

Abstract
Hepatic ischemia/reperfusion (IRI) injury remains a major challenge in clinical orthotopic liver transplantation (OLT). Tenascin-C (Tnc) is an extracellular matrix protein (ECM) involved in various aspects of immunity and tissue injury. Using a Tnc-deficient mouse model, we present data that suggest an active role for Tnc in liver IRI. We show that Tnc-deficient mice have a reduction in liver damage and a significant improvement in liver regeneration after IRI. The inability of Tnc−/− mice to express tenascin-C significantly reduced the levels of active caspase-3/TUNEL apoptotic markers and enhanced the expression of the proliferation cell nuclear antigen (PCNA) after liver IRI. The lack of Tnc expression resulted in impaired leukocyte recruitment and decreased expressions of IL-1β, IL-6 and CXCL2 after liver reperfusion. Tenascin-C deficient livers were characterized by altered expression patterns of vascular adhesion molecules, such as VCAM-1 and PECAM-1 post-IRI. Moreover, matrix metalloproteinase-9 (MMP-9) synthesis, which facilitates leukocyte transmigration across vascular barriers in liver IRI, was markedly downregulated in the absence of tenascin-C. We also show that tenascin-C is capable of inducing MMP-9 expression in isolated neutrophils through Toll-like receptor 4 (TLR4). Therefore, our data suggest that tenascin-C is a relevant mediator of the pathogenic events underlying liver IRI. The data also support the view that studies aimed at further understanding how newly synthesized ECM molecules, such as tenascin-C, participate in inflammatory responses are needed to improve therapeutic approaches in liver IRI.
Hepatic ischemia/reperfusion injury (IRI) occurs during trauma, shock, transplantation, and other surgical procedures where blood supply to liver is temporarily interrupted. In transplantation, IRI insult can lead to significantly higher incidence of acute and chronic rejections 1. Hepatocellular damage caused by IRI is the result of complex interactions between various inflammatory mediators, including leukocytes, pro-inflammatory cytokines, and free radicals 2, 3.
Extracellular matrix (ECM) proteins can act as inflammatory stimuli by promoting leukocyte migration and by inducing the expression of pro-inflammatory cytokines, and growth factors 4. These ECM molecules are considered to be endogenous danger signals, or damage-associated molecular patterns (DAMPs) 5. Tenascin-C (Tnc) is likely a prominent DAMP molecule within the ECM 6. Tenascin-C is a hexameric protein of 1,5 million Da consisting of a single fibrinogen-like domain (FBG), and multiple FN-type III and epidermal growth factor (EGF)-like domains 7. Tnc is often called an oncofetal molecule because of its unique expression 8; it is abundantly expressed during embryogenesis, and is not normally expressed in adult tissues, with the exception of specific hematopoetic and lymphoid tissues 9. Notably, Tnc emerges in adult tissues during conditions associated with high rates of cell turnover and migration, including wound repair, tumorigenesis, rheumatoid arthritis, multiple sclerosis, and hepatitis 5, 7, 10, 11
The action of Tnc is complex; it is accepted that Tnc is involved in cell-cell interactions, cell-adhesion and deadhesion events 12. Tnc can influence cell behavior through interactions with cell surface receptors, and by altering cell binding to other ECM proteins 13. Leukocytes can form low-avidity adhesions to Tnc, producing tethering and rolling under flow in a more efficient manner than on selectins 14. While in vitro assays have supported a role for Tnc in leukocyte migration and activation, its precise function at sites of tissue injury remains mostly unclear. It has been recently demonstrated that Tnc induces cytokine release and tissue destruction in arthritic join disease 6. The role of tenascin-C at sites of inflammation is defined by the type of tissue injury or by the participating cell types 13. In the present study, we use Tnc−/− mice to examine the significance of tenascin-C expression in hepatic IRI.
MATERIALS AND METHODS
Animals
Tnc deficient (Tnc−/−) knockout (KO) mice (C57BL/6N-TgH) were obtained from the RIKEN, Japan 15. TLR4 deficient (TLR4−/−) mice (C57Bl/10ScNJ-Tlr4) and C57Bl/10J controls (WT) were purchased from the Jackson Laboratory. Tnc−/− mice were rederived by sterile embryo transfer to surrogate mothers, and housed in the UCLA animal facility under specific pathogen-free conditions.
Model of Partial Lobar Liver Ischemia/Reperfusion Injury
A warm hepatic IRI model was performed in 10-week-old male Tnc−/− mice and matched Tnc+/+ wild-type (WT) littermates, as previously described 16. Briefly, the arterial and portal venous blood supplies were interrupted to the cephalad lobes of the liver for 90 minutes using an atraumatic clip. After 90 minutes of ischemia, the clip was removed thus initiating hepatic reperfusion.
Assessment of Liver Damage
Serum alanine transaminase (ALT) and aspartate transaminase (AST) levels were measured in blood samples with an autoanalyzer by ANTECH Diagnostics (Los Angeles, CA). Liver specimens were fixed in a 10% buffered formalin solution, embedded in paraffin, and processed for H&E staining. The degree of hepatic necrosis was assessed in H&E stained paraffin sections; hematoxylin-eosin stains were digitally photographed and the percent of necrotic was quantified using NIH IMAGE (Image-J) software in a blind manner to the different experimental groups, as previously described 17. Ten random sections per slide were evaluated in duplicate to determine the percentage of necrotic area.
Myeloperoxidase Assay
Myeloperoxidase (MPO) activity was evaluated as previously described 16. Frozen tissue was homogenized in an iced solution of 0.5% hexadecyltrimethyl-ammonium (Sigma, St Louis, MO) and 50 mmol/L of potassium phosphate buffer solution (Sigma) with pH adjusted to 5. After centrifugation, the supernatants were mixed in a solution of hydrogen peroxide-sodium acetate and tetramethyl benzidine (Sigma). The quantity of enzyme degrading 1 μmol/L of peroxide per minute at 25° C per gram of tissue was defined as 1U of MPO activity.
RNA Extraction and Reverse Transcriptase PCR
For evaluation of gene expression, livers were harvested and RNA was extracted with Trizol (Life Technologies Inc., Grand Island, NY), as previously described 16. Reverse transcription was performed using 5 μg of total RNA in a first-strand cDNA synthesis reaction with SuperScript III RNaseH Reverse Transcriptase (Invitrogen Life Technologies, CA), as recommended by the manufacturer. Densitometric quantifications were performed using the NIH image J software.
Immunohistology
Immunohistology was performed in cryostat sections as previously described 16. Antibodies against mouse macrophage antigen–1 (Mac-1; M1/70BD), Ly-6G (1A8), intercellular adhesion molecule (ICAM-1; 3E2), platelet endothelial cell adhesion molecule-1 (PECAM-1; MEC13.3), all from BD Biosciences (San Jose, CA); tenascin-C (Tnc; 6-6B; Calbiochem, San Diego, CA), vascular cell adhesion molecule–1 (VCAM-1; MVCAM A 429; Serotec Inc., Raleigh NC); MMP-9 (AF909; R&D Systems, Minneapolis, MN); and proliferating cell nuclear antigen (PCNA; PC10; Lab Vision Corp., Fremont, CA) were used at optimal dilutions. The sections were evaluated blindly by counting labeled cells in triplicates in 10 high-power fields per section.
Western Blots
Western blots were performed as previously described16. Briefly, proteins (50 μg/sample) in sodium dodecyl sulfate (SDS)-loading buffer were electrophoresed through 10%-15% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes (Thermo Fisher). Membranes were incubated with specific antibodies against BclxL, caspase-3, cleaved-caspase-3, all from Cell Signaling (Beverly, MA), Bcl2 (Abcam Inc., Cambridge, MA), and Cyclin D1(DCS-6; BD Biosciences). After development, membranes were stripped and reblotted with an antibody against actin (Santa Cruz Biotechnology). Bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce). Relative quantities of protein were determined by densitometry using the NIH image J software.
Zymography
Protein extraction and zymography analyses were performed as we have previously described 16. Briefly, gelatinolytic activity was detected in liver extracts at a final protein content of 100 μg by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) containing 1 mg/mL of gelatin (Invitrogen) under nonreducing conditions. After incubation in development buffer [50 mmol/L Tris-HCl, 5 mmol/L CaCl2, and 0.02% NaN3 (pH 7.5)], gels were stained with Coomassie brilliant blue R-250 (Bio-Rad, Hercules, CA), and destained with methanol/acetic acid/water (20:10:70). A clear zone indicated enzymatic activity. Positive controls for MMP-9 (BIOMOL International, Plymouth, PA), and prestained molecular weight markers (Bio-Rad Laboratories) served as standards.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling Assay
The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay was performed on 5-μm cryostat sections using the In Situ Cell Death detection kit (Roche Diagnostics, Temecula, CA) according to the manufacturer’s instructions, and as previously described 18. The sections were evaluated blindly by counting labeled cells in triplicates in 10 high-power fields per section.
Cell Culture
Isolation of adult murine neutrophils from bone marrow was performed as previously published 16, 18. Briefly, femurs and tibias were harvested from Tnc−/−, TLR-4−/−, or WT mice and stripped of all muscle and sinew, and bone marrow was flushed with 2.5 mL of Hanks' balanced saline solution (HBSS) containing 0.1% (w/v) BSA and 1% (w/v) Glucose on ice. Cells were pelleted, and erythrocytes were removed by hypotonic lysis. The bone marrow preparation was resuspended at 5×107 cells/mL in HBSS. Cells were layered on a Percoll (Sigma–Aldrich) gradient (55% Percoll, top; 65% Percoll, middle; 80% Percoll, bottom). Mature neutrophils were recovered at the interface of the 65% and 80% fractions and were more than 90% pure and more than 95% viable in the neutrophil-rich fraction. Isolated neutrophils were placed on 24-well tenascin-C or polylysine coated plates at 5×106 cells/well and incubated at 37C, 5% CO2 for 6 hours. In parallel, isolated neutrophils were stimulated with LPS (1ng/ml) or IL-6 (100U/ml) for 6h, as we have previously described 18. Gelatinolytic activity was detected in cell supernatant by zymography, as described above. Data are presented as fold increase over controls.
Statistical analysis
All values are expressed as the mean ± the standard deviations. Differences between groups were compared using the Student t-test and a two-tailed p value <0.05 was considered significant. Calculations were made using SPSS software (SPSS Inc., Chicago, IL, USA).
RESULTS
Tenascin-C Expression in Hepatic IRI
Tenascin-C mRNA expression was hardly detected in naive wild-type livers and it was significantly up-regulated in Tnc+/+ livers post-IRI (Fig. 1A). These results were correlated with our immunohistological observations, in which tenascin-C protein expression was virtually undetectable in naïve livers, and it was readily deposited in the vascular areas of wild-type livers after 6h and 24h post-IRI (Fig. 1C). Tenascin-C was undetectable in Tnc−/− livers before and after IRI (Fig. 1B–C).
Figure 1.

Tenascin-C expression in liver IRI. Tenascin-C mRNA expression (panel A) was barely detected in naïve wild-type livers and it was upregulated in wild-type livers at 6h and 24h after IRI. Panel B illustrates the presence and absence of Tenascin-C mRNA expression in wild-type livers (lanes 1 and 2) and in Tnc −/− deficient livers (lanes 3 and 4), respectively, after 24h of reperfusion. Tenascin-C protein deposition (panel C) was virtually undetected in wild-type naïve livers (a), and readily detected in the vascular areas of wild-type livers at 6h (b) and 24h (c) post-IRI; in contrast, tenascin-C deposition was absent in Tnc −/− liver before (d) and after IRI (e–f). Arrows denote tenascin-C staining, (Magnification bars are 50 μm; CV:central vein; n=4/group; *p<0.05, **p<0.01).
Tenascin-C Deficiency Improved Liver Function and Histology after Liver IRI
There were no apparent differences in either the very low transaminase levels or liver histology between naïve Tnc−/− and Tnc+/+ mice. Tnc−/− and Tnc+/+ mice showed also comparable serum transaminase levels (U/L), vascular congestion and necrosis at 6h (AST: 13,977±2,620 vs. 14,362±3,489; ALT: 44,400±17,154 vs. 32,200±10,563; n=4 mice/group) and 12h (AST: 6,170±6,028 vs. 11,825±8,384; ALT: 37,400±7,213 vs. 36,400±8,854; n=4 mice/group) post-IRI (Fig. 2A–B). In contrast, the transaminase levels (U/L) were profoundly depressed in Tnc−/− mice (AST: 1,902±1,435 vs. 9,008±1,774; p<0.001; and ALT: 2,067±1,436 vs. 27,340±6,834; p<0.01; n=4–5 mice/group) at 24h post-IRI, (Fig. 2A). A sustained effect was observed in the Tnc−/− mice with transaminase levels (AST: 545±270 vs. 1,445±544; p<0.05; and ALT: 786±335 vs. 5,060±2,022; p<0.05; n=3 mice/group) significantly decreased at 48h post-IRI (Fig. 2A). Moreover, improvement of liver function in the Tnc−/− mice was correlated with better histological preservation. While Tnc+/+ livers were characterized by elevated sinusoidal congestion and extensive necrosis, Tnc−/− livers showed relatively modest signs of vascular changes or necrosis 24h (Fig. 2B). Compared with Tnc+/+ controls, Tnc−/− mice demonstrated a five-fold lower level of hepatocellular necrosis at 24h (n=4/group; p<0.01) (Fig. 2C). These data strongly support an important role for Tnc expression in the perpetuation of liver damage after IRI.
Figure 2.

Liver transaminases and histological preservation in Tnc −/− and wild-type mice. sAST and sALT levels (IU/L) (panel A) were measured in the blood samples taken at 6h, 12h, 24h, and 48h after I/R injury. sAST and sALT levels in the Tnc −/− mice and wild-type littermates were comparable at 6h and 12h; in contrast, they were profoundly depressed in the Tnc −/− mice at 24h and 48h post-IRI. Representative H&E staining of livers (panel B) at 6h and 24h post-I/R injury. Tnc −/− (b) and control wild-type (a) livers were mostly characterized by significant sinusoidal congestion at 6 h; however, whereas wild-type livers (c) showed large necrotic areas, Tnc −/− livers (d) showed rather good histological preservation at 24h after liver I/R injury. The percentage of hepatocellular necrosis (panel C) was decreased by about fivefold in the Tnc −/− livers after 24h of reperfusion, (Magnification bars are 125 μm; CV:central vein; PV: portal vein; *p<0.001, **p<0.05, &p<0.001).
Tenascin-C Deficiency Impaired Caspase-3 Activation in Liver IRI
Tnc−/− livers showed significantly lower numbers of TUNEL+ cells with hepatocyte morphology (47.2±17.1 vs. 128.7±9.8, p<0.01) at 6h post- IRI (Fig. 3A–B). Caspase-3 is an apoptotic effector caspase 19, expressed in tissues as an inactive 32–35 kDa precursor, and cleaved during apoptosis generating the 19–20 kDa fragment and the mature active 17 kDa subunit. While the pro-caspase-3 was present in all liver samples, the active 17-kDa caspase-3 was predominantly detected in Tnc+/+ control livers at 6h post-IRI. The active caspase-3 was significantly depressed in Tnc−/− livers at 6h of IRI (p<0.05) and virtually undetected in naïve livers (Fig. 3C–D). Protection against apoptosis may also involve anti-apoptotic mechanisms; however, Bcl-2, an anti-apoptotic molecule, was rather enhanced in Tnc+/+ livers and modestly expressed in both naïve and Tnc−/− livers post-IRI (Fig. 3C). Our results are consistent with earlier observations that expression of Bcl-2 can be induced in livers as a response to the post-reperfusion apoptotic stress 20, which seems to be reduced in Tnc−/− mice. Indeed, it has been shown that ischemic preconditioning down-regulated caspase-3 activity and inhibited apoptosis in livers post-IRI, despite lower levels of Bcl-2 expression detected in the preconditioned livers 20. Morphologic alterations of apoptosis are considered to be mostly mediated by caspases and cell death can occur via caspase-dependent and Bcl2-independent pathways 19, 21, 22. Therefore, our results provide an indication that Tnc−/− mice are less sensitive to apoptosis induced by liver IRI, regardless of showing comparable transaminase levels at 6h post-reperfusion. While necrosis has been shown to correlate with serum transaminases 23, apoptosis can occur without altering transaminase levels 24; this can perhaps be explained by observations that in contrast to necrosis, apoptotic cells maintain their plasma membrane integrity until late 25.
Figure 3.
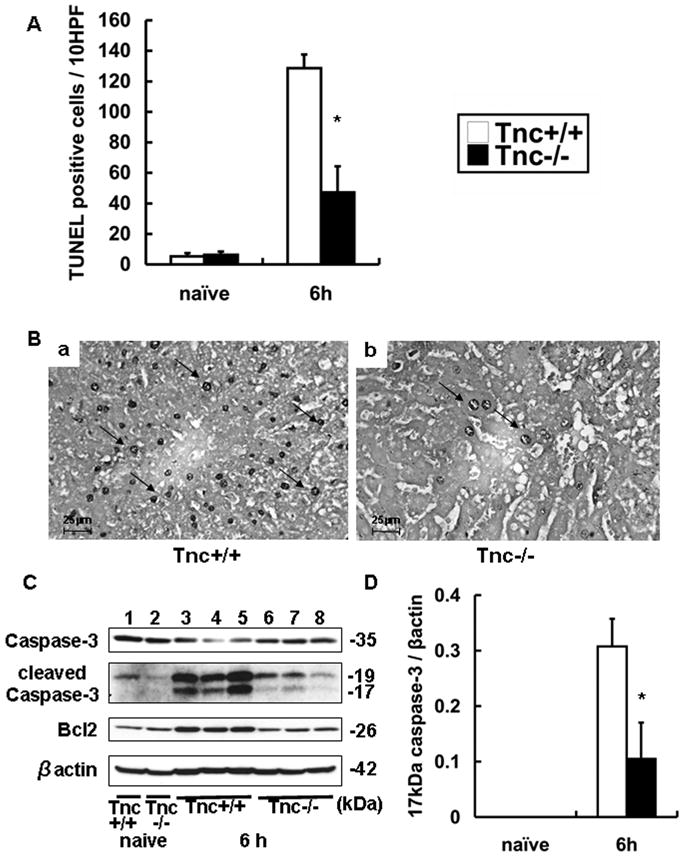
TUNEL staining and apoptotic markers in Tnc −/− and wild-type livers. TUNEL+ cells (panel A) were considerably detected in wild-type livers and significantly depressed in Tnc −/− livers at 6 h of hepatic IRI. Representative TUNEL staining (panel B) in WT livers (a) and Tnc −/− livers (b) at 6h post-IRI. Caspase-3 and Bcl2 expressions (panel C) in wild-type (lanes 1 and 3–5) and Tnc −/− (lanes 2 and 6–8) naïve livers (lanes 1 and 2) and livers 6h post-IRI (lanes 3–8). The densitometric ratio active caspase-3/β-actin (panel D) was significantly depressed in Tnc −/− livers at 6h post-IRI, when compared with respective controls, (Magnification bars are 25 μm; arrows denote TUNEL+ cells; n=4/group; *p<0.01).
Liver Regeneration after IRI Is Improved in the Absence of Tenascin-C
We evaluated the impact of Tnc deficiency on the hepatic regenerative response post-IRI. Cyclin D1 is normally expressed in livers 26, and reduced in impaired liver regeneration 27. Cyclin D1 expression was detected in significantly higher levels in Tnc−/−livers at 24h post-IRI (Fig. 4A and B). To determine whether Tnc expression interferes with proliferation after IRI, the number of S-phase cells was assessed by PCNA staining. Indeed, proliferating hepatocytes (PCNA Index %) were detected in increased numbers in the Tnc−/− livers (64.5±3.9 vs. 18.3±6.4, p<0.001; n=4/group) at 24h after IRI, suggesting that regeneration occurs earlier in the absence of Tnc (Fig. 4C–D).
Figure 4.

Cyclin D1 and PCNA labeling in Tnc −/− and wild-type livers. Cyclin D1 expression (panel A) was modestly detected in wild-type (lanes 1 and 9) and Tnc −/− (lane 2 and 10) naïve livers, depressed in wild type livers (lanes 3–5 and 11–13) and almost undisturbed in Tnc −/− (lanes 6–8 and 14–16) at 6h (lanes 3–8) and 24h (lanes 11–16) post-IRI. The densitometric ratio Cyclin D1/β-actin (panel B) was significantly increased in Tnc −/− livers at 24h post-IRI, when compared with respective wild-type control livers. The index of PCNA labeling (C) was significantly elevated in Tnc −/− livers at 24h of hepatic IRI, as compared with control. Representative PCNA staining (panel D) in WT livers (a) and Tnc −/− livers (b) at 24h post-IRI, (Magnification bars are 25 μm; arrows denote PCNA+ cells; n=4/group; *p<0.01, and **p<0.001).
Tenascin-C Deficiency Disrupted Leukocyte Accumulation and Expressions of IL-1β and IL-6 in Hepatic IRI
Myeloperoxidase activity was reduced in Tnc−/− livers at 6h (3.23±0.74vs. 7.03±1.71 U/g; p<0.05) and 24h (2.25±1.03 vs. 11.43±2.32 U/g; p<0.01) post-IRI, (Fig. 5A). Ly-6G neutrophil infiltration was clearly depressed in the portal areas of Tnc−/− livers at 6h (6.8±2.6 vs. 29.3±11.2; p<0.05) and 24h (21.3±8.4 vs. 64.7±7.3; p<0.05) post-IRI (Fig. 5B). Mac-1 leukocytes were also significantly reduced in the Tnc−/− livers at 6h (15.2±8.9 vs. 49.1±13.9; p<0.01) and 24h (29.2±13.7 vs. 85.9±8.7; p<0.05) post-IRI (Fig. 5C). Moreover, the expression of IL-1β was significantly depressed in Tnc−/− livers after 12h (p<0.04) and 24h (p<0.04) of reperfusion (Fig. 5D). Furthermore, the expressions of CXCL-2 (p<0.03), a potent neutrophil chemoattractant 28, and IL-6 (p<0.04) were significantly down-regulated in the Tnc−/− livers at 24h post-IRI (Fig. 5D).
Figure 5.

Intrahepatic MPO enzyme activity, leukocyte infiltration and IL-6/CXCL-2 expression in Tnc −/− and wild-type mice. MPO enzymatic activity (panel A), an index of neutrophil infiltration, was markedly reduced in the Tnc −/− mice at 6h and 24h of reperfusion. Ly-6G neutrophil (panel B) and Mac-1 macrophage (panel C) infiltration was predominantly detected in WT livers at 6h and 24h after IRI, contrasting with significantly less Ly-6G and Mac-1 leukocyte infiltration detected in Tnc −/− livers. Cytokine mRNA expression (panel D); IL-1β mRNA expression was significantly depressed in Tnc −/− livers at 12h and 24h post-reperfusion while IL-6 and CXCL-2 mRNA expressions were significantly depressed in Tnc −/− livers at 24h post-IRI. (Magnification bars are 25 μm; n=4–5/group; *p<0.05, **p<0.01, &p<0.04, &&p<0.03).
VCAM-1, ICAM-1, and PECAM-1 Showed Distinct Patterns of Expression in the Absence of Tenascin-C
VCAM-1 expression was virtually absent in naïve livers, and it was upregulated in the large vessels of Tnc+/+ livers at 6h and 24h post-IRI. In contrast, VCAM-1 was almost absent in Tnc−/− livers at 6h post-IRI, and it was significantly reduced in these livers at 24h post-reperfusion, suggesting that there was a disruption on VCAM-1 deposition in the absence of Tnc, (Fig. 6A). PECAM-1 is normally expressed on sinusoidal cells and downregulated under inflammatory conditions 30; in our settings, PECAM-1 expression was relatively high in naïve livers, and down-regulated in Tnc+/+ livers at both 6h and 24h post-IRI. On the other hand, PECAM-1 expression was fully restored in Tnc−/− livers at 24h after reperfusion, supporting that absence of Tnc slows, or even halts, the progression of inflammation (Fig. 6B). ICAM-1 showed a similar pattern of expression to PECAM-1. ICAM-1 expression was depressed in both Tnc−/− and Tnc+/+ livers at 6h post-IRI; however, while ICAM-1 expression remained depressed in the necrotic Tnc+/+ livers at 24h post-IRI, it was restored to almost normal levels in the Tnc−/− livers (Fig. 6C).
Figure 6.
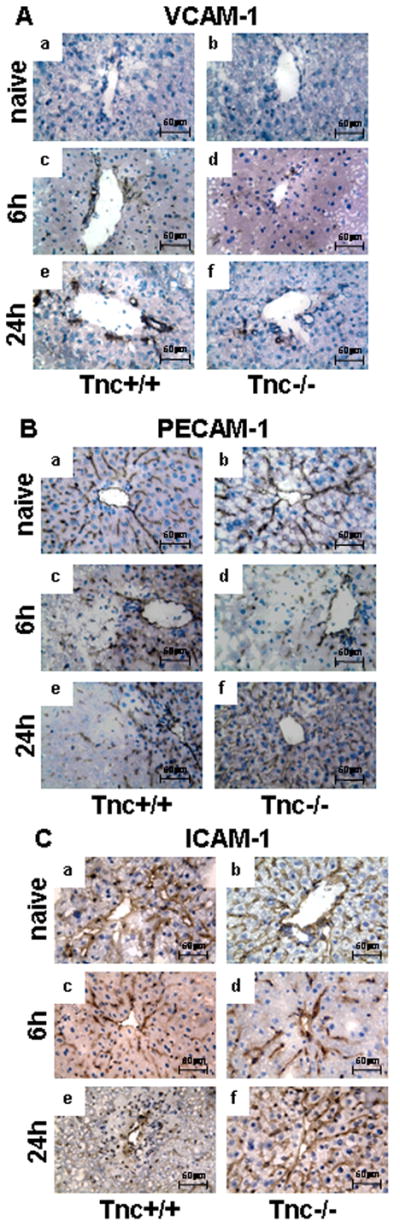
VCAM-1, PECAM-1 and ICAM-1 immunostaining in Tnc −/− and wild-type mice. VCAM-1 expression (panel A) was almost absent in wild-type (a) and Tnc−/− (b) naïve livers and readily upregulated in the vasculature of wild-type livers at 6h (c) and 24h (e) post-IRI; in contrast, Tnc −/− livers showed considerably less VCAM-1 deposition at 6h (d) and 24h (f) post-reperfusion when compared with respective controls. PECAM-1 (panel B) and ICAM-1 (panel C) showed similar patterns of expression, both adhesion molecules were eagerly detected in wild-type (a) and Tnc −/− (b) naïve livers and depressed in wild-type (c) and Tnc −/− (d) livers at 6h post-IRI; however, PECAM-1 and ICAM-1 expressions were restored in Tnc −/− livers (f) at 24h after IRI, while depressed in the respective wild-type controls (e) (Magnification bars are 50 μm; n=4/group).
TNC Deficiency Depressed the Expression/Activity of MMP-9 in Hepatic IRI
MMP-9 activity, assessed by zymography, was reduced in Tnc−/− deficient livers at 6h (~1.7-fold; n=4 mice/group; p<0.05) and 24h (~4.4-fold; p<0.003) post-IRI (Fig. 7A). Moreover, MMP-9 positive leukocytes were markedly depressed in Tnc−/− livers at 6h (11.8±5.8 vs. 31.8±9.4; p<0.05) and at 24h (18.8±7.4 vs. 59.8±7.3; p<0.05) after IRI, and contrasting with high levels of MMP-9+ leukocytes in controls (Fig. 7B–C).
Figure 7.

MMP-9 expression/activity in Tnc −/− and wild-type mice. MMP-9 activity detected by zymography (panel A) was virtually negative in WT (lanes 1 and 9), and in Tnc −/− (lanes 2 and 10) naïve livers. It was mildly detected in Tnc −/− deficient livers at 6h of IRI (lanes 6–8) and 24h (lanes 14–16) and highly upregulated in the respective WT controls (6h: lanes 3–5 and 24h:lanes 11–13). In addition, MMP-9+ leukocyte infiltration (panel B) was profoundly reduced in Tnc −/− livers as compared to respective WT controls at 6h and 24h post-IRI. Representative staining for MMP-9 in WT livers (a) and in Tnc −/− livers (b) is shown in panel C. Regulation of MMP-9 activity (expressed as mean ± SD of three independent experiments, panel D); conditioned media obtained from Tnc −/− and wild-type neutrophils stimulated with Tnc, IL-6, or LPS was subjected to a gelatin zymography assay and results are represented as fold increase in enzymatic activity over unstimulated neutrophils, (Magnification bars are 50 μm; n=4–5/group; *p<0.05).
To further support our observations, we assessed whether exogenous Tnc can regulate MMP-9 expression in isolated neutrophils. Tnc−/− and Tnc+/+ neutrophils cultured in Tnc-coated plates both showed about a 2-fold increase (p<0.05) in MMP-9 expression/activity, compared to controls (Fig. 7D). However, while IL-6 significantly stimulated MMP-9 activity in Tnc+/+ neutrophils, it was rather inefficient in upregulating MMP-9 activity in Tnc−/− neutrophils (Fig. 7D); future experimentation is needed to explain this result. Thus, these data evidence that tenascin-C favors a significant increase in MMP-9+ expression/activation in liver IRI.
Tenascin-C mediated MMP-9 expression is TLR4 dependent
Recent findings have identified tenascin-C as a ligand of Toll-like receptor-4 (TLR4) 6. TLR-4 is expressed by cells of the immune system and is considered to mediate inflammation and liver damage after IRI 32. Therefore, we attempted to investigate whether tenascin-C stimulates upregulation of MMP-9 activity in isolated neutrophils via TLR4. We found that the levels of MMP-9 activity were significantly depressed in neutrophils isolated from TLR4−/− mice and stimulated with tenascin-C, as compared to controls (Fig. 8). Moreover, while LPS induction of MMP-9 activity was TLR4 dependent, IL-6 stimulated similarly MMP-9 activity in neutrophils isolated from TLR-4−/− and from wild-type mice, suggesting that IL-6 regulates MMP-9 activity independently of TLR4 (Fig. 8). Therefore, our results provide evidence that tenascin-C is capable of regulating MMP-9 activity through TLR4.
Figure 8.
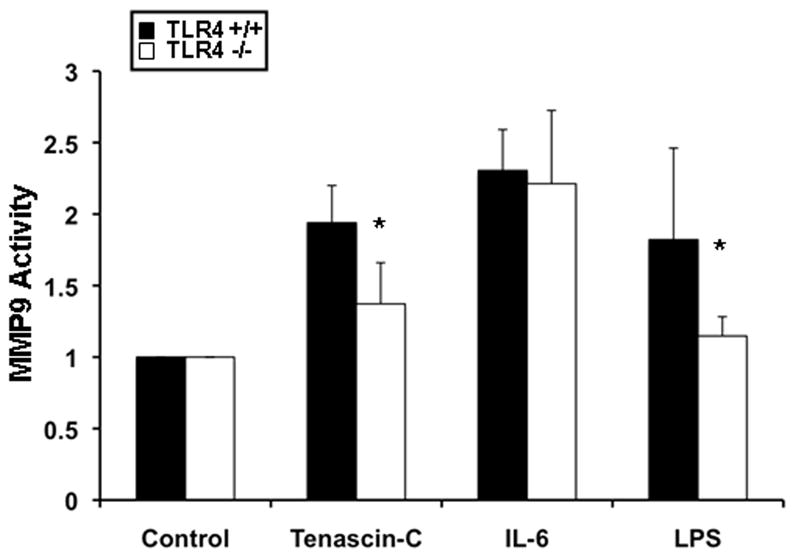
Tenascin-C mediated MMP-9 activity in TLR4 −/− and wild-type neutrophils. MMP-9 activity measured in neutrophils isolated from TLR4 −/− or wild-type (TLR4 +/+) mice that were either unstimulated or stimulated with tenascin-C, IL-6, or LPS. The conditioned media was subjected to a gelatin zymography assay and results are represented as fold increase in enzymatic activity over unstimulated neutrophils. MMP-9 activity was significantly decreased in neutrophils isolated from TLR4−/− mice that were either stimulated with Tnc or LPS. In contrast, MMP-9 activity was unaffected in neutrophils isolated from TLR4−/− mice that were stimulated with IL-6 (data expressed as mean ± SD of three independent experiments; *p<0.05).
DISCUSSION
In the present study, we investigate the functional significance of tenascin-C expression in liver IRI. We show that tenascin-C is virtually absent in naïve livers and readily detected in the vascular areas of damaged wild-type livers after IRI. Tenascin-C deficient mice showed (a) improved liver transaminases and histological outcomes, (b) decreased necrosis, (c) reduced caspase-3 activity and apoptotic cell labeling (d) enhanced liver regeneration, (e) impaired leukocyte infiltration and IL1β and IL-6 expressions, (f) altered patterns of expression of adhesion molecules, and (g) downregulated MMP-9 expression/activation after liver IRI. Moreover, in vitro investigations provided evidence that exogenous tenascin-C (h) regulated MMP-9 activity in wild-type and Tnc−/− neutrophils, and (i) induced MMP-9 expression through TLR-4.
Tenascin-C is virtually not expressed in most healthy adult tissues, but its expression is specifically and rapidly induced in response to injury 33. Tenascin-C has been identified as an endogenous ligand of TLR-4 6 and its expression has been linked to inflammatory condition, which include, rheumatoid arthritis 6, cancer 13, liver fibrosis 34 and chronic hepatitis 11, 35. Indeed, given the significant role that tenascin-C may have in inflammation, drugs targeting tenascin-C are currently being developed or undergoing clinical trials 13. In the present work, Tnc deposition was readily upregulated in the liver vascular areas of wild-type Tnc+/+ livers post-IRI, with a similar pattern of distribution to that of chronic hepatitis 11. We used Tnc−/− deficient mice to test the relevance of tenascin-C expression to liver IRI. Tnc−/− deficient mice have previously been reported to have no grossly phenotypic abnormalities 36. Tnc−/− mice, compared with their wild-type counterparts, were less susceptible to liver IRI. While tenascin-C deficiency did not particularly affect transaminase levels or histological preservation at 6h and 12h post-IRI, it was highly effective in ameliorating liver function/damage at 24h and 48h post-reperfusion. Tnc−/− livers exhibited limited liver necrosis at 24h post-IRI, contrasting with extensive necrosis in controls. Hepatocyte death by both necrosis and apoptosis is a prominent feature of liver IRI 37. Indeed, caspase-3 activation was significantly reduced in Tnc−/− livers as compared with control littermates after IRI, and it was accompanied by a reduced number of TUNEL-positive cells in the Tnc-deficient livers. ECM proteins can act either as survival or pro-apoptotic mediators 38. In this regard, it has been shown that the epidermal growth factor-like (EGF-L) domains of tenascin-C are capable of exerting caspase-dependent pro-apoptotic activities 39.
In addition to hepatocyte cell death, impaired liver regeneration/repair is common in acute liver failure. Here we show that the number of PCNA-positive hepatocytes appeared increased in the Tnc−/− mice after IRI, suggesting that hepatocytes progress faster into the S phase of the cell cycle in the absence of tenascin-C. Moreover, cyclin D1, an important in the transition from the G1 to S phase, was significantly depressed in controls and almost normally expressed in the Tnc−/− deficient livers post-IRI. Inhibition of cyclin D1 leads to growth arrest 40 and to impaired hepatic regeneration 27. Though, it is still not clear the role of PECAM-1 in liver IRI, PECAM-1 is constitutively expressed on sinusoidal endothelial cells and its loss is regarded as a marker of sinusoidal injury 41. In our settings, PECAM-1 expression was depressed in livers post-IRI and it was restored to normal levels sooner in the Tnc−/− livers. The intact expression of PECAM-1 along the sinusoids has been associated with less sinusoidal congestion/inflammation 30, suggesting that preventing PECAM downregulation may be valuable in the treatment of early stages of liver damage 42. These observations support the view that hepatic regeneration/repair post-IRI is enhanced in the absence of tenascin-C. Moreover, they are consistent with the reduction of liver necrosis observed earlier in the Tnc−/− deficient mice post-IRI. Therefore, our results agree with previous findings that Tenascin-C mediates a persistent inflammation 6, which possibly interferes with liver regeneration and contributes to the perpetuation and aggravation of necrosis post-IRI.
Leukocyte infiltration is a hallmark feature in hepatic IRI. Indeed, neutrophils, critical mediators in acute inflammatory liver injury 43, were significantly decreased in Tnc−/− livers post-IRI. Macrophages were also depressed in the Tnc−/− livers post-IRI. Tenascin-C is a ligand for several integrin receptors present on leukocytes, and it has been linked to diverse effects on cell migration that result from differences in cell type and in vitro assays 13. CXCL2, a cytokine-induced neutrophil chemoattractant 46, was rather down-regulated in the Tnc−/− livers post-IRI, suggesting its participation in neutrophil recruitment in this model. Notably, VCAM-1 expression, which we and others have detected on the portal track vessels of inflamed liver 16, 47, was significantly depressed in the Tnc−/− livers post-reperfusion.
One of the most striking effects observed in the Tnc−/− livers was a marked decrease in MMP-9 expression/activation. Leukocyte transmigration, across endothelial and ECM barriers, results from a complex series of adhesive and focal matrix degradation events. While adhesion molecules are essential to promote leukocyte attachment to the vascular endothelium, MMPs are important for facilitating leukocyte transmigration across vascular barriers. We have previously demonstrated that MMP-9 is predominantly expressed by leukocytes in damaged livers 16, and mediates leukocyte transmigration in liver IRI 16, 31. Our results showing that MMP-9 is upregulated by Tnc in leukocytes are in agreement with other reports that demonstrated the induction of MMP-9 by Tnc in RAW264.7-macrophages 48, fibroblasts 49 and cancer cells 50. Furthermore, our data also suggests that TLR4 signaling mediates tenascin-C-induced upregulation of MMP-9 activity in neutrophils. In this regard, studies using mice that lack TLR4 have shown that TLR4 mediates inflammation in hepatic IRI 32 and that MMP-9 expression is reduced in TLR4-deficient mice after experimental stroke 51. Though, Tnc mediated MMP-9 upregulation in liver IRI is possibly due to a combination of factors, which may also include the expression of pro-inflammatory cytokines. In our settings, IL-6, which regulates MMP-9 activity in neutrophils 18, was significantly depressed in Tnc−/− deficient livers post-reperfusion.
In summary, our studies demonstrate an active role for tenascin-C in liver IRI. Tnc deficiency interfered with VCAM-1 vascular deposition and downregulation of PECAM-1, disrupted MMP-9-positive leukocyte infiltration, hampered apoptosis and necrosis, and favored liver repair/regeneration after IRI. Thus, this work supports the view that further understanding how newly synthesized ECM molecules, such as tenascin-C, participate in inflammatory responses may lead to potential valuable therapies in liver IRI.
Acknowledgments
This work was supported by the following grants from the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID) R01AI057832, UCLA Academic Senate, and the Pfleger Foundation (to A.J.C.). S.D. was supported in part by a fellowship from the Fundação para a Ciência e Tecnologia (FCT), Portugal.
References
- 1.Howard TK, Klintmalm GB, Cofer JB, Husberg BS, Goldstein RM, Gonwa TA. The influence of preservation injury on rejection in the hepatic transplant recipient. Transplantation. 1990;49:103–7. doi: 10.1097/00007890-199001000-00023. [DOI] [PubMed] [Google Scholar]
- 2.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–73. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 3.Busuttil RW, Tanaka K. The utility of marginal donors in liver transplantation. Liver Transpl. 2003;9:651–63. doi: 10.1053/jlts.2003.50105. [DOI] [PubMed] [Google Scholar]
- 4.Coito AJ, Kupiec-Weglinski JW, Busuttil RW. Extracellular Matrix and Organ Transplantation. In: Wilkes DS, Burlingham WD, editors. Immunobiology of Organ Transplantation. New York: Kluwer Academic/Plenum Publishers; 2004. pp. 576–589. [Google Scholar]
- 5.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 6.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–80. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 7.Orend G. Potential oncogenic action of tenascin-C in tumorigenesis. Int J Biochem Cell Biol. 2005;37:1066–83. doi: 10.1016/j.biocel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka K, Hiraiwa N, Hashimoto H, Yamazaki Y, Kusakabe M. Tenascin-C regulates angiogenesis in tumor through the regulation of vascular endothelial growth factor expression. Int J Cancer. 2004;108:31–40. doi: 10.1002/ijc.11509. [DOI] [PubMed] [Google Scholar]
- 9.Hemesath TJ, Stefansson K. Expression of tenascin in thymus and thymic nonlymphoid cells. J Immunol. 1994;152:422–8. [PubMed] [Google Scholar]
- 10.Kanayama M, Kurotaki D, Morimoto J, Asano T, Matsui Y, Nakayama Y, Saito Y, Ito K, Kimura C, Iwasaki N, Suzuki K, Harada T, Li HM, Uehara J, Miyazaki T, Minami A, Kon S, Uede T. Alpha9 integrin and its ligands constitute critical joint microenvironments for development of autoimmune arthritis. J Immunol. 2009;182:8015–25. doi: 10.4049/jimmunol.0900725. [DOI] [PubMed] [Google Scholar]
- 11.El-Karef A, Yoshida T, Gabazza EC, Nishioka T, Inada H, Sakakura T, Imanaka-Yoshida K. Deficiency of tenascin-C attenuates liver fibrosis in immune-mediated chronic hepatitis in mice. J Pathol. 2007;211:86–94. doi: 10.1002/path.2099. [DOI] [PubMed] [Google Scholar]
- 12.Spring J, Beck K, Chiquet-Ehrismann R. Two contrary functions of tenascin: dissection of the active sites by recombinant tenascin fragments. Cell. 1989;59:325–34. doi: 10.1016/0092-8674(89)90294-8. [DOI] [PubMed] [Google Scholar]
- 13.Midwood KS, Orend G. The role of tenascin-C in tissue injury and tumorigenesis. J Cell Commun Signal. 2009;3:287–310. doi: 10.1007/s12079-009-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark RA, Erickson HP, Springer TA. Tenascin supports lymphocyte rolling. J Cell Biol. 1997;137:755–65. doi: 10.1083/jcb.137.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saga Y, Yagi T, Ikawa Y, Sakakura T, Aizawa S. Mice develop normally without tenascin. Genes Dev. 1992;6:1821–31. doi: 10.1101/gad.6.10.1821. [DOI] [PubMed] [Google Scholar]
- 16.Hamada T, Fondevila C, Busuttil RW, Coito A. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47:186–98. doi: 10.1002/hep.21922. [DOI] [PubMed] [Google Scholar]
- 17.Chen SW, Park SW, Kim M, Brown KM, D'Agati VD, Lee HT. Human heat shock protein 27 overexpressing mice are protected against hepatic ischemia and reperfusion injury. Transplantation. 2009;87:1478–87. doi: 10.1097/TP.0b013e3181a3c691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamada T, Duarte S, Tsuchihashi S, Busuttil RW, Coito AJ. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. Am J Pathol. 2009;174:2265–77. doi: 10.2353/ajpath.2009.080872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326 (Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yadav SS, Sindram D, Perry DK, Clavien PA. Ischemic preconditioning protects the mouse liver by inhibition of apoptosis through a caspase-dependent pathway. Hepatology. 1999;30:1223–31. doi: 10.1002/hep.510300513. [DOI] [PubMed] [Google Scholar]
- 21.Los M, Wesselborg S, Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity. 1999;10:629–39. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- 22.Bantel H, Lugering A, Poremba C, Lugering N, Held J, Domschke W, Schulze-Osthoff K. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology. 2001;34:758–67. doi: 10.1053/jhep.2001.28229. [DOI] [PubMed] [Google Scholar]
- 23.Dixon MF, Fulker MJ, Walker BE, Kelleher J, Losowsky MS. Serum transaminase levels after experimental paracetamol-induced hepatic necrosis. Gut. 1975;16:800–7. doi: 10.1136/gut.16.10.800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calabrese F, Pontisso P, Pettenazzo E, Benvegnu L, Vario A, Chemello L, Alberti A, Valente M. Liver cell apoptosis in chronic hepatitis C correlates with histological but not biochemical activity or serum HCV-RNA levels. Hepatology. 2000;31:1153–9. doi: 10.1053/he.2000.7123. [DOI] [PubMed] [Google Scholar]
- 25.Patel T, Gores GJ. Apoptosis and hepatobiliary disease. Hepatology. 1995;21:1725–41. doi: 10.1002/hep.1840210635. [DOI] [PubMed] [Google Scholar]
- 26.Kwon YH, Jovanovic A, Serfas MS, Tyner AL. The Cdk inhibitor p21 is required for necrosis, but it inhibits apoptosis following toxin-induced liver injury. J Biol Chem. 2003;278:30348–55. doi: 10.1074/jbc.M300996200. [DOI] [PubMed] [Google Scholar]
- 27.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci U S A. 2007;104:17081–6. doi: 10.1073/pnas.0704126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagano MB, Bartoli MA, Ennis TL, Mao D, Simmons PM, Thompson RW, Pham CT. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A. 2007;104:2855–60. doi: 10.1073/pnas.0606091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8 (Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramadori G, Moriconi F, Malik I, Dudas J. Physiology and pathophysiology of liver inflammation, damage and repair. J Physiol Pharmacol. 2008;59 (Suppl 1):107–17. [PubMed] [Google Scholar]
- 31.Coito AJ. Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Curr Opin Organ Transplant. 2011;16:34–40. doi: 10.1097/MOT.0b013e328342542e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vardanian AJ, Busuttil RW, Kupiec-Weglinski JW. Molecular mediators of liver ischemia and reperfusion injury: a brief review. Mol Med. 2008;14:337–45. doi: 10.2119/2007-00134.Vardanian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goh FG, Piccinini AM, Krausgruber T, Udalova IA, Midwood KS. Transcriptional regulation of the endogenous danger signal tenascin-C: a novel autocrine loop in inflammation. J Immunol. 2010;184:2655–62. doi: 10.4049/jimmunol.0903359. [DOI] [PubMed] [Google Scholar]
- 34.Lieber CS, Weiss DG, Paronetto F. Value of fibrosis markers for staging liver fibrosis in patients with precirrhotic alcoholic liver disease. Alcohol Clin Exp Res. 2008;32:1031–9. doi: 10.1111/j.1530-0277.2008.00664.x. [DOI] [PubMed] [Google Scholar]
- 35.El-Karef A, Kaito M, Tanaka H, Ikeda K, Nishioka T, Fujita N, Inada H, Adachi Y, Kawada N, Nakajima Y, Imanaka-Yoshida K, Yoshida T. Expression of large tenascin-C splice variants by hepatic stellate cells/myofibroblasts in chronic hepatitis C. J Hepatol. 2007;46:664–73. doi: 10.1016/j.jhep.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Settles DL, Kusakabe M, Steindler DA, Fillmore H, Erickson HP. Tenascin-C knockout mouse has no detectable tenascin-C protein. J Neurosci Res. 1997;47:109–17. [PubMed] [Google Scholar]
- 37.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: atale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 38.Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–62. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 39.Wallner K, Li C, Shah PK, Wu KJ, Schwartz SM, Sharifi BG. EGF-Like domain of tenascin-C is proapoptotic for cultured smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:1416–21. doi: 10.1161/01.ATV.0000134299.89599.53. [DOI] [PubMed] [Google Scholar]
- 40.Matsui T, Kinoshita T, Hirano T, Yokota T, Miyajima A. STAT3 down-regulates the expression of cyclin D during liver development. J Biol Chem. 2002;277:36167–73. doi: 10.1074/jbc.M203184200. [DOI] [PubMed] [Google Scholar]
- 41.Zhao X, Koshiba T, Nakamura T, Tsuruyama T, Li Y, Bando T, Wada H, Tanaka K. ET-Kyoto solution plus dibutyryl cyclic adenosine monophosphate is superior to University of Wisconsin solution in rat liver preservation. Cell Transplant. 2008;17:99–109. doi: 10.3727/000000008783906928. [DOI] [PubMed] [Google Scholar]
- 42.Neubauer K, Lindhorst A, Tron K, Ramadori G, Saile B. Decrease of PECAM-1-gene-expression induced by proinflammatory cytokines IFN-gamma and IFN-alpha is reversed by TGF-beta in sinusoidal endothelial cells and hepatic mononuclear phagocytes. BMC Physiol. 2008;8:9. doi: 10.1186/1472-6793-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006;26:912–9. doi: 10.1111/j.1478-3231.2006.01327.x. [DOI] [PubMed] [Google Scholar]
- 44.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brummer J, Rudolph V, Munzel T, Heitzer T, Meinertz T, Baldus S. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A. 2005;102:431–6. doi: 10.1073/pnas.0405193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–57. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–8. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Monzon C, Sanchez-Madrid F, Garcia-Buey L, Garcia-Arroyo A, Garcia-Sanchez A, Moreno-Otero R. Vascular adhesion molecule expression in viral chronic hepatitis: evidence of neoangiogenesis in portal tracts. Gastroenterology. 1995;108:231–41. doi: 10.1016/0016-5085(95)90029-2. [DOI] [PubMed] [Google Scholar]
- 48.Khan KM, Falcone DJ. Role of laminin in matrix induction of macrophage urokinase-type plasminogen activator and 92-kDa metalloproteinase expression. J Biol Chem. 1997;272:8270–5. doi: 10.1074/jbc.272.13.8270. [DOI] [PubMed] [Google Scholar]
- 49.Tremble P, Chiquet-Ehrismann R, Werb Z. The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol Biol Cell. 1994;5:439–53. doi: 10.1091/mbc.5.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalembeyi I, Inada H, Nishiura R, Imanaka-Yoshida K, Sakakura T, Yoshida T. Tenascin-C upregulates matrix metalloproteinase-9 in breast cancer cells: direct and synergistic effects with transforming growth factor beta1. Int J Cancer. 2003;105:53–60. doi: 10.1002/ijc.11037. [DOI] [PubMed] [Google Scholar]
- 51.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]