

Abstract
Alternative splicing allows a single gene to generate multiple mRNA transcripts, which can be translated into functionally diverse proteins. However, experimentally-determined structures of protein splice isoforms are rare, and homology modeling methods are poor at predicting atomic-level structural differences because of high sequence identity. Here we exploit the state-of-the-art structure prediction method I-TASSER to analyze the structural and functional consequences of alternative splicing of proteins differentially expressed in a breast cancer model. We first successfully benchmarked the I-TASSER pipeline for structure modeling of all seven pairs of protein splice isoforms which are known to have experimentally-solved structures. We then modeled three cancer-related variant pairs reported to have opposite functions. In each pair, we observed structural differences in regions where presence or absence of a motif can directly influence the distinctive functions of the variants. Finally, we applied the method to five splice variants over-expressed in mouse Her2/neu mammary tumor: anxa6, calu, cdc42, ptbp1, and tax1bp3. Despite > 75% sequence identity between the variants, structural differences were observed in biologically important regions of these protein pairs. These results demonstrate the feasibility of integrating proteomic analysis with structure-based conformational predictions of differentially-expressed alternative splice variants in cancers and other conditions.
Keywords: Alternative splice isoforms, 3D structure, I-TASSER, Functional motifs, Her2/neu breast cancer
Introduction
Alternative splicing and post-translational modifications provide functional diversity to genes and protein gene products. Splicing was once thought to be quite unusual. Recent analyses in which RNA sequence reads are mapped to exon–exon junctions indicate that 92–94% of human genes undergo alternative splicing, of which 86% have minor isoform frequency of ≥15% 1. Although the functions of a protein molecule and its isoforms depend on their three-dimensional structures, which are uniquely determined by the amino acid sequences, current knowledge of alternative splicing is derived mainly from mRNA transcripts 1; very little is known about the expression level and 3D structure of the proteins.
As of May 2011, there are only fifteen pairs of splice variants in the Alternative Splicing and Transcript Diversity (ASTD) database (http://www.ebi.ac.uk/asd/) which have the atomic structures of both protein isoforms experimentally solved in the Protein Data Bank (PDB); only seven are full-length. Despite significant progress in computational modeling of protein structures 2, 3, most current prediction methods are based on homologous templates which are unable to distinguish subtle differences between the isoforms because of high sequence identities. In contrast, the I-TASSER software program splits and reassembles multiple template fragments into atomic full-length structures, with the assembly simulations driven by optimized knowledge and physics-based force field analyses 4–6. It refines the template structure closer to the native state, and it has modeled functionally-important motifs in both benchmark tests (5, 7) and community-wide blinded experiments 7, 8. Its attributes in ab initio structural assembly and template refinement are essential for structural modeling of the atomic details of spliced protein variants. In this work, we used the I-TASSER pipeline to model the structures of spliced proteins and annotate the functional consequences arising due to subtle structural changes. We benchmarked the I-TASSER approach on the seven pairs of full-length splice variant proteins with experimentally-determined structures and on three pairs of cancer-related splice variants with known opposite functions, but without experimentally-determined structures.
We then applied our methods to characterize five alternative splice variants which we had identified in mouse Her2/neu mammary tumor 9: Annexin 6 (anxa6), Calumenin (calu), Cell division cycle 42 (cdc42), Polypyrimidine tract binding protein 1 (ptbp1), and Tax1-binding protein 3 (tax1bp3). By quantitative analysis these proteins were over-expressed in the tumor compared to normal samples (p < 0.001) (Text S1). The major aim of this study is to examine whether splice variants differentially expressed in tumors show evidence of altered functional characteristics. For each of these five tumor variants, we selected another variant derived from their parent gene to make a variant pair for structural comparison. Each variant pair has at least 75% sequence identity and also has homologous human proteins; these constitute an ideal, challenging test for the I-TASSER pipeline to initiate the characterization of the splice variant and provide clues for experiments. From distinct differences observed between the predicted structural models of the variant pairs, our integrated proteomics/structural biology approach reveals biologically interesting functional motifs which shed light on potential functional differences.
Materials and Methods
Three-dimensional Structural Modeling and Benchmark Analysis
The three-dimensional structures of the variants were generated using I-TASSER 6, a fully-automated structure prediction tool. We use TM-align 10 for protein structure comparisons; structural similarity is quantified using TM-score. A TM-score below 0.3 by TM-align corresponds to the similarity between random structure pairs. Detailed description of I-TASSER and the benchmark analysis is given in Text S1. We identified 15 pairs of alternative splice variants in ASTD which have both isoforms with solved structures in the PDB. Here, we focus our analysis on the six variant pairs of full-length solved structures; the other nine pairs either had only a part of a protein solved in PDB, or were solved without the spliced region, or the variant pairs were formed from totally exclusive exons. When searching the PDB database, we found the solved structures of Pyruvate Kinase (pkm2) variants which were not included in ASTD. Pkm2 is a well-known cancer biomarker implicated in many types of cancers 11. The PDB structure of pkm1 variant (1a49) is from rabbit muscle pkm1 protein which has 97% sequence identity to the human pkm1 variant, whereas the PDB structure of pkm2 variant (1t5a) is from human. The variation observed between the human pkm1 and pkm2 variants is same as that between rabbit muscle pkm1 and human pkm2 variants. With the purpose of benchmarking the I-TASSER structure predictions for alternative splice variants, we modeled the seven protein pairs (6 pairs found in ASTD plus the pkm2 variants) using the automated I-TASSER pipeline, after excluding both the query and the splice variant structures and their close homologs in the PDB from the threading template library. The accuracy of the I-TASSER models is assessed by the C-score 12, 13, which is a combination of the significance score of threading templates and the structural convergence of the I-TASSER simulations. It has a correlation coefficient of 0.91 with the actual TM-score of final models as reported in the large-scale benchmark tests 12. As a comparative measure, we also predicted structures of the seven protein pairs using MODELLER 14 and compared with experimentally determined structures.
It is important to note here that, in our process of searching alternative splice variant pairs in PDB, we found partial structures of many splice variant pairs (splicing due to exon insertion or deletion) where the alternatively-spliced regions were not resolved, indicating that splicing often occurs in inherently flexible locations. Wang et al 15 reported a similar observation. Hence, we were not able to use these pairs for our benchmark analysis. The PDB structures of the first domain of mouse ryr2 variants (3mi5 and 3qr5; in 3qr5 there is a deletion of 35 amino acids (aa)) were used to demonstrate the structural difference due to insertion/deletion. We predicted their structures using I-TASSER and compared them to the experimentally solved structures.
Alternative Splice Variants Differentially Expressed in Her2/neu Breast Cancer Selected for I-TASSER Modeling and Structure Comparison
The five criteria used in selecting variant pairs for this structural comparison study were:
The variants were differentially expressed in tumor samples compared to normal (see Text S1 for detailed information).
The variant is annotated as a known protein in the Ensembl database.
The parent genes of these splice variants had at least one other known variant in the Ensembl database with ≥ 75% sequence identity between the variant identified from tumor sample and the other known variant. If there is more than one known variant, the variant more similar to the tumor-associated variant in protein length and sequence content is selected for comparison.
There are known homologous variants of these protein pairs in Homo sapiens.
The C-score of the predicted models for both the variants is >−1.5.
The variants of anxa6, calu, cdc42, ptbp1, and tax1bp3 (Table 1) satisfied all of the above criteria. NCBI bla2seq was used to obtain the sequence similarities between the variants (http://blast.ncbi.nlm.nih.gov/). The sequence alignment information, along with the annotation of the variants provided in Ensembl (v 62), was used to explain the probable splicing event which created the variants from a single gene (Table 1). The sequences of the mouse variant pairs used in this analysis are given in Table S1.
Table 1.
The cancer-associated alternative splice variants selected for structural comparison study.
| Gene Symbol |
Description | Variant Name (Ensembl) |
Protein length |
Sequence identity between the variants |
Possible splicing mechanism |
Sequence identity with human variant |
|---|---|---|---|---|---|---|
| anxa6 | Annexin 6 | Anxa6-001 * | 673 | 99% | Deletion. Exon 2 found in anxa6-001 translates to 6 aa residues missing in anxa6-002 | 94% |
| Anxa6-002 | 667 | 94% | ||||
| calu | Calumenin | Calu-001 * | 315 | 92% | Exon swapping. Exon 2 is different. | 98% |
| Calu-002 | 315 | 99% | ||||
| cdc42 | Cell division cycle 42 homolog | Cdc42-001* | 191 | 95% | Exon swapping. Exon 5 is different. | 100% |
| Cdc42-002 | 192 | 100 | ||||
| ptbp1 | Polypyrimidine tract binding protein 1 | Ptbp1-001 * | 555 | 95% | Deletion. Exon 8 found in ptbp1-001 translates to 26 aa residues missing in ptbp1-002 | 96% |
| Ptbp1-002 | 529 | 96% | ||||
| tax1bp3 | Tax1 binding protein 3 | Tax1bp3-001 * | 124 | 79% | Deletion. Exon 3 found in tax1bp3-001 translates to 26 aa residues missing in tax1bp3-004 | 99% |
| Tax1bp3-004 | 98 | 99% |
The variants with an asterisk (*) were identified in the tumor sample as over-expressed compared to normal tissue by proteomic analysis. Each variant has a homologous human variant.
Functional Motif Analysis of Splice Variants
To functionally characterize the splice variants, tools including ELM and Motif Scan were used. ELM is a resource for predicting functional sites in eukaryotic proteins 16. MotifScan scans a sequence against protein profile databases 17.
Modeling of Known Alternative Splice Variants with Distinct Functions
With the intention of strengthening our findings from the structural comparison analyses of the selected five variant pairs, we modeled and compared the structures of human alternative splice variants of Bcl-X (bclx), Caspase 3 (casp3), and Odd-skipped related 2 (osr2) genes. The splice variants of these three genes have been reported by others to have distinct opposite functions (15–17). They represent additional benchmark examples for our analyses of new splice variant pairs.
Results
Benchmark of I-TASSER Modeling on Known Spliced Isoform Proteins
The results of the structural comparisons between I-TASSER models and experimentally- determined structures of the seven alternative splice variant pairs in PDB are presented in Table S2. The average RMSD between the experimentally-determined structure and the model predicted by I-TASSER for the 7 splice variant pairs was 1.72 Å. Generally, a structure model within 4–6 Å has roughly similar fold/topology to the native, which can be useful for domain definition and family assignment 18; models within 1–2 Å are considered of high-resolution comparable to the accuracy of medium-resolution NMR or low-resolution x-ray experiments 19, which can be used for high-resolution drug screening and detailed functional analysis 20, 21. All seven are due to exon swapping. Figures S1 & S2 show the experimentally-solved and predicted structures of three variant pairs: Ketohexokinase (khk), Acid phosphatase 1 (acp1), and Mitogen-activated protein kinase 8 (mapk8). Even alternative splice variants whose structures are very similar may have functional differences due to absence of a functionally critical residue or altered post-translational modification of residues in the swapped exon. For example, in the case of acp1 variants, the Mg2+ binding site is missing in the 1xwwA variant. Figures S3 and S4 show the side chains of the residues in the alternatively-spliced regions of acp1 and mapk8 variants.
The RMSD value between the experimentally determined structure and the corresponding predicted model by MODELLER for each splice variant studied in this benchmark analysis is also given in Table S2.
Since all of the seven variant pairs studied in this benchmark analysis were the outcome of exon swapping during RNA splicing, in order to investigate structural difference due to insertion/deletion of an exon, we compared the 3D structural models of the first domain from two mouse ryanodine receptor 2 (ryr2) splice variants to the experimentally-solved structures found in PDB. The domain sequences differ by 35 aa due to an exon deletion. Similar structural differences were observed in both experimentally-solved and predicted models. The average RMSD between the experimentally-solved and predicted models for ryr2 is 1.09 Å (Figure S5).
Structural Analysis of Known Alternative Splice Variants with Opposite Functions
Most alternative splice protein variants have high sequence identity and therefore have approximately similar structure and functions. Here, we focus on three cancer-related protein splice variant pairs from bclx, casp3, and osr2 which are known to have distinctly opposite functions, like pro- or anti-apoptotic or transcriptional activator or repressor activities 22–24. Compared with the longer variants, shorter variants all have sizeable deletions.
An additional 63 amino acids (aa 129–191) create an extra domain in the middle of the core structure of bclx-L (233 aa) compared to bclx-S (170 aa) (Figure 1a). The shorter bclx-S variant is missing the two Bcl-2 family motifs BH1 and BH2, whereas the longer variant contains all four Bcl-2 homology motifs (BH1-4); this results in a completely different topology of the structures. BclxL is anti-apoptotic, whereas bclx-S is pro-apoptotic 22. Indeed, the TM-align comparison of the I-TASSER models demonstrated structural changes at multiple locations in the bclx variant.
Figure 1.
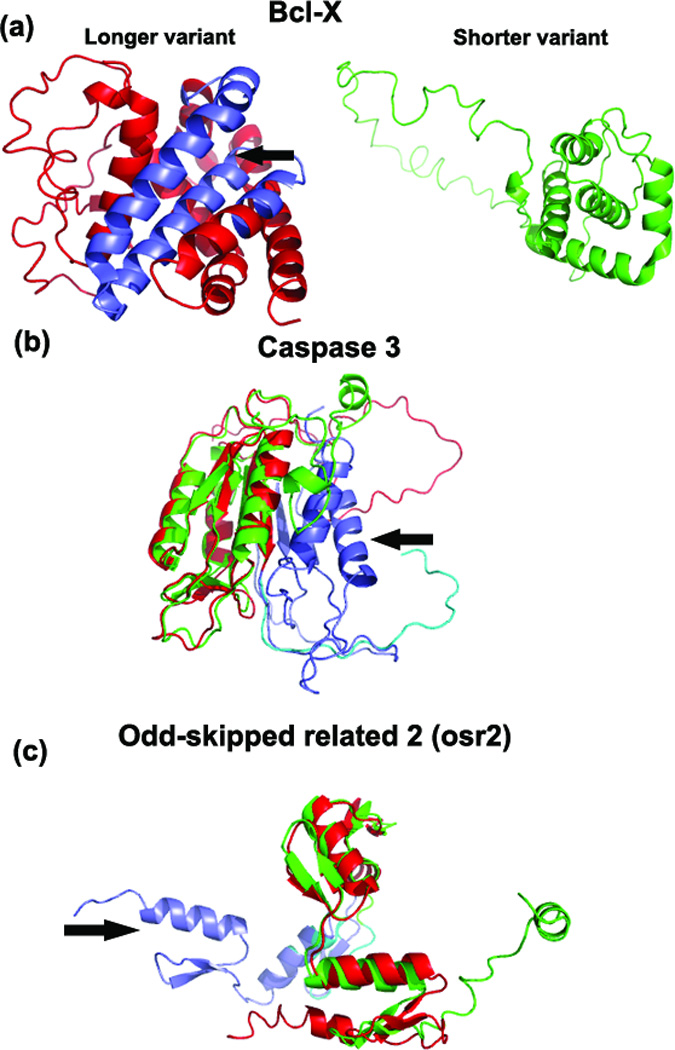
(a) The predicted models of bclx-L (red) and bclx-S (green). The bclx-S was created due to an alternative splice site in exon 1, which translates to a shorter peptide sequence (63 aa shorter), compared to that of bclx-L. The shorter variant lacks Bcl2-BH1 and Bcl2-BH2 motifs. The arrow points to the hydrophobic cleft formed by Bcl2 BH1, BH2 and BH3 domains in bclx-L. The absence of 63 aa in the shorter variant resulted in a different protein structure than the longer variant with an RMSD of 3.16 Å. (b) Superimposed predicted structures of casp3-L (red) and casp3-S (green). The RMSD between the casp3 predicted structures is 5.27 Å. (c) Superimposed predicted structures of osr2-L (red) and osr2-S (green). The RMSD between the osr2 predicted structures is 1.32 Å. In both (b) & (c), the alternatively-spliced regions are shown in purple and blue. In each case, the arrow points to the additional domain in the C-terminal end of the longer variant. In osr2-L, there are 2 additional C2H2 zinc finger additional domains, whereas in casp3-L caspase family p10 is the extra domain. The center part of the variant structures is aligned.
The casp3-L (277 aa) and casp3-S (182 aa) isoforms have identical 162 N-terminal residues, which corresponds to the highly conserved structure of the N-terminal domain in both molecules (Figure 1b). But pro-apoptotic casp3-L has a 115 aa long C-terminal region which has no sequence identity with the anti-apoptotic casp3-S whose C-terminal region is only 20 aa long. Alternative transcription gives rise to the shorter splice variant from a deletion of exon 6. The protein structures of the L and S isoforms in the C-terminal regions are indeed very different in t h e I-TASSER modeling (see Figure 1b). Nevertheless, both models have high confidence scores, since the multiple threading programs from LOMETS (see Text S1 for I-TASSER description) hit similar templates in the C-terminal domain, indicating reliability of the structural models in this domain. The caspase family p10 domain is missing in the casp3-S isoform.
Two alternatively-spliced transcripts of the osr2 gene encode osr2-L (312 aa) and osr2-S (276 aa), which have opposite transcriptional activities, activation and repression 24. The difference between the variants is in the C-terminal region translated from the third exon of the transcripts (60 aa in osr2-L and 24 aa in osr2-S). No significant sequence similarity was observed between these C-terminal regions. The 3D predicted structural models of the two variants (Figure 1c) show structural misalignment in the C-terminal ends. Osr2-L contains five C2H2 zinc finger domains, including two domains in the C-terminal spliced region, compared to three total C2H2 zinc finger domains in osr2-S.
Structural Comparisons and Functional Motif Analysis of the Selected Five Pairs of Alternative Splice Variants Differentially Expressed in Her2/neu-Induced Breast Cancer
I-TASSER was used to predict the structures of five tumor-associated alternative splice variant pairs (Table 1) identified by proteomic analysis (10). For each pair, we used TM-align 10 to compare the resultant I-TASSER models. TM-align is a sensitive algorithm to identify the optimal alignment of two protein structures by heuristic dynamic programming iterations. It returns a TM-score to assess the structural similarity, with TM-score=1 as identical structures, TM-score>0.5 as roughly the same fold, and TM-score < 0.3 as random structures. The TM-align comparisons show that the structures of the splice variants ranged from minor variations to large changes in the backbone structure. Table 2 shows the RMSD and TM-scores between the splice variants’ structures in each pair. Figure S6 shows the cartoon representation of the alternatively-spliced regions in these five pairs of variants. Next, we present alternative splicing-related structural changes in the models and plausible functional implications in each of these proteins.
Table 2.
Functional residue or motif found in the regions of structural variation between the breast cancer-related alternative splice variants. RMSD and TM-score are parameters of the I-TASSER analysis (see Methods and Text S1).
| Variant name |
Amino acid position |
Functional residue or motif | RMSD (Å) |
TM-score |
|---|---|---|---|---|
| anxa6-001 | 530–538 | Proline-Directed Kinase phosphorylation site (TP) and Serine phosphorylation; (Thr535, Ser537 in anxa6-001; Thr529, Ser531 in anxa6-002) | 0.38 | 0.99 |
| anxa6-002 | 529–531 | |||
| calu-001 | 33–53 | Ser-35 and Tyr-47 could be phosphorylated more readily in calu-001 | 2.66 | 0.87 |
| calu-002 | ||||
| cdc42-001 | No 3D-structural difference between the variants | C-terminal Leu191 in cdc42-001 preferred in prenylation; Ser185-Arg186-Arg187 is a potential protein kinase c phosphorylation site | 0.69 | 0.99 |
| cdc42-002 | ||||
| ptbp1-001 | Different structures | The positions of the 4 RNA recognition motifs shifted in ptbp1-002 | 3.13 | 0.86 |
| ptbp1-002 | ||||
| tax1bp3-001 | 43–73 | PDZ domain absent in tax1bp3-004 | 1.78 | 0.7 |
| tax1bp3-004 | 43–47 |
The only difference between anxa6-001 and anxa6-002 at the sequence level is the presence of six residues in anxa6-001 (‘VAAEIL’; 525–530) missing in anxa-002. Although the global topology of the I-TASSER models for the two isoforms is almost identical (with RMSD=0.38 Å and TM-score=0.99), there is an obvious structural variation identified by TM-align (Figure 2a). These six residues are in the end region of a helix (the blue-colored section in Figure 2a) which is followed by a loop. Because of the absence of ‘VAAEIL’ residues, the loop is smaller in the shorter variant. Interestingly, this region of structural variation harbors a proline-directed kinase phosphorylation ([ST]P) site followed by a serine phosphorylation site; these residues (Thr-535, Pro-536, Ser-537) are in the loop region in anxa6-001, whereas the threonine residue (now Thr-529) moves inside the helix region in anxa6-002 (Figure 2a inset), making phosphorylation less probable in anxa6-002.
Figure 2.
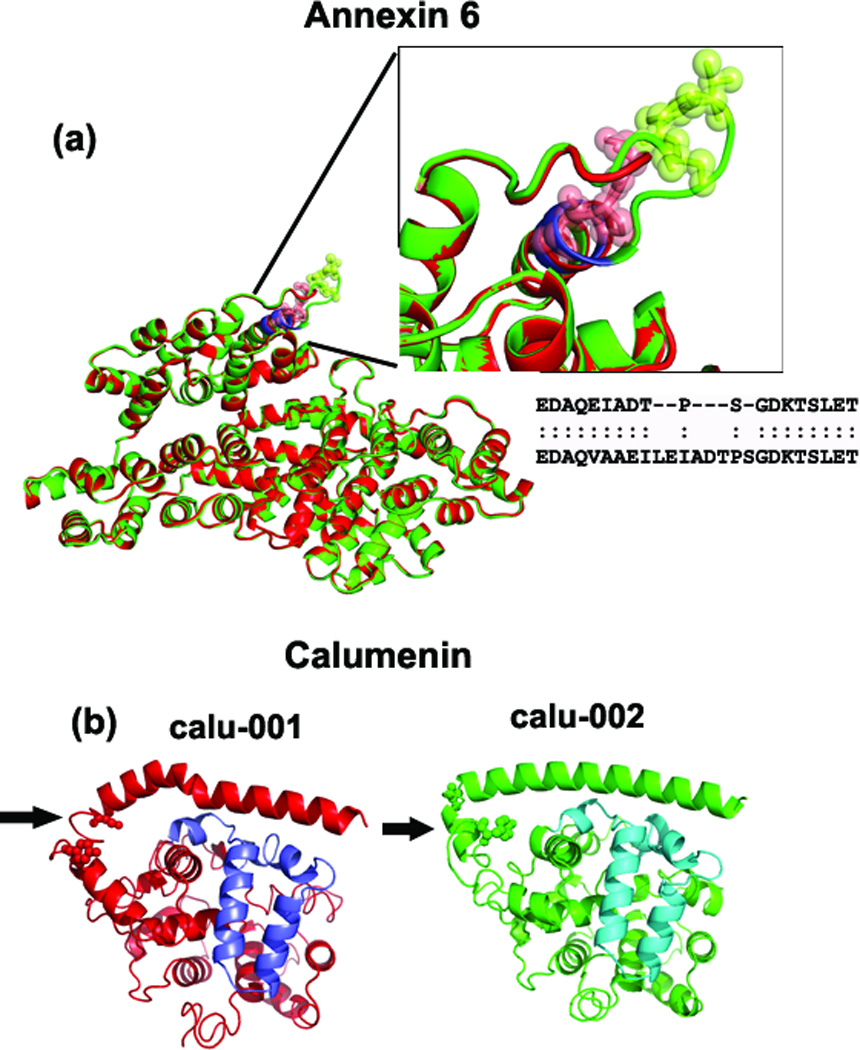
Superimposed predicted structures of anxa6 and calu. (a) Superimposed anxa6 models show perfect alignment except for one region (shown in the inset figure). Due to the absence of ‘VAAEIL’ residues (aa 525–530 in anxa6-001) in the anxa6-002 variant (shown in red), the loop region is smaller. The structural alignment shows that the ’TPS’ residues are not aligned between the anxa6 variants (see inset box), which could affect the post-translational modification of Thr & Ser residues (as shown in green and red spheres in the inset figure). (b) Calu-001 and calu-002 models with the region of structural variation indicated by arrow. This region in calu-001 (red) is in the loop instead of coil-helix-coil region found in calu-002; two potential phosphorylation sites (Ser-35, Tyr-47) are found here (shown in spheres). The purple and blue-colored regions show the alternatively-spliced regions in Calumenin variants.
The peptide sequences translated from the swapped exons of calu splice variant pairs are shown in purple and blue colors (Figure 2b). There is no structural variation in this region. Instead, we observed structural variation between residues 33 and 53 in calu-001 and calu-002 variants. The affected residues are in a coil region in calu-001 compared to coil-helix-coil in calu-002 (follow arrow in Figure 2b). While the sequence is the same, this structural difference (RMSD=2.66Å) could influence the binding affinity or post-translational modifications, especially 25 phosphorylation of Ser-35 and/or Tyr-47 (these two residues are shown as spheres in Figure 2b).
The structure models of the cdc42 variants are identical, with low RMSD and high TM-score (0.69/0.99) (see Table 2, Figure 3a). The C-terminal 29 amino acids constitute the alternatively-spliced region in both variants with sequence identity of 81% between them. The two variant models show different orientation in the C-terminal end; since C-terminal and N-terminal folding patterns are generally flexible, this difference in structure may not be reliable. However, there are important differences in the amino acid composition of the C-terminal spliced region of these cdc42 variants. The last four residues of cdc42-001 are Cys-Val-Leu-Leu, whereas cdc42-002 has Cys-Cys-Ile-Phe. C-terminal leucine is preferred over a phenylalanine during the prenylation of the nearby Cys residue 26. Prenyl groups play a role in anchoring proteins to cell membranes. In addition, our MotifScan analysis indicates Ser185-Arg186-Arg187 (SRR) residues as a potential protein kinase C (PKC) phosphorylation site.
Figure 3.
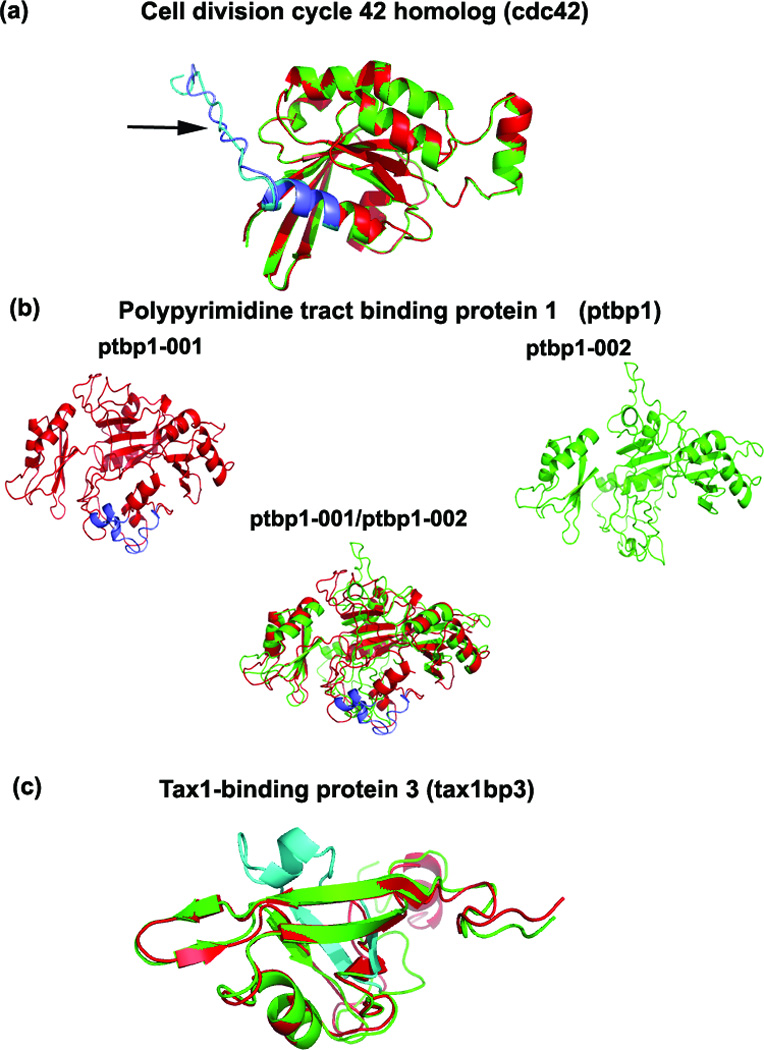
Superimposed predicted structures of cdc42, ptbp1 and tax1bp3. (a) No structural differences were observed between the cdc42 variants. The arrow points to the alternatively-spliced region. (b) The predicted pbp1 structures of the variants (see Results) show structural variation in multiple regions due to the insertion of 26 residues (purple color) in the ptbp1-001 (red). The RMSD between the two variants is 3.13Å (Table 2). (c) An additional domain is present (shown in blue) in the tax1bp3-001 (green) compared to the shorter tax1bp3-004 (red).
Ptbp1 is a large multi-domain protein (4 domains in each variant) with the two isoforms differing by the absence of 26 amino acids in the linker region between domains 2 and 3 in the shorter ptbp1-002 variant. Domain 1 of ptbp1 (residues 1–182) was not well aligned to the top templates by any of the I-TASSER threading programs and was highly flexible and structurally disordered during the I-TASSER simulations, yielding a low C-score (see Methods). Exclusion of residues 1–182 from the variant sequences during modeling led to a more ordered structure with high C-score. Since Domain 1 is distant from the splicing in the Domain 2–3 linker region, we truncated ptbp1 for modeling and functional analysis. The resulting RMSD/TM-score is 3.13 Å/0.86. These structural differences can be attributed to shift of the domains with respect to each other rather than changes in the local structures of the domains themselves (Figure 3b). Both ptbp1 variants have the four quasi-RNA recognition motif (RRM) domains that bind to RNA to enable splicing mechanisms. TM-align indicated multiple regions of structural misalignments between the variants, consistent with different positions of the RRM motifs in the two variants.
Due to the absence of 26 amino acids in its interior, the shorter tax1bp3–004 variant is missing a PDZ domain formed by these residues in the longer tax1bp3–001 variant (blue-colored region in Figure 3c). Except for the difference in the C-terminal end due to its flexible folding pattern, and the extra domain due to the spliced region in the longer variant, the remaining part of the protein structures aligned well. The PDZ domain extends from position 15 to 112 in the longer tax1bp3–001 protein.
Discussion
Alternative RNA splicing allows a single gene to generate multiple mRNA transcripts, which can be translated into functionally and structurally diverse proteins 27. Structural variations in alternative splice variants arising from amino acid sequence differences affect their functional roles in biological mechanisms. In this analysis, we observed alternative splice variant pairs with large structural changes and others with no structural difference. The Protein DataBase (PDB) has very few experimentally-solved structures of alternative splice variants 15. Our novel approach of functional inferences for alternative splice variants by comparing their predicted structures provides a relevant insight to explore the role of these variants in complex mechanisms of cancers or other diseases. It is important to note that we used a well-trained confidence score, C-score, to assess the quality of the I-TASSER structure predictions. In large-scale benchmark testing, C-score was highly correlated with accuracy of the I-TASSER models relative to the experimental structures 12, 13, 28. With C-score >-1.5 as the estimate of models of correct fold, for example, both false-positive and false-negative rates were below 0.1 (23). The majority of our models in this study had a C-score much higher than −1.5, reinforcing the reliability of the models for the splice variants. The structural variations due to splicing observed between the experimentally-determined PDB structures were captured well by the predicted models (Figure S2). In some cases, we observed structural differences between the variants in regions with complete sequence identity. This could be due to the influence of the sequence variation in the spliced region; in each case, we were able to find a functionally interesting motif or residue located where such structural differences were observed.
The structural comparisons of known splice variant pairs that have been reported to have distinctive opposite functions [bclx (13, 21), casp3 23 and osr2 24] further support the explanatory validity of our integrated approach. In each pair, we observed structural differences in regions where there is presence or absence of a motif that directly influences the distinct functions of the variants. The members of the Bcl-2 family share one or more of the four characteristic domains of homology named BH1, BH2, BH3 and BH4. The BH domains are known to be crucial for the function of the bcl2 family proteins. The anti-apoptotic bclx-L variant conserves all four BH domains, whereas the pro-apoptotic bclx-S protein lacks bcl2-BH1 and BH2 motifs. The structural comparison shows absence of the hydrophobic cleft formed by BH1, BH2 and BH3 domains in the bclx-S variant (shown in purple in Figure 1a). This bclx-L hydrophobic cleft is responsible for its interactions with BH3-containing death agonists and thus plays a role in inhibiting apoptosis 29. The absence of 63 aa in the interior of bclx-S resulted in completely different folding from bclx-L (Figure 1a). The models have a TM-score of 0.45 between the two variants suggesting that the bclx-L and bclx-S do not have similar protein folds (26).
Caspase 3 plays an essential role in apoptosis 30. The inactive procaspase 3 form is activated by proteolytic cleavage between p20 and p10 domains to yield two separate molecules that form heterodimers. This active form then cleaves other vital substrates in the cell during cell death. In the casp3-S variant, the caspase family p10 domain is absent, which prevents formation of the active heterodimer. Figure 1b shows the distinct structure of the p10 domain in the casp3-L variant (purple-colored region), which is absent in casp3-S. Vegran et al 23 showed that the casp3-S/casp3-L ratio might be used as a predictive marker to define a subset of patients with locally advanced breast cancer who are more likely to benefit from neoadjuvant cyclophosphamide-containing chemotherapy.
Osr2-L has five zinc-finger domains, whereas osr2B has three. The two extra zinc finger domains are found in the helix structure of the osr2-L variant, which is absent in osr2-S (the blue-colored helix in Figure 1c). C2H2 zinc finger domains are the most common DNA-binding motifs found in eukaryotic transcription factors. Transcription factors usually contain several zinc fingers capable of making multiple contacts along the DNA. The C2H2 zinc finger domains bind to the major groove of DNA via a short alpha-helix in the zinc finger 31. Kawai et al 24 showed that osr2-L and osr2-S variants have opposite transcriptional activities 24. They reported transcriptional activation or repression by osr2 variants expressed in cos-7 cells from different luciferase constructs. This opposing behavior seems to be caused by different numbers of zinc-finger domains, with different phosphorylation patterns and/or different affinity for DNA-binding 24.
One of the criteria used in selecting the breast cancer-related variants for this analysis was that the mouse variant should have a homologous human variant (for future biomarker purposes). We observed similar changes when we performed the structural comparisons of predicted models of human calu, cdc42 and ptpb1 variants (Table S3).
We now focus on annotation and interpretation of the structural differences found between the alternative splice variant pairs we selected based on our Her2/neu breast cancer proteomics data (see Methods and Results).
Annexin 6 has been implicated in several types of cancers (24–26) and could be a potential target for cancer immunoprevention strategies due to its distinct expression pattern in the HER-2/neu oncogene-driven mammary carcinogenesis model (26). We observed significant over-expression (p < 0.001) of anxa6 (anxa6-001) in Her2/neu tumor tissue compared to normal tissue. Post-translational phosphorylation of anxa6 is associated with cell growth 32. In 3T3 fibroblasts and human T-lymphoblasts, annexin 6 was not phosphorylated in quiescent cells, but serine and, to a lesser extent, threonine were phosphorylated several hours after cell stimulation 32. We found a single structural difference between the variants of anxa6; due to alternative splicing, the positions of both Thr-535 and Ser-537 are shifted to the inner part of the loop region in anxa6-001 compared to anxa6-002 (Figure 2a, inset figure), making anxa-001 more prone to undergo phosphorylation. Experimental validation was beyond the scope of this study; such validation of preferential phosphorylation of the Thr-535 and/or Ser-537 of anxa6-001 would be informative.
Recently, Lai et al reported calu as a potential breast cancer marker 33. Calu-001 and calu-002 variants were identified in our study of samples from mice with Her2/neu breast cancer versus wild-type mice 9. Calu-001 was identified only in the tumor sample whereas calu-002 was found in both normal and tumor samples. In the region of structural variation between the calu variants, we observed a helical unfolding in calu-001 (aa 33–53; arrow, Figure 2b), which we predict will enhance phosphorylation of Ser-35 and Tyr-47 in calu-001. Studies have suggested functional modification of Calumenin by phosphorylation 33, 34.
Cdc42 has been reported to affect cancer mechanisms in many ways 34. The spliced region of the two cdc42 variants differs in 11 C-terminal residues. However, the TM-align analysis of cdc42 variants did not show any structural differences (except the random orientation of the C-terminal coil region). The presence of a Leu as the last C-terminal residue in cdc42-001 compared to Phe in cdc42-002 has functional implications. The hydrophobic 20-carbon prenyl groups facilitate attachment to cell membranes and have been shown to be important for protein-protein binding through specialized prenyl-binding domains. During prenylation, it is the protein geranylgeranyltransferase-I (PGGT-I) that catalyzes the transfer of the 20-carbon prenyl group from geranylgeranyl pyrophosphate to the cysteine residue near the C-termini of a variety of eukaryotic proteins. Analysis of the substrate specificity of PGGT-I shows that peptides which contain a C-terminal leucine are preferred to those that end in serine or phenylalanine 27. The preferential prenylation, which, in turn, increases protein-protein interactions, may have a role in the over-expression of cdc42-001 in tumor. We also found a pkc phosphorylation site in cdc42-001 which is not present in cdc42-002 (Table 2).
The protein encoded by the ptbp1 gene has four repeats of quasi-RNA recognition motif (RRM) domains that bind RNA. Both variants studied here contain these four domains. The ptbp1-001 variant was over-expressed in the tumor tissue. The TM-align between the variant structures revealed multiple structural differences arising from exclusion of the 26 amino acids in the ptbp1-002 variant. Vitali et al35 reported the restricted positioning of ptbp RRM3 and RRM4 motifs in relation to each other and their tight interaction that induces the formation of RNA loops. Thus, ptbp1 could repress splicing by sequestering either a short alternative-exon or a branch point within these RNA loops 35. The shift in the positions of RRM motifs of ptbp1-002 variant may have an impact on its function.
Tax1-binding protein 3 consists almost entirely of a single PDZ domain with no other obvious protein interaction module. It is a general negative regulator of PDZ scaffolding, competing with other PDZ-containing proteins for binding to specific target proteins 36 such as beta catenin 37, a well known prognostic marker for breast cancer 38. The longer variant tax1bp3-001 was over-expressed in our tumor sample. The only structural difference between the variants was the absence of the PDZ domain in taxbp3-004 variant (Figure 3c). Based on what we observed in casp3 and osr2 variants, this shorter variant of tax1bp3 may have an opposite function to that of tax1bp3-001.
The splice variant pairs studied here show that alternative splicing produces structural diversity. Without experimental proof we can only speculate about the structures of these isoforms resulting from splicing events. However, our benchmark analyses indicate the high accuracy of I-TASSER structure predictions. Moreover, the RMSD values from both I-TASSER and MODELLER methods support this conclusion (Table S2). We also tried ROSETTA 39 for structure prediction of the seven splice variant pairs used in the benchmark analysis. ROSETTA’s ab-initio (template free) method may predict correct structures when the protein length is < 150 aa 40. Since six out of seven splice variant pairs in the benchmark analysis were of protein length > 150 aa (four > 250aa), the average RMSD value between the experimentally determined structure and the corresponding predicted structure by ROSETTA was much higher.
Many studies have reported cancer-specific alternative splicing. As examples, Thorsen et al 41 identified and validated cancer-specific splicing events in colon, bladder, and prostate cancer that may have diagnostic and prognostic implications. A splice variant of cholecystokinin-2 receptor (cck2r) contribute to the growth and spread of GI cancers through agonist-independent mechanisms that enhance tumor angiogenesis 42. Honda et al 43 identified a novel alternative splice variant RNA of actinin-4 in human small cell lung caner (SCLC). Studying these cancer-specific variants using our computational structural approach would give more insight to their functions.
Seeking structural differences between two splice variants may not be effective if the differences occur in a large loop region. The flexibility of a large loop increases the difficulty of resolving the structure of the region by either experimental determination or modeling approaches. In the case of anxa6 variants, we were able to predict the possible functional characteristics of the variants as the difference due to splicing was located in a very small loop region (Fig 2a). When the structural difference is in a large loop region or in cases where splice variants have the indistinguishable structures (cdc42 variants, Fig 3a), comparing the primary sequences of the variants can reveal potential functional differences.
The different types of splicing events, including exon skipping, exon swapping, intronic inclusion, and alternative splice sites all result in variants with sequence differences; even a small percentage of stable structures implies proteins with probable distinct functions. If a variant is able to fold into a stable structure similar to that of the canonical variant, it can mimic the structural features and interact with interaction partners without processing them further. Thus, alternative splicing could provide a mechanism for turning an activator into an effective inhibitor 44. As illustrated by ptbp1 variants that we analyzed in this study. In this case, both variants contained the 4 RRM motifs required for their RNA binding function; the absence of the linker region in the shorter variant resulted in the displacement of the third and fourth RRM motifs which may impact its role in RNA binding and splicing.
Conclusions
We have identified biologically interesting information about the alternative splice variants of anxa6, calu, cdc42, ptbp1, and tax1bp3, which were differentially expressed in mouse Her2/neu-induced breast cancer. Corresponding studies of the homologues of these splice variant proteins and others may have clinical relevance in human breast cancers. This study is the first to use 3D structural modeling to infer the functional characteristics of alternative splice variants discovered by proteomics. Splice variants discovered by RNA-sequencing will need to be confirmed at the protein level. The functional residues or motifs found in the regions showing structural differences provide clues to understand the specific functions of these proteins. Experimental studies will be necessary to elucidate and validate the specific functional mechanisms of these alternative splice variants.
Supplementary Material
Acknowledgements
The authors thank Dr. Dong Xu for helping with the benchmark analysis. The work is supported in part by grants from National Institutes of Health (GM083107, GM084222, U54DA21519, P30ES017885) and National Science Foundation (NSF 0746198).
Footnotes
Supporting Information
Text S1: This document contains the detailed description of the methods used in this study
Supplementary Table & Figures Menon et al.doc: This document contains the supplementary tables and figures (Table S# or Figure S#) mentioned in the main text.
References
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008;18(3):342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moult J, Fidelis K, Kryshtafovych A, Rost B, Hubbard T, Tramontano A. Critical assessment of methods of protein structure prediction-Round VII. Proteins. 2007;69 Suppl 8:3–9. doi: 10.1002/prot.21767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69 (S8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 5.Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Battey JN, Kopp J, Bordoli L, Read RJ, Clarke ND, Schwede T. Automated server predictions in CASP7. Proteins. 2007;69 (S8):68–82. doi: 10.1002/prot.21761. [DOI] [PubMed] [Google Scholar]
- 8.Cozzetto D, Kryshtafovych A, Fidelis K, Moult J, Rost B, Tramontano A. Evaluation of template-based models in CASP8 with standard measures. Proteins. 2009;77 Suppl 9:18–28. doi: 10.1002/prot.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menon R, Omenn GS. Proteomic characterization of novel aternative splice variant proteins in Human epidermal growth factor receptor 2/neu induced breast cancers. Cancer Research. 2010;70(9):3440–3449. doi: 10.1158/0008-5472.CAN-09-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haug U, Rothenbacher D, Wente MN, Seiler CM, Stegmaier C, Brenner H. Tumour M2-PK as a stool marker for colorectal cancer: comparative analysis in a large sample of unselected older adults vs colorectal cancer patients. Br J Cancer. 2007;96(9):1329–1334. doi: 10.1038/sj.bjc.6603712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl. Acad. Sci. USA. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Chapter 2. Curr Protoc Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Unit 2 9. [DOI] [PubMed] [Google Scholar]
- 15.Wang P, Yan B, Guo J-t, Hicks C, Xu Y. Structural genomics analysis of alternative splicing and application to isoform structure modeling. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(52):18920–18925. doi: 10.1073/pnas.0506770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gould CM, Diella F, Via A, Puntervoll P, Gemünd C, Chabanis-Davidson S, Michael S, Sayadi A, Bryne JC, Chica C, Seiler M, Davey NE, Haslam N, Weatheritt RJ, Budd A, Hughes T, Paś J, Rychlewski L, Travé G, Aasland R, Helmer-Citterich M, Linding R, Gibson TJ. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic acids research. 2009 doi: 10.1093/nar/gkp1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Hau J, Martin O, Kuznetsov D, Falquet L. MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic acids research. 2007;35 suppl_2:W433–W437. doi: 10.1093/nar/gkm352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y. Protein structure prediction: when is it useful? Curr Opin Struct Biol. 2009;19(2):145–155. doi: 10.1016/j.sbi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294(5540):93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 20.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. British Journal of Pharmacology. 2007;152(1):21–37. doi: 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brylinski M, Skolnick J. Q-Dock: Low-resolution flexible ligand docking with pocket-specific threading restraints. Journal of computational chemistry. 2008;29(10):1574–1588. doi: 10.1002/jcc.20917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Revil T, Toutant J, Shkreta L, Garneau D, Cloutier P, Chabot B. Protein kinase C-dependent control of bcl-x alternative splicing. Mol. Cell. Biol. 2007;27(24):8431–8441. doi: 10.1128/MCB.00565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Végran F, Boidot R, Oudin C, Riedinger J-M, Bonnetain F, Lizard-Nacol S. Overexpression of Caspase 3s Splice Variant in Locally Advanced Breast Carcinoma Is Associated with Poor Response to Neoadjuvant Chemotherapy. Clinical Cancer Research. 2006;12((19)):5794–5800. doi: 10.1158/1078-0432.CCR-06-0725. [DOI] [PubMed] [Google Scholar]
- 24.Kawai S, Kato T, Inaba H, Okahashi N, Amano A. Odd-skipped related 2 splicing variants show opposite transcriptional activity. Biochemical and Biophysical Research Communications. 2005;328(1):306–311. doi: 10.1016/j.bbrc.2004.12.173. [DOI] [PubMed] [Google Scholar]
- 25.Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, Jensen PR, Mann M. The Serine/Threonine/Tyrosine Phosphoproteome of the Model Bacterium Bacillus subtilis. Molecular & Cellular Proteomics. 2007;6(4):697–707. doi: 10.1074/mcp.M600464-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Yokoyama K, McGeady P, Gelb MH. Mammalian protein geranylgeranyltransferase-I: substrate specificity, kinetic mechanism, metal requirements, and affinity labeling. Biochemistry. 1995;34(4):1344–1354. doi: 10.1021/bi00004a029. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Hubbell E, Hu J-s, Mei G, Cline M, Lu G, Clark T, Siani-Rose MA, Ares M, Kulp DC, Haussler D. Gene structure-based splice variant deconvolution using a microarry platform. Bioinformatics. 2003;19 suppl 1:i315–i322. doi: 10.1093/bioinformatics/btg1044. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 2009;77 S9:100–113. doi: 10.1002/prot.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minn AJ, Kettlun CS, Liang H, Kelekar A, Vander Heiden MG, Chang BS, Fesik SW, Fill M, Thompson CB. Bcl-xL regulates apoptosis by heterodimerization-dependent and - independent mechanisms. Embo J. 1999;18(3):632–643. doi: 10.1093/emboj/18.3.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porter AG, Jänicke RU. Emerging roles of caspase 3 in apoptosis. Cell death and differentiation. 1999;6(2):99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 31.Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 32.Moss SE, Jacob SM, Davies AA, Crumpton MJ. A growth-dependent post-translational modification of annexin VI. Biochim Biophys Acta. 1992;1160(1):120–126. doi: 10.1016/0167-4838(92)90045-f. [DOI] [PubMed] [Google Scholar]
- 33.Lai T-C, Chou H-C, Chen Y-W, Lee T-R, Chan H-T, Shen H-H, Lee W-T, Lin S-T, Lu Y-C, Wu C-L, Chan H-L. Secretomic and proteomic analysis of potential breast cancer markers by two-dimensional differential gel electrophoresis. Journal of Proteome Research. 2010;9(3):1302–1322. doi: 10.1021/pr900825t. [DOI] [PubMed] [Google Scholar]
- 34.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Letters. 2008;582(14):2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 35.Vitali F, Henning A, Oberstrass FC, Hargous Y, Auweter SD, Erat M, Allain FHT. Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. Embo J. 2006;25(1):150–162. doi: 10.1038/sj.emboj.7600911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alewine C, Olsen O, Wade JB, Welling PA. TIP 1 has PDZ scaffold antagonist activity. Mol Biol Cell. 2006;17(10):4200–4211. doi: 10.1091/mbc.E06-02-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanamori M, Sandy P, Marzinotto S, Benetti R, Kai C, Hayashizaki Y, Schneider C, Suzuki H. The PDZ protein tax-interacting protein-1 inhibits β-catenin transcriptional activity and growth of colorectal cancer cells. Journal of Biological Chemistry. 2003;278(40):38758–38764. doi: 10.1074/jbc.M306324200. [DOI] [PubMed] [Google Scholar]
- 38.Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A. 2000;97(8):4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simons KT, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999 Suppl 3:171–176. doi: 10.1002/(sici)1097-0134(1999)37:3+<171::aid-prot21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 40.Simons KT, Strauss C, Baker D. Prospects for ab initio protein structural genomics. J Mol Biol. 2001;306(5):1191–1199. doi: 10.1006/jmbi.2000.4459. [DOI] [PubMed] [Google Scholar]
- 41.Thorsen K, Sorensen KD, Brems-Eskildsen AS, Modin C, Gaustadnes M, Hein AM, Kruhoffer M, Laurberg S, Borre M, Wang K, Brunak S, Krainer AR, Torring N, Dyrskjot L, Andersen CL, Orntoft TF. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol Cell Proteomics. 2008;7(7):1214–1224. doi: 10.1074/mcp.M700590-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Chao C, Goluszko E, Lee YT, Kolokoltsov AA, Davey RA, Uchida T, Townsend CM, Jr, Hellmich MR. Constitutively active CCK2 receptor splice variant increases Src-dependent HIF-1[alpha] expression and tumor growth. Oncogene. 2006;26(7):1013–1019. doi: 10.1038/sj.onc.1209862. [DOI] [PubMed] [Google Scholar]
- 43.Honda K, Yamada T, Seike M, Hayashida Y, Idogawa M, Kondo T, Ino Y, Hirohashi S. Alternative splice variant of actinin-4 in small cell lung cancer. Oncogene. 2004;23(30):5257–5262. doi: 10.1038/sj.onc.1207652. [DOI] [PubMed] [Google Scholar]
- 44.Birzele F, Csaba G, Zimmer R. Alternative splicing and protein structure evolution. Nucleic acids research. 2008;36(2):550–558. doi: 10.1093/nar/gkm1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.