

Abstract
Background
The citrus red mite is a worldwide citrus pest and a common sensitizing allergen of asthma and rhinitis. It has developed strong resistance to many registered acaricides, However, the molecular mechanisms of resistance remain unknown. we therefore used next generation sequencing technology to investigate the global transcriptomes between resistant strains and susceptible strains.
Results
We obtained 34,159, 30,466 and 32,217 unigenes by assembling the SS reads, RS reads and SS&RS reads respectively. There are total 17,581 annotated unigenes from SS&RS reads by BLAST searching databases of nr, the Clusters of Orthologous Groups (COGs) and Kyoto Encyclopedia of Genes and Genomes (KEGG) with an E-value ≤ 1e-5, in which 7,075 unigenes were annotated in the COG database, 12, 712 unigenes were found in the KEGG database and 3,812 unigenes were assigned to Gene ontology (GO). Moreover, 2,701 unigenes were judged to be the differentially expressed genes (DEGs) based on the uniquely mapped reads. There are 219 pathways in all annotated unigenes and 198 pathways in DEGs that mapped to the KEGG database. We identified 211 metabolism genes and target genes related to general insecticide resistance such as P450 and Cytochrome b, and further compared their differences between RS and SS. Meanwhile, we identified 105 and 194 genes related to growth and reproduction, respectively, based on the mode of action of Hexythiazox. After further analyses, we found variation in sequences but not in gene expression related to mite growth and reproduction between different strains.
Conclusion
To our knowledge, this is the first comparative transcriptome study to discover candidate genes involved in phytophagous mite resistance. This study identified differential unigenes related to general pesticide resistance and organism growth and reproduction in P. citri. The assembled, annotated transcriptomes provide a valuable genomic resource for further understanding the molecular basis of resistance mechanisms.
Introduction
The citrus red mite, Panonychus citri (McGregor), is a worldwide citrus pest causing a huge fruit yield loss every year. It has a short life cycle and high reproductive rate, infesting over 80 plant species such as citrus, rose, almond, pear, castor bean, and several broadleaf evergreen ornamentals [1]–[2]. It is also an important sensitizing allergen to asthma and rhinitis among people around citrus orchards [3]–[5]. Control of P. citri mainly depends on chemical applications, which lead to acaricide resistance in P. citri. Currently this situation is deteriorating because of the improper application of acaricides, leading to resistance to many registered acaricides [6]–[11].
Hexythiazox is a selective miticide that acts against various phytophagous mites. It has been widely used on various crops under integrated pest management programs, especially against citrus mites. However, phytophagous mites are capable of rapidly developing resistance to hexythiazox. A high level of resistance has already been reported in Tetranychus urticae (resistance factor > 1,000-fold) in Australia, in P. citri (resistance factor > 23,000-fold) in Japan, and in Brevipalpus phoenicis (resistance factor >10,000-fold) in Brazil [12]–[14]. Our previous study showed the resistance factor of P. citri increased 3532-fold after continuous selection with hexythiazox for 20 generations (Ran et al. unpublished data). Hexythiazox has little or no effect on survival of the adult mite [15], but interferes with its growth and reproduction [12]. Hexythiazox resistance may alter the target sites and increase the rate of metabolism. Studies on the resistance mechanism in P. citri have mainly focused on measuring the activity of detoxification enzymes [16]–[17]. Although its complete mitochondrial genome was sequenced recently [18]–[19], nuclear genome resources for P. citri are limited (only 234 records in NCBI in January 2011), which has severely hindered the study of resistance mechanisms at the molecular level.
A transcriptome is a complete set of transcripts in a cell or an organism. Various technologies have been developed to characterize transcriptomes, including serial analysis of gene expression (SAGE), cap analysis of gene expression (CAGE), massively parallel signature sequencing (MPSS) and RNA sequencing (RNA-Seq) [20]–[22]. Next generation sequencing technologies have dramatically accelerated genome-wide studies of transcriptomes and have been widely used to explore gene structure and gene expression even without a genome reference [23]–[27]. RNA-seq has made it increasingly possible to perform de novo transcriptome sequencing with the development of short read sequencing technologies such as the Roche 454, SOLiD and Solexa/Illumina platforms for various purposes [20], [28]–[29]. Illumina sequencing technology, generating large-scale sequence at a lower cost and greater time-saving, has recently been applied to transcriptome analysis of insects, such as Aedes aegypti, Anopheles funestus, Bemisia tabaci, Nilaparvata lugens and Locusta migratoria [30]–[34], but not of mites.
To gain detailed genetic information for resistance mechanism, we sequenced the transcriptomes of both a resistant strain (RS) and a susceptible strain (SS) of P. citri using a high-throughput sequencing platform – Illumina HiSeq™ 2000. We obtained over four billion bases of high-quality DNA sequence with an average read length of 90 bp. These sequences were assembled into 30,466 and 34,159 unigenes in the resistant and susceptible strains respectively. We then merged the above two groups of unigenes to produce longer unigenes; approximately 55% of these unigenes were already annotated, as identified by BLAST searches (E-value ≤ 1e-5) against the SwissProt (http://expasy.org/tools/blast), KEGG (http://www.genome.jp/kegg), COG (http://www.ncbi.nlm.nih.gov/COG/), nr (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and GO databases. We systematically examined differential gene expression in order to identify genes involved in general insecticide resistance. Furthermore, we developed a new method to compare more than 200 genes involved in growth and reproduction, based on the mode of action of hexythiazox. The assembled, annotated transcriptome sequences provide a valuable genomic resource for further understanding the molecular basis of resistance.
Results and Discussion
Illumina sequencing and reads assembly
In order to uncover the differences in P. citri gene expression between the resistant strains (RS) and susceptible strains (SS), a pooled cDNA sample from each strain, representing eggs, larvae and adults, was sequenced with the Illumina sequencing platform. 25 and 26 million clean reads of 90 bp (EMBL-EBI accession number: ERP000885) were obtained from RS and SS, respectively, after filtering out the dirty raw reads. To facilitate sequence assembly, the remaining clean raw reads were clipped into 21-mers randomly using SOAPdenovo software [35], and used to assemble 215,641 contigs with a mean length of 178 bp in RS, and 282,291 contigs with an average length of 151 bp in SS (Table 1). With paired-end read joining and gap-filling, the above contigs were further assembled into 38,178 scaffolds with a mean size of 453 bp in RS and 44,017 scaffolds with a mean size 406 bp in SS, where the number of contigs with a scaffold size of more than 1000 bp is 4,643 in RS and 4,270 in SS. With gap-filling and long sequence clustering, the scaffolds were assembled into 30,466 RS-unigenes with a mean length of 536 bp and 34,159 SS-unigenes with a mean length of 489 bp. In addition, using the same strategy we obtained 32,217 All-unigenes with a mean size of 631 bp from the RS clean reads (25 million) and SS reads (26 million) combined (All-unigene originated from both RS and SS reads; RS-unigene from RS reads; SS-unigene from SS reads.).
Table 1. Summary of reads in Susceptible Strains (SS) and Resistant Strains (RS) of Citrus red mite transcriptomes.
| SS | RS | SS&RS | |
| Total base pairs (bp) | 2,414,000,340 | 2,336,000,220 | - |
| Total number of Reads | 26,822,226 | 25,955,558 | - |
| GC percentage | 42.51% | 41.93% | - |
| Total number of contigs | 282,291 | 215,641 | - |
| Mean length of contigs (bp) | 151 | 178 | - |
| Total number of scaffolds | 44,017 | 38,178 | - |
| Mean length of scaffolds (bp) | 406 | 453 | - |
| The number of unigenes | 34,159 | 30,466 | - |
| Mean length of unigenes | 489 | 536 | - |
| The number of all-unigenes | - | - | 32,217 |
| Mean length of all-unigenes | - | - | 631 |
The sequence of unigene (the unique) represents that they cannot be extended on both ends and contains the least Ns in this paper. The length distribution and gap distribution of unigenes from the three combinations (RS, SS and RS&SS) are shown in Figure 1. RS-unigenes and SS-unigenes had a similar distribution in length, with 300-600-bp sequences representing the highest proportion, followed by 600-1,200-bp sequences. Interestingly, RS&SS-unigenes dramatically improved the length distribution with no more size of 200–300 bp region, and instead of the minimum of 300 bp compared to RS-unigenes or SS-unigenes. The gap distribution, however, had a slightly different tendency, showing more Ns compared to RS-unigenes or SS-unigenes at the same level (Figure 1). In total, All-unigenes are better than the other two kinds of unigenes in quality, and obtained higher identity scores when using BLASTx to search against the non-redundant (nr) NCBI nucleotide database. To validate the quality of sequencing data, 21 unigenes from RS-, SS- and All-unigenes were randomly selected, for which 21 pairs of primers were designed, respectively, for RT-PCR amplification. In this experiment, 18 out of 21 primer pairs resulted in an expected band size and were further confirmed by Sanger sequencing (data not shown).
Figure 1. The length and gap distribution of unigenes after de novo assembly using the reads from SS, RS and SS&RS samples.

A: Length distribution of unigenes. Numbers in square brackets (e.g., [200,300) and [1800,3000]) indicate the range of unigene length, while ‘>3000’ indicates unigenes longer than 3,000 bp. Numbers in parentheses (e.g., (5219) and (18827)) indicate the total number of unigenes falling in this length range. B: Gap distribution of unigenes. Values in square brackets indicate the ratio of the N number and all nucleotide base pair number for one unigene. Numbers in parentheses (e.g., (29321) and (2228)) indicate the number of unigenes that lie in the gap ratio (e.g., [0, 0.01) and [0.01, 0.05)).
Gene ontology (GO) and clusters of orthologous groups (COGs) classification
After functional annotation of unigenes (data not shown), the predicted P. citri genes were further classified by GO assignments. Based on homologous genes, 3,812 unigenes (Figure S1) from All-unigenes were categorized into 47 GO terms consisting of three domains: biological process, cellular component and molecular function (Figure 2). It was clear that the dominant distributions are from ‘Cellular process’, ‘Cell’, ‘Cell part’ and ‘Binding’ terms. We also observed a high percentage of genes assigned to ‘Biological regulation’, ‘Metabolic process’, ‘Organelle’, ‘Catalytic activity’ and a few genes assigned to ‘Cell killing’, ‘Synapse part’ and ‘Auxiliary transport protein activity’. In addition, we noticed some genes assigned to ‘Growth’, ‘Reproduction’, and ‘Developmental process’. However, no genes assigned to “Vinion”, “Vinior part” or “Electron carrier activity” were identified in this study (Fig. 2). To examine further the integrality of our transcriptome library and effectiveness of the annotation process, we calculated the unigene (with nr hits) numbers with COG classification. In total, there were 7,075 unigenes (Figure S1) identified from All-unigenes with a COG classification (Figure 3). Among the 25 COG categories, the cluster of ‘general function prediction’ occupied the highest number (2,480, 17%) followed by ‘Carbohydrate transport and metabolism’ (1,392, 9.4%), ‘Transcription’ (1,219, 8.2%) and ‘Translation, ribosomal structure and biogenesis’ (1,119, 7.5%). The categories of ‘Nuclear structure’ (4, 0.03%), ‘Extracellular structures’ (15, 0.1%) and ‘RNA processing and modification’ (71, 0.48%) had the fewest responding genes (Figure 3). To annotate further all-unigenes, we performed BLAST searches for all of them against the nr database of NCBI and obtained 17,457 unigenes, of which 7053 were present in the COG database (Figure S1).
Figure 2. Histogram representation of Gene Ontology classification.

GO categories, shown in the x-axis, are grouped into three main ontologies: biological process, cellular component and molecular function. The right y-axis indicates the number of genes in each category, while the left y-axis indicates the percentage of of total genes in that category. The ‘All-unigene’ indicates that the unigenes were those assembled from reads from the SS&RS sample.
Figure 3. Histogram representation of clusters of orthologous groups (COG) classification.

7,075unigenes were assigned to 25 categories in the COG classification. The y-axis ‘Frequency’ indicates the number of genes in a specific function cluster. The right legend shows a description of the 25 function categories.
Network of Unigene
To investigate further biological behavior, we analyzed the biological pathways that are active in our samples. 32217 All-unigenes were mapped to the reference canonical pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) [36] and 12712 of them obtained KEGG annotation, in which 102 unigenes were exclusively in the KEGG database (Figure S1). Further analyses found that 12,663 unigenes were assigned to the 219 KEGG pathways. There was a large number of unigenes restricted to only a single one pathway, such as metabolism pathway (1,983 unigenes), cancer pathway (623 unigenes) and lysosome pathway (477 unigenes). In constrast, we also found one unigene in one pathway, for example, polyketide sugar unit biosynthesis pathway (only one unigene), caffeine metabolism pathway (only one unigene). In terms of all above analyses, we had 17,581 unigenes annotated in the citrus red mite transcriptome (Figure S1).
Differential expression and pathway analyses in RS and SS
We developed a rigorous algorithm (see Methods) to identify genes differentially expressed between RS and SS by referring to “The significance of digital gene expression profiles” [37]. To explore further the gene expression levels, the Reads Per kb per Million reads (RPKM) method [38] was adapted to eliminate the influence of variation in gene length and the total reads number. 15,179 unigenes were up-regulated and 16,656 were down-regulated (Figure 4). Furthermore, the significance of gene expression differences was judged by using the threshold of false discovery rate (FDR ≤ 0.001) and the absolute value of log2Ratio (≥ 1). 2,701 significantly differentially expressed genes were obtained between the two samples, including 1,967 down-regulated and 734 up-regulated genes (Figure 4). The number of down-regulated genes was more than 2-fold that of up-regulated genes, which might be consistent with the observation that hexythiazox-poisoned individuals of P. citri are less active. To investigate further the expression difference, we examined the top ten up-regulated transcripts (Figure 5), in which the most up-regulated unigene (up to 15.24-fold) is homologous to F0F1-type ATP synthase.
Figure 4. Differences in gene expression profile between the Resistant Strain and Susceptible Strain.

‘ALL DEGs’ indicates those unigenes showing differential expression between the two samples, while ‘DEGs’ indicates those unigenes with FDR ≤ 0.001 and |log2Ratio| ≥ 1. The numbers of genes up-regulated and down-regulated in the Resistant Strain relative to the Susceptible Strain are indicated above the blue or red bars, respectively.
Figure 5. Analysis of expression levels of the top ten up-regulated transcripts between the two samples (RS vs. SS).

The y-axis indicate the fold-changes of up-regulated genes. Categories in the x-axis represent genes homologous to (A) ‘heme/copper-type cytochrome/quinol oxidases, subunit 1’, (B) ‘F0F1-type ATP synthase, subunit a’, (C) ‘heme/copper-type cytochrome/quinol oxidases, subunit 2’, (D) ‘cytochrome b subunit of the bc complex’, (F) ‘heme/copper-type cytochrome/quinol oxidase, subunit 3’ and (J) ‘NADH: ubiquinone oxidoreductase subunit 1 (chain H)’,respectively. The remaining genes (E, G, H, I) had no functional annotation.
To explore the biological function of the significantly differentially expressed genes, 2,701 of these were mapped to the KEGG database, and then enriched to important pathways such as metabolism and signal transduction based on the whole transcriptome background. There were 1,038 unigenes mapped to 198 pathways. Remarkably, specific pathways were observed that are involved in energy and reproduction, such as glycolysis, pentose phosphate pathway, TCA cycle, and cell cycle. Interestingly, all genes mapping to the above four pathways were dramatically down-regulated in the resistant strain. In contrast, genes involved in drug metabolism and steroid hormone biosynthesis pathways were substantially up-regulated, which may have biological relevance for survival under the selection of the pesticide for P. citri. Interestingly, we also noticed differentially expressed genes implicated in many disease pathways such as Parkinson’s disease, Huntington's disease, prion diseases and autoimmune thyroid disease.
Transcripts related to general pesticide detoxification and targets
To date, many genes involved in general insecticide resistance have been identified. Little is known as to whether these resistance genes are related to the resistance of phytophagous mites, such as P. citri, to special acaricides. We therefore blasted P. citri transcriptomes against the NCBI nucleotide database and obtained many genes related to general pesticide resistance in other insect species.
Numerous transcripts identified in the P. citri transcriptomes are homologous to genes encoding enzymes with previously identified roles in general pesticide detoxification. In total, there were 121, 30, 43, 2 and 4 unigenes related to cytochrome P450 monoxygenases (CYPs), Glutathione S-transferase (GSTs), Carboxylesterase, NADH dehydrogenase and Superoxide dismutase (SODs), respectively. We only selected 46 of the P450s for functional annotation according to their degree of sequence matching (E-value < le-5) (Table 2), owing to the low sequence similarity of other P450s. In addition, only a few unigenes have been sequenced so far, except for NADH dehydrogenases, in the mitochondrial genome of P. citri [18]–[19]. We found 17 genes including 9 of P450s, 4 GSTs, 3 carboxylesterases and 1 esterase that were greater than value of 2 based on a log2ratio formula (Figure 6). Particularly, 4 of the P450s belonging to the CYP3 clade (which includes CYP3, CYP6 and CYP9 members) were annotated to the xenobiotic metabolism pathway (ko00982 and ko00980), and are related to many general pesticides such as deltamethrin, permethrin, dichlorodiphenyltrichloroethane (DDT) and Imidacloprid [39]–[43]. One up-regulated GST was annotated to the xenobiotic metabolism pathway (ko00980). These data support the notion that the P450s, GSTs, and others, are likely to be involved in hexythiazox resistance in P. citri.
Table 2. Genes involved in insecticide metabolism.
| Candidate gene name | Number of sequences with nr1 hit | Number of known sequences from RefSeq2 |
| Cytochrome P450 | 46 | 2 |
| Glutathione S-transferase | 30 | 0 |
| Carboxylesterase | 43 | 0 |
| Catalase | 14 | 0 |
| Superoxide dismutase | 4 | 0 |
| NADH dehydrogenase | 2 | 21 |
| Esterase | 10 | 2 |
| Sodium channel | 41 | 0 |
| Cytochrome b | 2 | 11 |
| GABA receptor | 19 | 0 |
Number of unigenes obtained in our transcriptome database that had a hit with the corresponding proteins in non-redundant (nr) protein database of National Center for Biotechnology Information (NCBI).
Number of sequences from the Reference Sequence (RefSeq) database of National Center for Biotechnology Information (NCBI) that have homology with corresponding proteins (as of January 2011).
Figure 6. Pie chart representation of several genes, mainly implicated in insecticide metabolism.

The number preceding the each percentage indicates the number of unigenes encoding a specific metabolism enzyme with log2Ratio greater than 2. The percentages indicate the propotion of each specific unigenes in four kinds of known enzyme unigenes.
The majority of cases of pesticide resistance result from a point mutation in a single target gene. In the P. citri database, we identified a number of unigenes encoding pesticide resistance target proteins (Table 2), of which sodium channel and GABA receptor were first reported in P. citri. Sodium channels have been shown to be targets of many pesticides such as pyrethroids [44], organophosphates [45], while GABA receptors are targets of ivermectin and avermectins [46]. We also identified mitochondrial cytochrome b genes (Table 2) that have previously been demonstrated as targets of bifenazate in P. citri [19]. To our knowledge, cytochrome b is the only gene in which a mutation has been shown to mediate special acaricide resistance in P. citri. A sequence comparison of cytochrome b between our transcriptome data and the public database is shown in Text S1. In addition, a limited number of SNPs were found in different sequences that are related to the target resistance between resistant and susceptible strains (Table S1).
Analysis of genes related to the growth and reproduction process
The primary mode of action of hexyzhiazox has been proposed to involve interfering with the growth and reproduction of mites [8], [12]. However there are no available genes participated in above proposal in the public database in P. citri to date. After GO classification of the unigenes, we identified 105 and 194 unigenes related to growth and reproduction, respectively (Table S2, Text S2), and several of them are important targets of pesticides in insects. For example, azadirachtin, halofenazide, tebufenoxide and pyriproxyfen, which target growth, can explain the mode of action of hexyzhiazox due to a critical brain neuropeptide, prothoracicotropic hormone (PTTH) [47]. To elucidate the relationship between genetic changes and hexyzhiazox resistance in P. citri, we compared transcribed sequences between resistant and susceptible strains, and identified some unigenes related to growth and reproduction (Table S3). Because there are some false positive rates in RNA-Seq data, these different sequences need to be further examined by PCR. If these different sequences between different strains are confirmed, it is very likely that further functional studies will identify them as new hexyzhiazox resistance genes.
Conclusion
Illumina sequencing, a rapid and cost-effective method, provides an ideal means with which to analyze P. citri transcriptomes. It produced 32,217 unigenes with 17,581 unigenes annotated. These findings greatly extend the existing sequence resources for P. citri and will provide abundant genetic information for furthering understanding of the molecular mechanisms of acaricide resistance. In addition, this study identified 2,701 significantly differentially expressed genes between RS and SS. Most importantly, we identified and compared differences in genes related to both general pesticide resistance and to organism growth and reproduction. Many potential candidate genes related to special acaricide resistance in P. citri have been identified. The results indicated that acaricide resistance may be a complex biological process involving many genetic changes. To our knowledge, this is the first study to discover candidate genes involved in phytophagous mite resistance by comparative transcriptomics.
Materials and Methods
Citrus red mite rearing and resistant mite selection
A colony of Citrus red mite, Panonychus citri (McGregor), was collected from citrange next to citrus orchard fences (Citrus Research Institute, Chinese Academy of Agricultural Sciences), which had never been exposed to acaricides for many years. These mites were reared in citrange for 3 years under acaricide-free conditions (25±1°C, 80±5% RH and photoperiod of 14 hours light: 10 hours darkness) and considered as a susceptible strain. The susceptible strain was treated with hexyzhiazox (about 70% population mortality rate in every spraying) and continuously screened for 20 generations – this resultant strain was regarded as the resistant strain. The selection process and the resistant fold changes are shown in Table S4. The mixture of eggs, larvae and adults was collected in a PE (PerkinElmer) tube as one sample for SS and RS, respectively. The two samples were stored at −80°C until use.
RNA isolation, integrity examination and RNA-seq library preparation
Total RNA was extracted with TRIzol regent (Invitrogen, USA) according to the manufacturer's instruction and treated with RNase-free DNase I (Takara Biotechnology, China). RNA integrity was confirmed with a minimum RNA integrated number value of 8 by the 2100 bioanalyzer (Agilent). Poly(A) mRNA was isolated with oligo-dT beads and then treated with the fragmentation buffer. The cleaved RNA fragments were then transcribed into first-strand cDNA using reverse transcriptase and random hexamer primers. This was followed by second strand cDNA synthesis using DNA polymerase I and RNaseH. The double stranded cDNA was further subjected to end-repair using T4 DNA polymerase, Klenow fragment, and T4 Polynucleotide kinase followed by a single <A> base addition using Klenow 3′ to 5′ exo− polymerase. It was then ligated with adapter or index adapter using T4 quick DNA ligase. Adaptor ligated fragments were selected according to the size and the desired range of cDNA fragments were excised from the gel. PCR was performed to selectively enrich and amplify the fragments. Finally, after validating on an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System, the cDNA library was sequenced on a flow cell using Illumina HiSeq2000.
Assembly and functional annotation
De novo assembly of the short reads was carried out using SOAPdenovo [34] as described. After filtration of the low quality reads, the raw reads were cleaned up by removing adapter sequences. The reads obtained were randomly clipped to 21-bp k-mers, which were assembled using de Bruijin graph and SOPAdenovo software. After estimating the performance of different k-mer sizes, we found that the 21-mer was best to assemble the transcriptome, showing the de Bruijin graph complexity using small K-mers and the poor overlap regions using large K-mers. Furthermore, the reads producing large fragment without N was named contig. The contigs were joined into scaffolds using the paired-end reads. The paired-end reads were also used to fill the gaps in scaffolds, where the unigenes have the least Ns and cannot be extended on both ends. To obtain unique gene sequences, the unigenes were clustered using TGI Clustering tools [48]. In the last step, blastx (evalue < 0.00001) was employed to search for homologues of our obtained unigenes in protein databases such as nr, Swiss-Prot, KEGG and COG. The best results were used to further determine the sequence orientation of unigenes. If results from different databases conflicted with each other, a priority order (nr, Swiss-Prot, KEGG and COG) was followed to determine the sequence orientation. When a unigene aligned to none of the above databases, ESTScan software [49] was used to predict its coding regions and to determine its sequence orientation. Functional annotation by gene ontology terms (GO; http://www.geneontology.org) was analyzed with the program Blast2go. The COG and KEGG pathway annotations were performed using Blastall software against the Cluster of orthologous Groups database and the Kyoto Encyclopedia of Genes and Genomes database.
Differential expression of unigenes
The uniquely mapped reads for a specific transcript were counted by mapping reads to assembled sequences using SOAP [50]. Then the RPKM value for each transcript was measured in reads per kilobase of transcript sequence per million mapped reads [27]. The transcript fold change was then calculated by the formula of log2(RS_ RPKM/SS_RPKM). If the value of either RS_ RPKM or SS_RPKM was zero, we used 0.01 instead of 0 to calculate the fold change. We modified Audic's [36] method to analyze differential expression. The formula to calculate the probability of a specific gene being expressed equally between the two samples was defined as
where, N1 and N2 indicate the total number of clean reads in SS and RS, respectively, and x and y indicate the mapped clean read counts of the transcript in each sample respectively. We then used the FDR (False Discovery Rate) method to determine the threshold of the p value in multiple tests. In this study, we used ‘FDR ≤ 0.001 and the absolute value of log2Ratio ≥ 1’ as the threshold to judge the significance of differentiated gene expression.
Gene Ontology Functional Enrichment Analysis for differentially expressed genes (DEGs)
The analysis firstly maps all DEGs to GO terms in the database by virtue of calculating gene numbers for every term, followed by an ultra-geometric test to find significantly enriched GO terms in DEGs compared to the transcriptome background. The formula for calculation was
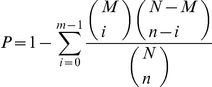 |
where: N is the number of all genes with GO annotation; n is the number of DEGs in N; M is the number of all genes that are annotated to the certain GO terms; m is the number of DEGs in M. The calculated p value was subjected to a Bonferroni Correction, taking a corrected p value of 0.05 as a threshold. GO terms fulfilling this condition were defined as significantly enriched GO terms in DEGs. This analysis is able to recognize the main biological functions that DEGs exercise. Our GO functional enrichment analysis also integrates the clustering analysis of expression patterns. Thus, we can easily obtain the expression patterns of DEGs annotated with a given GO-term.
DEG pathway analysis
We used the Blastall program to annotate the pathways of DEGs against the KEGG database. Subsequently, we adopted the same formula as that in GO analysis to carry out DEG pathway enrichment analysis. Here N is the number of all genes with KEGG annotation, n is the number of DEGs in N, M is the number of all genes annotated to specific pathways, and m is the number of DEGs in M.
Data deposition
Our sequencing data has been deposited in EMBL-EBI (accession: http://www.ebi.ac.uk/ena/data/view/ERP000885).
Supporting Information
17,581 all-unigenes were annotated with nr, Gene Ontology (GO), Clusters of Orthologous Groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG). The smallest circle, the second smallest circle, medium-sized circle and the largest circle represent the numbers, as shown in parentheses, of all-unigenes with GO, COG, KEGG and nr annotations, respectively. The areas of H, I and J reflect the numbers of all-unigenes exclusively with annotations from KEGG, COG and Nr databases. The numbers of all-unigenes that have overlapping annotations from GO, COG and KEGG database is represented by area A (annotation shared by GO and Nr), B (annotation shared by GO, KEGG, and Nr), D (annotation shared by GO, COG and Nr), E (annotation shared by Nr and KEGG), F (annotation shared by COG, Nr and KEGG) and G (annotation shared by Nr and COG). The last area of C indicates the number of all-unigenes with overlapping annotations from GO, COG, KEGG and nr.
(TIF)
SNPs and the blastx results of genes related to GABA receptor and sodium channel.
(XLS)
GOs of growth and reproduction related genes.
(XLS)
SNPs and the blastx results of genes related to growth and reproduction.
(XLS)
Resistant strain selection process.
(DOC)
The comparation of both cytochrome b (cyt b) nucleotide sequence and its protein sequence between public database and current transcriptome datum. The Unigene12480_All means the identity of a gene from the assembly of using the reads from both the resistant and susceptible strains. This gene is homology to the gene of cyt b (GenBank acc. no. HM367068) existing in the NCBI. The alignment was performed by a online software ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/); The marked in gray motifs are conserved residues of the cd1-helix of the Qo pocket of cyt b (GenBank acc. no. ADJ66666) of Panonchus citri (P. citri). The two arrows indicate the locations of two amino acid substitutions G126S and A133T related to bifenazate resistance in P. citri (Citrus red mite).
(DOC)
The list of full sequences of unigenes invovled in growth and reproduction: (The words in green in the list indicate the blastx results to the corresponding unigenes).
(DOC)
Acknowledgments
We thank Beijing Genomics Institute at Shenzhen (BGI Shenzhen) for assisting in sequencing and Hongshi Yu at University of Melbourne for editing the manuscript.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by the Science and Technique Foundation of CQ (CSTC, 2010AC1018) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT0976). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bolland HR, Flechtmann CH, Gutierrez J. Brill Academic Publishers; 1998. World catalogue of the spider mite family: Acari: Tetranychidae.392 [Google Scholar]
- 2.Zhang Z. CABI publishing Wallingford UK; 2003. Mites of greenhouses: identification, biology and control.240 [Google Scholar]
- 3.Lee MH, Cho SH, Park HS, Bahn JW, Lee BJ, et al. Citrus red mite (Panonychus citri) is a common sensitizing allergen among children living around citrus orchards. Annals of Allergy, Asthma & Immunology. 2000;85:200–204. doi: 10.1016/S1081-1206(10)62467-2. [DOI] [PubMed] [Google Scholar]
- 4.Kim YK, Son JW, Kim HY, Park HS, Lee MH, et al. New occupational allergen in citrus farmers: citrus red mite (Panonychus citri). Annals of Allergy, Asthma & Immunology. 1999;82:223–228. doi: 10.1016/S1081-1206(10)62601-4. [DOI] [PubMed] [Google Scholar]
- 5.Kim YK, Son JW, Kim HY, Park HS, Lee MH, et al. Citrus red mite (Panonychus citri) is the most common sensitizing allergen of asthma and rhinitis in citrus farmers. Clinical & Experimental Allergy. 1999;29:1102–1109. doi: 10.1046/j.1365-2222.1999.00607.x. [DOI] [PubMed] [Google Scholar]
- 6.Furuhashi K. Development of acaricide resistance in the citrus red mite, Panonychus citri (Mcgregor), in Shizuoka Prefecture. Proc Kansai Plant Prot Soc. 1994;41:267–269. [Google Scholar]
- 7.Masui S, Ohishi T, Kasuya K, Togawa M, Tatara A. Present status of resistance of the citrus red mite, Panonychus citri (McGregor), to acaricides in Shizuoka Prefecture. Proc Kanto Plant Proct Soc. 1995;42:245–246. [Google Scholar]
- 8.Yamamoto A, Yoneda H, Hatano R, Asada M. Studies on hexythiazox resistnace in phytophagous mites. Stability of hexythiazox resistance in the citurs red mite, Panonychus citri (McGregor) under laboratory and field conditions. J Pestic Sci. 1996;21:37–42. [Google Scholar]
- 9.Meng HS, Wang KY, Jiang XY, Yi MQ. Studies on the resistance of Panonychus citri to several acaricides. Agrochemicals. 2000;39:26–28. [Google Scholar]
- 10.Ran C, Chen Y, Yuan ML, Wang JJ, Liu HQ, Yao TS. Susceptibility of Panonychus citri field populations to different acaricides. Acta Phytophylacica Sinica. 2008;35:537–540. [Google Scholar]
- 11.Hu JF, Wang CF, Wang J, You Y, Chen F. Monitoring of resistance to spirodiclofen and five other acaricides in Panonychus citri collected from Chinese citrus orchards. Pest Manag Sci. 2010;66:1025–1030. doi: 10.1002/ps.1978. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto A, Yoneda H, Hatano R, Asada M. Genetic analysis of hexythiazox resistance in the citrus red mite, Panonychus citri (McGregor). J Pesticide Sci. 1995;20:513–519. [Google Scholar]
- 13.Gough N. Evaluation of miticides for the control of two-spotted mite Tetranychus urticae Koch on field roses in southern Queensland. Crop Protection. 1990;9:119–127. [Google Scholar]
- 14.Campos FJ, Omoto C. Resistance to hexythiazox in Brevipalpus phoenicis (Acari: Tenuipalpidae) from Brazilian citrus. Exp Appl Acaol. 2002;26:243–251. doi: 10.1023/a:1021103209193. [DOI] [PubMed] [Google Scholar]
- 15.Hoy MA, Ouyang Y. Selectivity of the acaricides clofentezine and hexythiazox to the predator Metaseiulus occidentalis (Acari: Phytoseiidae). J Econ Entomol. 1986;79:1377–1380. [Google Scholar]
- 16.Ran C, Chen Y, Wang JJ. Susceptibility and carboxylesterase activity of five field populations of Panonychus citri (McGregor) (Acari: Tetranychidae) to four acaricides. Int J Acarol. 2009;35:115–121. [Google Scholar]
- 17.Chen ZY, Ran C, Zhang L, Dou W, Wang JJ. Susceptibility and esterase activity in citrus red mite Panonychus citri (McGregor) (Acari: Tetranychidae) after selection with phoxim. Int J Acarol. 2009;35:33–40. [Google Scholar]
- 18.Yuan ML, Wei DD, Wang BJ, Dou W, Wang JJ. The complete mitochondrial genome of the citrus red mite Panonychus citri (Acari: Tetranychidae):high genome rearrangement and extremely truncated tRNAs. BMC Genomics. 2010;11:597. doi: 10.1186/1471-2164-11-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leeuwen TV, Nieuwenhuyse PV, Vanholme B, Dermauw W, Nauen R, et al. Parallel evolution of cytochrome b mediated bifenazate resistance in the citrus red mite Panonychus citri, Insect Mol Biol. 2011;20:135–140. doi: 10.1111/j.1365-2583.2010.01040.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faunes F, Sánchez N, Castellanos J, Vergara IA, Melo F, et al. Identification of novel transcripts with differential dorso-ventral expression in Xenopus gastrula using serial analysis of gene expression. Genome Biol. 2009; 2009;10:R15. doi: 10.1186/gb-2009-10-2-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yassoura M, Kaplana T, Fraser HB, Levin JZ, Pfiffner J, et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci USA. 2009;106:3264–3269. doi: 10.1073/pnas.0812841106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegedus Z, Zakrzewska A, Agoston VC, Ordas A, Racz P, et al. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol Immunol. 2009;46:2918–2930. doi: 10.1016/j.molimm.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Zhu H, Ruan J, Qian W, Fang X, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 26.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 28.Hudson HE. Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol Ecol Resour. 2008;8:3–17. doi: 10.1111/j.1471-8286.2007.02019.x. [DOI] [PubMed] [Google Scholar]
- 29.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, et al. De novo transcriptome sequencing in Anopheles funestus Using Illumina RNA-Seq Technology. PLoS ONE. 2010;5(12):e14202. doi: 10.1371/journal.pone.0014202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XW, Luan JB, Li JM, Bao YY, Zhang CX, et al. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics. 2010;11:400. doi: 10.1186/1471-2164-11-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue J, Bao YY, Li BL, Cheng YB, Peng ZY, et al. Transcriptome Analysis of the Brown Planthopper Nilaparvata lugens. PLoS ONE. 2010;5(12):e14233. doi: 10.1371/journal.pone.0014233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen S, Yang P, Jiang F, Wei Y, Ma Z, et al. De Novo Analysis of Transcriptome Dynamics in the Migratory Locust during the Development of Phase Traits. PLoS ONE. 2010;5(12):e15633. doi: 10.1371/journal.pone.0015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price DP, Nagarajan V, Churbanov A, Houde P, Milligan B, et al. The fat body transcriptomes of the yellow fever mosquito Aedes aegypti, pre- and post- blood meal. PLoS One. 2011;6(7):e22573. doi: 10.1371/journal.pone.0022573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Zhu H, Ruan J, Qian W, Fang X, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2009;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Audic S, Claverie JM. The Significance of Digital Gene Expression?Profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 38.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 39.Jiang HB, Tang PA, Xu YQ, An FM, Wang JJ. Molecular characterization of two novel deltamethrin-inducible P450 genes from Liposcelis bostrychophila Badonnel (Psocoptera: Liposcelididae). Arch Insect Biochem Physiol. 2010;74:17–37. doi: 10.1002/arch.20358. [DOI] [PubMed] [Google Scholar]
- 40.Komagata O, Kasai S, Tomita T. Overexpression of cytochrome P450 genes in pyrethroid-resistant Culex quinquefasciatus. Insect Biochem Mol Biol. 2010;40:146–152. doi: 10.1016/j.ibmb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Nikou D, Ranson H, Hemingway J. An adult-specific CYP6 P450 gene is overexpressed in a pyrethroid-resistant strain of the malaria vector, Anopheles gambiae. Gene. 2003;318:91–102. doi: 10.1016/s0378-1119(03)00763-7. [DOI] [PubMed] [Google Scholar]
- 42.Daborn PJ, Yen JL, Bogwitz MR, Goff GL, Feil E, et al. A Single P450 Allele Associated with Insecticide Resistance in Drosophila. Science. 2002;297:2253–2256. doi: 10.1126/science.1074170. [DOI] [PubMed] [Google Scholar]
- 43.Karunker I, Benting J, Lueke B, Ponge T, Nauen R. Over-expression of cytochrome P450 CYP6CM1 is associated with high resistance to imidacloprid in the B and Q biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae). Insect Biochem Mol Biol. 2008;38:634–44. doi: 10.1016/j.ibmb.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Zhou L, Lawrence GG, Vineis JH, Mcallister JC, Wirtz RA, et al. Detection of Broadly Distributed Sodium Channel Alleles Characteristic of Insect Pyrethroid Resistance in West Nile Virus Vector Culex pipiens Complex Mosquitoes in the United States. J Med Entomol. 2009;46:321–327. doi: 10.1603/033.046.0217. [DOI] [PubMed] [Google Scholar]
- 45.Morin S, Williamson MS, Goodson SJ, Brown JK, Tabashnik BE, et al. Mutations in the Bemisia tabaci para sodium channel gene associated with resistance to a pyrethroid plus organophosphate mixture. Insect Biochem Mol Biol. 2002;32:1781–1791. doi: 10.1016/s0965-1748(02)00137-6. [DOI] [PubMed] [Google Scholar]
- 46.Feng XP, Hayashi J, Beech RN, Prichard RK. Study of the nematode putative GABA type-A receptor subunits: evidence for modulation by ivermectin. J Neurochem. 2002;83:870–878. doi: 10.1046/j.1471-4159.2002.01199.x. [DOI] [PubMed] [Google Scholar]
- 47.Kataoka H, Nagasawa H, Isogai A, Ishizaki H, Suzuki A. Prothoracicotropic hormone of the silkworm, Bombyx mori: amino acid sequence and dimeric structure. Agric Biol Chem. 1991;55:73–86. [PubMed] [Google Scholar]
- 48.Pertea G, Huang X, Liang F, Antonescu V, Sultana R. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 49.Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999:138–148. [PubMed] [Google Scholar]
- 50.Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
17,581 all-unigenes were annotated with nr, Gene Ontology (GO), Clusters of Orthologous Groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG). The smallest circle, the second smallest circle, medium-sized circle and the largest circle represent the numbers, as shown in parentheses, of all-unigenes with GO, COG, KEGG and nr annotations, respectively. The areas of H, I and J reflect the numbers of all-unigenes exclusively with annotations from KEGG, COG and Nr databases. The numbers of all-unigenes that have overlapping annotations from GO, COG and KEGG database is represented by area A (annotation shared by GO and Nr), B (annotation shared by GO, KEGG, and Nr), D (annotation shared by GO, COG and Nr), E (annotation shared by Nr and KEGG), F (annotation shared by COG, Nr and KEGG) and G (annotation shared by Nr and COG). The last area of C indicates the number of all-unigenes with overlapping annotations from GO, COG, KEGG and nr.
(TIF)
SNPs and the blastx results of genes related to GABA receptor and sodium channel.
(XLS)
GOs of growth and reproduction related genes.
(XLS)
SNPs and the blastx results of genes related to growth and reproduction.
(XLS)
Resistant strain selection process.
(DOC)
The comparation of both cytochrome b (cyt b) nucleotide sequence and its protein sequence between public database and current transcriptome datum. The Unigene12480_All means the identity of a gene from the assembly of using the reads from both the resistant and susceptible strains. This gene is homology to the gene of cyt b (GenBank acc. no. HM367068) existing in the NCBI. The alignment was performed by a online software ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/); The marked in gray motifs are conserved residues of the cd1-helix of the Qo pocket of cyt b (GenBank acc. no. ADJ66666) of Panonchus citri (P. citri). The two arrows indicate the locations of two amino acid substitutions G126S and A133T related to bifenazate resistance in P. citri (Citrus red mite).
(DOC)
The list of full sequences of unigenes invovled in growth and reproduction: (The words in green in the list indicate the blastx results to the corresponding unigenes).
(DOC)