

Abstract
A familial form of lupus, termed exfoliative cutaneous lupus erythematosus (ECLE) has been recognized for decades in German shorthaired pointer dogs (GSP). Previous studies were suggestive of autosomal recessive inheritance. The disease presents as a severe dermatitis with age of onset between 16 and 40 weeks, and mirrors cutaneous lupus erythematosus (CLE) in humans. Lameness and, in advanced cases, renal disease may be present. Most affected dogs are euthanized before reaching the age of 4 years. The diagnosis is made by clinical observations and microscopic examination of skin biopsies. In humans, many different forms of CLE exist and various genes and chromosomal locations have been implicated. The large number of potential candidate loci combined with often weak association prevented in depth screening of the dog population thus far. During the course of our studies, we developed a colony of dogs with ECLE as a model for human CLE and the genetic analysis of these dogs confirmed the autosomal recessive mode of inheritance of CLE in GSPs. Using canine patient material, we performed a genome-wide association study (GWAS) to identify the genomic region harboring the gene involved in the development of the disease in GSPs. We identified a SNP allele on canine chromosome 18 that segregated with the disease in the 267 dogs tested. The data generated should allow identification of the mutant gene responsible for this form of cutaneous lupus erythematosus in dogs and assist in the understanding of the development of similar disease in humans.
Keywords: Exfoliative cutaneous lupus erythematosus (ECLE), Genome-wide association study (GWAS), Canine, Animal model
Introduction
Cutaneous lupus erythematosus (CLE) has been described in dogs as a localized skin disease specific to a few breeds but without a clear mode of inheritance (Gross et al. 2005). A hereditary form, termed exfoliative CLE (ECLE) had long been suspected in German shorthaired pointers (GSPs) with all the microscopic features of a cutaneous lupus disorder (Gross et al. 1992; Theaker and Rest 1992; Vroom et al. 1995; Bryden et al. 2005; Mauldin et al. 2010). Dogs typically develop clinical signs before 10 months of age, but age of onset has been reported as late as 2.75 years (White and Gross 1995; Scott and Miller 2001). No sex predilection has been reported (Scott and Miller 2001). Clinical signs consist of excessive scaling and crusting that first occur on the face, ears and back and then progress to a generalized form. Skin lesions consist of scales, crusts, papules, pustules, erythema and/or alopecia (Gross et al. 1992; Vroom et al. 1995; Scott and Miller 2001; Mauldin et al. 2010).
Diagnosis of ECLE is confirmed by histopathologic examination of a skin biopsy sample, in which histological features typical of cutaneous lupus are seen. The main change is a lymphocytic to cell-poor interface dermatitis with vacuoles in the basement membrane area. The stratum corneum is expanded by severe orthokeratotic hyperkeratosis (Gross et al. 1992; Vroom et al. 1995; Scott and Miller 2001; Mauldin et al. 2010). Sebaceous glands may be normal, small, or absent (Vroom et al. 1995). Histopathology of peripheral lymph nodes generally reveals a reactive lymphoid hyperplasia (Scott and Miller 2001; Mauldin et al. 2010). Similar microscopic findings are seen in dogs with systemic lupus erythematosus (SLE), discoid lupus erythematosus (DLE), and erythema multiforme, but these diseases typically show a later onset in life (Gross et al. 1992). In humans, discoid CLE has very similar histological findings as in GSPs with ECLE (Kuhn et al. 2007; Mauldin et al. 2010). About 10% of humans with CLE advance to systemic lupus (Patel and Werth 2002; Costner and Sontheimer 2003; Werth 2007). We have also found that affected dogs kept for several years develop mild to moderate lupus nephritis. As the skin disease progresses to a severity that necessitates humane euthanasia (Mauldin et al. 2010), the degree of renal disease that would develop over time is not known. As in humans with CLE, therapy for GSPs is based on empirical therapies with a variety of immunomodulatory drugs (Mauldin et al. 2010). However, there is no cure to date.
Studying human patients with CLE led to the identification of several genes and chromosomal regions contributing to the disease (Baechler et al. 2003; Remmers et al. 2007; Wenzel and Tuting 2007; Werth 2007). All of these are involved in immune processes that lead to inflammation and cell death, including those coding for components of the early complement cascade, type I interferons, toll-like receptors, STAT4, and MHC class II proteins (Baechler et al. 2003; Remmers et al. 2007; Wenzel and Tuting 2007; Werth 2007). While these studies provide potential genomic candidate regions, the clinical and histological features are not typical of any particular syndrome in which the genetic etiology has been elucidated in other species.
Genome-wide association studies (GWAS) have proven increasingly successful in identifying genomic regions associated with individual traits in both humans and dogs (Wiik et al. 2008; Salmon Hillbertz et al. 2007; Karlsson et al. 2007). They are particularly effective for diseases with an autosomal recessive mode of inheritance in combination with autozygosity mapping (Carr et al. 2009). In addition, inbred populations such as individual dog breeds require much smaller numbers of individuals than humans to obtain useful data, as linkage disequilibrium extends over larger distances than in humans (Sutter et al. 2004; Lindblad-Toh et al. 2005). This in turn reduces the number of single nucleotide polymorphisms (SNP) markers needed to cover the entire canine genome to between 5,000 and 30,000 versus the 200,000 to 500,000 needed in humans (Sutter et al. 2004; Lindblad-Toh et al. 2005). Thus, an initial assessment of the genome using a GWAS can significantly expedite the identification of a molecular change underlying ECLE in the GSP, by targeting a candidate gene approach to a specific chromosomal region.
The present study focuses on the genetic characteristics of ECLE and applies a GWAS to discover a genomic region linked to ECLE in the GSP. A marker or mutation-based test would assist in develop breeding programs that would eventually eliminate the disease from the GSP breed altogether while retaining valuable genetic lines. More so, identifying the molecular cause of ECLE in GSP could potentially provide an animal model of CLE in humans and further study may provide improved therapies in humans and dogs for these conditions.
Materials and methods
Animals and pedigree analysis
Samples from GSPs used in this study were recruited from 21 of the 50 states in the USA through veterinarians, breeders, and dog owners (Table 1). A clinical information questionnaire, pedigree, and written informed consent form were also obtained for each participant. The affected status of a dog was verified by review of a skin biopsy read by one of the authors, a board certified dermatopathologist (EAM). Pedigree information was entered into a database (PedDraw software; version 6.0, 2005 www.sfbr.org/pub/pedmgt) together with sex, numerical coded identification, and phenotype. After the pedigrees were drawn, they were scrutinized by eye. Segregation analysis was performed assuming incomplete ascertainment. For comparison, breeding studies were performed to verify the mode of inheritance using a normal unrelated beagle mix (Table 1) for the initial outcross. Samples from the first 13 dogs with ECLE seen at the University of Pennsylvania were used for the GWAS. Samples from the other affected dogs that were obtained later or through the breeding studies were used to evaluate the validity of the marker assay. All dogs were cared for according to the principles outlined in the National institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and in the International Guiding Principles for Biomedical Research Involving Animals.
Table 1.
Summary of dogs used in breeding studies, GWAS, and genotyping.
| Breed | German shorthaired pointers | Research mixed breeds | |||||
|---|---|---|---|---|---|---|---|
| Phenotype | Male | Female | Unknown | Total | Male | Female | Total |
| Normal | 97 | 102 | 16 | 215 | 14 | 11 | 25 |
| Affecteda | 11 | 9 | 0 | 20 | 4 | 3 | 7 |
| Total | 108 | 111 | 16 | 235 | 18 | 14 | 32 |
A total of 267 samples were evaluated
Confirmed by histopathology
DNA extraction, cDNA synthesis, and RT-PCR
Genomic DNA was extracted from EDTA blood, cheek swabs, or tissues from phenotypically normal and affected dogs using Ready PCR DNA Purification System (5 PRIME Co., Gaithersburg, MD) following the manufacturer’s instructions. DNA quantification was determined by Quant-iTrm dsDNA BR Assay kit together with a Qubit Fluorometer (Invitrogen, Carlsbad, CA). Purified DNA was stored at −80°C for further study.
Total RNA was extracted from skin and kidney from normal and affected dogs using TRIzol reagent® (Life Technologies, Grand Island, NY) according to the manufacturer’s protocol. cDNA was synthesized in a 50-μl reaction containing 10 μg total RNA 1× first-strand synthesis buffer (Invitrogen, Carlsbad, CA), 0.1 mM DTT (Invitrogen), 8 mM dNTPs (Promega, Madison, WI), 0.2 μg/μl BSA (New England Biolabs, Beverly, MA), 8 μM random hexamers (Promega), 8 μM oligod(T) (Promega), 80 U RNAsin (Promega), and 500 U Superscript II® reverse transcriptase (Invitrogen). DNA amplification was carried out according to the protocols described below, using 1 μl of cDNA as the template for each reaction.
Genome-wide association study
A genome-wide association study was used to locate the chromosomal location linked with the disease. Purified genomic DNA samples (2 μg) from 13 clinically confirmed ECLE dogs (seven males, six females) and 21 unaffected, clinically normal GSPs (eight males, 13 females) were sent to the Molecular Diagnosis and Genotyping Facility of the Hospital of the University of Pennsylvania for GWAS. The assay was conducted by using a canine Affymetrix 127 k SNP v2 full set chip (Broad Institute Scientists and Administrators, Cambridge, MA) following the manufacturer’s recommendations. The resulting signals were recorded by the Illumina Bead Array Reader, and the data transferred to the Illumina Bead Studio 3.2 software, which converted the fluorescence intensities into SNP genotypes based on the Bayesian robust linear modeling using Mahalanobis distance algorithm. After confirming high quality of data, loci with more than 10% missing scores, minor allele frequency less than 10%, and such loci that deviate from a relaxed Hardy-Weinberg equilibrium were excluded from further analysis. Data was analyzed using freely available software (PLINK; Purcell et al. 2007) for association studies by Chi-square test with and without Bonferroni correction. In addition, autozygosity mapping was performed based on manual calculation.
Sequencing
To sequence candidate genes, DNA was amplified by standard PCR and submitted to the University of Pennsylvania’s Sequencing Facility. Briefly, the PCR mixture contained 100 ng (2 μl) of canine genomic DNA as template, 5.0 μl of 10X PCR buffer, 1.5 μl of 50 mM MgCl2, 1.0 μl of 10 mM dNTPs (2.5 mM each), 1.0 μl of each 20 μM forward and reverse primers, 0.5 μl of platinum Taq polymerase (5 U/μl), and 38 μl of sterile de-ionized water (dIH2O; all reagents were from Invitrogen, Carlsbad, CA, except for the primers which were designed by the laboratory and synthesized by IDT Co., Newark, NJ). Thermocycling conditions were as follows: initial denaturing at 94°C for 5 min, followed by 35 cycles of 94°C, 30 s for denaturing; 62°C, 30 s for annealing; and 72°C, 45 s for extension. The primers and annealing temperatures used for the amplification and sequence of SSSCA1 and DPF2 genes are listed in Tables 2 and 3.
Table 2.
PCR primers and conditions for sequencing of SSSCA1
| Exon | Primer sequence (5′-3′) | Length (bp) |
Temp (°C) |
PCR temperature (°C) |
Product length (bp) |
|---|---|---|---|---|---|
| 1–4 | CAGGGCATCAAGGAGAGCAGAACA | 24 | 61 | 68 | 1,652 |
| genomic | GACCCCGCGCAGCCACTTTCAG | 22 | 65 | ||
| 1–4 | TCTCTCCCATCAACACTATG | 20 | 51 | 152 | |
| cDNA | GCTTGATCCGGGCTTGTG | 18 | 57 |
Table 3.
PCR primers and conditions for sequencing of DPF2
| Exon | Primer sequence (5′- 3′) | Length (bp) | Temp (°C) | PCR Temp.°C | Product length (bp) |
|---|---|---|---|---|---|
| 1 | AGTCGCAACAAGGCTTTCTCGGG | 23 | 62 | 62 | 306 |
| GCCCCTGTGGACAGTCACGAAGC | 23 | 64 | |||
| 2–4 | GAAGCCAGTCAGTATCCAGTGC | 22 | 58 | 63 | 1,516 |
| TCCCGGTCCCTTGAGCCTTCC | 21 | 64 | |||
| 5–6 | ATCGCTCCTATCCCCATGCCATTC | 24 | 61 | 63 | 1,269 |
| GTTTGATTCGGCCCCTTGGAGATG | 24 | 60 | |||
| 7–9 | CAAGCAAGTCCTCCCTGGGGTGTG | 24 | 64 | 63 | 720 |
| TGGCGGGGACATGGCACTCTT | 21 | 64 | |||
| 10 | AGTTAAACCGCTCCTTCTGG | 20 | 55 | 62 | 516 |
| ACGGCAGCCTGAATTTCCTATCCA | 24 | 61 | |||
| 11 and 3′UTR | CTTTACATATTGGGCTTTAGCA | 22 | 51 | 61 | 1,691 |
| TCAAGAGGGCCTAGGAGTGTATTT | 24 | 57 |
Marker assay
Once an informative polymorphism was detected, restriction fragment length polymorphisms were visualized on an 8% polyacrylamide gel after PCR amplification and digestion with Ddel. Briefly, the PCR was carried out in 25 μl reactions, which included 2.5 μl of 10X PCR buffer, 0.75 μl of 50 mM MgCl2 0.25 μl of 10 mM dNTPs (2.5 mM each), 0.50 μl of each 20 μM primers (forward: AACTCGCC CAGGCTGTCATCATCC, reverse: TGCCGAGTGTATT GAGTGTATTGG), 0.5 μl of Taq polymerase, 19 μl of diH2O, and 1 μl of template DNA. Thermocycling conditions were as followed: 95°C for 5 min, 35 cycles of 95°C for 30 s, 62°C for 30 s and 72°C for 30 s, then remained at 72°C for 10 min as final extension. The PCR product was then digested with 0.5 μl of Ddel (New England Biolabs, Beverly, MA) in 2.0 μl of 10X NEB buffer 3, and 7.5 μl of dH2O at 37°C overnight incubation.
Results
Pedigree analysis
Of the 235 purebred GSPs enrolled in this study, 20 were affected with ECLE (11 males, nine females; Table 1). Parental information was only available for 16 of the affected GSPs. These parents were clinically normal and had one ancestor in common (Fig. 1). Segregation analysis from those families with complete pedigree information is suggestive of an autosomal recessive trait, as 20.9% of dogs were affected. There was no statistically significant difference between observed and expected results (Chi-square=1.15). The mode of inheritance was further confirmed through breeding studies.
Fig. 1.

Partial pedigree of GSPs in which ECLE was segregating. Note the presence of a common ancestor (A); parents of affected dogs were not clinically affected; males and females were affected; and there were affected and normal dogs in the same litter. These are all hallmarks of autosomal recessive traits. Asterisk denotes animals that can be traced back to the common ancestor, A. The clinical phenotypes of all animals in this pedigree were known. ECLE was confirmed by histology in all affected dogs. Squares represent males, circles females, and in filled in symbols respresent dogs affected with ECLE
Breeding studies
An affected male (Dog 72) was outcrossed to a normal beagle producing seven clinically normal dogs (Dogs 100-106). Two of the F1 offspring (Dogs 101 and 106) were bred to each other producing nine F2 offspring. Two F1 females (Dogs 100 and 101) were bred back to a second affected male (Dog107) resulting in seven and ten offspring, respectively (BC1). By 16 to 24 weeks of age, a total of seven dogs (Dogs 112, 115, 116, 119, 128, 131, and 132) showed histological evidence of being affected with ECLE (Fig. 2). The mating between an affected dog (Dog 107) and the two F1 females resulted in four affected dogs from 17 dogs produced, which does not exclude an X-linked recessive or dominant trait. However, the mating between the two F1 dogs producing nine puppies containing three affected animals of both sexes rules out these modes of inheritance and is highly suggestive of an autosomal recessive trait (Fig. 2).
Fig. 2.
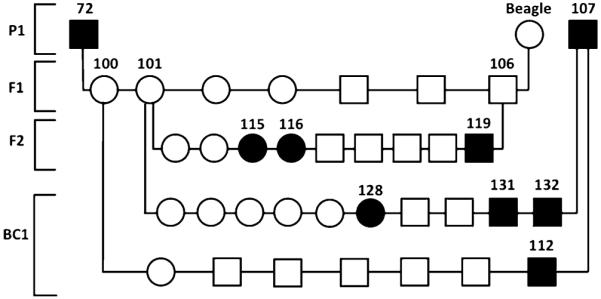
Test matings to examine the mode of inheritance. One affected ECLE GSP (72) was bred to a beagle mix (Beagle); two F1 offspring were bred together, and two females from the F1 litter were backcrossed (BC1) to an affected ECLE GSP. Squares represent males, circles females, and filled in symbols represent dogs affected with ECLE
Genome-wide association study
It has been shown that a sample size of ~20 dogs (usually 50% affected and 50% normal) was sufficient to find association for two Mendelian traits using only ~27,000 SNPs (Laird and Lange 2006). After excluding failed markers and low-frequency alleles (<10%), a total of 64,987 SNPs were evaluated and according to their p values. Results were graphically displayed by plotting the negative log10 of the respective p values against the chromosomal position of the SNP (Fig. 3). The highest supported signal that was associated with ECLE was observed on canine chromosome 18 (CFA18) at a p value of 1.36×10−12 (8.83×10−8 after correction), followed by strong signals on CFA6, 8, 15, 20, and 23 (Table 4).
Fig. 3.

Genome-wide association study. Genotyping results are displayed by plotting the negative log10 of the respective p values against the chromosomal (CFA18) position of the SNPs. The highest signal associated with ECLE was observed between positions 53,075,392–54,786,928. Percent homozygosity is shown in the lower panel
Table 4.
Forty-two SNPs with statistical significant association after Bonferroni correction identified through GWAS and their location
| SNP | Chromosome | Raw p value | p Value after Bonferroni correction |
|---|---|---|---|
| chr18.53913829 | 18 | 1.36×10−12 | 8.83×10−8 |
| chr6.9304579 | 6 | 1.33×10−11 | 8.65×10−7 |
| chr15.15788025 | 15 | 6.74×10−11 | 4.38×10−6 |
| chr23.46300532 | 23 | 6.74×10−11 | 4.38×10−6 |
| chr18.53029254 | 18 | 8.69×10−11 | 5.65×10−6 |
| chr20.39156399 | 20 | 1.01×10−10 | 6.56×10−6 |
| chr8.63289190 | 8 | 1.20×10−10 | 7.82×10−6 |
| chr23.45549471 | 23 | 1.21×10−10 | 7.87×10−6 |
| chr36.31445560 | 36 | 1.49×10−10 | 9.71×10−6 |
| chr17.18447826 | 17 | 1.75×10−10 | 1.14×10−5 |
| chr18.54940663 | 18 | 2.98×10−10 | 1.94×10−5 |
| chr18.54940784 | 18 | 2.98×10−10 | 1.94×10−5 |
| chr17.57743333 | 17 | 3.21×10−10 | 2.08×10−5 |
| chr23.17171959 | 23 | 3.21×10−10 | 2.08×10−5 |
| chr18.54758078 | 18 | 3.54×10−10 | 2.30×10−5 |
| chr17.17691257 | 17 | 4.13×10−10 | 2.69×10−5 |
| chr25.53132241 | 25 | 4.30×10−10 | 2.79×10−−5 |
| chr30.35617588 | 30 | 4.56×10−10 | 2.96×10−5 |
| chr18.53197305 | 18 | 5.10×10−10 | 3.32×10−5 |
| chr5.30948076 | 5 | 5.93×10−10 | 3.86×10−5 |
| chr6.9281668 | 6 | 5.93×10−10 | 3.86×10−5 |
| chr7.49872919 | 7 | 5.98×10−10 | 3.89×10−5 |
| chr5.30441037 | 5 | 6.36×10−10 | 4.13×10−5 |
| chr17.21160583 | 17 | 6.36×10−10 | 4.13×10−5 |
| chr35.6742308 | 35 | 6.36×10−10 | 4.13×10−5 |
| chr11.74120184 | 11 | 6.66×10−10 | 4.33×10−5 |
| chr36.31444641 | 36 | 9.49×10−10 | 6.17×10−5 |
| chr18.54724529 | 18 | 9.53×10−10 | 6.20×10−5 |
| chr18.54786928 | 18 | 9.53×10−10 | 6.20×10−5 |
| chr18.54796248 | 18 | 9.53×10−10 | 6.20×10−5 |
| chr18.54796618 | 18 | 9.53×10−10 | 6.20×10−5 |
| chr18.54809788 | 18 | 9.53×10−10 | 6.20×10−5 |
| chr18.54820915 | 18 | 9.53×10−10 | 6.20×10−5 |
| chr31.12385863 | 31 | 9.61×10−10 | 6.25×10−5 |
| chr18.52782734 | 18 | 1.16×10−9 | 7.52×10−5 |
| chr6.50817118 | 6 | 1.21×10−9 | 7.83×10−5 |
| chr8.63623084 | 8 | 1.44×10−9 | 9.35×10−5 |
| chr15.15630805 | 15 | 1.44×10−9 | 9.35×10−5 |
| chr15.15682422 | 15 | 1.44×10−9 | 9.35×10−5 |
| chr15.15720718 | 15 | 1.44×10−9 | 9.35×10−5 |
| chr15.18628813 | 15 | 1.44×10−9 | 9.35×10−5 |
| chr15.20218076 | 15 | 1.44×10−9 | 9.35×10−5 |
The largest group of SNPs with significant p values were located on CFA18
As pedigree analysis and test breeding results were highly suggestive of an autosomal recessive mode of inheritance, candidate regions were manually screened for homozygosity of SNP markers in the affected animals. Only the highly supported signal on CFA18 also showed an area of homozygosity in affected animals, which was not present in clinically normal animals. Thus, the defective gene region to be associated with ECLE disease was narrowed to a 0.5-MB region on CFA18 (roughly 54,500,000 to 55, 000,000 bp). The critical region was compared to the publicly available human genome and found to be homologous to HSA11q13. 1–13.2 and to HSA12q24 (NCBI Map Viewer, Ensembl, and UCSC Genome Browser). Genes of interest in the candidate region are listed in sequential order of their arrangement on CFA18 in Table 5 (Fig. 4).
Table 5.
Candidate genes in the critical region identified by GWAS on CFA18
| Gene | Chromosomal location | Protein encoded | Relation to skin disease |
|---|---|---|---|
| RELAa | CFA18: 54,576,239-54,583,367 | Protein in the NF-kappa B complex |
Regulation of epidermal development |
| SIPA1 | CFA18: 54,586,278-54,596,558 | Signal-induced proliferation- associated protein |
Involved in cell cycle progression; lupus-like nephritis in knockout mice |
| MAP3K11 | CFA18: 54,619,769-54,634,056 | Mitogen-activated protein kinase kinase kinase |
Activation of JNK, necessary for RAF/ERK signaling |
| SSSCA1 | CFA18: 54,660,354-54,661,601 | Sjogren's syndrome/scleroderma autoantigen 1 |
p27, Contributes to anticentromeric antibodies |
| DPF2 | CFA18: 54,859,255-54,872,054 | D4, zinc and double PHD fingers family 2 |
Required for apoptosis |
| CDC42EP2 | CFA18: 54,882,334-54,882,978 | Cdc42 effector protein (Rho GTPase binding) 2 |
Expressed in keratinocytes |
RELA is also named A1XFH4
Fig. 4.
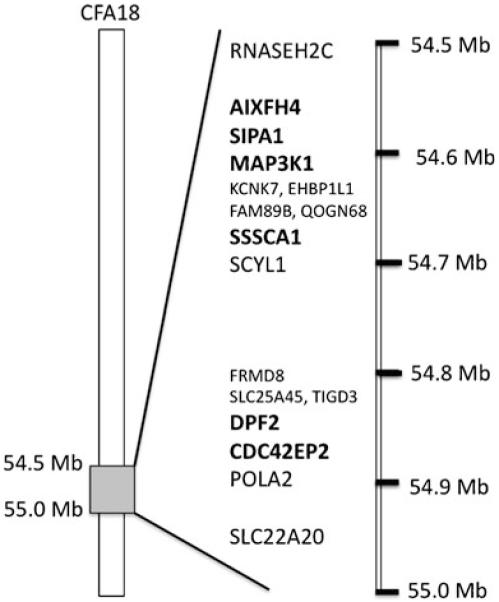
Arrangement of predicted genes in the candidate region on CFA18. Genes in bold represent candidates for ECLE as they are genes involved in cell cycle progression or immunity
Gene sequencing
Six genes are located within the genomic region identified by GWAS (Table 5) and, therefore, were considered potential candidates for ECLE in the GSP. The gene coding for SSSCA1 was sequenced first in two affected and two unrelated normal dogs. SSSCA1 was 1.2-kb long and contained four small predicted exons. Both the cDNA and the genomic DNA were sequenced, but did not contain informative polymorphisms. However, SSSCA1 cannot be entirely excluded as a candidate gene for ECLE in the GSP, as mutations in regulatory regions such as the promoter region may be present.
A single SNP (rs22638189) within an intron of DPF2 was identified by GWAS that appeared to segregate with the disease. Therefore, we focused on sequencing this gene, even though it was not the most likely candidate. The complete 16,498 bp genomic region, harboring all 10 DPF2 predicted coding exons and 3’UTR, was sequenced in eight affected dogs, eight unaffected relatives, and two unrelated normal dogs. Although there was no sequence variation found within the exons, a single base substitution of a guanine (G) to adenine (A) at position 54,866,704 (May 2005, Broad/canFam2 Assembly) was observed in intron 3 of DPF2 in the affected dogs, which was not present in unrelated normal dogs. Subsequently this region was sequenced from ten phenotypically affected dogs, ten obligate carriers, and four unrelated normal dogs. All of the affected dogs were homozygous for the A allele, obligate carriers were heterozygous (A/G), and all unrelated normal dogs were homozygous for the G allele.
Restriction Fragment Length Polymorphism
As the G→A substitution eliminates the recognition site of the restriction enzyme Ddel, an RFLP assay was developed to efficiently screen large numbers of dogs. Amplification of a fragment 292 bp including the restriction enzyme site was performed, and subsequent Ddel restriction enzyme digestion resulted in cleaving of DNA into 122 and 170-bp fragments in the presence of the G allele, whereas the DNA containing the A allele was not cut.
Utilizing this SNP marker as a genotyping tool, all 267 samples (Table 1) were examined, which included 27 histologically confirmed affected, 17 normal control dogs from a GSP sub-population, which had never been affected by the disease, 31 obligate carriers, and 192 dogs who were phenotypically normal but related to affected dogs. All ECLE affected dogs were homozygous for the A allele, including those produced in the experimental matings (mixed breed F2s; Fig. 2). Seventeen normal control dogs were homozygous for the G allele, and all 31 obligate carriers were heterozygous having both an A and a G allele. Among the 192 phenotypically normal dogs, 56% (108 dogs) were homozygous for the G allele, and 44% (84 cases) were heterozygous (A/G genotype) (Table 6).
Table 6.
DPF2 genotyping results of 267 ECLE affected, obligate carrier, normal control dogs, and phenotypically normal GSPs that were related to affected dogs. These numbers include the mixed breeds produced in the experimental matings
| Phenotype | Number | SNP genotype | Number |
|---|---|---|---|
| ECLE affected | 27 | A/A | 27 |
| Obligate carrier | 31 | A/G | 31 |
| Confirmed normal control | 17 | G/G | 17 |
| Clinically normal-related dogs | 192 | G/G | 108 |
| A/G | 84 | ||
| Total dogs | 267 | 267 |
Discussion
Exfoliative cutaneous lupus erythematosus (ECLE) has only been described in the GSP (Gross et al. 1992; Vroom et al. 1995; Bryden et al. 2005; Mauldin et al. 2010). Besides the typical skin lesions consisting of excessive scaling, erythema, erosions, and alopecia, arthralgia is a common finding in almost all affected dogs and in our study, some of the older affected GSPs developed renal disease with histological hallmarks of systemic LE (Mauldin et al. 2010). Immunohistochemical analysis of the skin reveals IgG deposition present at the dermoepidermal junction, which may be accompanied by IgM, IgA, or C3 deposition (Bryden et al. 2005). Immunohistochemical staining reveals CD3+ lymphocytes throughout the epidermis, the infundibulum of the hair follicles and around the sweat glands (Bryden et al. 2005). Therapy is not often rewarding and the progressively debilitating nature of the disease in dogs leads in all but rare cases to euthanasia (Vroom et al. 1995; Bryden et al. 2005; Mauldin et al. 2010).
In humans, lupus erythematosus (LE) can be classified as being chronic localized, chronic disseminated, subacute, and acute (Tan et al. 1982; Sontheimer 1997; Werth 2007). While chronic CLE affects mainly the skin, humans with disseminated discoid LE (DLE) have an increased risk of developing systemic manifestations of LE. The distribution of the lesions seen in our dogs is fairly symmetrical as they are in humans with SCLE (Kuhn et al. 2007). No sex predilection has been reported in dogs (Vercelli and Schiavi 1998) in contrast to humans with SCLE where about 8 times more females are affected than males (Kuhn et al. 2007). While we observed slightly more affected males than females (15 males versus 12 females), in our experience, females show more severe clinical signs (Mauldin et al. 2010). Comparable to arthritis, arthralgia, and myalgia commonly seen in human SCLE patients (Kuhn et al. 2007), the ECLE dogs also exhibited lameness and a hunched stance due to arthralgia (Mauldin et al. 2010). However, we were not able to demonstrate radiographic or histological lesions that could explain the joint pain. While, to date, the molecular basis of ECLE is unknown, clinical data securely established the similarity of these disorders in dog and man (Bryden et al. 2005; Mauldin et al. 2010).
We began to establish a colony of these dogs 7 years ago to study disease progression, perform controlled treatment trials, and investigate the genetic basis of the disease (Mauldin et al. 2010). Through these breeding studies and the evaluation of the GSP pedigrees, we were not only able to confirm that ECLE is a genetic disease, but also demonstrate an autosomal recessive mode of inheritance (Figs. 1 and 2). The simple Mendelian inheritance allowed us pursue a GWAS approach to narrow the genomic region linked to this disorder using a much smaller number of dogs than if the disease had been inherited in a complex manner. A combination of association analysis (Table 4) and homozygosity mapping successfully located the disease locus to a ~1.5 MB interval on CFA18 (Fig. 3). In addition, the confirmed inheritance pattern allowed to reject those chromosomal regions that resulted in significant SNPs in the association study, but were not confirmed by autozygosity mapping (e.g. CFA15). Thus, potential candidate genes within this area were identified (Table 5), and assessed using findings from comparable human disease.
Several genes and chromosomal regions have been associated with CLE in man (Millard and McGregor 2001; Baechler et al. 2003; Remmers et al. 2007; Wenzel and Tuting 2007; Werth 2007; Jarvinen et al. 2010). Two genes were of particular interest in the dog, the gene for complement subcomponent, C1r, (C1r) which is associated with inherited DLE in humans, arthralgia, autoimmune disease, arthritis, mild nephritis, and recurrent rhinobronchitis, and TREX1, which is associated with Chilblain lupus, another monogenetic form of cutaneous lupus (Day et al. 1972; Moncada et al. 1972; Rich et al. 1979; Gunther et al. 2009). C1r maps on CFA 27, and was excluded from major causative association with ECLE based on the GWAS data. However, TREX1 maps to CFA20 where a signal was found, but did not yield a significant score. We cannot reject the possibility that this implicates TREX1 as interacting gene or modifier locus for this disease, and this will become of particular interest once the ECLE causing mutation is identified.
The region discovered in our studies aligned to HSA11q13.1–13.2 and to HSA12q24. A number of genes and regions associated with SLE are located within these regions and include APIP, DDX6, PHRF1, IRF7, SH2B3, KIAA1853, TAOK3, SPPL3 and SLEB4, many of which are involved in immune regulatory functions, (Gateva et al. 2009; Graham et al. 2009). However, detailed analysis of the canine GWAS excluded many of these genes as candidates based on their individual locations on other canine chromosomes. Comparison to human genomic data did not yield an immediate candidate for ECLE in the GSP. Thus, one might assume that the gene responsible for the disease is either not yet known to contribute to this group of disorders, or constitutes a novel gene, which may have a similar function or interact with previously identified genes.
For the initial candidate gene discovery we therefore focused on genes that are involved in immune regulation or cell cycle processes. SSSCA1 was considered first, as the corresponding disease (Sjogren Syndrome/Scleroderma) has a familial component and is described as an autoimmune disease (Reveille et al. 1984), even though the clinical signs in humans were not the same in affected dogs. SSSCA1 could not be entirely excluded in the GSP, as neither a mutation nor an informative SNP was located. DPF2 plays an important role in apoptosis of myeloid cells and rapid cell turnover, and thus might be involved in ECLE (Gabig et al. 1998). Sequencing of DPF2 revealed an informative SNP in an intron, but no mutation was identified within the gene’s coding sequence. However, there remains the possibility that mutations in the regulatory regions of SSSCA1 or DPF2 could lead to ECLE through as of yet unknown mechanisms.
Association of the presented CFA18 region of interest linked to ECLE in GSP has been confirmed through complete linkage of the DPF2 SNP with disease status in a total of 267 individuals (Table 6). Thus, remaining genes in the area will be further investigated to identify the causative mutation. The next candidate is CDC42EP2, a Rho GTPase, regulates downstream effector proteins for the assembly of the actin cytoskeleton (Hirsch et al. 2001). RELA, also in the candidate region, is part of the NFkB complex, which is involved in a variety of immune processes and has been shown to cause epidermal defects in mice (Gugasyan et al. 2004). SIPA1, a gene involved cell cycle progression, is of interest, as its absence causes lupus-like nephritis in knockout mice (Ishida et al. 2006). Finally, MAP3K11 is required for activation of JNK, p38, and ERK (Chadee and Kyriakis 2004). However, it is more likely to be involved in neoplastic processes. While none of the genes described here have been implicated in CLE in humans, we will continue to focus on this region.
A recent paper used a GWAS to elucidate the genetic background of the SLE complex in the Nova Scotia duck tolling retriever (NSDTR) (Wilbe et al. 2010). These dogs are predisposed to immune-mediated rheumatic disease and steroid responsive meningitis/arteritis. The fact that ECLE in our dogs appears to be inherited as a simple autosomal recessive trait as opposed to the autoimmune disease in the NSDTRs could be due to a popular sire effect coupled with intensive line breeding, which resulted in homozygosity of modifier genes accentuating the major gene involved in ECLE. The simple Mendelian inheritance of ECLE in the GSP also made it possible to perform a GWAS with very few animals (13 affected) to obtain reasonable and useful results, where as 81 samples from affected NSDTRs were needed for the GWAS to obtain useful data (Wilbe et al. 2010). However, the results of the GWAS in the NSDTR demonstrate the power of using dogs to unravel the genetic basis of complex inherited diseases.
Here we present a naturally occurring model of ECLE in the GSP with a simple autosomal recessive mode of inheritance in which the GWAS allowed for a rapid identification of a critical chromosomal region. The development of a genetic marker-based diagnostic test allows identification of disease status for ECLE in GSPs, and significantly improves our ability to screen animals for being affected or at risk for disease. This test is an alternative for the currently performed biopsy. In addition, carrier animals can be identified for the first time, and breeders can use this information to prevent the birth of affected puppies and plan future breeding strategies. Future studies will now focus on elucidating the causal gene(s) and further developing this canine model to better understand the disease process in humans and ultimately to develop effective treatment modalities.
Acknowledgements
The study was supported by grants from the NIH (National Center for Research Resources, RR02512), the American Kennel Club (AKC) Canine Health Foundation, and generous German shorthaired pointer breeders.
Contributor Information
Ping Wang, School of Veterinary Medicine, Section of Medical Genetics, University of Pennsylvania, Philadelphia, PA, USA.
Barbara Zangerl, School of Veterinary Medicine, Section of Ophthalmology, University of Pennsylvania, Philadelphia, PA, USA.
Petra Werner, Division of Cardiology, Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Elizabeth A. Mauldin, Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, USA
Margret L. Casal, School of Veterinary Medicine, Section of Medical Genetics, University of Pennsylvania, Philadelphia, PA, USA
References
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryden SL, White SD, Dunston SM, Burrows AK, Olivry T. Clinical, histopathological and immunological characteristics of exfoliative cutaneous lupus erythematosus in 25 German short haired pointers. Vet Dermatol. 2005;16:239–252. doi: 10.1111/j.1365-3164.2005.00468.x. [DOI] [PubMed] [Google Scholar]
- Carr IM, Szymanska K, Sheridan E, Markham AF, Bonthron DT, Johnson CA. Shadow autozygosity mapping by linkage exclusion (SAMPLE): a simple strategy to identify the geneticbasis of lethal autosomal recessive disorders. Hum Mutat. 2009;30:1642–1649. doi: 10.1002/humu.21105. [DOI] [PubMed] [Google Scholar]
- Chadee DN, Kyriakis JM. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat Cell Biol. 2004;6:770–776. doi: 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- Costner MI, Sontheimer RD. In: Lupus erythematosus. Fitzpatrick’s dermatology in general medicine. Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. McGraw-Hill; New York: 2003. pp. 1677–1693. [Google Scholar]
- Day NK, Geiger H, Stroud R, DeBracco M, Mancaido B, Windhorst D, Good RA. C1r deficiency: an inborn error associated with cutaneous and renal disease. J Clin Invest. 1972;51:1102–1108. doi: 10.1172/JCI106902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabig TG, Crean CD, Klenk A, Long H, Copeland NG, Gilbert DJ, Jenkins NA, Quincey D, Parente F, Lespinasse F, Carle GF, Gaudray P, Zhang CX, Calender A, Hoeppener J, Kas K, Thakker RV, Farnebo F, Teh BT, Larsson C, Piehl F, Lagercrantz J, Khodaei S, Carson E, Weber G. Expression and chromosomal localization of the Requiem gene. Mamm Genome. 1998;9:660–665. doi: 10.1007/s003359900840. [DOI] [PubMed] [Google Scholar]
- Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jonsen A, Bengtsson AA, Rantapaa-Dahlqvist S, Baechler EC, Brown EE, Alarcon GS, Edberg JC, Ramsey-Goldman R, McGwin G, Jr, Reveille JD, Vila LM, Kimberly RP, Manzi S, Petri MA, Lee A, Gregersen PK, Seldin MF, Ronnblom L, Criswell LA, Syvanen AC, Behrens TW, Graham RR. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RR, Hom G, Ortmann W, Behrens TW. Review of recent genome-wide association scans in lupus. J Intern Med. 2009;265:680–688. doi: 10.1111/j.1365-2796.2009.02096.x. [DOI] [PubMed] [Google Scholar]
- Gross TL, Ihrke PJ, Walder EJ. Veterinary dermatopathology: a macroscopic and microscopic evaluation of canine and feline skin diseases. Mosby Year Book; St. Louis: 1992. Hereditary lupoid dermatosis of the German shorthaired pointer; pp. 26–28. [Google Scholar]
- Gross TL, Ihrke PJ, Walder EJ, Affolter VK. In: Interface diseases of the dermal-epidermal junction. Skin diseases of the dog and cat. Clinical and histopathologic diagnosis. Gross TL, editor. Blackwell Science, Ltd; Ames: 2005. pp. 49–74. [Google Scholar]
- Gugasyan R, Voss A, Varigos G, Thomas T, Grumont RJ, Kaur P, Grigoriadis G, Gerondakis S. The transcription factors c-rel and RelA control epidermal development and homeostasis in embryonic and adult skin via distinct mechanisms. Mol Cell Biol. 2004;24:5733–5745. doi: 10.1128/MCB.24.13.5733-5745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Meurer M, Stein A, Viehweg A, Lee-Kirsch MA. Familial chilblain lupus—a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in TREX1. Dermatology. 2009;219:162–166. doi: 10.1159/000222430. [DOI] [PubMed] [Google Scholar]
- Hirsch DS, Pirone DM, Burbelo PD. A new family of Cdc42 effector proteins, CEPs, function in fibroblast and epithelial cell shape changes. J Biol Chem. 2001;276:875–883. doi: 10.1074/jbc.M007039200. [DOI] [PubMed] [Google Scholar]
- Ishida D, Su L, Tamura A, Katayama Y, Kawai Y, Wang SF, Taniwaki M, Hamazaki Y, Hattori M, Minato N. Rap1 signal controls B cell receptor repertoire and generation of self-reactive B1a cells. Immunity. 2006;24:417–427. doi: 10.1016/j.immuni.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Jarvinen TM, Hellquist A, Koskenmies S, Einarsdottir E, Koskinen LL, Jeskanen L, Berglind L, Panelius J, Hasan T, Ranki A, Kere J, Saarialho-Kere U. Tyrosine kinase 2 and interferon regulatory factor 5 polymorphisms are associated with discoid and subacute cutaneous lupus erythematosus. Exp Dermatol. 2010;19:123–131. doi: 10.1111/j.1600-0625.2009.00982.x. [DOI] [PubMed] [Google Scholar]
- Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NH, Zody MC, Anderson N, Biagi TM, Patterson N, Pielberg GR, Kulbokas EJ, 3rd, Comstock KE, Keller ET, Mesirov JP, von Euler H, Kampe O, Hedhammar A, Lander ES, Andersson G, Andersson L, Lindblad-Toh K. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet. 2007;39:1321–1328. doi: 10.1038/ng.2007.10. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124–1137. doi: 10.1111/j.1610-0387.2007.06554.x. [DOI] [PubMed] [Google Scholar]
- Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet. 2006;7:385–394. doi: 10.1038/nrg1839. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, 3rd, Zody MC, Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, Ponting CP, Galibert F, Smith DR, DeJong PJ, Kirkness E, Alvarez P, Biagi T, Brockman W, Butler J, Chin CW, Cook A, Cuff J, Daly MJ, DeCaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M, Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli KP, Parker HG, Pollinger JP, Searle SM, Sutter NB, Thomas R, Webber C, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-Zahra M, Aldredge T, Allen N, An P, Anderson S, Antoine C, Arachchi H, Aslam A, Ayotte L, Bachantsang P, Barry A, Bayul T, Benamara M, Berlin A, Bessette D, Blitshteyn B, Bloom T, Blye J, Boguslavskiy L, Bonnet C, Boukhgalter B, Brown A, Cahill P, Calixte N, Camarata J, Cheshatsang Y, Chu J, Citroen M, Collymore A, Cooke P, Dawoe T, Daza R, Decktor K, DeGray S, Dhargay N, Dooley K, Dooley K, Dorje P, Dorjee K, Dorris L, Duffey N, Dupes A, Egbiremolen O, Elong R, Falk J, Farina A, Faro S, Ferguson D, Ferreira P, Fisher S, FitzGerald M, Foley K, Foley C, Franke A, Friedrich D, Gage D, Garber M, Gearin G, Giannoukos G, Goode T, Goyette A, Graham J, Grandbois E, Gyaltsen K, Hafez N, Hagopian D, Hagos B, Hall J, Healy C, Hegarty R, Honan T, Horn A, Houde N, Hughes L, Hunnicutt L, Husby M, Jester B, Jones C, Kamat A, Kanga B, Kells C, Khazanovich D, Kieu AC, Kisner P, Kumar M, Lance K, Landers T, Lara M, Lee W, Leger JP, Lennon N, Leuper L, LeVine S, Liu J, Liu X, Lokyitsang Y, Lokyitsang T, Lui A, Macdonald J, Major J, Marabella R, Maru K, Matthews C, McDonough S, Mehta T, Meldrim J, Melnikov A, Meneus L, Mihalev A, Mihova T, Miller K, Mittelman R, Mlenga V, Mulrain L, Munson G, Navidi A, Naylor J, Nguyen T, Nguyen N, Nguyen C, Nguyen T, Nicol R, Norbu N, Norbu C, Novod N, Nyima T, Olandt P, O’Neill B, O’Neill K, Osman S, Oyono L, Patti C, Perrin D, Phunkhang P, Pierre F, Priest M, Rachupka A, Raghuraman S, Rameau R, Ray V, Raymond C, Rege F, Rise C, Rogers J, Rogov P, Sahalie J, Settipalli S, Sharpe T, Shea T, Sheehan M, Sherpa N, Shi J, Shih D, Sloan J, Smith C, Sparrow T, Stalker J, Stange-Thomann N, Stavropoulos S, Stone C, Stone S, Sykes S, Tchuinga P, Tenzing P, Tesfaye S, Thoulutsang D, Thoulutsang Y, Topham K, Topping I, Tsamla T, Vassiliev H, Venkataraman V, Vo A, Wangchuk T, Wangdi T, Weiand M, Wilkinson J, Wilson A, Yadav S, Yang S, Yang X, Young G, Yu Q, Zainoun J, Zembek L, Zimmer A, Lander ES. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Mauldin EA, Morris DO, Brown DC, Casal ML. Exfoliative cutaneous lupus erythematosus in German shorthaired pointer dogs: disease development, progression and evaluation of three immunomodulatory drugs (ciclosporin, hydroxychloroquine, and adalimumab) in a controlled environment. Vet Dermatol. 2010;21(4):373–382. doi: 10.1111/j.1365-3164.2010.00867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard TP, McGregor JM. Molecular genetics of cutaneous lupus erythematosus. Clin Exp Dermatol. 2001;26:184–191. doi: 10.1046/j.1365-2230.2001.00793.x. [DOI] [PubMed] [Google Scholar]
- Moncada B, Day NK, Good RA, Windhorst DB. Lupus-erythematosus-like syndrome with a familial defect of complement. N Engl J Med. 1972;286:689–693. doi: 10.1056/NEJM197203302861304. [DOI] [PubMed] [Google Scholar]
- Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin. 2002;20:373–385. doi: 10.1016/s0733-8635(02)00016-5. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reveille JD, Wilson RW, Provost TT, Bias WB, Arnett FC. Primary Sjogren’s syndrome and other autoimmune diseases in families. Prevalence and immunogenetic studies in six kindreds. Ann Intern Med. 1984;101:748–756. doi: 10.7326/0003-4819-101-6-748. [DOI] [PubMed] [Google Scholar]
- Rich KC, Jr, Hurley J, Gewurz H. Inborn C1r dificiency with a mild lupus-like syndrome. Clin Immunol Immunopathol. 1979;13:77–84. doi: 10.1016/0090-1229(79)90022-9. [DOI] [PubMed] [Google Scholar]
- Salmon Hillbertz NH, Isaksson M, Karlsson EK, Hellmen E, Pielberg GR, Savolainen P, Wade CM, von Euler H, Gustafson U, Hedhammar A, Nilsson M, Lindblad-Toh K, Andersson LAndersson G. Duplication of FGF3, FGF4, FGF19 and ORAOV1 causes hair ridge and predisposition to dermoid sinus in Ridgeback dogs. Nat Genet. 2007;39:1318–1320. doi: 10.1038/ng.2007.4. [DOI] [PubMed] [Google Scholar]
- Scott DW, Miller WH. Small animal dermatology. WB Saunders; Philadelphia: 2001. Hereditary lupoid dermatosis of German shorthair pointers; pp. 948–949. [Google Scholar]
- Sontheimer RD. The lexicon of cutaneous lupus erythematosus—a review and personal perspective on the nomenclature and classification of the cutaneous manifestation in lupus erythematosus. Lupus. 1997;8:84–95. doi: 10.1177/096120339700600203. [DOI] [PubMed] [Google Scholar]
- Sutter NB, Eberle MA, Parker HG, Pullar BJ, Kirkness EF, Kruglyak L, Ostrander EA. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 2004;14:2388–2396. doi: 10.1101/gr.3147604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- Theaker AJ, Rest JR. Lupoid dermatosis in a German short haired pointer. Vet Rec. 1992;131:495. doi: 10.1136/vr.131.21.495. [DOI] [PubMed] [Google Scholar]
- Vercelli A, Schiavi S. A case report of lupoid dermatosis in a German short-haired pointer. Adv Vet Derm. 1998;3:466–467. [Google Scholar]
- Vroom MW, Theaker AJ, Rest JR, White SD. Case report: lupoid dermatosis in 5 German short-hair pointer. Vet Dermatol. 1995;6:93–98. doi: 10.1111/j.1365-3164.1995.tb00049.x. [DOI] [PubMed] [Google Scholar]
- Wenzel J, Tuting T. Identification of type I interferon-associated inflammation in the pathogenesis of cutaneous lupus erythematosus opens up options for novel therapeutic approaches. Exp Dermatol. 2007;16:454–463. doi: 10.1111/j.1600-0625.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- Werth VP. Cutaneous lupus: insights into pathogenesis and disease classification. Bull NYU Hosp Jt Dis. 2007;65:200–204. [PubMed] [Google Scholar]
- White SD, Gross TL. Hereditary lupoid dermatosis of the German short hair pointer. In: Kirk RW, Bonagura JD, editors. Current veterinary therapy small animal practice. WB Saunders; Philadelphia: 1995. pp. 605–606. [Google Scholar]
- Wiik AC, Ropstad EO, Bjerkas ELingaas F. A study of candidate genes for day blindness in the standard wire haired dachshund. BMC Vet Res. 2008;4:23. doi: 10.1186/1746-6148-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbe M, Jokinen P, Truve K, Seppala EH, Karlsson EK, Biagi T, Hughes A, Bannasch D, Andersson G, Hansson-Hamlin H, Lohi H, Lindblad-Toh K. Genome-wide association mapping identifies multiple loci for a canine SLE-related disease complex. Nat Genet. 2010;42:250–254. doi: 10.1038/ng.525. [DOI] [PubMed] [Google Scholar]