

Abstract
Disruption of cell cycle checkpoints and interference with the normal cell cycle progression frequently result in cell death or malignant transformation. Hexavalent chromium [Cr(VI)] is a well-known carcinogen that has been implicated in the occurrence of many types of human malignancies, including lung cancer. However, the exact mechanism by which Cr(VI) causes malignant transformation in the lung remains unknown. We have demonstrated that chronic exposure to a noncytotoxic concentration of Cr(VI) induced a variety of chromosomal abnormalities, including premature sister chromatid separation, chromosomal breakage and the presence of lagging/misaligned chromosomes. After treatment with nocodazole, both HeLa and normal lung bronchial epithelial cells were arrested at mitosis. However, Cr(VI) significantly compromised M-phase arrest induced by nocodazole. Cr(VI) suppressed BubR1 activation and reduced expression of Emi1, leading to an unscheduled activation of APC/C. Consistent with this observation, Cr(VI) treatment caused enhanced polyubiquitination of geminin during mitotic release, while it deregulated the activity of Cdt1, a DNA replication licensing factor. Combined, these results suggest that Cr(VI)-induced chromosomal instability is partly due to a perturbation of APC/C activities, leading to chromosomal instability.
Key words: chromium, checkpoint, chromosome instability, APC/C, BubR1, Emi1
Introduction
Given that the integral mission of the cell division cycle is a faithful and error-free completion of genome duplication, a disruption in a cell cycle checkpoint and subsequent perturbation of the normal cell cycle progression can often result in unforeseen and often detrimental cellular consequences, including neoplastic transformations and carcinogenesis. Hexavalent chromium [Cr(VI)] is an established human carcinogen and has been associated with the occurrence of a number of cancers.1–3 Although not the only viable route of exposure, inhalation remains one of the primary exposure means,4 and Cr(VI)-induced tumors as well as elevated levels of the metal are often found at lung bifurcation sites.5 Despite the existence of vast epidemiologic and experimental evidence regarding its adverse effects on human health,6 the exact mechanism by which Cr(VI) causes neoplastic transformation remains largely unclear.
Chromium is capable of inducing DNA damage in the form of double- and single-strand breaks, Cr adducts and oxidized nucleotide bases.1–3,7 These genotoxic events are all very likely to trigger the action of cell cycle checkpoints that monitor cell cycle progression in normal cells.8–10 However, the mutagenic properties of Cr can lead to either inactivation or altered expression of key genes involved in the checkpoint response. Compromised checkpoint responses frequently cause genomic instability during cell division, resulting in neoplastic transformation.11 We have previously reported that arsenic trioxide [As(III)] treatment significantly affects S-phase function and inhibits the activation of the spindle checkpoint.12 Given that Cr(VI) induces cell cycle arrest,10 we asked whether it plays a role in the perturbation of the spindle checkpoint activation, DNA replication and chromosomal stability.
Using a combination of cellular and molecular approaches, we observed that chronic exposure to Cr(VI) at non-cytotoxic concentrations induced a variety of chromosomal abnormalities, including chromosome breaks, premature sister chromatid separation and the presence of lagging and misaligned chromosomes. Mitotic arrest induced by nocodazole was significantly attenuated when the cells were co-treated with Cr(VI), leading to an enrichment of S-phase cells. Moreover, Cr(VI) suppressed expression of Emi1 and activation of the spindle checkpoint by nocodazole, which was correlated an increased APC/C activity. Cr(VI) also perturbed the level and/or activity of geminin and Cdt1, the latter being a licensing factor for DNA replication. Our combined studies strongly suggest that chromosomal instability caused by Cr(VI) exposure is likely due to an unscheduled activation of APC/C as the result of BubR1-dependent spindle checkpoint suppression and Emi1 expression.
Results
In order to study the effect of Cr(VI) exposure on chromosomal stability during cell division, we chronically exposed HeLa cells by culturing them in medium containing a non-cytotoxic concentration (0.25 µM) of the chemical. After exposure to Cr(VI) in culture for one month, HeLa cells were subjected to mitotic chromosome spread analysis. We observed that cells exposed to Cr(VI) contained major chromosomal abnormalities, including premature sister chromatid separation and chromosome breaks (Fig. 1A and B) as compared with the control cells cultured in the absence of Cr(VI). Fluorescence microscopy revealed that acute treatment of cells with Cr(VI) (10 µM) also significantly increased misaligned or lagging chromosomes in mitotic cells (Fig. 1C). These data suggest that Cr(VI) induces major manifestations of chromosome instability.
Figure 1.

Chronic exposure to non-cytotoxic concentrations of Cr(VI) induced chromosomal instabilities. (A and B) HeLa cells were cultured in the medium containing Cr(VI) at a final concentration of 0.25 µM for one month. Cells were then prepared for the analysis of metaphase chromosomes. Major chromosomal abnormalities, including premature sister chromatid separation and chromosome breakage, were examined. Representative images (A) were shown. Insets show magnified images. Percent of cells with premature sister chromatid separation and chromosome breakage were summarized (B). (C) HeLa cells were acutely exposed to Cr(VI) at a final concentration of 10 µM for 24 hours. Cells were then collected and stained with the antibody to phosphorylated H3 serine 10 (p-H3S10, Red). DNA was stained with DAPI (Blue). Representative images are shown.
To understand how Cr(VI) increased chromosomal instability, we first examined whether Cr(VI) would affect mitotic progression. Overnight (16 hours) treatment with 10 µM Cr(VI) did not cause overt morphological differences compared with the vehicle-treated control (Fig. 2A). As expected, the majority of cells treated with nocodazole, a mitotic inducer, arrested at mitosis, characterized by their rounded-up appearance. Intriguingly, co-treatment with Cr(VI) significantly suppressed the rounding-up of HeLa cells induced by nocodazole, suggesting that this metal toxicant may interfere with mitotic arrest.
Figure 2.

Cr(VI) suppresses mitotic arrest induced as the result of microtubule disruption. (A) HeLa cells were treated with nocodazole (50 ng/ml) and/or Cr(VI) (10 µM) for 16 hours. At the end of treatment, cells were examined under a light microscope. Representative cell images are shown. (B) HeLa cells seeded in chamber slides were treated with nocodazole (Noc) and/or Cr(VI) for 16 hours. At the end of treatment, cells were fixed and stained with an antibody to phosphorylated serine 10 of histone H3 (p-H3S10). DNA was stained with DAPI. Representative images are shown. (C) HeLa cells were treated with Cr(VI) (10 µM) and/or nocodazole for 24 hours. Cell viability was evaluated by the MTT assay. The data were summarized from three independent experiments.
To confirm that Cr(VI) suppressed mitotic arrest, we treated HeLa cells with Cr(VI) in the presence or absence of nocodazole. Cells with various treatments were fixed and stained with an antibody to phosphorylated serine 10 of histone H3 (p-H3S10), a known mitotic marker.13 Whereas a low percentage of p-H3S10-positive cells was present in the vehicle group or the Cr(VI)-treated control group, the vast majority of HeLa cells were positive for p-H3S10 when they were treated with nocodazole (Fig. 2B). However, co-treatment with Cr(VI) significantly reduced the fraction of p-H3S10-positive cells induced by nocodazole (Fig. 2B). To eliminate the possibility that Cr(VI) might preferentially induce cells to undergo apoptosis or mitotic catastrophe, we determined the viability of cells treated with nocodazole and/or Cr(VI). MTT assays revealed that cells treated with Cr(VI) and nocodazole did not exhibit any noticeable decrease in the cell viability (Fig. 2C), suggesting that Cr(VI) does not promote mitotic cell death. Combined, our studies strongly suggest that Cr(VI) is capable of compromising mitotic arrest.
We next measured the cell cycle distribution of HeLa cells with various treatments by flow cytometry. Cr(VI) treatment alone significantly enriched S-phase cell population (Fig. 3A), which is consistent with the notion that this metal toxicant can form DNA adducts and cause DNA strand breaks, thus potentially leading to the activation of S-phase checkpoint. As expected, nocodazole treatment alone arrested the majority of cells at the G2/M phase, based on the DNA content analysis. However, nocodazole-induced enrichment of the G2/M population was significantly suppressed when the cells were co-treated with Cr(VI). In the meantime, the S-phase cell population remained high when the cells were co-treated with nocodazole and Cr(VI). These observations thus suggest that Cr(VI) may attenuate the effect of nocodazole-induced mitotic arrest by either blocking cells at the S phase or promoting mitotic exit.
Figure 3.
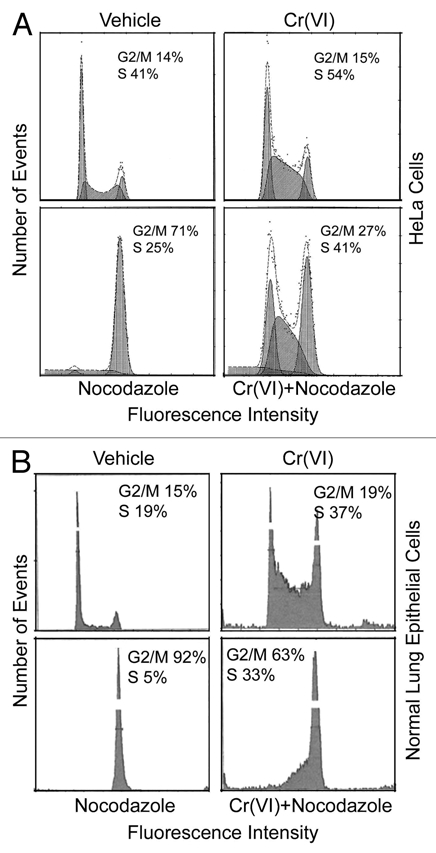
Suppression of mitotic arrest by Cr(VI) is correlated with an increase in S-phase population. (A) HeLa cells treated with Cr(VI) (10 µM) and/or nocodazole (50 ng/ml) for 16 hours were then collected, fixed and processed for cell cycle distributions by flow cytometry. Representative data are shown. (B) Normal human lung epithelial cells were treated with Cr(VI) and/or nocodazole for 48 h. The treated cells were fixed and processed for analysis for their DNA content by flow cytometry.
To study whether Cr(VI) would have a similar effect on perturbing cell cycle distributions in normal cells, we treated human primary lung epithelial cells with nocodazole in the presence or absence of Cr(VI) for 48 h. Compared with that of vehicle-treated control, Cr(VI) and nocodazole treatment significantly enriched S-phase and G2/M cells, respectively (Fig. 3B). Again, Cr(VI) treatment significantly compromised the G2/M arrest induced by nocodazole, resulting in a reduction in G2/M cells with a concomitant increase in the S-phase cell population. Combined, these studies suggest that Cr(VI) suppresses cell cycle progression of both normal lung epithelial cells and transformed cells and compromises their mitotic arrest, likely by blocking them in the S phase.
Cr(VI) induces DNA damage in the form of strand breaks,3,14 which should provoke the activation of the S-phase checkpoint. The observation that Cr(VI) enriches S-phase population is consistent with an activated DNA damage checkpoint. We next determined if co-treatment of HeLa cells with Cr(VI) and caffeine would have an effect on modulating cell cycle distributions, because caffeine is capable of weakening the DNA damage checkpoint by inhibiting activities of ATM/ATR.15,16 Caffeine treatment alone did not have any noticeable effects on perturbing cell cycle distribution, whereas Cr(VI) greatly enriched S-phase cells (Figs. 3A and 4A). On the other hand, caffeine significantly reduced the percentage of S-phase cells induced by Cr(VI) with a concomitant increase in the G2/M population (Figs. 3A and 4A). Moreover, the addition of nocodazole further increased the mitotic fraction when HeLa cells were also co-treated with Cr(VI) and caffeine (Fig. 4A).
Figure 4.
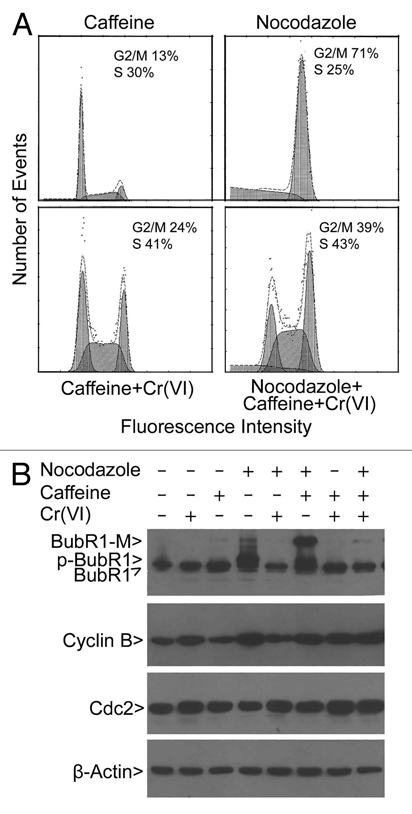
Suppression of nocodazole-induced mitotic arrest and BubR1 activation by Cr(VI) is attenuated by caffeine. (A) HeLa cells were treated with Cr(VI) (10 µM), caffeine (3 mM) and/or nocodazole (50 ng/ml) for 16 hours. Cells were then collected, fixed and processed for cell cycle distributions by flow cytometry. (B) HeLa cells were treated with Cr(VI), caffeine and/or nocodazole for 16 hours. Equal amounts of the lysates from different treatments were blotted for BubR1, cyclin B1, Cdc2 and β-actin. Arrow p-BubR1 denotes the activated, phosphorylated form. Arrow modified denotes a BubR1 band (BubR1-M), the nature of which remains unclear.
We next examined the status of the spindle checkpoint in HeLa cells treated with nocodazole, Cr(VI) and/or caffeine. BubR1 is a key component of the spindle checkpoint, and compromised BubR1 activity causes chromosomal instability and malignant transformation.17,18 BubR1 is known to be phosphorylated (p-BubR1) after treatment with nocodazole, and its phosphorylation is a reliable indicator of checkpoint activation.19 Neither Cr(VI) nor caffeine induced activation of BubR1 (p-BubR1); however, Cr(VI) induced a low level of accumulation of cyclin B1 (Fig. 4B). As expected, nocodazole treatment resulted in a marked increase of p-BubR1 as well as cyclin B1 (Fig. 4B). However, Cr(VI) co-treatment, but not caffeine co-treatment, significantly decreased p-BubR1 levels. Consistent with the deregulation of mitotic arrest by Cr(VI), cyclin B1 levels induced by nocodazole were also significantly reduced in cells co-treated with the metal (Fig. 4B), suggesting the activation of APC/C. Intriguingly, co-treatment with nocodazole and caffeine, but not chemicals of other combinations, strongly induced a modified BubR1 (BubR1-M), the nature of which remained unknown. As a control, we also examined expression of Cdc2. No changes in the level of Cdc2 were observed in cells treated with nocodazole, caffeine and/or Cr(VI).
Compromised BubR1 activity results in the premature activation of APC/C,17 which negatively regulates, by polyubiquitination, several substrates, including geminin, securin and cyclin B. Because Emi1 also suppresses APC/C activity, and its expression is modulated during the cell cycle,20 we determined whether Cr(VI) affected its steady-state levels in the cells. Mitotic shake-off cells (collected after treatment with nocodazole for 16 h) were released into medium with or without Cr(VI) for various times. Immunoblotting reveals that Emil levels were greatly reduced by Cr(VI) (Fig. 5). Because reduced levels of Emi1 as well as suppressed BubR1 activation would predict an increase in APC/C activity, we determined the ubiquitination status of geminin, an APC/C substrate. Consistent with the prediction, geminin polyubiquitination was enhanced in cells treated with Cr(VI) compared with the control cells (Fig. 5).
Figure 5.
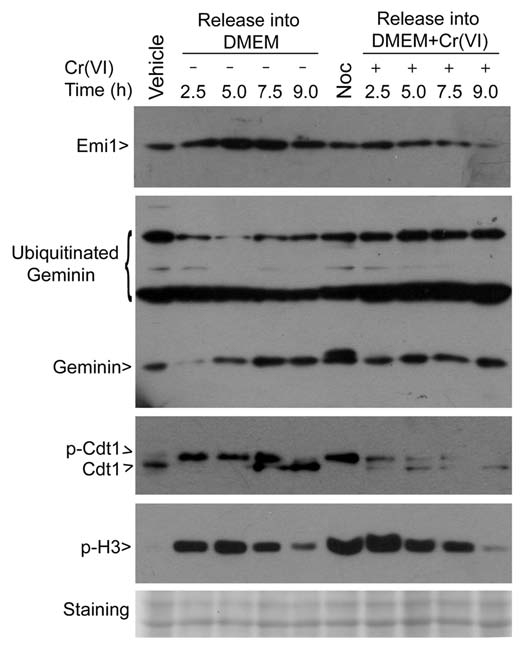
Cr(VI) reduces expression of Emi1 and perturbs regulation of geminin and Cdt1. HeLa cells were treated with nocodazole (50 ng/ml) for 16 hours. Mitotic cells collected by shake-off were released into fresh medium supplemented with or without Cr(VI) (10 µM) for various times. Cell lysates were prepared at the end of treatment. Interphase (vehicle) and mitotic (Noc) cell lysates were also prepared. Equal amounts of cell lysates were blotted for emi1, geminin, Cdt1 and phorsphorylated histone H3. Ubiquitinated geminin and phosphorylated Cdt1 were also indicated. The blot was stained as a loading control.
Cdt1 is a critical component of the pre-replicative complex and licenses DNA for replication.20 Geminin directly interacts with and inhibits Cdt1 activity, thereby blocking re-firing of DNA replication origin.20,21 It is conceivable that APC/C plays an important role in regulating DNA replication through control of activities as well as levels of geminin and Cdt1. Immunoblotting revealed that Cdt1 was heavily phosphorylated in mitotic shake-off cells, whereas it remained largely unphosphorylated in asynchronous cells (Fig. 5, vehicle lane), which is consistent with the report that phosphorylation by Cdks is a mechanism that negatively regulates Cdt1.22 During mitotic release, Cdt1 was converted to the active (dephosphorylated) form much earlier (e.g., 2.5 and 5 h time points) in cells treated with Cr(VI) than the untreated control cells (Fig. 5). By 9 h post release, Cdt1 was completely converted to its dephosphorylated form, either with or without Cr(VI), and these cells were apparently no longer mitotic, as very little p-H3S10 signal was left (Fig. 5). Interestingly, Cr(VI) treatment also caused significant reduction of Cdt1 during mitotic release.
Discussion
APC/CCdc20 is inhibited by the spindle checkpoint of which Bub1 and BubR1 are major checkpoint components.23–25 Haploinsufficiency of BubR1 results in the activation of APC/CCdc20 and the reduction of securing,17 a major substrate of the ubiquitin E3 ligase complex. APC/CCdh1 activity is primarily inhibited by Emi1 during S, G2 and early mitosis, so that its key substrates, including cyclin B and geminin, can be stabilized. Premature activation of APC/CCdh1 by depletion of Emi1 during S, G2 and early mitosis has a positive effect on DNA re-replication due to an increased activity of Cdt1, a major DNA replication licensing factor, following polyubiquitination and subsequent degradation of its inhibitor geminin.20 Based on our data as well as those reported in the literature, we propose the following model to explain the mode of action of Cr(VI) in the induction of chromosomal instability during cell division (Fig. 6). Cr(VI) exposure suppresses expression and activation of Emi1 and BubR1, respectively, which lead to unscheduled activation of APC/C activities. The enhanced APC/CCdc20 activity causes polyubiquitination and degradation of securin, leading to premature sister chromatid separation. APC/CCdh1 activity also polyubiquitinates geminin, which inevitably perturbs phosphorylation and activation status of Cdt1. Aberrant DNA re-replication, including repeated firing of the origin of DNA replication, frequently results in DNA replication fork collision and double-strand breaks. Cells, if they survive, are frequently aneuploid.
Figure 6.

A model depicting the proposed mode of action of Cr(VI) in the induction of chromosomal instability during cell division. Arrows (→) denote positive regulation and blocks (┤) indicate negative regulation. APC/C denotes anaphase-promoting complex/cyclosome; PSS denotes premature sister chromatid separation.
It is of interest to observe that Cr(VI)-induced DNA damage checkpoint can significantly countermand the activation of the spindle checkpoint. The co-treatment with caffeine and Cr(VI) significantly reduced the percentage of S-phase cells, with a concomitant increase in mitotic cells in the presence of nocodazole, thus suggesting that the suppression of the DNA damage checkpoint by a cell cycle inhibitor, such as caffeine, can lead to the release of previously arrested cells into mitosis. Moreover, nocodazole is a potent inducer of the spindle checkpoint characterized by activating/phosphoyrlating BubR1.19 Cr(VI) treatment completely blocks phosphorylation of BubR1 in cells exposed to nocodazole (Fig. 4B), indicating its ability to override the spindle checkpoint activation by the microtubule disrupter. Given that human exposure to Cr(VI) is not only limited to the occupational setting (i.e., environmental contamination), it is essential to elucidate the mechanisms by which Cr(VI) affects the spindle checkpoint activation and diminishes mitotic arrest induced by microtubule poisons.
Cr(VI) is known to have a plethora of effects on cells, including activation of the mismatch repair system26,27 and induction of oxidative stress.28,29 Cr(VI)-induced oxidative stress appears to perturb the activities of transcription factors, including NFκB, AP-1, p53 and HIF-1.29 However, the molecular basis by which Cr(VI) causes deregulation of cell cycle checkpoints and chromosomal instability remains poorly understood. Given that chromosomal instability is believed to be a major driving force of malignant transformation and tumor development,30,31 additional studies on the regulation of the APC/C substrate geminin should shed light on chromium carcinogenesis. It is of importance to note that chronic exposure to a non-cytotoxic concentration of Cr(VI) can induce a variety of chromosomal abnormalities. The presence of premature sister chromatid separation and chromosome breaks will compromise cell division and survival of these cells. One frequent outcome is the formation of bi-nucleated/polyploid cells. It is generally believed that aneuploid cells are derived from unstable tetraploid cells. Recent studies strongly suggest that there is a synergistic interaction between chromosome instability and p53 inactivation during malignant transformation.30,32 Whereas p53 is required for the maintenance of chromosomal stability during tetraploidization,32 chromosome missegregation due to checkpoint deficiency favors the selection of cells with mutated/null p53 allele during colonic tumorigenesis.30 It is conceivable that p53 mutations or loss of its activity is likely to accelerate the development of aneuploid cells, which, in turn, facilitates the selection of rare clones with growth advantages, leading to neoplastic transformation.
Materials and Methods
Cell culture and treatment.
HeLa cells were obtained from the American Type Culture Collection (Manassas, VA). HeLa cells were cultured in dishes or on Lab-Tek II chamber slides (Fisher Scientific, Pittsburgh, PA) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) with 5% CO2 at 37°C. Na2CrO4 (Sigma Chemical Co., St. Louis, MO) was dissolved in 0.9% (w/v) NaCl at the concentration of 10 mM as a stock solution. Nocodazole (Sigma Chemical Co.) was dissolved in dimethyl sulphoxide (DMSO). Maximal final concentration of DMSO used in cell culture was kept below 0.1%. Caffeine (Sigma Chemical Co.) was prepared in DMEM without serum at a concentration of 80 mM. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT,) was dissolved in PBS at a concentration of 5 mg/ml. For chronic exposure, HeLa cells were cultured in medium containing Cr(VI) at a final concentration of 0.25 µM for one month. For acute exposure, HeLa cells were treated with different chemicals, including Cr(VI), nocodazole, caffeine or their combinations for 16 hours. Human lung bronchial epithelial cells (NHBE) obtained from ATCC were maintained in bronchial epithelial growth media (Lonza, Walkersville, MD) in tissue culture flasks/dishes in an ambient condition as described above. NHBE cells of either the third or the fourth passage were treated with Cr(VI) at a concentration of 10 µM and/or nocodazole (or vehicle) for 2 days. At the end of the treatment, cells were collected and processed for DNA content analysis by flow cytometry.
Cytogenetics.
To obtain mitotic chromosome spreads, HeLa cells cultured in the presence or absence of Cr(VI) (0.25 µM) for one month were treated with nocodazole (100 ng/ml) for 4 hours. Cells were collected and incubated in 3.7 mM KCl at 37°C for 15 min. Cells were finally suspended in a fixative solution (glacial acetic acid/methanol in a ratio of 1:3) prior to spreading onto microscope slides (Fisher Scientific). Chromosome spreads were counter-stained with DAPI. Chromosomal images were captured with a Leica TCS SP5 confocal microscope or a Leica AF6000 fluorescence microscope. For each treatment, at least 100 metaphase spreads were examined.
Fluorescence microscopy.
Fluorescence microscopy was essentially performed as described in reference 33 and 34. Following the respective chemical treatments, cells were fixed at room temperature in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min. After fixation, they were permeabilized with 0.1% Triton for 10 min., followed blocking with 2% bovine serum albumin (BSA) at room temperature. The cells were then incubated with anti-human phosphorylated serine 10 of histone H3 (p-H3S10) monoclonal antibody (PharMingen, BD Bioscience, San Jose, CA) in PBS containing 2% BSA overnight at 4°C. Afterwards, they were washed and stained with Alexa Fluor 555-labeled secondary antibody (Invitrogen) at room temperature for 1 h in the dark; cells were finally stained with 4′,6-diamidino-2-phenylindole (1 µg/ml, Fluka, St. Louis, MO). Images were captured with a Leica TCS SP5 confocal microscope or a Leica AF6000 fluorescnce microscope.
Flow cytometry analysis.
Cells were initially fixed in 75% ethanol, then suspended in a solution of PBS containing 100 µg/ml of RNase A (Sigma Chemical Co., St. Louis, MO) and 10 µg/ml of propidium iodide (Molecular Probes, Eugene, OR) and kept at room temperature for 1 h. Cellular fluorescence was then measured using Beckman Coulter® Epics XL-MCL™ Flow Cytometer (Fullerton, CA). DNA frequency distribution histograms were deconvoluted using Muticycle software (Phoenix Flow System, San Diego, CA) to estimate percent of cells in different phases of the cell cycle.
Protein gel blot analysis.
Cells were harvested and lysed in a lysis buffer as previously described in reference 35. Cell lysates were then centrifuged at 12,000 g for 15 min at 4°C, and their supernatants were collected. Approximately equal amounts of protein were subjected to a sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by an electrotransfer to PVDF membranes. Protein blots were probed with antibodies to BubR1, cyclin B, Cdc2, Emi1, Cdt1, Geminin, phosphorylated histone H3 and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA). Specific signals were detected using horseradish peroxidaseconjugated goat-anti-rabbit (or anti-mouse) secondary antibodies (Sigma) and enhanced chemiluminescence reagents (Amersham Pharmacia Biotech, Pittsburgh, PA).
Cell viability assay.
Cell viability was assayed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay method. HeLa cells (1 × 104 cells/well) were seeded in a 96-well plate in sextuplicates. Cr(VI) was added to the cell cultures at indicated concentrations for 24 h. MTT (15 µl) was supplied to each well. Following an additional 3 h incubation at 37°C, the medium was removed. MTT formazan precipitates were dissolved in 150 µl of dimethyl formamide solution. The optical density of the dissolved samples was measured at 570 nm using a plate reader.
Statistical analysis.
The Student's t-test was used to evaluate the significance of difference between two groups. A value of p < 0.05 was considered to be statistically significant.
Acknowledgments
This study was supported in part by US Public Service Awards to W.D. (CA090658). We thank Dr. Frank Traganos at New York Medical College for his assistance in flow cytometry and coworkers for various assistances and helpful discussions. This work is also supported in part by HIEHS center fund and Superfund grants (ES000260 and ES010344).
References
- 1.Singh J, Carlisle DL, Pritchard DE, Patierno SR. Chromium-induced genotoxicity and apoptosis: Relationship to chromium carcinogenesis (review) Oncol Rep. 1998;5:1307–1318. doi: 10.3892/or.5.6.1307. [DOI] [PubMed] [Google Scholar]
- 2.Beyersmann D, Hartwig A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch Toxicol. 2008;82:493–512. doi: 10.1007/s00204-008-0313-y. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 2003;533:3–36. doi: 10.1016/j.mrfmmm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Friess SL. Carcinogenic risk assessment criteria associated with inhalation of airborne particulates containing chromium (VI/III) Sci Total Environ. 1989;86:109–112. doi: 10.1016/0048-9697(89)90198-8. [DOI] [PubMed] [Google Scholar]
- 5.Ishikawa Y, Nakagawa K, Satoh Y, Kitagawa T, Sugano H, Hirano T, et al. “Hot spots” of chromium accumulation at bifurcations of chromate workers' bronchi. Cancer Res. 1994;54:2342–2346. [PubMed] [Google Scholar]
- 6.Chiu A, Katz AJ, Beaubier J, Chiu N, Shi X. Genetic and cellular mechanisms in chromium and nickel carcinogenesis considering epidemiologic findings. Mol Cell Biochem. 2004;255:181–194. doi: 10.1023/b:mcbi.0000007274.25052.82. [DOI] [PubMed] [Google Scholar]
- 7.Zecevic A, Menard H, Gurel V, Hagan E, DeCaro R, Zhitkovich A. WRN helicase promotes repair of DNA double-strand breaks caused by aberrant mismatch repair of chromium-DNA adducts. Cell Cycle. 2009;8:2769–2778. doi: 10.4161/cc.8.17.9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- 9.Vilcheck SK, Ceryak S, O'Brien TJ, Patierno SR. FANCD2 monoubiquitination and activation by hexavalent chromium [Cr(VI)] exposure: activation is not required for repair of Cr(VI)-induced DSBs. Mutat Res. 2006;610:21–30. doi: 10.1016/j.mrgentox.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wakeman TP, Xu B. ATR regulates hexavalent chromium-induced S-phase checkpoint through phosphorylation of SMC1. Mutat Res. 2006;610:14–20. doi: 10.1016/j.mrgentox.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lal MA, Bae D, Camilli TC, Patierno SR, Ceryak S. AKT1 mediates bypass of the G1/S checkpoint after genotoxic stress in normal human cells. Cell Cycle. 2009;8:1589–1602. doi: 10.4161/cc.8.10.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan Q, Komissarova E, Dai W. Arsenic trioxide suppresses paclitaxel-induced mitotic arrest. Cell Prolif. 2009;42:404–411. doi: 10.1111/j.1365-2184.2009.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan Q, Chen H, Costa M, Dai W. Phosphorylation of H3S10 blocks the access of H3K9 by specific antibodies and histone methyltransferase. Implication in regulating chromatin dynamics and epigenetic inheritance during mitosis. J Biol Chem. 2008;283:33585–33590. doi: 10.1074/jbc.M803312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reynolds M, Stoddard L, Bespalov I, Zhitkovich A. Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res. 2007;35:465–476. doi: 10.1093/nar/gkl1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Y, Black CP, Cowan KH. Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene. 2007;26:4689–4698. doi: 10.1038/sj.onc.1210268. [DOI] [PubMed] [Google Scholar]
- 16.Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst) 2009;8:1009–1017. doi: 10.1016/j.dnarep.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, et al. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 2004;64:440–445. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- 18.Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, et al. Colonic tumorigenesis in BubR1+/− ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci USA. 2005;102:4365–4370. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li W, Lan Z, Wu H, Wu S, Meadows J, Chen J, et al. BUBR1 phosphorylation is regulated during mitotic checkpoint activation. Cell Growth Differ. 1999;10:769–775. [PubMed] [Google Scholar]
- 20.Sivaprasad U, Machida YJ, Dutta A. APC/C—the master controller of origin licensing? Cell Div. 2007;2:8. doi: 10.1186/1747-1028-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786:49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Liu E, Li X, Yan F, Zhao Q, Wu X. Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J Biol Chem. 2004;279:17283–17288. doi: 10.1074/jbc.C300549200. [DOI] [PubMed] [Google Scholar]
- 23.Pesin JA, Orr-Weaver TL. Regulation of APC/C activators in mitosis and meiosis. Annu Rev Cell Dev Biol. 2008;24:475–499. doi: 10.1146/annurev.cellbio.041408.115949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell. 2007;27:3–16. doi: 10.1016/j.molcel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Yu H, Tang Z. Bub1 multitasking in mitosis. Cell Cycle. 2005;4:262–265. [PubMed] [Google Scholar]
- 26.Zhitkovich A, Peterson-Roth E, Reynolds M. Killing of chromium-damaged cells by mismatch repair and its relevance to carcinogenesis. Cell Cycle. 2005;4:1050–1052. [PubMed] [Google Scholar]
- 27.Peterson-Roth E, Reynolds M, Quievryn G, Zhitkovich A. Mismatch repair proteins are activators of toxic responses to chromium-DNA damage. Mol Cell Biol. 2005;25:3596–3607. doi: 10.1128/MCB.25.9.3596-3607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Hudson LG, Liu KJ. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic Biol Med. 2004;37:582–593. doi: 10.1016/j.freeradbiomed.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Yao H, Guo L, Jiang BH, Luo J, Shi X. Oxidative stress and chromium(VI) carcinogenesis. J Environ Pathol Toxicol Oncol. 2008;27:77–88. doi: 10.1615/jenvironpatholtoxicoloncol.v27.i2.10. [DOI] [PubMed] [Google Scholar]
- 30.Baker DJ, van Deursen JM. Chromosome missegregation causes colon cancer by APC loss of heterozygosity. Cell Cycle. 2010;9:1711–1716. doi: 10.4161/cc.9.9.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClelland SE, Burrell RA, Swanton C. Chromosomal instability: A composite phenotype that influences sensitivity to chemotherapy. Cell Cycle. 2009;8:3262–3266. doi: 10.4161/cc.8.20.9690. [DOI] [PubMed] [Google Scholar]
- 32.Ho CC, Hau PM, Marxer M, Poon RY. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget. 2010;1:583–595. doi: 10.18632/oncotarget.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y, Yao Y, Xu HZ, Wang ZG, Lu L, Dai W. Defects in chromosome congression and mitotic progression in KIF18A-deficient cells are partly mediated through impaired functions of CENP-E. Cell Cycle. 2009;8:2643–2649. doi: 10.4161/cc.8.16.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Yang Y, Dai W. Differential subcellular localizations of two human Sgo1 isoforms: Implications in regulation of sister chromatid cohesion and microtubule dynamics. Cell Cycle. 2006;5:635–640. [PubMed] [Google Scholar]
- 35.Ouyang B, Pan H, Lu L, Li J, Stambrook P, Li B, et al. Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J Biol Chem. 1997;272:28646–28651. doi: 10.1074/jbc.272.45.28646. [DOI] [PubMed] [Google Scholar]