediately adjacent to A836. Mutation of L835 to lysine (L835K) had no effect on the voltage or pH sensitivity of CLH-3b while mutating I837 to lysine (I837K) gave rise to CLH-3a-like behavior (Fig. 8A–D). Both amino acids are predicted to face away from the R-helix linker (Fig. 6D). Crystal structures of various tandem CBS domain containing proteins indicate that that α1 and α2 lie close together and may physically interact.13–15,35,36 We examined the CLH-3b homology model to assess the relationship of α2 amino acids to α1 in CBS2. As shown in Figure 6D, L835 faces away from α1 while I837 faces towards it and could interact with amino acids on this helix. This suggests that the I837K mutation may disrupt putative α1-α2 interactions. To test this possibility, we mutated L833, which also lies close to α1 (Fig. 6D), to glutamate (L833E). The L833E mutant exhibited CLH-3a-like behavior (Fig. 8A–D). Data shown in Figures 2–4, 7 and 8, and our previous findings18,19 indicate that mutations that disrupt CBS domain structure and/or putative interactions of CBS2 with the R-helix linker give rise to gating characteristics resembling those of CLH-3a.
Discussion
The CBS domain is a highly conserved motif found in diverse proteins including channels, transporters, kinases and metabolic enzymes of archeabacteria, eubacteria and eukaryotes.16,17 Mutations in CBS domains cause diverse inherited diseases such as cardiomyopathy, homocystinuria, retinitis pigmentosa, osteopetrosis and myotonia.16,37,38
The precise function of CBS domains is poorly understood.16 In several diverse proteins, CBS domains have been shown to bind adenosyl compounds such as ATP, ADP and AMP that in turn regulate protein function.39–42 CBS domains in the MgtE Mg2+ transporter likely bind Mg2+ and function as part of an intracellular Mg2+ sensor that regulates transporter activity.43 Recent studies on the bacterial osmoregulatory organic osmolyte transporter OpuA have shown that CBS motifs regulate the transporter on/off state by binding in an ionic strength dependent manner to charged membrane domains.44,45
CBS motifs are typically present in proteins as two or four copies. Pairs of CBS motifs associate to form a dimeric structure known as a Bateman module or domain.46–48 The monomer-monomer interface between two CBS motifs occurs at β-sheets formed by β-strands 2 and 3.13–15,35,36,43,47
CBS dimers also interact to form oligomeric structures. The interface between Bateman domains is formed by the α-helices of CBS motifs in a head-to-tail or head-to-head arrangement.35,40,43,47 Meyer and Dutzler14 postulated a head-to-tail interaction between Bateman domains of ClC-0 cytoplasmic C-termini (i.e., CBS1 of one Bateman domain interacts with CBS2 of the second domain). However, X-ray crystallography studies on dimers of ClC-5 and ClC-Ka cytoplasmic C-termini revealed a novel interface formed by the CBS2 motifs of each C-terminal monomer.13,15 This “tail-to-tail” arrangement of Bateman domains is shown in the CLH-3b homology model in Figure 6B.
Large deletion mutations in either CBS1 or CBS2 are expected to dramatically disrupt CBS domain tertiary structure and this in turn likely disrupts oligomerization. Our results demonstrate that disruption of CLH-3b CBS domain structure (Fig. 2)18,19 gives rise to channels with CLH-3a-like biophysical properties while disruption of CLH-3a CBS structure has little effect on channel gating (Fig. 3). These findings indicate (1) that the overall CBS domain architecture of CLH-3b plays a critical role in determining the functional properties of the channel and (2) that CLH-3a gating appears to be largely insensitive to changes in CBS domain conformation.
The C-terminal end of CBS2 is alternatively spliced in CLH-3a and CLH-3b (Fig. 1). This spliced region forms the second α-helix domain, α2. Splice variation alters not only the primary sequence, but also is predicted to alter α2 secondary structure. Rosetta modeling suggested that α2 in CLH-3b is longer than that of CLH-3a (Fig. 5). Consistent with this, we found that insertion of the last six amino acids of the CLH-3a CBS2 domain (i.e., VCFLIS) into CLH-3b CBS2 gives rise to CLH-3a-like voltage- and pH-sensitivity (Fig. 4). Inserting the analogous six amino acids of CLH-3b (i.e., LRLAIE) into CLH-3a had little effect on activation voltage or pH sensitivity and had an inconsistent effect on sensitivity to depolarized holding potentials. However, when the nine alternatively spliced amino acids comprising the longer α2 of CLH-3b (i.e., LRLAIEYLQ) were inserted into CLH-3a, the mutant channels were fully insensitive to depolarized voltages (Fig. 4C and D).
Given that large scale disruption of CBS structure by deletion mutations has no effect on CLH-3a gating, we suggest that splice variation of α2 in CBS2 of this channel disrupts critical interactions between CBS domains and other regions within the C-terminus. The absence of these interactions gives rise to the gating characteristics of CLH-3a. In contrast, both the primary sequence and predicted altered secondary structure of CLH-3b α2 mediate important functional interactions. Disruption of these interactions by mutation of either CBS1 or CBS2 gives rise to channels with CLH-3a-like properties.
It is important to note that the CLH-3a + LRLAIEYLQ chimeric channel did not fully recapitulate CLH-3b gating. Activation voltage and pH sensitivity of this chimera resembled those of wild-type CLH-3a (Fig. 4A and B). These results indicate that other cytoplasmic domains unique to CLH-3b play a role in determining these gating characteristics. Our previous studies have shown that deletion of the 101 amino acid linker domain between CBS1 and CBS2 in CLH-3b (Fig. 1) gives rise to channels with a strongly hyperpolarized activation voltage.18 In addition, deletion of the 160 amino acids immediately C-terminal to LRLAIEYLQ (Fig. 1) increases pH sensitivity and hyperpolarizes channel activation voltage (Dave and Strange, unpublished observations).
The molecular mechanisms by which cytoplasmic structures and intracellular signaling events regulate ClC channel/transporter function are poorly understood. Dutzler and colleagues have suggested that the membrane-associated R-helix may play an important role in this regulation.7 By virtue of its direct connection to the large cytoplasmic C-terminus, the R-helix may provide a pathway by which conformational changes in intracellular domains induce rearrangements of the pore that in turn alter channel/transporter activity.
A short, cytoplasmic linker connects the R-helix to CBS1. Interestingly, despite significant differences in primary sequence, part of the R-helix linkers of ClC-0, ClC-5 and ClC-Ka have similar and well ordered crystal structures.13–15 The conservation of this structure suggests that the linker likely plays an important role in channel function.
Crystal structures of ClC-0, ClC-5 and ClC-Ka13–15 indicate that α2 of CBS2 lies close to the cytoplasmic R-helix linker. Our homology models of CLH-3a and CLH-3b suggested putative interactions between α2 amino acids and an R-helix linker phenylalanine residue that could be altered by splice variation. Consistent with this, we found that mutation of CLH-3b F559 to aspartate or mutation of a closely apposed alanine residue in α2 gave rise to channels with gating properties and extracellular cysteine reactivity that fully recapitulated those of CLH-3a (Figs. 7 and 8, and Results). In contrast, mutation of the analogous R-helix linker phenylalanine residue in CLH-3a had no effect on channel activity.
Our data are consistent with the hypothesis that amino acids in α2 of CBS2 and the R-helix linker interact, and suggest that this putative interaction could be important for determining the conformation of the R-helix and other associated regions that control channel gating. We postulate that splice variation of α2 in CLH-3a disrupts its ability to interact with the R-helix linker, giving rise to the channel's unique gating characteristics. Mutation of CBS1, CBS2 or the R-helix linker of CLH-3b also disrupts this putative interaction, giving rise to CLH-3a-like voltage- and pH-sensitivity. Further experiments such as biophysical measurements and mutant cycle analysis will be needed to test directly for a physical interaction between CBS2 and the R-helix linker.
In summary, our studies have provided unique insights into the functional roles of ClC CBS domains and the R-helix linker in regulating channel properties. We propose a mechanism in which the R-helix linker interacts with CBS2 α2 and provides a structural pathway by which C-terminus conformational changes could modulate the conformation of the R-helix and associated structures and lead to changes in channel function. This hypothesis is supported by recent studies by Martinez and Maduke,24 which demonstrated that a point mutation in the R-helix linker of ClC-Kb abolishes extracellular Ca2+ activation of the channel and alters bromide permeability. Additional mutagenesis, modeling, biochemical and structural studies are needed to directly test this hypothesis. It will be particularly important to determine the role played by the R-helix linker and CBS domain conformation in mediating the regulation of ClC channels and transporters by intracellular signaling events such as phosphorylation49,50 and ATP binding.51,52
Methods
Transfection and whole cell patch clamp recording.
HEK293 (human embryonic kidney) cells were cultured in 35 mm diameter tissue culture plates in Eagle's minimal essential medium (MEM; Gibco, Gaithersburg, MD) containing 10% fetal bovine serum (Hyclone Laboratories, Inc., Logan, UT), nonessential amino acids, sodium pyruvate, 50 U/ml penicillin and 50 µg/ml streptomycin. After reaching 50–70% confluency, cells were transfected using FuGENE 6 (Roche Diagnostics Corporation, Indianapolis, IN) with 1 µg GFP and 1–8 µg of channel cDNAs ligated into pcDNA3.1. Point mutants were generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and deletion mutants were generated by polymerase chain reaction (PCR)-based fusion strategies. For single amino acid substitutions, local structure was analyzed using a homology model (Fig. 6) to identify mutations that would be most disruptive to putative local interactions. All mutants were confirmed by DNA sequencing. Experimental protocols were performed on at least two independently transfected groups of cells.
Following transfection, cells were incubated at 37°C for 24–30 h. Approximately 2 h prior to patch clamp experiments, cells were detached from growth plates by exposure to Hank's balanced salt solution containing 0.25% trypsin and 1 mM EDTA (Gibco) for 45 sec. Detached cells were suspended in MEM and then plated onto poly-L-lysine coated cover slips. Plated cover slips were placed in a bath chamber mounted onto the stage of an inverted microscope. Cells were visualized by fluorescence and differential interference contrast microscopy.
Transfected cells were identified by GFP fluorescence and patch clamped using a bath solution that contained 90 mM NMDG-Cl, 5 mM MgSO4, 1 mM CaCl2, 12 mM HEPES free acid, 8 mM Tris, 5 mM glucose, 80 mM sucrose and 2 mM glutamine and was titrated to pH 7.4 with HEPES free acid and Tris (297–303 mOsm), and a pipette solution containing 116 mM NMDG-Cl, 2 mM MgSO4, 20 mM HEPES titrated to pH 7.0 with CsOH, 6 mM CsOH, 1 mM EGTA titrated to pH 8.3–8.5 with CsOH, 2 mM ATP, 0.5 mM GTP and 10 mM sucrose (pH 7.2, 274–280 mOsm). Low pH bath solution contained 90 mM NMDG-Cl, 5 mM MgSO4, 1 mM CaCl2, 5 mM glucose, 80 mM sucrose and 2 mM glutamine and was titrated to pH 5.9 using Hepes free acid, (297–303 mOsm).
Patch electrodes with resistances of 3–7 Ω were pulled from 1.5 mm outer diameter silanized borosilicate microhematocrit tubes. Whole cell currents were measured with an Axopatch 200B (Axon Instruments, Foster City, CA) patch clamp amplifier. Electrical connections to the patch clamp amplifier were made using Ag/AgCl wires and 3 M KCl/agar bridges. Data acquisition and analysis were performed using pClamp 10 software (Axon Instruments).
Voltage clamp protocols and data analysis.
Whole cell currents were evoked by stepping membrane voltage for 1 sec between −120 mV and +100 mV in 20 mV increments from a holding potential of 0 mV. Test pulses were followed by a 1 sec interval at 0 mV. Current-to-voltage relationships were constructed from mean current values recorded over the last 25 msec of each test pulse. For pre-depolarization studies, cells were held at holding voltages of −20 mV to 60 mV for 3 sec and then stepped to −120 mV for 3 sec to activate channels. Peak hyperpolarization-induced current amplitude was measured over a 100 msec interval. Pseudo-steady-state current was measured over the last 20 msec of the −120 mV test pulse.
Channel activation voltages were estimated from current-to-voltage relationships. A line was drawn by linear regression analysis of currents measured between 20 mV and 100 mV. A second line was drawn by linear regression analysis of currents measured between the first voltage at which inward current was detected and a second voltage 20 mV more negative. The intercept of these two lines is defined as the activation voltage.
Homology modeling.
A homology model for the CLH-3b ytoplasmic C-terminus was generated based on the crystal structure of the CLC-Ka C-terminus (Protein Data Bank accession code 2PFI). We chose CLC-Ka as a template because biochemical data indicate that this crystal structure represents the native dimeric state of the channel.13 BCL::Align software53 with manual adjustment of the linker region between CBS1 and CBS2 was used to align amino acids 549–615 and 783–841 of CLH-3b with the corresponding amino acids 542–681 of CLC-Ka. Modeller software54 version 9.3 was used to develop 120 candidate structures for CLH-3b. The best-scoring structure by Modeller's molpdf objective function was chosen and side chain orientations optimized from rotamer libraries were determined using SCWRL 3.55 The model was refined with AMBER 9 using 500 steps of energy minimization.56,57
As shown in Figure 1, CBS1 and CBS2 of CLH-3a and CLH-3b have identical sequences except for the last six amino acids of CBS2, which encompasses part of the second α helix (α2). To investigate how splice variation affects α2 helical structure, we used Rosetta software33,34 and the lowest energy structure generated by Modeller. The modeled domain includes amino acids 620–686 and 753–811 for CLH-3a and amino acids 549–615 and 783–841 for CLH-3b. Rosetta was used to predict de novo 20,000 candidate structures of amino acids 803–811 for CLH-3a and 833–841 for CLH-3b. These nine amino acids include the C-terminus of CBS2 and the following three amino acids, which are also alternatively spliced in CLH-3a and CLH-3b (Fig. 1). The remainder of the modeled region surrounding these nine amino acids was included for structural context and held in a fixed conformation.
MTSET inhibition experiments.
[2-(Trimethylammonium) ethyl] methanethiosulfonate bromide (MTSET; Toronto Research Chemicals, Toronto, Ontario, Canada) was dissolved in water as a 400 mM stock and stored in 40 µl aliquots at −80°C until use. Working solutions with an MTSET concentration of 1 mM were made immediately before each experiment. Whole cell current amplitude in MTSET experiments was recorded by stepping membrane voltage to −100 mV for 500 msec every 1 sec from a holding potential of 0 mV.
Statistical analysis.
Data are presented as means ± S.E. Statistical significance was determined using Student's two-tailed test for unpaired means. p values of ≤0.05 were taken to indicate statistical significance.
Acknowledgements
This work was supported by NIH grants R01 GM080403 to Jens Meiler and R01 DK51610 to Kevin Strange. Experiments described in this paper were proposed and designed by Sonya Dave, Jens Meiler and Kevin Strange. Sonya Dave and Jonathan H. Sheehan performed modeling studies. Mutagenesis and electrophysiological experiments were carried out by Sonya Dave. All authors participated in the analysis and interpretation of data, in the writing of the manuscript and in the approval of the final version of the manuscript for publication. We thank Rebekah Karns and Stacey Ells for technical assistance and Dr. Jerod Denton for helpful discussions and for critical reading of the manuscript.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/12445
References
- 1.Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:39–42. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- 3.Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- 4.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 5.Zifarelli G, Pusch M. CLC chloride channels and transporters: a biophysical and physiological perspective. Rev Physiol Biochem Pharmacol. 2007;158:23–76. doi: 10.1007/112_2006_0605. [DOI] [PubMed] [Google Scholar]
- 6.Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 7.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 8.Estevez R, Schroeder BC, Accardi A, Jentsch TJ, Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 2003;38:47–59. doi: 10.1016/s0896-6273(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 9.Accardi A, Pusch M. Conformational changes in the pore of CLC-0. J Gen Physiol. 2003;122:277–294. doi: 10.1085/jgp.200308834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Traverso S, Elia L, Pusch M. Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic inhibitor. J Gen Physiol. 2003;122:295–306. doi: 10.1085/jgp.200308784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engh AM, Maduke M. Cysteine accessibility in ClC-0 supports conservation of the ClC intracellular vestibule. J Gen Physiol. 2005;125:601–617. doi: 10.1085/jgp.200509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estevez R, Pusch M, Ferrer-Costa C, Orozco M, Jentsch TJ. Functional and structural conservation of CBS domains from CLC chloride channels. J Physiol. 2004;557:363–378. doi: 10.1113/jphysiol.2003.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markovic S, Dutzler R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure. 2007;15:715–725. doi: 10.1016/j.str.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 14.Meyer S, Dutzler R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure. 2006;14:299–307. doi: 10.1016/j.str.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Meyer S, Savaresi S, Forster IC, Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat Struct Mol Biol. 2007;14:60–67. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 16.Ignoul S, Eggermont J. CBS domains: structure, function and pathology in human proteins. Am J Physiol Cell Physiol. 2005;289:1369–1378. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- 17.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 18.He L, Denton J, Nehrke K, Strange K. Carboxy terminus splice variation alters ClC channel gating and extracellular cysteine reactivity. Biophys J. 2006;90:3570–3581. doi: 10.1529/biophysj.105.078295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denton J, Nehrke K, Yin X, Beld A, Strange K. Altered gating and regulation of a carboxy-terminus ClC channel mutant expressed in the C. elegans oocyte. Am J Physiol (Cell Physiol) 2006;290:1109–1118. doi: 10.1152/ajpcell.00423.2005. [DOI] [PubMed] [Google Scholar]
- 20.Bennetts B, Rychkov GY, Ng HL, Morton CJ, Stapleton D, Parker MW, et al. Cytoplasmic ATPsensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J Biol Chem. 2005;280:32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- 21.Wu W, Rychkov GY, Hughes BP, Bretag AH. Functional complementation of truncated human skeletal-muscle chloride channel (hClC-1) using carboxyl tail fragments. Biochem J. 2006;395:89–97. doi: 10.1042/BJ20050966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yusef YR, Zúñiga L, Catalán M, Niemeyer MI, Cid LP, Sepúlveda FV. Removal of gating in voltage-dependent ClC-2 chloride channel by point mutations affecting the pore and C-terminus CBS-2 domain. J Physiol. 2006;572:173–181. doi: 10.1113/jphysiol.2005.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennetts B, Parker MW, Cromer BA. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J Biol Chem. 2007;282:32780–32791. doi: 10.1074/jbc.M703259200. [DOI] [PubMed] [Google Scholar]
- 24.Martinez GQ, Maduke M. A cytoplasmic domain mutation in ClC-Kb affects long-distance communication across the membrane. PLoS ONE. 2008;3:2746–2741. doi: 10.1371/journal.pone.0002746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petalcorin MI, Oka T, Koga M, Ogura K, Wada Y, Ohshima Y, et al. Disruption of clh-1, a chloride channel gene, results in a wider body of Caenorhabditis elegans. J Mol Biol. 1999;294:347–355. doi: 10.1006/jmbi.1999.3241. [DOI] [PubMed] [Google Scholar]
- 26.Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- 27.Schriever AM, Friedrich T, Pusch M, Jentsch TJ. CLC chloride channels in Caenorhabditis elegans. J Biol Chem. 1999;274:34238–34244. doi: 10.1074/jbc.274.48.34238. [DOI] [PubMed] [Google Scholar]
- 28.Nehrke K, Begenisich T, Pilato J, Melvin JE. C. elegans ClC-type chloride channels: novel variants and functional expression. Am J Physiol. 2000;279:2052–2066. doi: 10.1152/ajpcell.2000.279.6.C2052. [DOI] [PubMed] [Google Scholar]
- 29.Denton J, Nehrke K, Rutledge E, Morrison R, Strange K. Alternative splicing of N- and C-termini of a C. elegans ClC channel alters gating and sensitivity to external Cl− and H+ J Physiol. 2004;555:97–114. doi: 10.1113/jphysiol.2003.053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:247–251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 32.Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Falquet L. MyHits: a new interactive resource for protein annotation and domain identification. Nucleic Acids Res. 2004;32:332–335. doi: 10.1093/nar/gkh479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 34.Bonneau R, Strauss CE, Rohl CA, Chivian D, Bradley P, Malmstrom L, et al. De novo prediction of threedimensional structures for major protein families. J Mol Biol. 2002;322:65–78. doi: 10.1016/s0022-2836(02)00698-8. [DOI] [PubMed] [Google Scholar]
- 35.Miller MD, Schwarzenbacher R, von Delft F, Abdubek P, Ambing E, Biorac T, et al. Crystal structure of a tandem cystathionine-β-synthase (CBS) domain protein (TM0935) from Thermotoga maritima at 1.87 Å resolution. Proteins. 2004;57:213–217. doi: 10.1002/prot.20024. [DOI] [PubMed] [Google Scholar]
- 36.Zhang R, Evans G, Rotella FJ, Westbrook EM, Beno D, Huberman E, et al. Characteristics and crystal structure of bacterial inosine-5Δ-monophosphate dehydrogenase. Biochem. 1999;38:4691–700. doi: 10.1021/bi982858v. [DOI] [PubMed] [Google Scholar]
- 37.Sasaki R, Ichiyasu H, Ito N, Ikeda T, Takano H, Ikeuchi T, et al. Novel chloride channel gene mutations in two unrelated Japanese families with Becker's autosomal recessive generalized myotonia. Neuromuscul Disord. 1999;9:587–592. doi: 10.1016/s0960-8966(99)00050-4. [DOI] [PubMed] [Google Scholar]
- 38.Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum Mutat. 2002;19:423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- 39.Scott JW, Hawley SA, Green KA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Townley R, Shapiro L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science. 2007;315:1726–1729. doi: 10.1126/science.1137503. [DOI] [PubMed] [Google Scholar]
- 41.Jin X, Townley R, Shapiro L. Structural insight into AMPK regulation: ADP comes into play. Structure. 2007;15:1285–1295. doi: 10.1016/j.str.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 42.Day P, Sharff A, Parra L, et al. Structure of a CBSdomain pair from the regulatory γ1 subunit of human AMPK in complex with AMP and ZMP. Acta Crystallogr D Biol Crystallogr. 2007;63:587–596. doi: 10.1107/S0907444907009110. [DOI] [PubMed] [Google Scholar]
- 43.Hattori M, Tanaka Y, Fukai S, Ishitani R, Nureki O. Crystal structure of the MgtE Mg2+ transporter. Nature. 2007;448:1072–1075. doi: 10.1038/nature06093. [DOI] [PubMed] [Google Scholar]
- 44.Biemans-Oldehinkel E, Mahmood NA, Poolman B. A sensor for intracellular ionic strength. Proc Natl Acad Sci USA. 2006;103:10624–10629. doi: 10.1073/pnas.0603871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahmood NA, Biemans-Oldehinkel E, Poolman B. Engineering of ion sensing by the cystathionine betasynthase module of the ABC transporter OpuA. J Biol Chem. 2009;284:14368–14376. doi: 10.1074/jbc.M901238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams J, Chen ZP, Van Denderen BJ, Morton CJ, Parker MW, Witters LA, et al. Intrasteric control of AMPK via the γ1 subunit AMP allosteric regulatory site. Protein Sci. 2004;13:155–165. doi: 10.1110/ps.03340004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudolph MJ, Amodeo GA, Iram SH, Hong SP, Pirino G, Carlson M, et al. Structure of the Bateman2 domain of yeast Snf4: dimeric association and relevance for AMP binding. Structure. 2007;15:65–74. doi: 10.1016/j.str.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 48.Kemp BE. Bateman domains and adenosine derivatives form a binding contract. J Clin Invest. 2004;113:182–184. doi: 10.1172/JCI20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falin R, Morrison R, Ham A, Strange K. Identification of regulatory phosphorylation sites in a Ste20 kinase regulated cell cycle- and volume-senstive ClC anion channel. J Gen Physiol. 2009;133:29–42. doi: 10.1085/jgp.200810080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denton J, Nehrke K, Yin X, Morrison R, Strange K. GCK-3, a newly identified Ste20 kinase, binds to and regulates the activity of a cell cycle-dependent ClC anion channel. J Gen Physiol. 2005;125:113–125. doi: 10.1085/jgp.200409215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Angeli A, Moran O, Wege S, Filleur S, Ephritikhine G, Thomine S, et al. ATP binding to the C terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J Biol Chem. 2009;284:26526–26532. doi: 10.1074/jbc.M109.005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zifarelli G, Pusch M. Intracellular regulation of human ClC-5 by adenine nucleotides. EMBO Rep. 2009;10:1111–1116. doi: 10.1038/embor.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong E, Smith J, Heinze S, Alexander N, Meiler J. BCL::Align-Sequence alignment and fold recognition with a custom scoring function online. Gene. 2008;422:41–46. doi: 10.1016/j.gene.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 55.Canutescu AA, Shelenkov AA, Dunbrack RL., Jr A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponder JW, Case DA. Force fields for protein simulations. Adv Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]