

Abstract
The histone deacetylase inhibitors (HDIs) have shown promise in the treatment of a number of hematologic malignancies, leading to the approval of vorinostat and romidepsin for the treatment of cutaneous T-cell lymphoma and romidepsin for the treatment of peripheral T-cell lymphoma by the U. S. Food and Drug Administration. Despite these promising results, clinical trials with the HDIs in solid tumors have not met with success. Examining mechanisms of resistance to HDIs may lead to strategies that increase their therapeutic potential in solid tumors. However, relatively few examples of drug-selected cell lines exist, and mechanisms of resistance have not been studied in depth. Very few clinical translational studies have evaluated resistance mechanisms. In the current review, we summarize many of the purported mechanisms of action of the HDIs in clinical trials and examine some of the emerging resistance mechanisms.
Keywords: histone deacetylase inhibitor, resistance, romidepsin, vorinostat, panobinostat
Introduction
In the nucleus, DNA is wound around four core histone proteins (H2A, H2B, H3 and H4, see Figure 1) to form the nucleosomes, which, when compacted, form the condensed structure of chromatin. Each histone protein in the nucleosome has a lysine-rich tail that extends outside of the nucleosome and the accessibility of DNA within the nucleosome is, in part, controlled by modifications of the tail. Histones can be modified in a number of ways, including acetylation, methylation, phosphorylation, ubiquitination, sumoylation and citrullination. As shown in Figure 1, the lysines of the histone tails can be modified by methylation and acetylation. Acetylation is controlled by two enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs transfer acetyl groups to the lysine residues of the histones, which neutralizes the positively charged lysines, decreasing attraction of the negatively charged DNA, thereby resulting in greater access by transcription factors and RNA polymerase. HDACs, on the other hand, remove acetyl groups, resulting in decreased access to DNA1. Changes in global histone acetylation were found to be associated with tumorigenesis in some disease models2–4.
Figure 1.

Modification of lysines in histone tails. DNA is wound around four core histone proteins: H2A, H2B, H3 and H4. Each of the histones possess lysine-rich tails and accessibility of the DNA is controlled by modifications to the tail. Lysines can either be multiply methylated or acetylated. Methylation and deacetylation of lysines both contribute to a more condensed chromatin structure, preventing transcription of genes. Demethylation and acetylation promote a more open chromatin structure allowing for increased gene transcription.
Most of the human HATs function as co-activators of transcription2. The most studied function of this group of proteins is the acetylation of the histone tails, although they can also acetylate various cellular transcription factors2. Similarly, while HDACs are primarily known for their ability to deacetylate histones, they have also been shown to deacetylate other proteins such as tubulin, p53, Hsp90, Bcl-2, and Ku705, 6. Based on their similarity to yeast HDAC proteins, HDACs are divided into four classes (compiled from references1, 7):
Class I
HDACs 1-3, and 8, 40–55 kD proteins, belong to this class and are ubiquitously expressed in human tissues. HDACs 1, 2, and 3 are localized in the nucleus while HDAC 8 is located in both the cytoplasm and the nucleus. This class of HDACs shares a structural similarity with the yeast transcription factor Rpd-3.
Class IIA, IIB
HDACs 4, 5, 7, and 9, make up class IIA and HDACs 6 and 10 make up the class IIB HDACs, all of which are 70–130 kD proteins. They share a structural similarity with the yeast HDA1 deacetylase. HDAC6 is unique in that it is a cytoplasmic HDAC that does not deacetylate histones.
Class III
This group of HDACs, known as the sirtuins, is made up of HDACs structurally similar to the yeast SirT2, and requires NAD+ as a cofactor for enzymatic activity.
Class IV
The only known HDAC of this class is HDAC 11. Relatively little is known about this HDAC, which is localized to the nucleus.
Histone deacetylase inhibitors
The histone deacetylase inhibitors (HDIs) are a promising class of chemotherapeutic agents that have been added to the anticancer armamentarium. HDIs prevent the deacetylase activity of HDACs, leading to unrestricted HAT activity and increased gene transcription. Several HDAC inhibitors are currently in clinical trials both in monotherapy and in combination therapy with other anti-tumor drugs. The HDIs currently in clinical trials fall primarily into the short-chain fatty acid class, the hydroxamate class, the cyclic peptide class and the benzamide class. A partial listing of these HDIs is provided in Table 1, and structures for several are provided in Figure 2. The HDACs that are targets for the HDIs discussed are all zinc-dependent enzymes. To date, most of the responses using HDAC inhibitors as single agents were observed in advanced hematological cancers and only very few were observed in solid tumors. In hematological malignancies, clinical efficacy has been observed in cutaneous T-cell lymphoma (CTCL), peripheral T cell-lymphoma (PTCL), Hodgkin and non-Hodgkin lymphoma, while only a few responses were observed in patients with myeloid malignancies8.
Table 1.
Partial list of histone deacetylase inhibitors currently in clinical trials for the treatment of cancer.
| Class | Compound |
|---|---|
| Aliphatic acids | Valproic acid |
| AR-42 (OSU-HDAC42) | |
| Hydroxamic Acids | *Vorinostat (suberoylanilide hydroxamic acid, SAHA) |
| Belinostat (PXD101) | |
| Dacinosat (LAQ824) | |
| Panobinostat (LBH589) | |
| Resminostat (4SC-201) | |
| PCI-24781 | |
| SB939 | |
| CHR2845 | |
| CHR3996 | |
| JNJ-26481585 | |
| Benzamides | Entinostat (MS-275) |
| Mocetinostat (MGCD0103) | |
| 4SC-202 | |
| Cyclic peptides | *Romidepsin (depsipeptide, FK228, FR901228) |
Compounds marked with an * are currently FDA approved. Belinostat is currently in registration trials.
Figure 2.

Structures of some of the histone deacetylase inhibitors currently in clinical trials.
Some of the HDAC inhibitors that are being studied in clinical trials have demonstrated therapeutic potential in CTCL and other malignancies and are detailed below:
Vorinostat (suberoylanilide hydroxamic acid, SAHA)
An orally-available, pan-HDAC inhibitor, vorinostat was found in in vitro studies to facilitate transcription of genes associated with growth arrest, differentiation, and apoptosis9, 10. Responses in patients with refractory CTCL led to the approval of vorinostat in 2006 by the Food and Drug Administration (FDA) for the treatment of patients with relapsed or refractory CTCL11. In the registration trial, patients were treated with 400 mg daily of oral vorinostat, with an overall response rate of approximately 30% and a response duration of over 6 months11. Promising results have also been observed in follicular lymphoma and marginal zone lymphoma12. Combinations with the proteasome inhibitor bortezomib in the treatment of multiple myeloma have also seen clinical success13. In contrast, single agent trials with vorinostat for the treatment of most solid tumors have not met with success14.
Romidepsin (FK228, FR901228, NSC630176, depsipeptide)
Romidepsin is unique among the HDIs in that it is actually a prodrug; the disulfide bond of romidepsin must be reduced to yield the active form15. Most studies seem to suggest that romidepsin is an inhibitor of class I HDACs16, but some studies also find romidepsin treatment leads to Hsp90 acetylation, leading to speculation that it might somehow affect HDAC617. In 2001, we first reported the efficacy of romidepsin in a phase I trial where partial responses (PRs)were observed in three patients with CTCL and a complete response(CR) was observed in one patient with PTCL18. These early successes in T-cell lymphoma led to two registration trials, culminating in the approval of romidepsin in November 2009 for the treatment of CTCL patients who had received at least one prior systemic therapy8. In the trial sponsored by the National Cancer Institute, among 71 patients with CTCL, the overall response rate was 34% with a median duration of response of 13.7 months19. In a second, independent, international trial of 96 patients with CTCL, the overall response rate was 38% and the median duration of response was 15 months20. In PTCL, an overall response rate of 38% was observed with a median duration of response of 8.9 months in a number of subtypes21. Romidepsin was recently approved by the FDA for the treatment of patients diagnosed with PTCL. As with vorinostat, results in solid tumors have been disappointing22–24.
Panobinostat (LBH589)
The first clinical trials with this pan-HDAC inhibitor were conducted in patients with acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and myelodysplastic syndrome (MDS)25. Panobinostat is currently in phase I and II clinical trials, with the most significant anti-tumor activity of this drug observed in patients with refractory CTCL and other hematologic malignancies26, 27. As with the other HDIs, panobinostat has not been successful in solid tumor clinical trials26, so that the overall activity in T-cell lymphoma and inactivity in solid tumors appears to be a class effect.
Belinostat (PXD101)
Belinostat is another pan-HDAC inhibitor that has been studied in multiple clinical trials as a single agent or in combination with chemotherapeutic agents. A phase I trial of this drug reported disease stabilization in patients with hematological malignancies28. Like romidepsin, belinostat has produced responses in patients diagnosed with PTCL29. Preliminary results from a phase II trial in lymphoma have been promising30.
Other HDIs
Less clinical data are available for some of the other HDIs. Some clinical activity was observed with valproic acid in pediatric patients with central nervous system tumors31, but not in a phase I trial for prostate cancer32. In a phase I trial, entinostat, an HDAC1-specific inhibitor, produced a partial remission in a patient with melanoma33; however, a subsequent phase II trial did not yield any objective responses34. Mocetinostat is a class I selective HDI that has demonstrated clinical activity in non-Hodgkin lymphoma and in relapsed or refractory Hodgkin lymphoma35. One patient with myelodysplastic syndrome and two patients with AML responded to single-agent mocetinostat in a phase I trial in leukemia36, while a phase II trial of mocetinostat in patients with chronic lymphocytic leukemia did not yield any responses37. A phase I trial of dacinostat in solid tumors demonstrated degradation of CRAF, an Hsp90 client protein, and increased histone acetylation in circulating peripheral blood mononuclear cells, but no responses were observed38.
Mechanisms of action
One global effect of HDI treatment is an increase in histone acetylation. In clinical trials of HDIs, increased histone acetylation has been monitored in circulating peripheral blood mononuclear cells as a surrogate marker for the inhibition of HDACs39. However, this effect alone is apparently inadequate to confer activity, as solid tumor trials have demonstrated increased histone acetylation in tumor samples despite little clinical effect40, 41 In the search for the mechanism of action of HDIs, several have been suggested, including cell cycle arrest, activation of apoptotic pathways, induction of autophagy, reactive oxygen species generation, Hsp90 inhibition, and disruption of the aggresome pathway1, 7, 14. We elaborate on several of these potential mechanisms of action below:
Alteration of gene expression
Studies with cDNA arrays have shown that treatment with HDIs such as sodium butyrate, entinostat, vorinostat or romidepsin leads to a two-fold or greater change in the expression of approximately 7–10% of the genes examined1. HDI treatment was found to induce about as many genes as were repressed. The histone deacetylase inhibitors induce p21 expression42, leading to G1 cell cycle arrest, and frequently downregulate cyclin D and c-myc. Whether or not these gene expression changes result in cell death probably depends upon cellular context. For example, decreased expression of c-myc will probably be more important for myc-dependent cancers, such as Burkitt’s lymphoma, than for cancers that do not rely on c-myc for survival43. Vorinostat treatment has been shown to lead to decreased cyclin D1 expression and cell death in mantle cell lymphoma cell lines44.
Degradation of Hsp90 client proteins
Hsp90 is required to stabilize a number of “client” proteins that play a role in cancer cell proliferation, such as the human epidermal growth factor receptor 2 (ErbB-2, Her-2) and epidermal growth factor receptor (ErbB-1, EGFR); the fusion oncogene Bcr-Abl; and signaling transduction molecules such as Akt45. HDAC6 has been shown to be a deacetylase of both tubulin and Hsp9046. Treatment of cells with compounds that are known to inhibit HDAC6 leads to increased tubulin acetylation, increased Hsp90 acetylation and degradation of Hsp90 client proteins, having much the same mechanism of action as Hsp90 inhibitors.
Romidepsin was one of the first HDIs found to induce Hsp90 acetylation and cause degradation of the Hsp90 client proteins EGFR, Her-2 and Raf-117. In breast cancer cell lines overexpressing Her-2, treatment with vorinostat or panobinostat has been shown to lead to increased Hsp90 acetylation, Her-2 degradation and cell death, and synergy has been demonstrated when the HDIs were combined with Hsp90 inhibitors or Her-2 inhibitors such as trastuzumab or lapatinib47, 48. Similarly, treatment of lung cancer cell lines expressing mutant EGFR with panobinostat resulted in decreased EGFR expression and synergized with the EGFR inhibitors erlotinib or lapatinib48, 49. Leukemia cells expressing the Bcr-Abl fusion protein are particularly sensitive to treatment with vorinostat, dacinostat, romidepsin or panobinostat and cotreatment with imatinib or nilotinib results in synergistic cell death50–53. Again, the ability of an HDI to inhibit HDAC6 would probably be only effective in cancers where proliferation is driven by Hsp90 client proteins, although cell death could also occur as a result of deacetylation of other HDAC6 substrates54.
It is not clear, however, whether HDAC6 is the sole deacetylase of Hsp90. Romidepsin is considered a rather weak inhibitor of HDAC6, as romidepsin treatment does not result in acetylation of tubulin42, yet has still been shown to cause acetylation of Hsp9017. As seen in Figure 3, treating HEK293 (Flp-In-293) human embryonic kidney cells with varying concentrations of romidepsin, vorinostat, panobinostat or valproic acid results in increased histone H3 acetylation, but only results in tubulin acetylation in cells treated with vorinostat and panobinostat. Additionally, entinostat treatment has been shown to result in apoptosis due to degradation of mutant FLT-3, another Hsp90 client protein, in leukemia cells that express the mutant protein despite the fact that entinostat does not inhibit HDAC655. It has been suggested the HDAC1 mediates this effect56. One study has suggested that acetylation of Hsp70 mediates degradation of the Bcr-Abl fusion protein57. Further study is needed to determine how HDIs that are weak inhibitors of HDAC6 mediate apoptosis in cell lines where proliferation is driven by Hsp90 client proteins.
Figure 3.
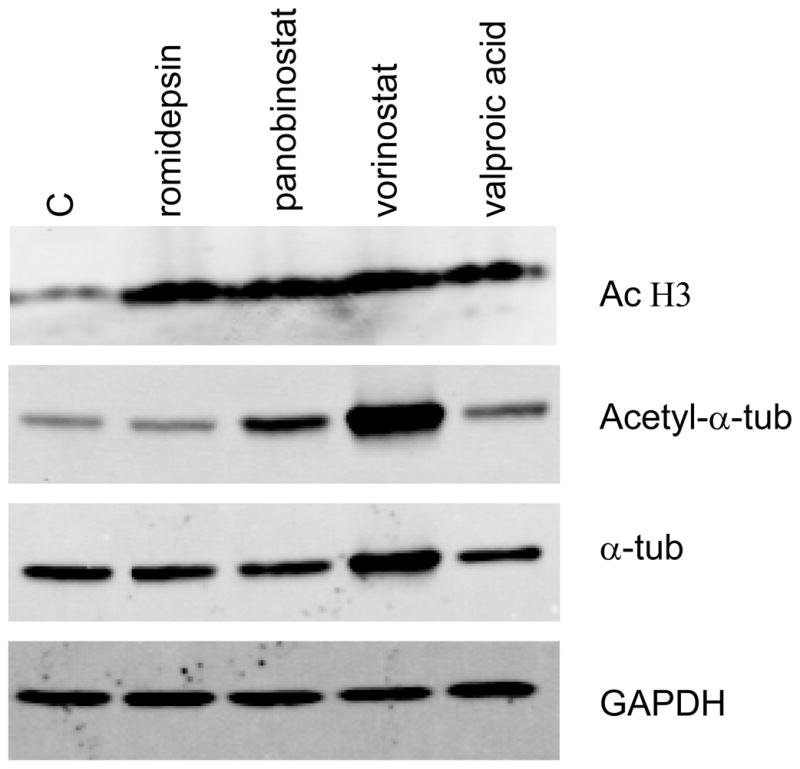
Histone deacetylase inhibitors all cause increased histone acetylation but differentially cause tubulin acetylation. HEK293 (Flp-In-239) cells were treated with 46 nM romidepsin, 100 nM panobinostat, 10 μM vorinostat or 1 mM valproic acid for 24 h after which protein was extracted, subjected to electrophoresis, and transferred to a PVDF membrane. The membrane was subsequently probed for acetylated histone H3 (AcH3), acetylated α-tubulin (acetyl-α-tub), total α-tubulin (α-tub) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). While all of the HDIs were able to induce histone acetylation, only panobinostat and vorinostat were able to cause increased tubulin acetylation, suggesting that these HDIs also target HDAC6
Increased production of reactive oxygen species (ROS)
Several studies have suggested that the generation of reactive oxygen species (ROS) is a key event in HDI-induced cell death. ROS generated by HDIs leads to DNA damage and the addition of free radical scavengers such as N-acetyl cysteine during the time of HDI treatment has been shown to result in decreased ROS generation and decreased HDI-mediated cell death58–61. HDI treatment has also been shown to prevent repair of DNA damage62, 63. Another mechanism by which HDIs increase ROS production is through downregulation of thioredoxin and upregulation of thioredoxin-binding protein 2 (TBP-2). Thioredoxin is a thiol reductase that acts as a scavenger of ROS, and TBP-2 has been shown to be a negative regulator of thioredoxin, decreasing its reducing activity64. Vorinostat treatment has been shown to not only increase TBP-2 expression, but also to suppress thioredoxin expression65.
Alterations in the apoptotic pathway
Apoptosis proceeds either via the extrinsic or cell death receptor-mediated pathway or the intrinsic or mitochondria-mediated pathway. Several HDIs have been shown to facilitate death by the extrinsic pathway by causing an increase in the expression of TRAIL, DR-4, DR-5, Fas and FasL as well as a decrease in c-FLIP, a protein associated with resistance to TRAIL-mediated apoptosis66–68. Activation of the apoptotic machinery associated with the intrinsic pathway, by decreasing expression of the anti-apoptotic proteins Bcl-2, Bcl-XL, Mcl-1 and survivin and increasing expression of the pro-apoptotic proteins Bax and Bim, seems to be a class effect of the HDIs47, 49, 58, 69.
Mechanisms of resistance
As the clinical course of the HDIs is pursued, it becomes important to identify potential mechanisms of resistance so as to increase efficacy and identify potential drug combinations. Despite the fact that HDIs have been in development for several years, relatively few drug-selected cell lines have been developed. Most of these in vitro selections have been with romidepsin alone, where emergence of P-glycoprotein (Pgp) seems to be the dominant mechanism of resistance70–72. Non-Pgp mechanisms of resistance have only been observed when cells are selected with romidepsin in the presence of a Pgp inhibitor71. However, some mechanisms of resistance have been identified by transfection with purported resistance mechanisms. Summarized below are some of the major mechanisms of resistance to HDIs that have been characterized.
As will be noted below, the search for resistance mechanisms has primarily centered on laboratory models, and sparse clinical data have been gathered. Most studies have detected histone acetylation, whether in peripheral blood mononuclear cells or in biopsy samples, and some have concluded that the presence of histone acetylation does not correlate with response to therapy73. Our data with romidepsin suggested otherwise, when we compared histone acetylation analyzed by an immunodot blot assay with response on a clinical trial in cutaneous and peripheral T-cell lymphoma. It appeared that higher and more durable levels of histone acetylation in peripheral blood mononuclear cells were associated with better clinical response39. These findings may have been unique to romidepsin, which as a prodrug, requires reduction of the disulfide bond to an active form15.
ATP-binding cassette transporters
Treatment with histone deacetylase inhibitors, such as sodium butyrate, was found several years ago to induce a more differentiated phenotype, accompanied by increased expression of the multidrug-resistance gene, MDR1 (ABCB1), and its product, Pgp74, 75. Trichostatin A treatment was also found to increase Pgp expression 76. Romidepsin was first brought to our attention after it was identified as a Pgp substrate based on rhodamine efflux patterns in the NCI Anticancer Drug Screen and was also found to induce expression of Pgp77. Valproic acid and apicidin have also been shown to increase ABCB1 expression78, 79; thus upregulation of ABCB1 and increased expression is believed to be a class effect of histone deacetylase inhibitors.
Romidepsin is unique among the HDIs in that it is also a substrate of Pgp, while other HDIs, such as vorinostat and belinostat, are not 80, 81. Cell lines expressing the multidrug resistance-associated protein-1 (MRP1/ABCC1) have also been found to be resistant to romidepsin, although less so than cells that express Pgp82. Cell lines selected for resistance to romidepsin express Pgp and are also resistant to other Pgp substrates such as vincristine or taxol; resistance to romidepsin can be overcome by Pgp inhibitors70, 72, 83. Despite the major role of Pgp in romidepsin resistance in cancer cell lines, ABCB1 gene expression in tumor biopsy samples from patients with CTCL enrolled on the NCI phase II trial did not appear to correlate with resistance to romidepsin treatment39, even though increased ABCB1 expression has been observed in peripheral blood mononuclear cells and circulating tumor cells obtained from patients receiving depsipeptide70. Thus, other mechanisms of resistance are likely to play a role in the intrinsic resistance observed in clinical trials with HDIs.
In an attempt to identify non-Pgp mechanisms of resistance, we selected the HuT 78 cell line, derived from a patient with Sézary syndrome, with romidepsin in the presence of the Pgp inhibitor verapamil71. One of the resulting romidepsin-resistant cell lines, HuT DpVp50, is maintained in 50 ng/mL romidepsin in the presence of 5 μg/ml verapamil. As seen in Figure 4, although the DpVp50 cells express some Pgp, this is not the dominant mechanism of resistance; treatment of the resistant line with 10 ng/ml romidepsin for 48 h in the presence of the Pgp inhibitor tariquidar does not result in increased cell death compared to romidepsin treatment without tariquidar. HuT 78 parental cells, which do not express Pgp are exquisitely sensitive to romidepsin; accordingly, the addition of tariquidar does not have any effect on cytotoxicity. We are currently working to characterize the mechanism of resistance to romidepsin in these cell lines.
Figure 4.

Resistance to romidepsin is not mediated by Pgp expression in HuT78 DpVp50 cells. HuT78 parental and DpVp50 cells were incubated with the Pgp-specific antibody, MRK-16 (blue histogram), or IgG negative control antibody (red histogram) for 30 min after which cells were washed and incubated with phycoerythrin-labeled secondary antibody (top row, Pgp). While HuT78 parental cells are Pgp negative, the DpVp50 cells express low but detectable levels. Cells were also left untreated (second row, C) or were incubated with 50 ng/mL romidepsin for 48 h in the presence (third row, DP) or absence (bottom row DP + TAR) of 250 nM of the Pgp inhibitor tariquidar, after which cells were incubated with annexin V antibody and propidium iodide. Cells in the lower left quadrant are viable cells, while cells in the lower right quadrant are early apoptotic cells, and cells in the upper right quadrant are late apoptotic or necrotic cells. HuT parental cells readily undergo apoptosis after incubation with romidepsin either in the presence or absence of tariqudiar, so shown by the increase of cells in the upper and lower right quadrants. DpVp50 cells are resistant to romidepsin whether the inhibitor is added or not, suggesting a resistance mechanism that does not involve Pgp.
Cell cycle proteins
It has been postulated that the induction of p21, which is responsible for the G1 arrest caused by HDI treatment, might serve a protective role. We reported that, when p21-deficient HCT116 cells were treated with romidepsin, cells arrested only in G2 and were more sensitive to treatment compared to wild-type cells84. In accordance with that result, U937 leukemia cells transfected with a p21 antisense construct were found to be more sensitive to vorinostat treatment when compared to untransfected cells85. Additionally, cotreatment with flavopiridol has been shown to potentiate the cytotoxicty of romidepsin, sodium butyrate and vorinostat, due in part to the prevention of p21 upregulation86–88. Temisirolimus treatment has also been shown to decrease p21 expression in mantle cell lymphoma cell lines and to synergize with sublethal concentrations of vorinostat89. Clinical trials with vorinostat and temisirolimus or sirolimus are currently ongoing.
Increased thioredoxin levels
As mentioned above, thioredoxin is a scavenger of reactive oxygen and expression of TBP-2 has been shown to decrease its reductive capacity. Ungerstedt and colleagues found that high levels of thioredoxin in normal cells served to protect cells from ROS induced by HDI treatment90. Additionally, when HDI-sensitive transformed cells were transfected with a small-interfering RNA (siRNA) against thioredoxin, the cells exhibited higher ROS levels and increased cell death compared to untransfected cells90. Similarly, Chen and colleagues reported in romidepsin-treated, human lung cancer cells, that thioredoxin expression negatively correlated with ROS generation and apoptosis91, supporting the idea that HDIs in combination with compounds that decrease thioredoxin expression or function may lead to increased activity.
Apoptosis-related proteins
Enforced expression of anti-apoptotic proteins has been shown to prevent HDI-mediated cell death. High levels of Bcl-2 or Bcl-XL have been shown to confer resistance to treatment with vorinostat, dacinostat, panobinostat or oxamflatin, while only high levels of Bcl-2 were found to confer resistance to romidepsin treatment85, 92, 93. Bcl-2 expression has also been linked to sensitivity to panobinostat treatment in CTCL cell lines94. Increased apoptosis has been observed when HDIs are combined with Bcl-2 inhibitors such as ABT-737 or others in several model systems94–98, suggesting that the combination should be tested in the clinical setting.
When Shao and colleagues knocked down expression of the pro-apopototic Bax in panobinostat-sensitive, T-cell lymphoma cell lines, toxicity was diminished94. Similarly, knockdown of anti-apoptotic Mcl-1 was found to potentiate HDI-mediated apoptosis in primary chronic lymphocytic leukemia cells and K562 cells99. Expression of Bim has also been shown to be required for romidepsin-mediated apoptosis in lung cancer cells100. Thus, in cells where these proapoptic proteins are silenced, HDI-mediated cell death might be blunted. Treatment with targeted therapeutics such as erlotinib or imatinib has been shown to increase levels of Bim in some model systems101, 102, suggesting that combination with an HDI might be advantageous.
Alterations in HDAC protein levels
Another cell line selected for resistance to HDIs was generated by selection of the HL-60 leukemia cell line with dacinostat, resulting in the HL-60/LR cell line maintained in 200 nM dacinostat103. In addition to dacinostat, the resistant line was also highly cross-resistant to vorinostat, panobinostat and sodium butyrate103. While levels of BCL-XL and XIAP were attenuated, levels of Bim and Bax remained unchanged103. Interestingly, HL-60/LR cells expressed higher levels of HDAC1, 2, and 4, but lacked expression of HDAC6 and had higher levels of Hsp90 acetylation compared to the parental line103. The resistant line also demonstrated collateral sensitivity to Hsp90 inhibitors103. A separate study demonstrated that increased HDAC1 expression prevented sodium butyrate-mediated toxicity in a melanoma cell line104. It is not clear whether this mechanism of resistance has clinical relevance; no studies have yet linked altered HDAC protein expression levels with clinical response to HDIs. However, HDAC2 expression levels were found to correlate with histone acetylation in a phase I trial of doxorubicin and vorinostat105 in solid tumors and combined tamoxifen and vorinostat treatment in a phase II trial in breast cancer106.
Signaling proteins
Activation of the mitogen-activated protein kinase (MAPK) or phosphoinositide 3-kinase (PI3K) pathways has increasingly been associated with resistance to HDIs. Combination of romidepsin with mitogen-activated protein kinase kinase (MEK) inhibitors has been shown to increase cell death, suggesting that activation of the MAPK pathway is an important mechanism of resistance to romidepsin107, 108. In agreement with this hypothesis, Yu and colleagues found that enforced expression of constitutively active MEK1, but not Akt, in lung cancer cell lines reduced romidepsin-mediated cytotoxicity109. Other studies, however, seem to suggest that phosphorylated Akt is, in fact, an important resistance mechanism to romidepsin, as combination of romidepsin with inhibitors of Akt results in synergistic cytotoxicity110. Combination of panobinostat with compounds that abrogate MAPK and PI3K signaling has also been shown to result in synergistic cytotoxicity, possibly due to increased ROS111. Similarly, Jane and colleagues found that treatment of glioma cells with vandetanib inhibited both the MAPK and PI3K pathways and was synergistic with vorinsotat treatment112. Vorinostat in combination with PI3K inhibitors has shown promise in T-cell lymphoma cell line models113. These combination studies, while targeting different signaling molecules, do suggest that activation of one or more of these pathways may confer clinical resistance.
Activation of the signal transducer and activator of transcription (STAT) pathway has also been linked to vorinostat resistance. In a panel of almost 40 lymphoma cell lines, expression of STAT1, 3 and 5 was higher in cell lines that were more resistant to vorinostat compared to sensitive lines; phosphorylation levels of the STAT proteins were also higher in the resistant lines114. In a series of skin biopsy samples, patients with higher nuclear staining of phosphorylated STAT3 were more likely to be resistant to vorinostat treatment114, again implicating activation of signaling pathways in resistance to HDIs. Combined treatment with vorinostat and compounds shown to inhibit STAT3 phosphorylation, such as lestaurtinib115, may therefore increase efficacy in T-cell lymphoma.
NFκB activation
Activation of the NFκB pathway is a hallmark of a number of cancers and leads to deactivation of the apoptotic pathway and increased cell survival116. Acetylation of the p65, or RelA, subunit of NFκB has been shown to increase the acitivity of NFκB117, and HDI treatment has been shown to result in increased p65 acetylation118, 119. Constitutive activation of NFκB has been linked to resistance to HDIs in cell line models94, as has p65 acetylation caused by HDI treatment; combination of an HDI with an inhibitor of NFκB activation leads to synergistic cytotoxocity120. The increased cytotoxicity observed when HDIs are combined with proteasome inhibitors is believed to be due, at least in part, to decreased NFκB activity mediated by proteasome inhibitors121, 122. However, while a phase II trial with vorinostat and bortezomib in multiple myeloma had a 42% response rate and elicited some responses from patients whose disease was refractory to bortezomib, response did not correlate with levels of NFκB or IκB13 in CD138+ bone marrow cells, casting some doubt on the relevance of NFκB status.
Conclusion
The histone deacetylase inhibitors have shown promise in the treatment of peripheral and cutaneous T-cell lymphomas. Their lack of success in clinical trials for solid tumors has been disappointing. As noted above, a long list of mechanisms of action has been compiled through in vitro studies. It can be concluded from this list that we still do not really understand how the HDIs work when they are effective, as in T-cell lymphomas. We have yet to validate a marker that predicts clinical response to HDI treatment. We also do not understand why they work in T-cell lymphomas and not in solid tumors. Perhaps in the context of the overall sensitivity of lymphomas to anticancer therapy, a decades-old observation, this is not so surprising. But the activity in T-cell lymphoma seems beyond that in other hematologic malignancies and is a class effect. It is likely that a dominant mechanism of action, not necessarily on the list of mechanisms outlined above, is responsible for the efficacy.
The insensitivity of solid tumors also appears to be a class effect. Unfortunately, in vitro models do not typically reflect the insensitivity of solid tumors in the clinic. This may be due in part to the rapid doubling time of cell lines in culture, and the epigenetic alterations that accompany that cell growth rate. Combined with the duration of exposure typically used in cell culture sensitivity assays, responses to HDIs in the laboratory are homogenous across different tumor types. The negative impact of this ubiquitous responsiveness is to divert those working in the field from focusing on a few candidate solid tumor types for targeted drug development.
One approach to overcoming resistance in solid tumors is to exploit the diverse effects of HDIs in solid tumors in combination therapies. Thus, if altered gene expression does not itself prompt cytotoxicity, it may allow promising combination therapies to be developed. Induction of the sodium iodide symporter and increased sensitivity of thyroid cancer cells to radioiodine uptake is one such example123. If altered handling of reactive oxygen species or reduced DNA repair are insufficient to induce cell death following HDI exposure, it may still allow for effective combination therapies with DNA damaging agents, whether chemotherapy or radiotherapy124, 125. It is critical, however, in developing the rationale for combination studies that attention be paid to sequence of administration. For example, in small cell lung cancer cells, simultaneous exposure to an HDI and cisplatin or etoposide was more effective than sequential exposure126. Among the plethora of HDI effects on cells are likely to be some that are detrimental to cytotoxicity (e.g. p21 induction).
Further, the in vitro paradox also has the potential to yield misleading clinical directions from combination assays that require durations of exposure for efficacy that are not achievable in the clinic by any HDI developed to date. The length of the list of mechanisms of resistance generated from laboratory models confirms that we do not understand resistance, either. We can only hope that somewhere on those lists are the answers and that translational studies will help us separate the important mechanisms from those that are trivial. What we do know is that development of the HDIs is a major step in bringing epigenetic therapy to the anticancer armamentarium. We need to figure out how to exploit them more fully and in many more tumor types.
Table 2.
Summary of resistance mechanisms to HDIs
|
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
References
- 1.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 2.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–99. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 3.Bianco-Miotto T, Chiam K, Buchanan G, Jindal S, Day TK, Thomas M, Pickering MA, O’Loughlin MA, Ryan NK, Raymond WA, Horvath LG, Kench JG, Stricker PD, Marshall VR, Sutherland RL, Henshall SM, Gerald WL, Scher HI, Risbridger GP, Clements JA, Butler LM, Tilley WD, Horsfall DJ, Ricciardelli C, BioResource APC. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol Biomarkers Prev. 2010;19(10):2611–22. doi: 10.1158/1055-9965.EPI-10-0555. [DOI] [PubMed] [Google Scholar]
- 4.Elsheikh S, Green A, Rakha E, Powe D, Ahmed R, Collins H, Soria D, Garibaldi J, Paish C, Ammar A, Grainge M, Ball G, Abdelghany M, Martinez-Pomares L, Heery D, Ellis I. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009;69(9):3802–9. doi: 10.1158/0008-5472.CAN-08-3907. [DOI] [PubMed] [Google Scholar]
- 5.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 6.Marks P, Xu W. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107(4):600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schrump D. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res. 2009;15(12):3947–57. doi: 10.1158/1078-0432.CCR-08-2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol Res. 2010;62(1):18–34. doi: 10.1016/j.phrs.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Rasheed W, Bishton M, Johnstone RW, Prince HM. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther. 2008;8(3):413–32. doi: 10.1586/14737140.8.3.413. [DOI] [PubMed] [Google Scholar]
- 10.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25(1):84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 11.Olsen E, Kim Y, Kuzel T, Pacheco T, Foss F, Parker S, Frankel S, Chen C, Ricker J, Arduino J, Duvic M. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 12.Kirschbaum M, Frankel P, Popplewell L, Zain J, Delioukina M, Pullarkat V, Matsuoka D, Pulone B, Rotter AJ, Espinoza-Delgado I, Nademanee A, Forman SJ, Gandara D, Newman E. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(9):1198–203. doi: 10.1200/JCO.2010.32.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Badros A, Burger A, Philip S, Niesvizky R, Kolla S, Goloubeva O, Harris C, Zwiebel J, Wright J, Espinoza-Delgado I, Baer M, Holleran J, Egorin M, Grant S. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15(16):5250–7. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lane A, Chabner B. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27(32):5459–68. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 15.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62(17):4916–21. [PubMed] [Google Scholar]
- 16.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dümpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29(3):255–65. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 17.Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, Schrump DS. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94(7):504–13. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 18.Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood. 2001;98(9):2865–8. doi: 10.1182/blood.v98.9.2865. [DOI] [PubMed] [Google Scholar]
- 19.Piekarz R, Frye R, Turner M, Wright J, Allen S, Kirschbaum M, Zain J, Prince H, Leonard J, Geskin L, Reeder C, Joske D, Figg W, Gardner E, Steinberg S, Jaffe E, Stetler-Stevenson M, Lade S, Fojo A, Bates S. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–7. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, Scarisbrick J, Reddy S, Robak T, Becker JC, Samtsov A, McCulloch W, Kim YH. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485–91. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 21.Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, Jaffe ES, Ling A, Turner M, Peer CJ, Figg WD, Steinberg SM, Smith S, Joske D, Lewis I, Hutchins L, Craig M, Fojo AT, Wright JJ, Bates SE. Phase II trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011 doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, Gardner ER, Figg WD, Bates SE. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther. 2010;10(7):997–1008. doi: 10.1586/era.10.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molife L, Attard G, Fong P, Karavasilis V, Reid A, Patterson S, Riggs CJ, Higano C, Stadler W, McCulloch W, Dearnaley D, Parker C, de Bono J. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC) Ann Oncol. 2010;21(1):109–13. doi: 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- 24.Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clinical Genitourinary Cancer. 2006;5(1):57–60. doi: 10.3816/CGC.2006.n.018. [DOI] [PubMed] [Google Scholar]
- 25.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12(15):4628–35. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 26.Prince H, Bishton M, Johnstone R. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009;5(5):601–12. doi: 10.2217/fon.09.36. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson M, Ritchie D, DeAngelo D, Spencer A, Ottmann O, Fischer T, Bhalla K, Liu A, Parker K, Scott J, Bishton M, Prince H. Preliminary evidence of disease response to the pan deacetylase inhibitor panobinostat (LBH589) in refractory Hodgkin Lymphoma. Br J Haematol. 2009;147(1):97–101. doi: 10.1111/j.1365-2141.2009.07837.x. [DOI] [PubMed] [Google Scholar]
- 28.Gimsing P, Hansen M, Knudsen L, Knoblauch P, Christensen I, Ooi C, Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur J Haematol. 2008;81(3):170–6. doi: 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- 29.Howman RA, Prince HM. New drug therapies in peripheral T-cell lymphoma. Expert Rev Anticancer Ther. 2011;11(3):457–72. doi: 10.1586/era.11.4. [DOI] [PubMed] [Google Scholar]
- 30.Zain JM, O’Connor O, Zinzani PL, Norman A, de Nully Brown P. Multicenter, open-label trial of PXD 101 in patients with relapsed/refractory peripheral T-cell lymphoma. ASCO Meeting Abstracts. 2010;28(15_suppl):e18565. [Google Scholar]
- 31.Su JM, Li XN, Thompson P, Ou CN, Ingle AM, Russell H, Lau CC, Adamson PC, Blaney SM. Phase 1 study of valproic acid in pediatric patients with refractory solid or CNS tumors: a children’s oncology group report. Clin Cancer Res. 2011;17(3):589–97. doi: 10.1158/1078-0432.CCR-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma S, Symanowski J, Wong B, Dino P, Manno P, Vogelzang N. A Phase II Clinical Trial of Oral Valproic Acid in Patients with Castration-Resistant Prostate Cancers Using an Intensive Biomarker Sampling Strategy. Transl Oncol. 2008;1(3):141–7. doi: 10.1593/tlo.08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gore L, Rothenberg M, O’Bryant C, Schultz M, Sandler A, Coffin D, McCoy C, Schott A, Scholz C, Eckhardt S. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14(14):4517–25. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauschild A, Trefzer U, Garbe C, Kaehler K, Ugurel S, Kiecker F, Eigentler T, Krissel H, Schott A, Schadendorf D. Multicenter phase II trial of the histone deacetylase inhibitor pyridylmethyl-N-{4-[(2-aminophenyl)-carbamoyl]-benzyl}-carbamate in pretreated metastatic melanoma. Melanoma Res. 2008;18(4):274–8. doi: 10.1097/CMR.0b013e328307c248. [DOI] [PubMed] [Google Scholar]
- 35.Boumber Y, Younes A, Garcia-Manero G. Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin Investig Drugs. 2011;20(6):823–9. doi: 10.1517/13543784.2011.577737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112(4):981–9. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C, Dubay M, Dumouchel J, Drouin M, Lucas DM, Martell RE, Byrd JC. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol. 2009;147(4):507–14. doi: 10.1111/j.1365-2141.2009.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, Karavasilis V, Mita M, Shaw H, Workman P, Kaye S, Rowinsky EK, Aherne W, Atadja P, Scott JW, Patnaik A. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008;14(20):6663–73. doi: 10.1158/1078-0432.CCR-08-0376. [DOI] [PubMed] [Google Scholar]
- 39.Bates S, Zhan Z, Steadman K, Obrzut T, Luchenko V, Frye R, Robey R, Turner M, Gardner E, Figg W, Steinberg S, Ling A, Fojo T, To K, Piekarz R. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br J Haematol. 2010;148(2):256–67. doi: 10.1111/j.1365-2141.2009.07954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Kruchin E, Wright JJ, Rosing DR, Sparreboom A, Figg WD, Steinberg SM. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res. 2008;14(1):188–98. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 41.Siu L, Pili R, Duran I, Messersmith W, Chen E, Sullivan R, MacLean M, King S, Brown S, Reid G, Li Z, Kalita A, Laille E, Besterman J, Martell R, Carducci M. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol. 2008;26(12):1940–7. doi: 10.1200/JCO.2007.14.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blagosklonny MV, Robey R, Sackett DL, Du L, Traganos F, Darzynkiewicz Z, Fojo T, Bates SE. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther. 2002;1(11):937–41. [PubMed] [Google Scholar]
- 43.Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, Jove R, Forman SJ, Yen Y, Kirschbaum MH. Combining Histone Deacetylase Inhibitor Vorinostat with Aurora Kinase Inhibitors Enhances Lymphoma Cell Killing with Repression of c-Myc, hTERT, and microRNA Levels. Cancer Res. 2011;71(11):3912–20. doi: 10.1158/0008-5472.CAN-10-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawamata N, Chen J, Koeffler HP. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood. 2007;110(7):2667–73. doi: 10.1182/blood-2005-11-026344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–49. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011;2011:875824. doi: 10.1155/2011/875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bali P, Pranpat M, Swaby R, Fiskus W, Yamaguchi H, Balasis M, Rocha K, Wang H, Richon V, Bhalla K. Activity of suberoylanilide hydroxamic Acid against human breast cancer cells with amplification of her-2. Clin Cancer Res. 2005;11(17):6382–9. doi: 10.1158/1078-0432.CCR-05-0344. [DOI] [PubMed] [Google Scholar]
- 48.Labonte MJ, Wilson PM, Fazzone W, Russell J, Louie SG, El-Khoueiry A, Lenz HJ, Ladner RD. The Dual EGFR/HER2 Inhibitor Lapatinib Synergistically Enhances the Antitumor Activity of the Histone Deacetylase Inhibitor Panobinostat in Colorectal Cancer Models. Cancer Res. 2011;71(10):3635–48. doi: 10.1158/0008-5472.CAN-10-2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edwards A, Li J, Atadja P, Bhalla K, Haura E. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol Cancer Ther. 2007;6(9):2515–24. doi: 10.1158/1535-7163.MCT-06-0761. [DOI] [PubMed] [Google Scholar]
- 50.Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, Atadja P, Bhalla K. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63(16):5126–35. [PubMed] [Google Scholar]
- 51.Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101(8):3236–9. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- 52.Okabe S, Tauchi T, Nakajima A, Sashida G, Gotoh A, Broxmeyer H, Ohyashiki J, Ohyashiki K. Depsipeptide (FK228) preferentially induces apoptosis in BCR/ABL-expressing cell lines and cells from patients with chronic myelogenous leukemia in blast crisis. Stem Cells Dev. 2007;16(3):503–14. doi: 10.1089/scd.2007.9994. [DOI] [PubMed] [Google Scholar]
- 53.Fiskus W, Pranpat M, Bali P, Balasis M, Kumaraswamy S, Boyapalle S, Rocha K, Wu J, Giles F, Manley PW, Atadja P, Bhalla K. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl-expressing human leukemia cells. Blood. 2006;108(2):645–52. doi: 10.1182/blood-2005-11-4639. [DOI] [PubMed] [Google Scholar]
- 54.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci U S A. 2008;105(28):9633–8. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler H, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leuk Res. 2008;32(9):1382–92. doi: 10.1016/j.leukres.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 56.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6(5):873–83. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y, Wang S, Zhang X, Zhao M, Hou C, Xu Y, Du Z, Yu X. FK228 inhibits Hsp90 chaperone function in K562 cells via hyperacetylation of Hsp70. Biochem Biophys Res Commun. 2007;356(4):998–1003. doi: 10.1016/j.bbrc.2007.03.076. [DOI] [PubMed] [Google Scholar]
- 58.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63(13):3637–45. [PubMed] [Google Scholar]
- 59.Yu C, Subler M, Rahmani M, Reese E, Krystal G, Conrad D, Dent P, Grant S. Induction of apoptosis in BCR/ABL+ cells by histone deacetylase inhibitors involves reciprocal effects on the RAF/MEK/ERK and JNK pathways. Cancer Biol Ther. 2003;2(5):544–51. doi: 10.4161/cbt.2.5.454. [DOI] [PubMed] [Google Scholar]
- 60.Rosato RR, Maggio SC, Almenara JA, Payne SG, Atadja P, Spiegel S, Dent P, Grant S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol Pharmacol. 2006;69(1):216–25. doi: 10.1124/mol.105.017145. [DOI] [PubMed] [Google Scholar]
- 61.Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107(33):14639–44. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471(7336):74–9. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17(9):1144–51. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marks PA. Thioredoxin in cancer--role of histone deacetylase inhibitors. Semin Cancer Biol. 2006;16(6):436–43. doi: 10.1016/j.semcancer.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, Richon VM. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A. 2002;99(18):11700–5. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo F, Sigua C, Tao J, Bali P, George P, Li Y, Wittmann S, Moscinski L, Atadja P, Bhalla K. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004;64(7):2580–9. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- 67.Kauh J, Fan S, Xia M, Yue P, Yang L, Khuri FR, Sun SY. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One. 2010;5(4):e10376. doi: 10.1371/journal.pone.0010376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yeh CC, Deng YT, Sha DY, Hsiao M, Kuo MY. Suberoylanilide hydroxamic acid sensitizes human oral cancer cells to TRAIL-induced apoptosis through increase DR5 expression. Mol Cancer Ther. 2009;8(9):2718–25. doi: 10.1158/1535-7163.MCT-09-0211. [DOI] [PubMed] [Google Scholar]
- 69.Jiang X, Tsang YH, Yu Q. c-Myc overexpression sensitizes Bim-mediated Bax activation for apoptosis induced by histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through regulating Bcl-2/Bcl-xL expression. Int J Biochem Cell Biol. 2007;39(5):1016–25. doi: 10.1016/j.biocel.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 70.Robey RW, Zhan Z, Piekarz RL, Kayastha GL, Fojo T, Bates SE. Increased MDR1 expression in normal and malignant peripheral blood mononuclear cells obtained from patients receiving depsipeptide (FR901228, FK228, NSC630176) Clin Cancer Res. 2006;12(5):1547–55. doi: 10.1158/1078-0432.CCR-05-1423. [DOI] [PubMed] [Google Scholar]
- 71.Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH, Torrico S, Bates SE. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood. 2004;103(12):4636–43. doi: 10.1182/blood-2003-09-3068. [DOI] [PubMed] [Google Scholar]
- 72.Xiao JJ, Huang Y, Dai Z, Sadee W, Chen J, Liu S, Marcucci G, Byrd J, Covey JM, Wright J, Grever M, Chan KK. Chemoresistance to Depsipeptide FK228 [(E)-(1S,4S,10S,21R)-7-[(Z)-Ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithi a-5,8,20,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone] Is Mediated by Reversible MDR1 Induction in Human Cancer Cell Lines. J Pharmacol Exp Ther. 2005;314(1):467–75. doi: 10.1124/jpet.105.083956. [DOI] [PubMed] [Google Scholar]
- 73.Prince H, Bishton M, Harrison S. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res. 2009;15(12):3958–69. doi: 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- 74.Mickley LA, Bates SE, Richert ND, Currier S, Tanaka S, Foss F, Rosen N, Fojo AT. Modulation of the expression of a multidrug resistance gene (mdr-1/P-glycoprotein) by differentiating agents. J Biol Chem. 1989;264(30):18031–40. [PubMed] [Google Scholar]
- 75.Frommel TO, Coon JS, Tsuruo T, Roninson IB. Variable effects of sodium butyrate on the expression and function of the mdr-1 (P-glycoprotein) gene in colon carcinoma cell lines. Int J Cancer. 1993;55:297–302. doi: 10.1002/ijc.2910550221. [DOI] [PubMed] [Google Scholar]
- 76.Jin S, Scotto KW. Transcriptional regulation of the MDR1 gene by histone acetyltransferase and deacetylase is mediated by NF-Y. Mol Cell Biol. 1998;18(7):4377–84. doi: 10.1128/mcb.18.7.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute Drug Screen. Mol Pharmacol. 1994;46:627–638. [PubMed] [Google Scholar]
- 78.Cerveny L, Svecova L, Anzenbacherova E, Vrzal R, Staud F, Dvorak Z, Ulrichova J, Anzenbacher P, Pavek P. Valproic acid induces CYP3A4 and MDR1 gene expression by activation of constitutive androstane receptor and pregnane X receptor pathways. Drug Metab Dispos. 2007;35(7):1032–41. doi: 10.1124/dmd.106.014456. [DOI] [PubMed] [Google Scholar]
- 79.Kim YK, Kim NH, Hwang JW, Song YJ, Park YS, Seo DW, Lee HY, Choi WS, Han JW, Kim SN. Histone deacetylase inhibitor apicidin-mediated drug resistance: involvement of P-glycoprotein. Biochem Biophys Res Commun. 2008;368(4):959–64. doi: 10.1016/j.bbrc.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 80.Peart MJ, Tainton KM, Ruefli AA, Dear AE, Sedelies KA, O’Reilly LA, Waterhouse NJ, Trapani JA, Johnstone RW. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003;63(15):4460–71. [PubMed] [Google Scholar]
- 81.Qian X, LaRochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, Sehested M, Lichenstein HS, Jeffers M. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther. 2006;5(8):2086–95. doi: 10.1158/1535-7163.MCT-06-0111. [DOI] [PubMed] [Google Scholar]
- 82.Xiao JJ, Foraker AB, Swaan PW, Liu S, Huang Y, Dai Z, Chen J, Sadee W, Byrd J, Marcucci G, Chan KK. Efflux of depsipeptide FK228 (FR901228, NSC-630176) is mediated by P-glycoprotein and multidrug resistance-associated protein 1. J Pharmacol Exp Ther. 2005;313(1):268–76. doi: 10.1124/jpet.104.072033. [DOI] [PubMed] [Google Scholar]
- 83.Yamada H, Arakawa Y, Saito S, Agawa M, Kano Y, Horiguchi-Yamada J. Depsipeptide-resistant KU812 cells show reversible P-glycoprotein expression, hyper-acetylated histones, and modulated gene expression profile. Leuk Res. 2006;30(6):723–34. doi: 10.1016/j.leukres.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 84.Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83(6):817–25. doi: 10.1054/bjoc.2000.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vrana J, Decker R, Johnson C, Wang Z, Jarvis W, Richon V, Ehinger M, Fisher P, Grant S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene. 1999;18(50):7016–25. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- 86.Nguyen DM, Schrump WD, Tsai WS, Chen A, Stewart JHt, Steiner F, Schrump DS. Enhancement of depsipeptide-mediated apoptosis of lung or esophageal cancer cells by flavopiridol: activation of the mitochondria-dependent death-signaling pathway. J Thorac Cardiovasc Surg. 2003;125(5):1132–42. doi: 10.1067/mtc.2003.180. [DOI] [PubMed] [Google Scholar]
- 87.Rosato RR, Almenara JA, Yu C, Grant S. Evidence of a functional role for p21WAF1/CIP1 down-regulation in synergistic antileukemic interactions between the histone deacetylase inhibitor sodium butyrate and flavopiridol. Mol Pharmacol. 2004;65(3):571–81. doi: 10.1124/mol.65.3.571. [DOI] [PubMed] [Google Scholar]
- 88.Almenara J, Rosato R, Grant S. Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) Leukemia. 2002;16(7):1331–43. doi: 10.1038/sj.leu.2402535. [DOI] [PubMed] [Google Scholar]
- 89.Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, Kondo S, Younes A. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36(4):443–50. doi: 10.1016/j.exphem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 90.Ungerstedt J, Sowa Y, Xu W, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks P. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102(3):673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen G, Li A, Zhao M, Gao Y, Zhou T, Xu Y, Du Z, Zhang X, Yu X. Proteomic analysis identifies protein targets responsible for depsipeptide sensitivity in tumor cells. J Proteome Res. 2008;7(7):2733–42. doi: 10.1021/pr7008753. [DOI] [PubMed] [Google Scholar]
- 92.Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, Scott CL, Strasser A, Atadja P, Lowe SW, Johnstone RW. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. 2009;114(2):380–93. doi: 10.1182/blood-2008-10-182758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Newbold A, Lindemann RK, Cluse LA, Whitecross KF, Dear AE, Johnstone RW. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol Cancer Ther. 2008;7(5):1066–79. doi: 10.1158/1535-7163.MCT-07-2256. [DOI] [PubMed] [Google Scholar]
- 94.Shao W, Growney JD, Feng Y, O’Connor G, Pu M, Zhu W, Yao YM, Kwon P, Fawell S, Atadja P. Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: Defining molecular mechanisms of resistance. Int J Cancer. 2010;127(9):2199–208. doi: 10.1002/ijc.25218. [DOI] [PubMed] [Google Scholar]
- 95.Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks KM, Ralli R, Scott CL, Frenzel A, Villunger A, Johnstone RW. Deciphering the Molecular Events Necessary for Synergistic Tumor Cell Apoptosis Mediated by the Histone Deacetylase Inhibitor Vorinostat and the BH3 Mimetic ABT-737. Cancer Res. 2011;71(10):3603–15. doi: 10.1158/0008-5472.CAN-10-3289. [DOI] [PubMed] [Google Scholar]
- 96.Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci U S A. 2006;103(42):15540–5. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Whitecross KF, Alsop AE, Cluse LA, Wiegmans A, Banks KM, Coomans C, Peart MJ, Newbold A, Lindemann RK, Johnstone RW. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood. 2009;113(9):1982–91. doi: 10.1182/blood-2008-05-156851. [DOI] [PubMed] [Google Scholar]
- 98.Wei Y, Kadia T, Tong W, Zhang M, Jia Y, Yang H, Hu Y, Tambaro FP, Viallet J, O’Brien S, Garcia-Manero G. The combination of a histone deacetylase inhibitor with the Bcl-2 homology domain-3 mimetic GX15–070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Clin Cancer Res. 2010;16(15):3923–32. doi: 10.1158/1078-0432.CCR-10-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Inoue S, Walewska R, Dyer MJ, Cohen GM. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia. 2008;22(4):819–25. doi: 10.1038/leu.2008.1. [DOI] [PubMed] [Google Scholar]
- 100.Yang Y, Zhao Y, Liao W, Yang J, Wu L, Zheng Z, Yu Y, Zhou W, Li L, Feng J, Wang H, Zhu WG. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11(4):313–24. doi: 10.1593/neo.81358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, Pao W. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4(10):e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW, Strasser A. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103(40):14907–12. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fiskus W, Rao R, Fernandez P, Herger B, Yang Y, Chen J, Kolhe R, Mandawat A, Wang Y, Joshi R, Eaton K, Lee P, Atadja P, Peiper S, Bhalla K. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood. 2008;112(7):2896–905. doi: 10.1182/blood-2007-10-116319. [DOI] [PubMed] [Google Scholar]
- 104.Bandyopadhyay D, Mishra A, Medrano EE. Overexpression of histone deacetylase 1 confers resistance to sodium butyrate-mediated apoptosis in melanoma cells through a p53-mediated pathway. Cancer Res. 2004;64(21):7706–10. doi: 10.1158/0008-5472.CAN-03-3897. [DOI] [PubMed] [Google Scholar]
- 105.Munster P, Marchion D, Thomas S, Egorin M, Minton S, Springett G, Lee J, Simon G, Chiappori A, Sullivan D, Daud A. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer. 2009;101(7):1044–50. doi: 10.1038/sj.bjc.6605293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104(12):1828–35. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matsubara H, Watanabe M, Imai T, Yui Y, Mizushima Y, Hiraumi Y, Kamitsuji Y, Watanabe K, Nishijo K, Toguchida J, Nakahata T, Adachi S. Involvement of extracellular signal-regulated kinase activation in human osteosarcoma cell resistance to the histone deacetylase inhibitor FK228 [(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-bis(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone] J Pharmacol Exp Ther. 2009;328(3):839–48. doi: 10.1124/jpet.108.147462. [DOI] [PubMed] [Google Scholar]
- 108.Ozaki K, Minoda A, Kishikawa F, Kohno M. Blockade of the ERK pathway markedly sensitizes tumor cells to HDAC inhibitor-induced cell death. Biochem Biophys Res Commun. 2006;339(4):1171–7. doi: 10.1016/j.bbrc.2005.11.131. [DOI] [PubMed] [Google Scholar]
- 109.Yu X, Wang S, Chen G, Hou C, Zhao M, Hong J, Nguyen D, Schrump D. Apoptosis induced by depsipeptide FK228 coincides with inhibition of survival signaling in lung cancer cells. Cancer J. 2007;13(2):105–13. doi: 10.1097/PPO.0b013e318046eedc. [DOI] [PubMed] [Google Scholar]
- 110.Kodani M, Igishi T, Matsumoto S, Chikumi H, Shigeoka Y, Nakanishi H, Morita M, Yasuda K, Hitsuda Y, Shimizu E. Suppression of phosphatidylinositol 3-kinase/Akt signaling pathway is a determinant of the sensitivity to a novel histone deacetylase inhibitor, FK228, in lung adenocarcinoma cells. Oncol Rep. 2005;13(3):477–83. [PubMed] [Google Scholar]
- 111.Yu C, Friday BB, Lai JP, McCollum A, Atadja P, Roberts LR, Adjei AA. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res. 2007;13(4):1140–8. doi: 10.1158/1078-0432.CCR-06-1751. [DOI] [PubMed] [Google Scholar]
- 112.Jane EP, Premkumar DR, Addo-Yobo SO, Pollack IF. Abrogation of mitogen-activated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J Pharmacol Exp Ther. 2009;331(1):327–37. doi: 10.1124/jpet.109.155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martínez Leal JF, de La Cueva P, Rodriguez ME, Herreros B, Martin-Perez D, Longo MI, Herrera M, Piris MA, Ortiz-Romero PL. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010;95(4):613–21. doi: 10.3324/haematol.2009.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fantin V, Loboda A, Paweletz C, Hendrickson R, Pierce J, Roth J, Li L, Gooden F, Korenchuk S, Hou X, Harrington E, Randolph S, Reilly J, Ware C, Kadin M, Frankel S, Richon V. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68(10):3785–94. doi: 10.1158/0008-5472.CAN-07-6091. [DOI] [PubMed] [Google Scholar]
- 115.Diaz T, Navarro A, Ferrer G, Gel B, Gaya A, Artells R, Bellosillo B, Garcia-Garcia M, Serrano S, Martínez A, Monzo M. Lestaurtinib inhibition of the Jak/STAT signaling pathway in hodgkin lymphoma inhibits proliferation and induces apoptosis. PLoS One. 2011;6(4):e18856. doi: 10.1371/journal.pone.0018856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2(6):a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen Lf, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293(5535):1653–7. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 118.Duan J, Friedman J, Nottingham L, Chen Z, Ara G, Van Waes C. Nuclear factor-kappaB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2007;6(1):37–50. doi: 10.1158/1535-7163.MCT-05-0285. [DOI] [PubMed] [Google Scholar]
- 119.Dai Y, Rahmani M, Dent P, Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25(13):5429–44. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rundall BK, Denlinger CE, Jones DR. Combined histone deacetylase and NF-kappaB inhibition sensitizes non-small cell lung cancer to cell death. Surgery. 2004;136(2):416–25. doi: 10.1016/j.surg.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 121.Domingo-Domènech J, Pippa R, Tápia M, Gascón P, Bachs O, Bosch M. Inactivation of NF-kappaB by proteasome inhibition contributes to increased apoptosis induced by histone deacetylase inhibitors in human breast cancer cells. Breast Cancer Res Treat. 2008;112(1):53–62. doi: 10.1007/s10549-007-9837-8. [DOI] [PubMed] [Google Scholar]
- 122.Dai Y, Chen S, Kramer LB, Funk VL, Dent P, Grant S. Interactions between bortezomib and romidepsin and belinostat in chronic lymphocytic leukemia cells. Clin Cancer Res. 2008;14(2):549–58. doi: 10.1158/1078-0432.CCR-07-1934. [DOI] [PubMed] [Google Scholar]
- 123.Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, Bates S, Fojo T. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(-) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab. 2001;86(7):3430–5. doi: 10.1210/jcem.86.7.7621. [DOI] [PubMed] [Google Scholar]
- 124.Bruzzese F, Rocco M, Castelli S, Di Gennaro E, Desideri A, Budillon A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol Cancer Ther. 2009;8(11):3075–87. doi: 10.1158/1535-7163.MCT-09-0254. [DOI] [PubMed] [Google Scholar]
- 125.Nguyen T, Dai Y, Attkisson E, Kramer L, Jordan N, Nguyen N, Kolluri N, Muschen M, Grant S. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin Cancer Res. 2011;17(10):3219–32. doi: 10.1158/1078-0432.CCR-11-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Luchenko VL, Salcido CD, Zhang Y, Agama K, Komlodi-Pasztor E, Murphy RF, Giaccone G, Pommier Y, Bates SE, Vartikovski L. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle. 2011;18 doi: 10.4161/cc.10.18.17190. [DOI] [PMC free article] [PubMed] [Google Scholar]